

Abstract
Cerebral amyloid angiopathy (CAA) is common in Alzheimer's disease (AD) and may contribute to dementia and cerebral hemorrhage. Parenchymal β-amyloid deposition is dependent on the activity of zinc transporter 3 (ZnT3), a neocortical synaptic vesicle membrane protein that causes enrichment of exchangeable Zn2+ in the vesicle, which is externalized on neurotransmission. However, the contribution of zinc to vascular β-amyloid deposition remains unclear. Here, we identify for the first time an exchangeable pool of Zn2+ in the cerebrovascular wall of normal mice. This histochemically reactive Zn2+ is enriched in CAA in a transgenic mouse model of AD (Tg2576), and a dramatic reduction of CAA occurs after targeted disruption of the Znt3 gene in these mice. Also, in Znt3 knock-out mice, the amount of exchangeable Zn2+ [detected by N-(6-methoxy-8-quinolyl)-p-carboxybenzoylsulphonamide (TFL-Zn)] in the perivascular space was significantly decreased in the neocortex but not in peripheral organs. ZnT3 was not detected in the cerebral vessel walls or in blood components of wild-type mice. Thus, synaptic ZnT3 activity may promote CAA by indirectly raising exchangeable Zn2+ concentrations in the perivascular spaces of the brain.
Keywords: Alzheimer, blood–brain, neuropathology, synapse, zinc, amyloid, cerebrovasculature, congophilic angiopathy
Introduction
β-amyloid (Aβ) is a ubiquitous 4 kDa peptide that accumulates characteristically in the brain in Alzheimer's disease (AD) as parenchymal plaque deposits (Masters et al., 1985) and cerebral amyloid angiopathy (CAA) (Glenner and Wong, 1984). CAA occurs in vessel walls of leptomeningeal (i.e., pia and arachnoid mater) and cerebral arterioles, arteries, and capillaries, is associated with smooth muscle cell degeneration, microinfarction, and hemorrhage (Coria et al., 1987; Kawai et al., 1992; Jellinger and Attems, 2003), and may contribute more than plaque amyloid to cognitive loss (Pfeifer et al., 2002).
CAA is not simply a product of bulk Aβ accumulation, because subregions of the brain (e.g., occipital lobe, thalamus) are especially vulnerable to CAA, whereas others (e.g., superior temporal cortex) are vulnerable to parenchymal Aβ deposition (Braak and Braak, 1996). Some Aβ mutations cause profound CAA but minimal parenchymal amyloid deposition (Van Broeckhoven et al., 1990).
These observations suggest that Aβ aggregation is triggered by reaction with microregional neurochemical factors, and two major promoters of Aβ deposition in amyloid β protein precursor (APP) transgenic (Tg) mice are apolipoprotein E (ApoE) and Zn2+. Parenchymal amyloid and CAA are markedly inhibited by genetic ablation of mouse ApoE (Holtzman et al., 2000; Fryer et al., 2003). ApoE, the isoforms of which modify the risk for AD, is expressed in brain and enriches within parenchymal amyloid and CAA (Fryer et al., 2003). However, it is not yet clear how this protein behaves to foster Aβ deposition.
Zn2+ also plays a major role in the reversible precipitation of Aβ (Bush et al., 1994b; Huang et al., 1997). Zn2+ is concentrated in amyloid plaques in AD and in Tg2576 mice (Lee et al., 1999; Suh et al., 2000). Cu2+ and Zn2+ bind directly to Aβ subunits in brain plaque amyloid in AD through histidine bridges (Dong et al., 2003), selectively copurify with Aβ from AD affected brain (Opazo et al., 2002), and Cu2+/Zn2+ chelation resolubilizes Aβ from postmortem AD tissue (Cherny et al., 1999). Treatment of Tg2576 mice with the orally bioavailable Cu2+/Zn2+ chelator clioquinol markedly decreases brain amyloid burden (Cherny et al., 2001).
Genetic ablation of the zinc transporter 3 (Znt3) is similar to ApoE ablation in inhibiting amyloid deposition in Tg mice (Lee et al., 2002). ZnT3 inserts Zn2+ in a chemically exchangeable form into synaptic vesicles and is only found in neocortical synaptic vesicles (Palmiter et al., 1996). Zn2+ [detected by N-(6-methoxy-8-quinolyl)-p-toluensulfonamide (TSQ)] is concentrated within CAA in AD (Suh et al., 2000) but unlike plaque amyloid, which accumulates in the vicinity of exchangeable Zn2+ discharged into the synaptic space (Lee et al., 2002), the origin of Zn2+ within CAA is not obvious. Therefore, we investigated the effect of Znt3 gene ablation on cerebrovascular Zn2+ levels in non-Tg mice and on CAA in Tg2576 mice to determine whether the activity of synaptic ZnT3 may affect Aβ deposition at a site remote from the synapse. Here, we report evidence that synaptic ZnT3 activity promotes CAA by indirectly raising exchangeable Zn2+ levels in the perivascular spaces of cerebral and leptomeningeal vessel walls.
Materials and Methods
Transgenic mice. Human Swedish mutant amyloid precursor protein transgenic Tg2576 mice (APP+; C57Bl6/B6SJL hybrid) were crossed with Znt3-deficient mice (Znt3–/–; C57Bl6/Sv129 hybrid), and the resulting F1 generation of APP+/Znt3+/– mice were interbred to produce APP+/Znt3+/+, APP+/Znt3+/–, and APP+/Znt3–/– mice. The official gene name of the mouse Znt3 gene is now slc30a3 (Palmiter and Huang, 2003). Genotyping was performed as described previously (Hsiao et al., 1995; Cole et al., 1999). All animal experiments were performed in accordance with the National Institutes of Health guideline for welfare and use of laboratory animals.
