

Abstract
Sanfilippo syndrome is a mucopolysaccharidosis (MPS) caused by a lysosomal enzyme defect interrupting the degradation pathway of heparan sulfates. Affected children develop hyperactivity, aggressiveness, delayed development, and severe neuropathology. We observed relevant behaviors in the mouse model of Sanfilippo syndrome type B (MPSIIIB), in which the gene coding for α-N-acetylglucosaminidase (NaGlu) is invalidated. We addressed the feasibility of gene therapy in these animals. Vectors derived from adeno-associated virus serotype 2 (AAV2) or 5 (AAV5) coding for NaGlu were injected at a single site in the putamen of 45 6-week-old MPSIIIB mice. Normal behavior was observed in treated mice. High NaGlu activity, far above physiological levels, was measured in the brain and persisted at 38 weeks of age. NaGlu immunoreactivity was detected in neuron intracellular organelles, including lysosomes. Enzyme activity spread beyond vector diffusion areas. Delivery to the entire brain was reproducibly obtained with both vector types. NaGlu activity was higher and distribution was broader with AAV5-NaGlu than with AAV2-NaGlu vectors. The compensatory increase in the activity of various lysosomal enzymes was improved. The accumulation of gangliosides GM2 and GM3 present before treatment and possibly participating in neuropathology was reversed. Characteristic vacuolations in microglia, perivascular cells, and neurons, which were prominent before the age of treatment, disappeared in areas in which NaGlu was present. However, improvement was only partial in some animals, in contrast to high NaGlu activity. These results indicate that NaGlu delivery from intracerebral sources has the capacity to alleviate most disease manifestations in the MPSIIIB mouse model.
Keywords: lysosomal storage disease, α-N-acetylglucosaminidase, adeno-associated virus vectors, gangliosides, hyperactivity, elevated plus-maze
Introduction
Mucopolysaccharidosis type III [MPSIII (Sanfilippo syndrome)] is characterized by early-onset neuropathology, in contrast to mild somatic manifestations. Affected children show intense hyperactivity, intractable behavioral problems, sleep disturbance, and progressive mental retardation. Histopathology reveals vacuolation in neurons, glia, and perivascular cells. The disease results from a genetic defect in a lysosomal enzyme required for heparan sulfate (HS) catabolism. The accumulation of abnormal HS species likely affects signaling by members of the fibroblast growth factor family (Turnbull et al., 2001), which have important functions in the development and plasticity of neural cells (Reuss and von Bohlen und Halbach, 2003). HS storage presumably induces the accumulation of ganglioside GM2 in lysosomes as well as ganglioside GM3 and cholesterol accumulation in late endosomes and other cell membranes (Haust and Gordon, 1986; Gondre-Lewis et al., 2003). Ganglioside accumulation in membranes affects calcium uptake (Pelled et al., 2003), cell recognition, synaptogenesis (Walkley et al., 1991), cell survival (Huang et al., 1997), and neurite growth (Walkley et al., 1988).
The deficient enzyme in MPSIII type B (MPSIIIB) is α-N-acetylglucosaminidase [NaGlu (EC 3.2.1.50)]. NaGlu is targeted to lysosomes through interaction with the mannose-6-phosphate receptors (Ullrich et al., 1979). In contrast to other lysosomal hydrolases, recombinant NaGlu produced in Chinese hamster ovary (CHO) cells is not efficiently captured by MPSIIIB cells, both in vitro (Zhao and Neufeld, 2000; Weber et al., 2001) and in vivo (Yu et al., 2000). These results suggest that mechanisms involved in the intracellular trafficking of the enzyme may have been only partially elucidated and raise questions with respect to enzyme substitution therapy. Treatment is rendered even more challenging by the necessity to deliver the missing enzyme to the entire CNS. The creation of focal sources of NaGlu in the brain through gene therapy currently appears as the only potential therapeutic approach.
A mouse model of MPSIIIB has been created (Li et al., 1999). Animals show an accumulation of HS in tissues, an elevation of GM2 and GM3 gangliosides, a widespread activation of microglia (Ohmi et al., 2003), and vacuolation in brain cells at early stages of the disease. Abnormal behavior was reported (Li et al., 1999). Local improvement in pathology was described in areas surrounding the injection of adeno-associated virus (AAV) vectors coding for NaGlu (Fu et al., 2002). These authors suggested that delivery of the missing enzyme to the brain through gene therapy might be less efficient in MPSIIIB mice than in other previously investigated mouse models of lysosomal storage disease (for review, see Bosch and Heard, 2003). We observed that MPSIIIB mice were more active in home cages and showed a reduced propensity for anxiety in the elevated plus-maze than normal mice, two behaviors that might be relevant to clinical manifestations in affected children. We examined the effect of gene therapy on these symptoms. AAV2 or AAV5 vectors coding for human NaGlu were injected at a single site at 6 weeks of age in 45 animals. Neuropathology and ganglioside accumulation were already installed at this time. Improved behavior, efficient NaGlu delivery to the entire brain, decreased ganglioside overload, and complete or partial disappearance of cell vacuolation suggested that intracerebral AAV-NaGlu vector injections might represent an effective treatment for MPSIIIB.