Histochemistry and immunocytochemistry. At 15 months of age (n = 7 for APP+/Znt3+/+; n = 7 for APP+/Znt3+/–; n = 7 for APP+/Znt3–/–) or 18 months of age (n = 8 for APP+/Znt3+/+; n = 7 for APP+/Znt3+/–; n = 7 for APP+/Znt3–/–), coronal brain sections (bregma plus 3.0 mm) were processed for Congo Red histochemistry and anti-Aβ immunocytochemistry.
For Congo Red staining, 10 μm thick sections were first stained in Gill's hematoxylin solution (Sigma, St. Louis) for 10 min and then rinsed in tap water for 5 min and incubated in alkaline sodium chloride solution for 20 min. Sections were then stained with alkaline Congo Red solution (0.2% in 80% ethanol saturated with sodium chloride; Sigma) and washed in absolute ethanol (Lee et al., 1999).
For Aβ immunocytochemistry, 10 μm thick sections were treated with 3% hydrogen peroxide in methanol for 30 min. After blocking with 1% bovine serum albumin (BSA) and 3% goat serum in PBS, the sections were incubated for 24 hr at 4°C with the primary antibody (Ab) (anti-human amyloid β Ab; PharMingen, San Diego, CA; 1:500) in PBS containing 1% BSA and 3% goat serum. The sections were sequentially incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), avidin–horseradish peroxide solution (Vector Laboratories), 0.015% diaminobenzidine plus 0.001% hydrogen peroxide (Vector Laboratories), and finally counterstained with hematoxylin. For anti-ZnT3 immunostaining, the sections were reacted with affinity-purified ZnT-3 Ab (1:50) as described previously (Palmiter et al., 1996).
The identification and quantification of exchangeable Zn2+ in cerebral and peripheral blood vessels (Fig. 6, Table 2) was performed by a blinded observer. Tissue samples were handled uniformly and systematically, as in our previous studies using TSQ and N-(6-methoxy-8-quinolyl)-p-carboxybenzoylsulphonamide (TFL-Zn) to monitor fluorescence in the brains of Tg2576 mice (Lee et al., 1999, 2002). Immediately after cryostat preparation, unfixed frozen brain sections (12 μm) were incubated for 90 sec with 0.25 mm TFL-Zn (Budde et al., 1997) (Tef Labs, Austin, TX) dissolved in Tris-Cl buffer (0.1 mm, pH 8.0). Immediately after washing in 0.9% NaCl solution, pH 7.2, for 15 sec, the sections were photographed with a digital camera (Camedia Z-2000; Olympus Optical, Tokyo, Japan) under a fluorescence microscope (BX60; Olympus Optical) using a U-MWU cube, dichroic mirror (400 nm), excitation filter (330–385 nm), and barrier filter (420 nm). The barrier filter used (BA420) has a steep exclusion slope for wavelengths of light <420 nm but permits transmission of wavelengths >420 nm. Therefore, the excitation–barrier filter combinations that we used were suitable for the evaluation of TFL-Zn fluorescence, which has excitation and emission peaks of 360 and 498 nm, respectively (Budde et al., 1997).
Figure 6.

Residual amyloid plaque deposits and CAA in APP+/Znt3–/– mice still contain histochemically reactive zinc. A–D, The Zn2+-specific fluorescent dye TFL-Zn (A) reacts with amyloid plaque and CAA (B) present in APP+/Znt3–/– mice, similar to that seen in APP+/Znt3+/+ mice (C,D). Scale bar, 50 μm.
Table 2.
Quantification of relative TFL-Zn fluorescent intensities in blood vessel (major axis; 30-100 μm) walls of Znt3-/- brain (cortex or hippocampus, excluding leptomeningeal vessels), heart, liver, and kidney compared with Znt3+/+ (n = 6 in each group)
|
Tissue |
ZnT3+/+ |
ZnT3-/- |
p |
|---|---|---|---|
| Cerebral cortex | 100.00 ± 0.48 | 79.80 ± 0.63 | 5.8 × 10-10 |
| (98.73-101.86) | (77.97-82.02) | ||
| Heart | 100.00 ± 1.26 | 97.55 ± 1.20 | 0.19 |
| (94.54-103.61) | (94.01-100.71) | ||
| Liver | 100.00 ± 2.88 | 97.60 ± 3.21 | 0.59 |
| (93.21-111.70) | (94.55-110.81) | ||
| Kidney | 100.00 ± 1.23 | 97.58 ± 1.47 | 0.24 |
|
|
(96.05-104.04) |
(92.17-101.47) |
|
The values are means ± SEM (range in brackets) normalized to the mean fluorescent value (in arbitrary units) for the Znt3+/+ tissue. p values are for t tests (two-tailed).
After the conversion of the fluorescent images into a black and white mode, the fluorescence intensity in vessel walls was determined with a computer-assisted image analysis program (Image-Pro; Media Cybernetics, Silver Spring, MD). The vessel walls were traced, and fluorescence was quantified within the trace lines. Because of inaccuracies in tracing small vessels with this technique, only larger vessels (30–100 μm) were traced. To control for vessel wall autofluorescence, intensities from unstained sections were quantified as described above and determined to not contribute to >5% of assayed fluorescence in all organs and did not differ between the two groups. For each organ in each mouse, the average fluorescence intensity measured from 10 blood vessels (with major axis; 30–100 μm) on one tissue section was calculated. The average intensity per group was then determined (n = 7 for Znt3+/+; n = 6 for Znt3–/–), and the value of TFL-Zn fluorescence was normalized to the mean fluorescence in Znt3+/+ mice (100%).