Materials and Methods
AAV vector construction and production
Inverted terminal repeats from AAV2 or AAV5 were obtained from pSUB201 (Samulski et al., 1987) and rAAV5LacZ (Chiorini et al., 1999), respectively. They flanked a transcription unit (identical in AAV2-NaGlu and AAV5-NaGlu vectors) consisting of the mouse phosphoglycerate kinase (mPGK)-1 promotor (552 bp; position -524 to -20 bp from the first translated codon) (Adra et al., 1987), human NaGlu cDNA isolated from pBS-NaGlu (Zhao et al., 1996), the post-transcriptional regulatory element of woodchuck hepatitis virus (Zufferey et al., 1999), and the bovine hormone gene polyadenylation unit (Goodwin and Rottman, 1992). AAV2-NaGlu vectors were produced by transient transfection of human embryonic kidney 293 (HEK 293) cells with AAV2-NaGlu and pDG (Grimm et al., 1998) plasmid DNA (Brument et al., 2002), and particles were purified using iodixanol gradients and heparin affinity resin (Zolotukhin et al., 1999). Recombinant AAV5 particles were produced by transient transfection of HEK 293 with the helper plasmid AAV-5.2 DNA (Chiorini et al., 1999), the adenoviral helper function pXX6 plasmid DNA (Xiao et al., 1998), and the AAV5-NaGlu vector plasmid DNA (12.5, 25, and 12.5 μg, respectively for a 15 cm cell dish). AAV5-NaGlu particles were purified through two successive CsCl gradients (Salvetti et al., 1998), and purified fractions were pooled and dialyzed against PBS-Ca2+-Mg2+ (Biowhitaker). The amount of vector genome was measured by dot blot (Salvetti et al., 1998).
MPSIIIB mice and treatment
All experiments were approved by the institutional committee on animal experimentation and performed by authorized investigators (Ministère de l'Agriculture et de la Pêche, authorization 75-268). C57BL/6NaGlu+/ - breeders were obtained from Prof. E. Neufeld (University of California Los Angeles, Los Angeles, CA) (Li et al., 1999). NaGlu- / - MPSIIIB mutants were genotyped at weaning by PCR on DNA from tail-clipped samples, amplifying both the mouse NaGlu gene exon 6 and the inserted neor sequences.
Six-week-old MPSIIIB mice were anesthetized with ketamine/xylazine (0.1/0.01 mg per gram of body weight) and installed on a stereotactic frame (David Kopf Instruments, Tujunga, CA). Injection in the striatum was performed at coordinates +0.7 mm anterior to bregma, 2.0 mm lateral to midline, and at 4.5 mm depth. Five microliters of the AAVNaGlu vector preparation (109 vector genomes) were delivered with an ultramicropump (World Precision Instruments, Hertfordshire, UK) through a 30 gauge blunt-tip needle with a flow of 0.5 μl/min. The needle was brought up 0.5 mm after the injection of 2.5 μl, and the rest of the volume was injected. The needle was slowly withdrawn after 5 min, the scalp was closed, and the animals were returned to recovery cages.
Behavioral testing
Animals had access ad libitum to food and drink. Circadian rhythm was set on light from 7:30 A.M. to 7:30 P.M.
Open field testing. Testing was performed between 8:00 and 12:00 A.M. The open field was a brightly illuminated (240 lux) empty white circular plastic arena (diameter, 110 cm; height, 35 cm). Movements were analyzed with a Videotrack System (View-point, Lyon, France). The system was set up to break down each minute of the recording session (10 min) into three types of displacement: navigation (movements at a speed of ≥11.82 cm/sec), fast exploration (11.82 cm/sec < speed < 6.72 cm/sec) and slow exploration (speeds of ≤6.72 cm/sec and resting periods). Cumulative time spent and distances for each type of displacement were recorded. Habituation was investigated by analyzing the time-course evolution of navigation time minute by minute within a session (Thinus-Blanc, 1996). The difference between times spent at 5 weeks (before treatment) and 29 weeks for navigation, fast exploration, and slow exploration was recorded.
Elevated plus-maze. Testing was performed between 2:00 and 6:00 P.M. We used a cross-maze built with opaque Plexiglas panels consisting of two opposing open arms (length, 30.5 cm; width, 7 cm) and two walled arms (same size with 18-cm-high walls) extending a central square platform (7 × 7 cm). The maze is 54 cm above floor level. Mice were tested according to Hogg (1996). Testing was performed under red-light illumination (dark condition) with intensity set up to 1 lux. Animals entered the maze on the central platform, facing an open arm. Each trial (10 min) was videotaped for off-line analyses. Time spent in arms was scored when animals put two forepaws within an arm and continued until they exited the arm (Vasar et al., 1993). To reduce activity-induced artifacts, data were expressed as ratio of time spent in open arms relative to total time spent in arms during a 10 min session.
Home cage activity test. Global activity and circadian rhythm were analyzed in home cages (52 × 26 × 32 cm) by continuous monitoring over a 24 hr period (start, 2:30 P.M.; sampling period of 30 min; 0 sec interval between adjacent periods) (Zhuang et al., 2001) using a Videotrack System. Sampling periods were broken down into very fast or fast movements (speeds of ≥6.72 cm/sec) and slow movements or inactivity periods (speeds of <6.72 cm/sec). The cumulative time spent for these movements was recorded. Red-light illumination was used for dark periods (7:30 P.M. to 7:30 A.M.).