Timm's stain. Protocols were as published previously (Danscher and Zimmer, 1978; Danscher, 1981; Perez-Clausell and Danscher, 1985). Mice were transcardiac perfused under deep anesthesia with buffered sodium sulfide solution (12 gm/l Na2S.H2O, 12 gm/l NaH2PO4.H2O, pH 7.4) (1 min), 4% paraformaldehyde solution (10 min), and then buffered sodium sulfide solution (10 min). For Neo-Timm's (Holm, 1989), DBA mice were administered 20 mg/kg Na2SeO3 (2 mg/ml in normal saline) by intraperitoneal injection 2 hr before perfusion. Paraffin sections were mounted on gelatinized slides and dried. The slides were rehydrated (96% alcohol for 15 min, 70% alcohol for 2 min, 50% alcohol for 2 min, and then 3 dH2O) for 3 min. The slides were placed in developing solution in the water bath at 26°C in the dark for 60 min. Developing solution is made by mixing 60 ml of gum Arabic solution (500 gm/l) with 10 ml of citrate buffer (255 gm/l citric acid.H2O, 217 gm/l tri-sodium citrate.2H2O) and then adding 15 ml of hydroquinone solution (at 40°C) (56.67 gm/l hydoquinone) and 15 ml of silver lactate solution (at 40°C) (0.121 gm per 15 ml of dH2O). Components were prepared freshly. The slides were then washed in water for 15 min, placed in 70% ethanol for 30 min, washed four times in dH2O (5 min each), and then dehydrated in an ethanol series and cleared with xylene and coverslipped.
Western blotting. Wild-type mouse hippocampus served as a positive control for anti-ZnT3 Western blotting. From mouse, blood was collected from 12-month-old female mice from the inferior vena cava into a heparinized syringe. Platelets, erythrocytes, and plasma were separated as described previously (Bush et al., 1990). Cell pellets or solid tissue were homogenized in eight volumes of sample buffer (2% SDS/5% 2-mercaptoethanol/50 mm Tris-HCl, pH 7.0) and boiled. Western blotting was performed as described previously (Goldstein et al., 2003), except 1:500 anti-ZnT3 primary Ab was used.
Inductively coupled plasma mass spectroscopy. Blood was collected from 12-month-old female mice from the inferior vena cava into an EDTA-treated syringe and transferred into plastic tubes prewashed with 1% HNO3. HNO3 was inductively coupled plasma mass spectroscopy (ICPMS) grade “Aristar” (BDH Chemicals, Poole, UK). Plasma was immediately separated and later analyzed by ICPMS using an Ultramass 700 (Varian, Victoria, Australia) in peak-hopping mode with one point per peak, 50 scans per replicate, and three replicates per sample. Plasma flow was 15 L/min with auxiliary flow of 1.5 L/min. Radio frequency power was 1.2 kW. Samples were introduced using a glass nebulizer at a flow rate of 0.88 L/min. The apparatus was calibrated using a 1% HNO3 solution containing Cu and Zn at 5, 10, 50, and 100 ppb, with 89Y as the internal standard for all isotopes of Cu and Zn.
Statistical analysis. Differences between groups were assessed by individual analysis of difference in the means as unpaired t tests with unequal variance. A p value of <0.05 was considered significant.
Results
Genetic ablation of Znt3 markedly decreased CAA in the brains of Tg2576 mice (Figs. 1, 2, 3). In parallel with reduced interstitial Aβ deposition, the numbers of Congo Red birefringent and anti-Aβ-immunopositive vessels in the neocortex were markedly and significantly reduced in the APP+/Znt3+/– and APP+/Znt3–/– mice at both 15 and 18 months of age (Figs. 1, 3). In agreement with previous reports (Cole et al., 1999), histochemically reactive Zn2+ (which is contained in vesicles) in the hippocampal mossy fibers was decreased by ZnT3 genetic ablation (Fig. 4A–D). We also noted that TFL-Zn fluorescence was frequently detected in the perivascular space of small and medium sized neocortical, thalamic, and leptomeningeal vessels in wild-type mice and was significantly diminished by ≈20% in Znt3-deficient mice (Fig. 4E–L; Table 2). These results imply that, somehow, CAA formation is linked to a pool of Zn2+ that communicates between the synaptic boutons and the cerebrovascular wall.
Figure 1.
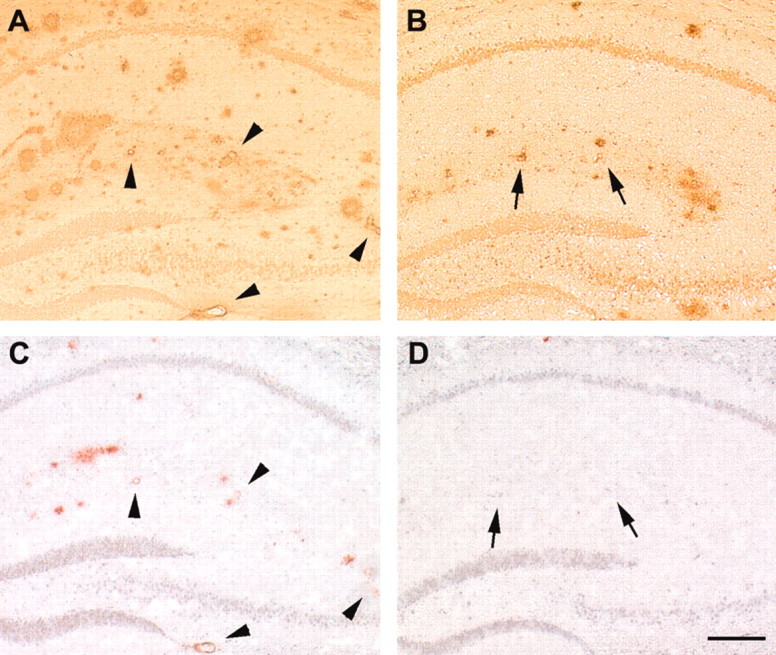
Vascular and parencymal amyloid burden are both markedly decreased by Znt3 genetic ablation. A–D, Compared with 18-month-old APP+/Znt3+/+ mice (A, C), 18-month-old APP+/Znt3–/– mice (B, D) demonstrate decreased numbers of Aβ-immunopositive and congophilic vessels. Arrowheads indicate Aβ-immunopositive vessels in APP+/Znt3+/+ mice (A) that are also congophilic (C). Arrows indicate Aβ-immunopositive vessels (B) in APP+/Znt3–/– mice that are not congophilic (D). Anti-Aβ immunostaining from hippocampus is shown in A and B and, from serial sections, Congo Red staining is shown in C and D. Data are typical of n = 8 APP+/Znt3+/+, and n = 7 APP+/Znt3–/–. Scale bar, 200 μm.