NaGlu and other lysosomal enzyme assays
Deeply anesthetized animals were perfused with PBS alone (assays on whole hemispheres) or with PBS followed by 4% paraformaldehyde (PFA) in PBS (assays on brain slices). Lysosomal enzyme activity levels were equivalent in both conditions when assayed in control mice. Tissue was homogenized in water, submitted to 10 freeze-thaw cycles, and clarified by centrifugation. Enzyme assays for NaGlu (Marsh and Fensom, 1985), total hexosaminidase (Saifer and Perle, 1974), hexosaminidase A (Inui et al., 1985), β-galactosidase (Van Hoof and Hers, 1968), α-fucosidase (Wood, 1975), β-glucosidase (Peters et al., 1976), α-glucosidase (Salafsky and Nadler, 1973), β-glucuronidase (Gehler et al., 1974), and α-l-iduronidase (Young, 1992) were performed as described. Activity units correspond to the hydrolysis of 1 nmol of substrate per hour and are expressed per milligram of protein in extracts.
Quantitative PCR
AAV-NaGlu vector genome copy numbers were measured by Taqman quantitative PCR. Genomic DNA was prepared from mouse brain tissues. Vector woodchuck post-transcriptional regulatory element (WPRE) sequences and mouse genomic CC chemokine receptor 5 (CCR5) sequences were simultaneously amplified from serial DNA dilutions (0.039-10 ng/reaction). Copy numbers were calculated for each Ct through the simultaneous amplification of serial dilutions of plasmid DNA containing the WPRE sequence (38-6 × 105 copies of the AAV2-NaGlu plasmid) or the mouse CCR5 cDNA sequence (43.6-6.9 × 105 copies of the pRc/CMV-CCR5 plasmid) (Picard et al., 1997). Values determined for each Ct allowed the determination of mean copy numbers per 2N genomes, according to CCR5 copy numbers. SDs were always <20% of mean values and are not indicated. The following primers and probes were used: WPRE, 5′-TTGCTCCTTTTACGCTATGTGGAT-3′ (forward), 5′-AACCAGGA-TTTATACAAGGAGGAGAA-3′ (reverse), 5′-FAM-TCATGCTATTGCTTCCCGTATGGCTTTC-TAMRA (probe); mouse CCR5, 5′-ACAC-CCTGTTTCGCTGTAGGAA-3′ (forward), 5′-TGTAGGGAGTCCAGAAGAGAAAGTAGAC-3′ (reverse), 5′-FAM-AGGGCTGTGAGGCTCATCTTTGCCAT-TAMRA-3′ (probe). Amplification parameters were 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 sec and 60°C for 1 min in a Model 5700 Sequence Detector (PerkinElmer Life Sciences, Emeryville, CA). Ct values of >37 were considered as background. The assay sensitivity threshold was <0.1 copy per diploid genome.
Analysis of gangliosides
Total lipids were extracted from water homogenates (Fujita et al., 1996), isolated, and desalted using reverse-phase Bond Elut C18 columns (Varian, Palo Alto, CA) (Kyrklund, 1987). Gangliosides were separated on G60 high-performance thin-layer chromatography plates (Merck, Darmstadt, Germany), visualized by resorcinol-HCl, and quantified by densitometry. Data (mean of duplicates) were expressed as a molar percentage of total gangliosides according to N-acetylneuraminic acid (NeuAc) moiety numbers.
Pathology
Fragments (1 mm edge cubes) were taken from coronal brain slices that were postfixed with 4% PFA, 1% glutaraldehyde, and 1% osmium tetroxide and then embedded in Epon-Araldite. Semithin sections (1 μm) further stained with toluidine blue were examined by light microscopy. Eight to ten sections from each fragment were examined completely. A slice was declared affected when at least one neuron, glial cell, or perivascular cell with distended lysosomes was observed in at least one section.
Immunofluorescence
Brain tissue was fixed with 4% PFA in PBS. Cryosections (20 μm) were blocked with 5% goat serum (Invitrogen, San Diego, CA), 2% bovine serum albumin (Sigma, St. Louis, MO), and 0.3% Triton X-100 in PBS for 30 min at room temperature and then incubated with one of the following antibodies: rabbit anti-human NaGlu serum (1:1000; a kind gift from Prof. E. Neufeld), anti-mouse neuronal-specific nuclear protein (NeuN) monoclonal antibody (1:500; mAb 377; Chemicon, Temecula, CA), anti-mouse glial fibrillary acidic protein (GFAP) (1:200; clone GA-5; Sigma), anti-mouse CD11b (1:200; clone MRC OX-42/OBT0036; Immunological Direct, Kidlington, UK), and anti-mouse lysosomal-associated membrane glycoprotein-1 (Lamp1) (1:500; clone P3-NSI/1Ag4-1; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Anti-NaGlu and anti-CD11b were incubated overnight at 4°C. Anti-NeuN, anti-GFAP, and anti-Lamp1 were incubated for 1 hr at room temperature. Anti-NaGlu was revealed with a goat anti-rabbit tetramethylrhodamine isothiocyanate (1:600; Jackson ImmunoResearch, West Grove, PA) for 1 hr at room temperature. Other primary antibodies were revealed with a biotinylated goat anti-mouse antibody (1:200; RPN 1177; Amersham Pharmacia Biotech, Arlington Heights, IL) for 1 hr followed by streptavidin Alexa 488 (1:200; Molecular Probes, Eugene, OR).
Statistics
Statistics were performed using Statistical Program for the Social Sciences (SPSS) software (SPSS, Chicago, IL). Nonparametric tests were used when the Kolmogorov-Smirnov or the Shapiro-Wilk test for normal distribution of samples indicated a significance level that was <0.05.
Results
MPSIIIB mice are more active and less prone to anxiety than normal mice
At onset, clinical manifestations in Sanfilippo B children include hyperactivity and loss of inhibition tending to aggressiveness. These components can be assessed in the mouse. Recording of animal behavior in the open field provided an initial glimpse of locomotion and anxiety level of MPSIIIB mice. We scored the performances of MPSIIIB mice (n = 10) and normal littermates (n = 12) at 5 and 29 weeks of age during a 10 min session in the open field. Time devoted to navigation decreased during sessions, indicating normal habituation at both time points. In 5-week-old animals, mean time spent in navigation, exploration, and resting did not differ between control and MPSIIIB mice. Although mean time spent in navigation was equivalent at 5 and 29 weeks of age for control mice (time at 29 weeks - time at 5 weeks = +6 sec), it increased with age in MPSIIIB mice (time at 29 weeks - time at 5 weeks = +67 sec; p = 0.04; Student's t test), suggesting that affected animals might be more active (Fig. 1A).
Figure 1.
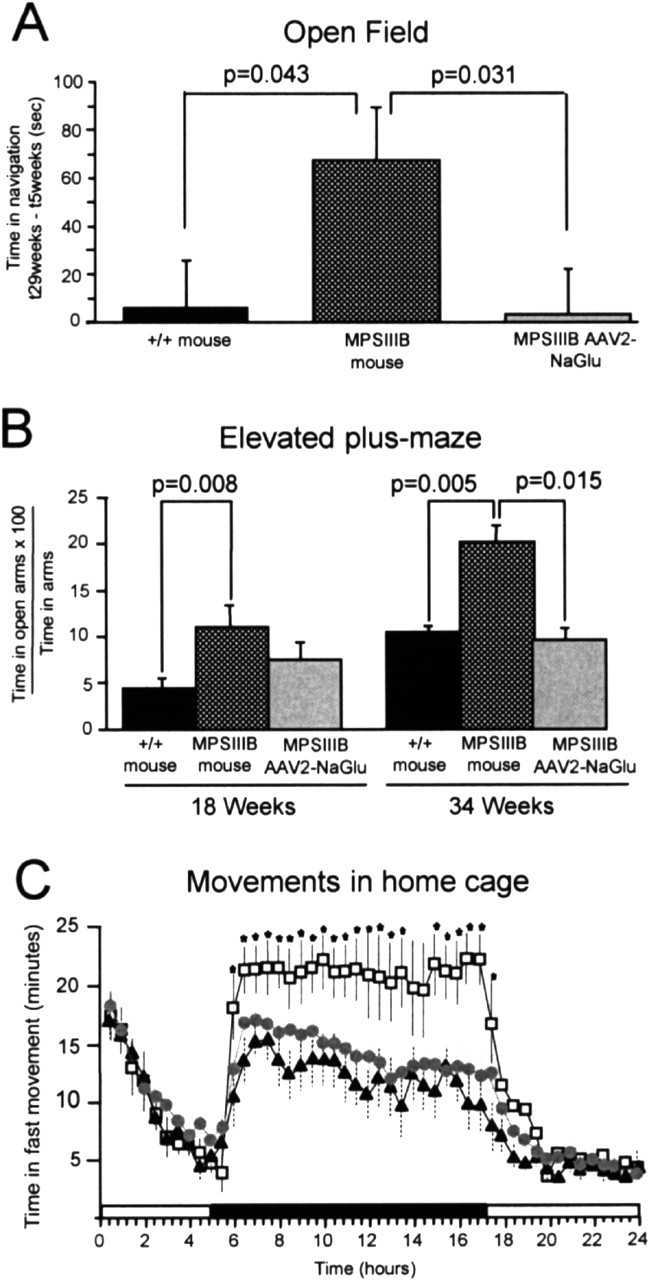
Abnormal behaviors were prevented in treated MPSIIIB mice. A, Time spent in navigation was recorded in the open field during 10 min sessions. Normal mice (+/+; n = 12), untreated MPSIIIB mice (MPSIIIB; n = 10), and AAV2-NaGlu-treated MPSIIIB mice (MPSIIIB-AAV2-NaGlu; n = 9) were tested at 5 and 29 weeks of age. Data are means ± SEM of differences between times spent in navigation at 29 and 5 weeks (t29weeks to t5weeks). Significance was determined using the Student's t test. B, The same animals were tested in the elevated plus-maze at 18 and 34 weeks of age in a dark environment. Data are means ± SEM of ratios of time spent in open arms relative to total time spent in arms. Significance was determined using the Student's t test. C, Movements of normal mice (black triangles; n = 6), MPSIIIB mice treated with mock AAV5-GFP vectors (open squares; n = 6), untreated MPSIIIB mice (equivalent to MPSIIIB mice treated with mock AAV5-GFP vectors and not shown here; n = 6), or MPSIIIB mice treated with AAV5-NaGlu vectors (gray circles; n = 15) were recorded in 18-week-old mice over 24 hr in home cages. Data are means ± SEM of times spent in fast movements during 30 min periods. The shadowed area indicates the dark period. Significance: stars indicate p < 0.05; one-way ANOVA; Bonferroni's post hoc analysis.