Figure 2.

CAA is markedly decreased by Znt3 genetic ablation (high power). A–F, Congo Red staining of different brain regions in 18-month-old APP+/Znt3+/+ mice (A, C,E) compared with 18-month-old APP+/Znt3–/– mice (B, D,F), demonstrating the impact of Znt3 ablation on CAA in vascular walls in the leptomeninges (A, B), hippocampus (C,D), and cortex (E, F). The insets (A, B) depict the vessels indicated by the arrowheads when viewed under polarized light. The congophilic vessel in A demonstrates the green birefringence characteristic of amyloid. Scale bars: A, B, 100 μm; A, B, insets, 200 μm; C–F, 50 μm. Data are typical of n = 8 APP+/Znt3+/+, and n = 7 APP+/Znt3–/–.
Figure 3.

Quantitative analysis of the inhibitory effect of Znt3 genetic ablation on CAA abundance in Tg2576 mice. A–C, Coronal sections at bregma plus 3.0 mm were processed for Congo Red staining or anti-Aβ immunocytochemistry. In the neocortex of each section, the number (no.) of congophilic (A) or anti-Aβ-immunopositive vessels (B) was counted at 15 (15 mo) and 18 (18 mo) months of age by an operator blinded to the phenotype of the mouse. Small vessels (<30 μm in diameter) were particularly prone to develop CAA (C). Asterisks denote significant differences between groups (*p < 0.05; **p < 0.01; ***p < 0.001).
Figure 4.

Disrupted synaptic Znt3 expression decreases Zn2+ detected by TFL-Zn in both hippocampal neurons and vessel walls in the neocortex and leptomeninges. A–D, Coronal sections of hippocampi of 15-month-old Znt3+/+ and Znt3–/– mice stained with TFL-Zn (A, C) or with anti-Znt3 Ab (B, D). Zn2+ (TFL-Zn fluorescence) is detectable in the mossy fibers (mf) localizing with Znt3 immunoreactivity in Znt3+/+ (A, B) but not Znt3–/– (C,D) mice. The same vessels in 15-month-old mice were immunoreactive for von Willebrand factor (F, J, arrows), a marker for endothelial cells in vessel walls (Theilen and Kuschinsky, 1992), but lacked Znt3 immunoreactivity (G, K, arrows). Zn2+ (TFL-Zn fluorescence) was detected in small cortical vessels in Znt3+/+ but not in Znt3–/– mice (E, I, arrows). Zn2+ (TFL-Zn fluorescence) was more abundant in leptomeningeal vessels of Znt3+/+ (H) as compared with Znt3–/– mice (L). Scale bars: A–D, 100 μm; E–L, 50 μm. M, ZnT3 was detected by Western blot in homogenate of mouse hippocampus but was absent from plasma, erythrocytes, and platelets in the mouse (20 μg of protein/lane).
To confirm the presence of such a pool, we performed Timm's stains on normal mice and examined brain areas that are most prone to CAA deposition (pial, thalamic, cortical, and hippocampal vessels) (Preston et al., 2003). Neocortical neuropil stains so darkly with the Timm's stain (because of zinc in synaptic vesicles) (Danscher, 1981; Holm et al., 1987) that the vessel walls were hard to differentiate in the neocortex and on the pial surface. However, we visualized distinct cerebral vessel wall staining by Timm's stain in hippocampal and thalamic areas (where there are regions of little background, because the synaptic boutons do not contain exchangeable Zn2+) (Fig. 5). Timm's staining revealed numerous casts of small vessels with puncta that could represent endothelial cell nuclei (Danscher, 1981) or possibly astrocytic foot processes (Fig. 5B,D). The intravital trapping of free zinc by the Timm's procedure excludes the possibility that the vessel wall zinc detected is because of artifactual release by slicing.
Figure 5.
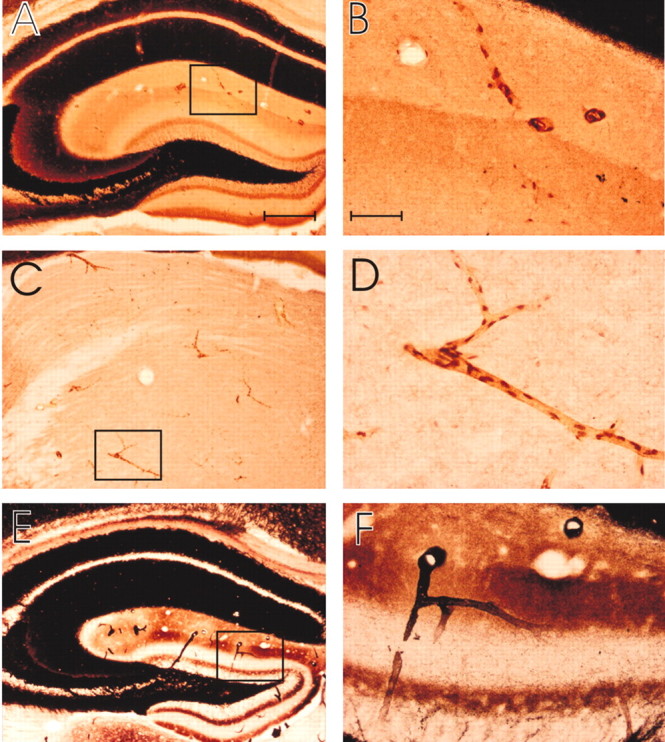
Timm's staining of cerebral vessel wall zinc. A–F, Low-power (A, B, C) and high-power (B, D, F) photomicrographs of coronal sections through the brain of a mouse (A, B,E, and F are from hippocampus; C and D are from thalamus), stained with Timm's (A–D) or neo-Timm's (E, F) method. A–D are C57BL/6 mice, and E and F are DBA; all are 3 months of age. The photomicrographs demonstrate the staining of blood vessels. B, D, and F are the high-power images of the box indicated in A, C, and E, respectively. Scale bars: A, C, E (in A), 250 μm; B, D, F (in B), 50 μm. Data are representative of n = 15 mice.