Animal anxiety was measured in the elevated plus-maze. Tests were performed at 18 and 34 weeks of age. At both ages, MPSIIIB mice (n = 10) visited more often and spent more time in the open arms of the maze than normal mice (n = 12; p < 0.008; Student's t test) (Fig. 1B). This result provided evidence for a reduced propensity to anxiety in MPSIIIB mice than in normal mice. The overall activity of the animals was investigated at 18 weeks of age by a 24 hr recording of movements in home cages. Normal mice (n = 6) were compared with untreated MPSIIIB mice (n = 6). Animals showed equivalent periods of activity in light conditions. In contrast, MPSIIIB mice spent significantly more time in motion (Fig. 1C) and less time resting (data not shown) than normal mice in the dark.
Treatment improved behavior scores in MPSIIIB mice
MPSIIIB mice were scored in the open field before treatment and 25 weeks after a single injection of AAV2-NaGlu vector in the striatum (animal age, 5 and 29 weeks, respectively; n = 9). Time devoted to navigation decreased during sessions, indicating normal habituation at both time points. Mean time spent in navigation was equivalent at 5 and 29 weeks of age for treated mice (t29weeks to t5 weeks, +3 sec) (Fig. 1A). The scores of AAV2-NaGlu vector-treated MPSIIIB mice differed from those of untreated mice (p = 0.031; Student's t test) but were equivalent to those of normal mice (p = 0.92). Eight of these mice were tested in the elevated plus-maze at 18 and 34 weeks of age (i.e., 12 and 28 weeks after vector injection). Treated mouse scores at 18 weeks of age were equally distant and not significantly different from those of normal and untreated MPSIIIB mice (Fig. 1B). Treated MPSIIIB mouse scores differed from those of untreated mice (p = 0.005) but were equivalent to those of normal mice (p = 0.75).
The activity of MPSIIIB mice treated with a single injection of AAV5-NaGlu vectors (n = 15) was recorded over 24 hr in home cages and compared with that of untreated MPSIIIB mice (n = 6) or MPSIIIB mice injected with a mock AAV5-green fluorescent protein (GFP) vector (n = 6). Animals were 18 weeks of age. Time spent in motion was equivalent for all animal groups in the light. Recording in the dark showed equivalent time spent in motion for normal and AAV5-NaGlu vector-treated MPSIIIB mice, whereas untreated MPSIIIB mice and AAV5-GFP vector-treated MPSIIIB mice were more active (Fig. 1C).
These results showed that behavioral scores assessing anxiety and activity levels were improved in MPSIIIB mice after a single injection of AAV2-NaGlu or AAV5-NaGlu vectors.
A single AAV vector injection allowed widespread NaGlu delivery in brains of MPSIIIB mice
Animals that underwent behavioral investigations were part of a study including 45 MPSIIIB mice. All animals received a single intrastriatal injection of AAV2-NaGlu or AAV5-NaGlu vectors at 6 weeks of age. Thirty mice received AAV2-NaGlu vectors and were killed at 8 (n = 3), 12 (n = 6), 22 (n = 6), 32 (n = 7), or 38 (n = 8) weeks of age, which corresponds to 2, 6, 16, 26, or 32 weeks after vector injection, respectively. Animals killed at 38 weeks of age underwent testing in the open field and the elevated plus-maze. Fifteen mice received AAV5-NaGlu. They all underwent testing in home cages at 18 weeks of age and were killed at 22 (n = 7) or 38 (n = 8) weeks of age, which correspond to 16 or 32 weeks after vector injection, respectively.