Although CAA and parenchymal amyloid are both much less abundant in Tg2576 after Znt3 gene ablation, the amount of CAA in APP+/Znt3–/– mice still increased between 15 and 18 months of age (Figs. 1, 3). We hypothesized that Zn2+ from sources other than ZnT3 activity could contribute to this residual amyloid accumulation. We determined that residual CAA and interstitial amyloid that is deposited by 18 months of age in APP+/Znt3–/– mice still contains an enrichment of histochemically reactive zinc, qualitatively similar to that of APP+/Znt3+/+ mice (Fig. 6). This suggests that some of the Zn2+ that induces CAA and plaque formation can originate from sources other than the synapse, but that most of the Zn2+ for CAA pathogenesis is ZnT3 dependent.
We next determined whether the vessel wall could receive Zn2+ from ZnT3 expression in other tissue components. Previous studies have localized ZnT3 solely to presynaptic vesicle membranes (Palmiter et al., 1996; Wenzel et al., 1997; Cole et al., 1999; Kay, 2003), and ZnT3 mRNA and protein are known not to be present in cortical vessels (Palmiter et al., 1996). We confirmed the absence of ZnT3 immunoreactivity in cerebral blood vessels of wild-type mice (Fig. 4G) and excluded the presence of ZnT3 in major blood components (platelets, erythrocytes, and plasma) by Western blot (Fig. 4M). Therefore, the ZnT3-mediated pool of exchangeable Zn2+ detected in cerebral vessel walls appears to be derived solely from synaptic ZnT3 activity.
We examined bulk metal levels in organs of Znt3 knock-out mice and their wild-type littermates (Table 1). Plasma zinc was significantly reduced by 13.6% in Znt3 knock-out mice compared with wild-type littermates, suggesting that synaptic ZnT3 activity contributes to peripheral Zn2+ levels. Bulk tissue Zn2+ levels were otherwise significantly decreased by 12.6% only in cerebral cortex but not different in cerebellum, kidney, heart, or pancreas of Znt3–/– mice compared with Znt3+/+ littermates. Liver zinc levels were elevated by 12.4% in Znt3–/– mice near the margin of statistical significance. Tissue Cu levels were unaffected by Znt3 knock-out.
Table 1.
Tissue Cu and Zn levels in Znt3 wild-type (n = 6) and knock-out (n = 6) mice
|
|
Metal |
|
|
|
|
|
|---|---|---|---|---|---|---|
| Zinc |
|
|
|
|
|
|
| Tissue |
ZnT3+/+
|
ZnT3-/-
|
p
|
ZnT3+/+
|
ZnT3-/-
|
p
|
| Plasma | 16.74 ± 0.58 | 14.47 ± 0.39 | 0.011 | 10.95 ± 0.50 | 10.14 ± 0.19 | 0.155 |
| (15.60-19.39) | (12.82-15.34) | (9.62-12.24) | (9.41-10.58) | |||
| Cerebral cortex | 11.77 ± 0.24 | 10.29 ± 0.14 | 0.0007 | 3.99 ± 0.11 | 3.88 ± 0.14 | 0.548 |
| (11.12-12.56) | (9.79-10.68) | (3.58-4.29) | (3.47-4.42) | |||
| Cerebellum | 9.72 ± 0.51 | 10.93 ± 0.46 | 0.106 | 3.73 ± 0.90 | 4.65 ± 0.44 | 0.617 |
| (7.70-11.33) | (9.49-12.44) | (3.61-9.51) | (3.70-6.66) | |||
| Heart | 13.79 ± 0.99 | 12.24 ± 0.89 | 0.273 | 4.65 ± 0.33 | 4.39 ± 0.26 | 0.547 |
| (10.04-17.40) | (9.22-14.85) | (3.63-5.51) | (3.52-5.45) | |||
| Liver | 24.27 ± 0.84 | 27.28 ± 1.18 | 0.066 | 6.50 ± 1.46 | 5.85 ± 0.92 | 0.716 |
| (21.45-27.18) | (23.83-31.58) | (4.07-13.50) | (3.88-10.19) | |||
| Kidney | 16.18 ± 0.80 | 15.06 ± 0.28 | 0.233 | 3.57 ± 0.17 | 3.33 ± 0.07 | 0.217 |
| (14.32-18.93) | (13.98-15.87) | (3.22-4.19) | (3.09-3.51) | |||
| Pancreas | 21.96 ± 0.82 | 23.17 ± 1.61 | 0.521 | 1.06 ± 0.04 | 1.04 ± 0.02 | 0.740 |
|
|
(20.12-25.74) |
(17.62-28.38) |
|
(0.91-1.16) |
(0.97-1.09) |
|
Values are means ± SEM (range); microgram per gram of wet weight, except for plasma, which is in micromoles per liter. p values are for t tests (two-tailed).
In peripheral organs that do not express ZnT3, including heart, liver, and kidney, histochemically reactive zinc was also detected in the perivascular space but was not decreased in Znt3 knock-out animals (Fig. 7, Table 2). Therefore, perivascular dissociable Zn2+ in the neocortex is a physiologically unique pool with a significant proportion of its concentration (Table 2) dependent on Zn2+ transport at the remote synapse.
Figure 7.
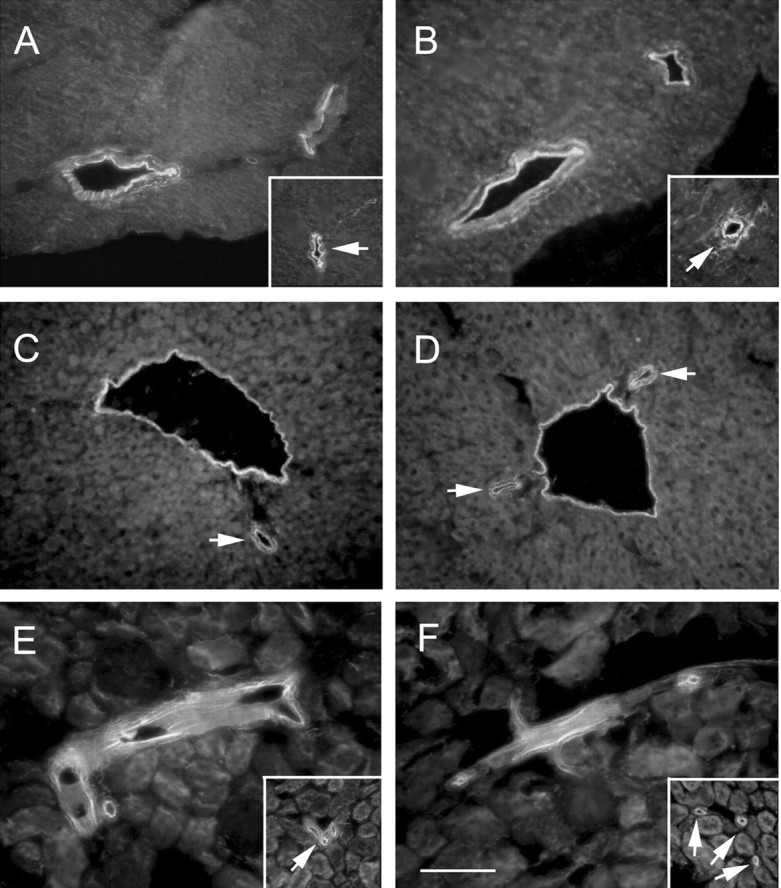
Synaptic Znt3 expression mediates perivascular zinc sequestration in brain but not in peripheral organs. A–F, TFL-Zn staining in the heart (A, B), liver (C,D), or kidney (E, F) reveals no significant difference between Znt3+/+ (A, C,E) and Znt3–/– (B, D,F) mice in either large caliber vessels (>30 μm) or small vessels (<30 μm; arrows). Scale bar, 100 μm. See Table 2 for quantifications.
Discussion
We demonstrated that CAA is markedly decreased after targeted disruption of the Znt3 gene. This may be explained by a remote effect of ZnT3 activity located in synaptic vesicles in raising exchangeable perivascular Zn2+ concentrations in the brain neocortex (Fig. 4, Table 2). Exchangeable Zn2+ was also identified by Timm's stain in vessel walls of the hippocampus and thalamus (Fig. 5). These findings are consistent with the sites of CAA formation in APP transgenic mice (CAA in the thalamus and parts of the hippocampus is known to form at a distance from the sites of plaque deposition), even when neuronal-specific expression of Aβ occurs (i.e., no vascular origin of Aβ) (Calhoun et al., 1999).
Most tissue Zn2+ is tightly held in proteins like metallothionein and transcription factors and not readily available for chemical exchange with the exterior (Frederickson and Bush, 2001). Synaptic vesicular Zn2+ is far more loosely ligated and exteriorizes during synaptic transmission, where Zn total concentration has been reported to rise to 300 μm (Howell et al., 1984), although only a small fraction of this may be freely ionic (Kay, 2003). Unlike tightly complexed Zn2+ in the neuronal soma (representing 70–80% of parenchymal Zn), exchangeable Zn2+ in synaptic vesicles is detectable with Timm's stain (a well established intravital stain) (Danscher and Zimmer, 1978; Danscher, 1981) and various fluorescent dyes (e.g., TSQ and TFL-Zn) (Budde et al., 1997; Frederickson and Bush, 2001).
The Zn2+ fluorophores TSQ and TFL-Zn strongly detect Zn2+ in amyloid plaques in Tg2576 mice and in AD cases (Lee et al., 1999, 2002, 2004; Suh et al., 2000; Bush, 2003). The Zn2+ binding sites on Aβ have affinity constants of ≈100 nm to ≈10 μm (Bush et al., 1994a); affinities that are low enough to surrender Zn2+ for TSQ and TFL-Zn to either make ternary Aβ-Zn-dye complexes or release Zn-dye complexes that embed in the pathology on the basis of lipophilicity (Snitsarev et al., 2001). The detection of Zn2+ in vessel walls by TFL-Zn (Fig. 4) and Timm's (Fig. 5) stains implies that this Zn2+ is in the proximity of perivascular space contents.
The neocortical perivascular spaces may function as an efflux or drainage system joining the brain interstitial spaces to the capillary walls and then larger arteries, arterioles, and leptomeningeal vessels. The efflux of Aβ from the brain to peripheral circulation may be mediated by this route (Weller et al., 1998; Preston et al., 2003). Our data indicate that neuronal Zn2+ may also communicate with the periphery through this route. Therefore, CAA may be promoted by neuronal Zn2+ that is externalized by synaptic transmission, abnormally reacting with Aβ that is exiting the brain. This may explain why the synaptic cleft and the neocortical perivascular spaces (Yamaguchi et al., 1992) are the two most likely sites of Aβ deposition, because they are the sites with the highest fluxes of exchangeable Zn2+ in the brain.
ApoE is also essential for CAA formation in the perivascular spaces of the cerebral vessels in APP transgenic mice (Holtzman et al., 2000; Fryer et al., 2003). ApoE binds transition metal ions, including Zn2+ (Miyata and Smith, 1996). We previously found that the ability of ApoE to buffer Zn2+ protects Aβ from precipitation by Zn2+, and that the E4 isoform is the worst Zn2+ buffer (<E3 <E2) (Moir et al., 1999), paralleling the association of the ApoE isoform with the risk for AD. Because ApoE may colocalize with exchangeable Zn2+ in the perivascular space of cerebral vessels, we hypothesize that Aβ may be precipitated at this site by Zn2+, which is in exchange with ApoE. One hypothesis that may unify the findings is that ApoE may normally ferry some Zn2+ from the liver (where ApoE is synthesized) to neocortical ZnT3 and therefore, in ZnT3 knock-out mice, liver Zn2+ levels rise (and plasma zinc levels fall) (Table 1), because the liver pool of zinc is not summoned.