Figure 2 shows mean NaGlu activities in the injected hemisphere, the noninjected hemisphere, and a combination of the cerebellum and brainstem. All treated mice exhibited NaGlu activity in the injected hemisphere. Mean values were 50- to 500-fold higher than in normal mice (normal mice, 0.72 ± 0.12 U per milligram of protein, n = 18; AAV2-NaGlu, 37.3 ± 4.9 U per milligram of protein; AAV5-NaGlu, 346 ± 76 U per milligram of protein). NaGlu activity was detected in the noninjected hemisphere of 63% of AAV2-NaGlu and 100% of AAV5-NaGlu vector-treated mice. Mean values in the noninjected hemisphere of AAV2-NaGlu vector-treated mice were equivalent to normal levels (0.96 ± 0.42 U per milligram of protein). They were 30-fold higher in AAV5-NaGlu vector-treated mice (22 ± 7 U per milligram of protein). Activity was detected in the cerebellum and brainstem in only 10% of AAV2-NaGlu-treated mice (mean, 0.19 ± 0.14 U per milligram of protein), whereas most AAV5-NaGlu-treated mice (80%) showed higher NaGlu activity than normal mice (2.48 ± 1.07 vs 0.90 ± 0.19 U per milligram of protein). Activity in the injected hemisphere, the noninjected hemisphere, and the cerebellum and brainstem were significantly higher in AAV5-NaGlu-treated than in AAV2-NaGlu-treated mice (p ≤ 0.005; Student's t test). Neither the mean NaGlu activity nor the proportion of positive mice varied between 12 and 38 weeks of age in AAV2-NaGlu treated mice (p > 0.1; one-way ANOVA), showing that vectors persisted and maintained expression over time. In contrast, NaGlu activities increased between 22 and 38 weeks of age in the injected and noninjected hemispheres of AAV5-NaGlu vector-treated mice (p < 0.02; Student's t test).
Figure 2.

AAV2-NaGlu and AAV5-NaGlu vectors induced NaGlu activity in the MPSIIIB mouse brain. NaGlu activity was measured in tissue homogenates prepared from the brain hemispheres or the cerebellum and brainstem of normal mice (+/+), heterozygous MPSIIIB mice (+/-), untreated MPSIIIB mice (-/-), or MPSIIIB mice injected in the right putamen at 6 weeks of age with AAV2-NaGlu or AAV5-NaGlu vectors. The age of mice at analysis and the number of mice in each group are indicated. Data are means ± SEM of NaGlu activities measured in homogenates.
These results showed that a single injection of AAV-NaGlu vectors in the putamen led to robust, sustained, and widespread enzyme delivery in the MPSIIIB mouse brain. Enzyme activity at locations distant from the vector injection site was more often detected and higher with AAV5-NaGlu than with AAV2-NaGlu vectors.
Widespread NaGlu delivery resulted from both vector and enzyme spreading
To determine whether wide NaGlu delivery resulted from the dispersion of the vector or from the spreading of the enzyme, we measured the number of vector genomes and the amount of active NaGlu in 1-mm-thick serial coronal brain slices.
The number of vector genomes was determined by quantitative PCR in AAV2-NaGlu (n = 5) and AAV5-NaGlu (n = 8) vector-treated mice (Fig. 3). Vector genomes were concentrated at the injection site. Mean copy number was higher in AAV5-NaGlu-treated (0.90 ± 0.18 copies per 2N genomes) than in AAV2-NaGlu-treated (0.16 ± 0.05 copies per 2N genomes) mice (p < 0.000; Student's t test). Vectors spread in the injected hemisphere, transducing cells over distances of 5.0 ± 0.6 mm (AAV2-NaGlu) or 7.0 ± 0.5 mm (AAV5-NaGlu; rostrocaudal dimension, 12 mm). Only four mice treated with AAV5-NaGlu vectors showed vector genomes in the noninjected hemisphere.
Figure 3.

Analysis of AAV-NaGlu vector genome dispersion in mouse brains by quantitative PCR. MPSIIIB mice received a single injection of AAV2-NaGlu or AAV5-NaGlu vectors in the right putamen at 6 weeks of age and were analyzed at the indicated time. One-millimeter-thick coronal brain slices were prepared from the injected (IL) and noninjected (CL) hemispheres. Brain maps of AAV vector dispersion for each analyzed mouse are shown. Strips on brain maps schematically represent coronal slices (slice number is shown on the left). Values shown in the strips are numbers of AAV-NaGlu vector genomes measured in DNA extracted from the slice, which are expressed as copies per diploid cell genome. Zero indicates values of ≤0.1 copies per 2N genomes. Strips corresponding to slices in which vector genomes were detected are shadowed in gray according to the code shown at the bottom. Strips without values correspond to brain slices that were not analyzed. Mouse reference numbers are indicated.
NaGlu activity was measured in brain slice extracts from AAV2-NaGlu (n = 15) and AAV5-NaGlu (n = 15) vector-treated mice (Fig. 4). Highest activities were detected near the vector injection site (96 ± 11 and 661 ± 144 U per milligram of protein in AAV2-NaGlu and AAV5-NaGlu vector-treated mice, respectively). NaGlu activity was detected in all PCR-positive slices and was proportional to the number of vector genomes measured in the slice (n = 155; r = 0.713; p < 0.01; Pearson correlation). However, NaGlu activity extended far beyond PCR-positive regions, including to the noninjected hemisphere, encompassing the entire brain in eight AAV5-NaGlu vector-treated mice. The number of NaGlu-positive slices, and thus the spreading of the enzyme, was directly proportional to the number of vector genome copies in PCR-positive slices and to the mean NaGlu activity in these slices (n = 13; r = 0.648 and 0.804; p = 0.02 and 0.01, respectively; Pearson correlation). This result indicates that active enzyme synthesized by transduced cells in regions surrounding the injection site was released to distant regions where transduced cells were absent. Efficient gene transfer and high expression level favored widespread NaGlu delivery.
Figure 4.