It is not yet clear whether the presence of Zn2+ in the cerebral vessel wall represents influx or efflux. The presence of Zn2+ in CAA in APP+/Znt3–/– mice (Fig. 6), as well as the presence (albeit attenuated) of Zn2+ in the cerebrovascular walls of Znt3–/– mice (Fig. 4L), suggests that components of the vessel walls themselves could also offer exchangeable Zn2+.
The small but significant decrease in cerebral bulk Zn levels induced by ZnT3 ablation (≈13%) (Table 1) confirms our previous results in younger mice (6–12 months of age) (Lee et al., 2002, 2004) and is consistent with previous estimates of the contribution of synaptic bouton zinc to total brain zinc content (Holm et al., 1987; Frederickson et al., 1992). The marked diminution in congophilic angiopathy in APP+/Znt3–/– mice (Fig. 3A,C) appears disproportionately greater than the ≈20% loss in Zn2+ fluorescence measured in the neocortical vessel walls of nontransgenic mice (Table 2). This may be explained by a degree of unavoidable loss of some extracellular Zn2+ during histological preparation, decreasing our estimation of the differences in Zn2+ concentration in the vessel walls. Also, the TFL-Zn and TSQ fluorescent dyes may only be semiquantitative indicators of Zn2+ concentration, because the lipid environment of the neuronal membranes may make the relationship between Zn2+ binding to the TFL-Zn indicator nonlinear (Snitsarev et al., 2001). Therefore, although the decrease in TFL-Zn fluorescence in ZnT3 knock-out mice indicates that a significant decrease in detectable Zn2+ has occurred as a result of the ZnT3 genetic ablation, this decrease may be >20% in absolute value.
There is evidence for increased soluble Aβ concentrations in both AD and APP-transgenic brain tissue (Lue et al., 1999; McLean et al., 1999). We proposed that Alzheimer amyloid formation is the product of an abnormal elevation of soluble Aβ, perhaps liberated from its normal membrane-associated cellular compartment by an abnormal oxidation (Barnham et al., 2003), that then drifts into microanatomical regions (synapse and perivascular space) possessing exceptionally high Zn2+ concentrations that induce Aβ precipitation (Bush, 2003). Once the Aβ accumulates, however, it may impede the flow of Aβ and Zn2+ through the perivasculature. ZnT3 activity contributes to a flux of brain Zn2+ that is likely to subserve important physiologic functions and contributes ∼14% of plasma Zn levels in mice (Table 1). Therefore, it is possible that the trapping of essential Zn2+ in the amyloid mass in AD may contribute to central and peripheral Zn2+ deficiency. A recent phase 2 clinical trial of the Cu2+ and Zn2+ chelator clioquinol induced a significant ≈25% increase in plasma Zn2+ levels in AD subjects over a 9 month treatment regimen (Ritchie et al., 2003). On the basis of our current findings, this increase in plasma Zn2+ may be attributable to the dissolution of CAA, leading to increased Zn2+ flux between the pool of brain synaptic Zn2+ and the peripheral circulation. Our findings of decreased CAA in Tg2576 mice after Znt3 gene ablation suggest that the pathways involved in ZnT3-dependent zinc transport may have therapeutic relevance in AD.
Footnotes
This work was supported by Alzheimer's Association, National Institute on Aging Grant 5RO1AG12686–09, National Health and Medical Research Council (A.I.B.), National Institute of Neurological Disorders and Stroke Grant F32NS10874 (A.L.F.), and Creative Research Initiatives of the Korean Ministry of Science and Technology (J.Y.K.). We thank Karen Hsiao-Ashe for the Tg2576 mice.
Correspondence should be addressed to Dr. Ashley I. Bush, Laboratory for Oxidation Biology, Genetics and Aging Research Unit, Massachusetts General Hospital, and Department of Psychiatry, Harvard Medical School, Building 114, 16th Street, Charlestown, MA 02129-4404. E-mail: bush@helix.mgh.harvard.edu.
DOI:10.1523/JNEUROSCI.0297-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243453-07$15.00/0
A.L.F. and J.-Y.L. contributed equally to this work.