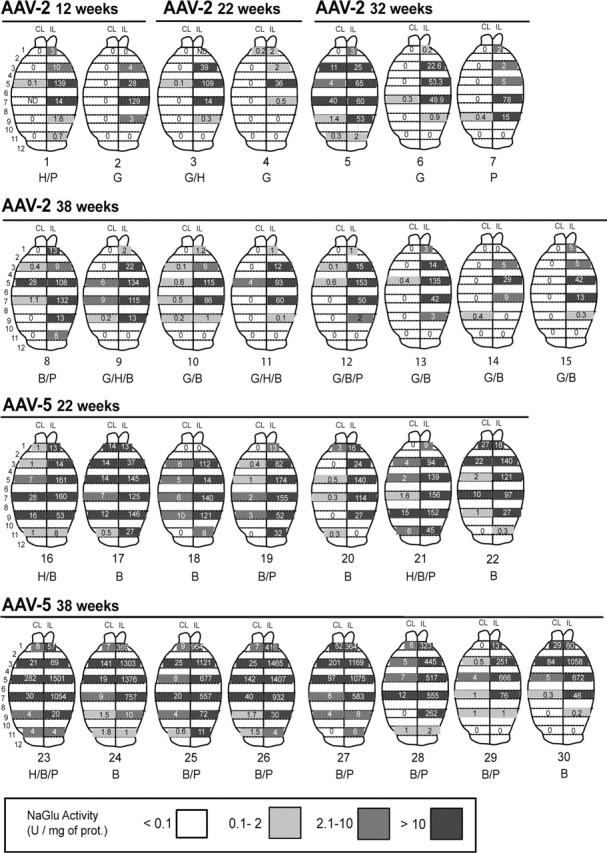
Widespread NaGlu activity in treated MPSIIIB mouse brains. MPSIIIB mice received a single injection of AAV2-NaGlu or AAV5-NaGlu vectors in the right putamen at 6 weeks of age and were analyzed at the indicated time. One-millimeter-thick coronal brain slices were prepared from the injected (IL) and noninjected (CL) hemispheres. Brain maps of NaGlu activities for each analyzed mouse are shown. Strips on brain maps schematically represent coronal slices (slice number is shown on the left). Values shown in the strips are NaGlu activities measured in the slice homogenate (in units per milligram of protein). Zero indicates that activity was <0.1 U per milligram of protein. Strips corresponding to slices in which NaGlu activity was detected are shadowed in gray according to the code shown at the bottom. Strips without values correspond to brain slices not used for NaGlu assay. Mouse reference numbers are indicated, as well as a letter code indicating other investigations performed on the same mouse: H, Histopathology; G, ganglioside assay (not all mice in which gangliosides were investigated are shown); B, behavioral testing (1 mouse tested in the open field is not shown here); P, quantitative PCR amplification of vector genomes.
Cryosections were prepared from a normal mouse brain and from slice 6 of the injected hemisphere of mouse 23 (AAV5-NaGlu; age, 38 weeks). Because very high NaGlu activities were detected in adjacent slices (1505 and 1053 U per milligram of protein in slices 5 and 7, respectively; see Fig. 4), we presumed that slice 6 also contained very high NaGlu activity. NaGlu was revealed by immunofluorescence using a rabbit anti-human NaGlu serum. This antibody did not detect signal in the normal mouse brain (data not shown). In contrast, many cells were stained with the anti-human NaGlu antibody in slice 6 of mouse 23 (Fig. 5A,D). Confocal microscopy showed that few NaGlu-stained organelles were also labeled with an antibody against the lysosomal marker Lamp1, although most signals did not colocalize (Fig. 5B,C). NaGlu-expressing cells coexpressed the neuronal marker NeuN (Fig. 5E,F). We did not observe cells coexpressing NaGlu and the astrocyte marker GFAP or the microglia marker CD11b (data not shown). These results indicate that NaGlu accumulated in neuron intracellular organelles, including lysosomes.
Figure 5.

NaGlu accumulated in neurons. Slice 6 from the injected hemisphere of mouse 23 was processed for cryosection (slice IL6, mouse 23 on Fig. 3). NaGlu was revealed by confocal fluorescence microscopy using a rabbit anti-human NaGlu serum (A, D). The anti-NaGlu (A) and lysosome-specific anti-Lamp1 (B) antibodies essentially stained different cytoplasmic organelles, although signals might occasionally colocalize (arrow in C). Cells showing cytoplasmic NaGlu signals (D) coexpressed the nuclear neuronal marker NeuN (E, F). Scale bar, 25 μm.
NaGlu delivery improved the activity of various lysosomal enzymes
A significant augmentation of total hexosaminidase, hexosaminidase A, and α-fucosidase activities was detected in untreated MPSIIIB mice compared with controls (p < 0.001; Student's t test) (Fig. 6), as described previously (Li et al., 1999). These enzyme activities were closer to normal values in treated mice. β-glucuronidase, β-galactosidase, and α-l-iduronidase activities were less affected and did not significantly differ between untreated and treated animals. These results indicated that the correction of the NaGlu defect tended to normalize the activity of several other lysosomal enzymes. To assess the effect of NaGlu overexpression on lysosomal functions, enzyme activities were assayed in samples containing NaGlu activities ranging from 1-5 to >1000 U per milligram of protein. Equivalent values were measured in the various samples (p = 0.97; one-way ANOVA), indicating that NaGlu overexpression was not deleterious to these lysosomal functions.