References
- Barnham KJ, Ciccotosto GD, Tickler AK, Ali FA, Smith DG, Williamson NA, Lam Y-H, Carrington D, Tew D, Kocak G, Volitakis I, Separovic F, Barrow CJ, Wade JD, Masters CL, Cherny RA, Curtain CC, Bush AI, Cappai R (2003) Neurotoxic, redox-competent Alzheimer's β-amyloid is released from lipid membrane by methionine oxidation. J Biol Chem 278: 42959–42968. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand [Suppl] 165: 3–12. [DOI] [PubMed] [Google Scholar]
- Budde T, Minta A, White JA, Kay AR (1997) Imaging free zinc in synaptic terminals in live hippocampal slices. Neuroscience 79: 347–358. [DOI] [PubMed] [Google Scholar]
- Bush AI (2003) The metallobiology of Alzheimer's disease. Trends Neurosci 26: 207–214. [DOI] [PubMed] [Google Scholar]
- Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P (1990) The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem 265: 15977–15983. [PubMed] [Google Scholar]
- Bush AI, Pettingell Jr WH, Paradis MD, Tanzi RE (1994a) Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem 269: 12152–12158. [PubMed] [Google Scholar]
- Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE (1994b) Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science 265: 1464–1467. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96: 14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny RA, Legg JT, McLean CA, Fairlie D, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI (1999) Aqueous dissolution of Alzheimer's disease Aβ amyloid deposits by biometal depletion. J Biol Chem 274: 23223–23228. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y-S, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30: 665–676. [DOI] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD (1999) Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci USA 96: 1716–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coria F, Castaño EM, Frangione B (1987) Brain amyloid in normal aging and cerebral amyloid angiopathy is antigenically related to Alzheimer's disease β-protein. Am J Pathol 129: 422–428. [PMC free article] [PubMed] [Google Scholar]
- Danscher G (1981) Histochemical demonstration of heavy metals. A revised version of the sulphide silver method suitable for both light and electronmicroscopy. Histochemistry 71: 1–16. [DOI] [PubMed] [Google Scholar]
- Danscher G, Zimmer J (1978) An improved Timm sulphide silver method for light and electron microscopic localization of heavy metals in biological tissues. Histochemistry 55: 27–40. [DOI] [PubMed] [Google Scholar]
- Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR (2003) Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: raman microscopic evidence. Biochemistry 42: 2768–2773. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Bush AI (2001) Synaptically released zinc: physiological functions and pathological effects. Biometals 14: 353–366. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Rampy BA, Reamy-Rampy S, Howell GA (1992) Distribution of histochemically reactive zinc in the forebrain of the rat. J Chem Neuroanat 5: 521–530. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003) Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23: 7889–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885–890. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X, Mavros C, Coccia JA, Faget KY, Fitch KA, Masters CL, Tanzi RE, Chylack LT, Bush AI (2003) Cytosolic β-amyloid deposition in Alzheimer's disease lens. Lancet 361: 1258–1265. [DOI] [PubMed] [Google Scholar]
- Holm IE (1989) Neo-Timm and selenium stainable glial cells of the rat telencephalon. Histochemistry 91: 133–141. [DOI] [PubMed] [Google Scholar]
- Holm IE, Andreasen A, Danscher G, Perez-Clausell J, Nielsen H (1987) Quantification of vesicular zinc in the rat brain. Histochem 89: 289–293. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT (2000) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol 47: 739–747. [PubMed] [Google Scholar]
- Howell GA, Welch MG, Frederickson CJ (1984) Stimulation-induced uptake and release of zinc in hippocampal slices. Nature 308: 736–738. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G (1995) Age related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15: 1203–1218. [DOI] [PubMed] [Google Scholar]
- Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel J-P, Tanzi RE, Bush AI (1997) Zinc-induced Alzheimer's Aβ1–40 aggregation is mediated by conformational factors. J Biol Chem 272: 26464–26470. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J (2003) Incidence of cerebrovascular lesions in Alzheimer's disease: a postmortem study. Acta Neuropathol 105: 14–17. [DOI] [PubMed] [Google Scholar]
- Kawai M, Cras P, Richey P, Tabaton M, Lowery DE, Gonzalez-DeWhitt PA, Greenberg BD, Gambetti P, Perry G (1992) Subcellular localization of amyloid precursor protein in senile plaques of Alzheimer's disease. Am J Pathol 140: 947–958. [PMC free article] [PubMed] [Google Scholar]
- Kay AR (2003) Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J Neurosci 23: 6847–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Mook-Jung I, Koh J-Y (1999) Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J Neurosci 19: RC10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Cole TB, Palmiter RD, Suh SW, Koh J-Y (2002) Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA 99: 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Kim J-H, Hong SH, Lee JY, Cherny RA, Bush AI, Palmiter RD, Koh J-Y (2004) Estrogen decreases zinc transporter 3 expression and synaptic vesicle zinc levels in mouse brain. J Biol Chem, in press. [DOI] [PubMed]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82: 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C, Cherny R, Fraser F, Fuller S, Smith M, Beyreuther K, Bush A, Masters C (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860–866. [DOI] [PubMed] [Google Scholar]
- Miyata M, Smith JD (1996) Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and β-amyloid peptides. Nat Genet 14: 55–61. [DOI] [PubMed] [Google Scholar]
- Moir RD, Atwood CS, Romano DM, Laurans MH, Huang X, Bush AI, Smith JD, Tanzi RE (1999) Differential effects of apolipoprotein E isoforms on metal-induced aggregation of Aβ using physiological concentrations. Biochemistry 38: 4595–4603. [DOI] [PubMed] [Google Scholar]
- Opazo C, Huang X, Cherny R, Moir R, Roher A, White A, Cappai R, Masters C, Tanzi R, Inestrosa N, Bush A (2002) Metalloenzyme-like activity of Alzheimer's disease β-amyloid: Cu-dependent catalytic conversion of dopamine, cholesterol and biological reducing agents to neurotoxic H2O2 J Biol Chem 277: 40302–40308. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Huang L (2004) Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflügers Arch 447: 744–751. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD (1996) ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci USA 93: 14934–14939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Clausell J, Danscher G (1985) Intravesicular localization of zinc in rat telencephalic boutons. A histochemical study. Brain Res 337: 91–98. [DOI] [PubMed] [Google Scholar]
- Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58: 1629–1634. [DOI] [PubMed] [Google Scholar]
- Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO (2003) Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol 29: 106–117. [DOI] [PubMed] [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny RA, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer's disease: a pilot phase 2 clinical trial. Arch Neurol 60: 1685–1691. [DOI] [PubMed] [Google Scholar]
- Snitsarev V, Budde T, Stricker TP, Cox JM, Krupa DJ, Geng L, Kay AR (2001) Fluorescent detection of Zn(2+)-rich vesicles with Zinquin: mechanism of action in lipid environments. Biophys J 80: 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G, Frederickson CJ (2000) Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Res 852: 274–278. [DOI] [PubMed] [Google Scholar]
- Theilen H, Kuschinsky W (1992) Fluorescence labeling of the capillary network in rat brains. J Cereb Blood Flow Metab 12: 347–350. [DOI] [PubMed] [Google Scholar]
- Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA (1990) Amyloid β protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248: 1120–1122. [DOI] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Cole TB, Born DE, Schwartzkroin PA, Palmiter RD (1997) Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl Acad Sci USA 94: 12676–12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ (1992) Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer's disease. An immunoelectron microscopic study. Am J Pathol 141: 249–259. [PMC free article] [PubMed] [Google Scholar]