Figure 6.

NaGlu delivery was associated with improved lysosomal functions. The activity of various lysosomal enzymes was assayed in brain homogenates from normal mice (n = 5), untreated MPSIIIB mice (n = 5), or MPSIIIB mice treated with either AAV2-NaGlu or AAV5-NaGlu vector (n = 2 or 3). Homogenates were prepared from tissues containing various levels of NaGlu activity (1-5, 10-50, 50-100, or >1000 U per milligram of protein). Values were not significantly different among these four groups (Kruskal-Wallis one-way ANOVA). The data shown are ratios of enzyme activity relative to normal mice. Enzymes with values that were significantly improved in treated mice are indicated by an asterisk in the untreated mouse panel (significance: p < 0.05; Mann-Whitney U test). Tot. hexo, Total hexosaminidase; Hexo A, hexosaminidase A; α-fuco, α-fucosidase; β-glucu, β-glucuronidase; β-gal, β-galactosidase; IDUA, α-l-iduronidase.
NaGlu delivery reversed GM2 and GM3 ganglioside overload
The accumulation of GM2 and GM3 gangliosides in a 5-month-old MPSIIIB mouse brain has been reported previously (Li et al., 1999). We measured these compounds in brain lipid extracts at different time points in normal and MPSIIIB mice. A marked accumulation of GM3 (and accumulation of GM2 to a lesser extent) was observed in MPSIIIB mice as early as 5 weeks of age (Fig. 7). Values increased further with time. According to these data, it seems likely that GM2 and GM3 levels were already elevated in the brains of MPSIIIB mice at the age of treatment.
Figure 7.

NaGlu delivery was associated with a decreased accumulation of GM2 and GM3 gangliosides. Gangliosides were measured in total lipid extracts from the brains of normal mice and untreated MPSIIIB mice and from the injected or noninjected hemisphere of MPSIIIB mice that received a single injection of AAV2-NaGlu vectors in the putamen at 6 weeks of age (AAV2-NaGlu vector-treated MPSIIIB mice). The age of mice at analysis and the number of mice in each group are indicated. Data are expressed as molar percentages of total gangliosides according to NeuAc moiety numbers. Asterisks indicate mean values in treated mice that are significantly lower than those in untreated mice at 5 weeks of age (p ≤ 0.04; Mann-Whitney U test). Mean GM2 and GM3 values at 12 weeks of age in the injected hemisphere of treated mice were equivalent to mean values at the same age in normal mice.
The proportion of GM2 and GM3 gangliosides was measured in the injected and noninjected brain hemispheres of AAV2-NaGlu vector-treated mice at 8, 12, 22, 32, or 38 weeks of age, which corresponded to delays of 2, 6, 16, 26, or 32 weeks after vector injection (NaGlu activity in some of the analyzed mice is indicated in Fig. 4). At 8 and 12 weeks of age, mean GM2 and GM3 values in the injected hemisphere were lower than in untreated MPSIIIB mice at 5 weeks of age or at later time points (Fig. 7) and equivalent to those of age-matched normal mice. These results provided evidence for a reversion of the accumulation of GM2 and GM3 gangliosides in treated mouse brains, presumably as a consequence of NaGlu delivery. The reduction of GM2 and GM3 overload was less marked in the noninjected hemisphere, where mean NaGlu activity was lower. This observation suggests that the delivery of the missing enzyme interfered with the pathological process(es) that induced ganglioside accumulation.
Surprisingly, GM2 and GM3 levels tended to increase at later time points (22, 32, or 38 weeks of age), although mean NaGlu activities were equivalent or higher than at 8 or 12 weeks of age (Fig. 7). GM3 values in the injected hemispheres were still significantly reduced compared with age-matched untreated mice at 22 or 32 weeks of age. GM3 values in the noninjected hemisphere and GM2 values in both hemispheres reached untreated MPSIIIB mouse levels. These observations showed that NaGlu delivery improved but did not normalize ganglioside turnover in treated MPSIIIB mouse brains.
Lysosomal storage lesions disappeared or were drastically reduced in enzyme-positive regions of mouse brains
NaGlu deficiency induces cell vacuolation that is visible by light microscopy on semithin sections. We examined the brains of untreated MPSIIIB mice at 6 weeks of age (the age at which vector was injected in treated littermates) and at 3, 6, and 8 months of age. Widespread lesions were observed in the striatum, cortex, and cerebellum in 6-week-old animals (Fig. 8A-C). They consisted of white inclusions in microglia, fine granular white vacuoles in perivascular cells, and more polymorphic inclusions in neurons, astrocytes, and Purkinje cells. Inclusions were larger at 3 months of age (Fig. 8D-F). At 6 and 8 months of age, a high proportion of neurons and astrocytes contained large vacuoles. They were often juxtaposed to microglia, as described previously in MPS related neuropathology (Bhaumik et al., 1999). Few neurons showed picnotic nuclei (Fig. 8G-I).
Figure 8.
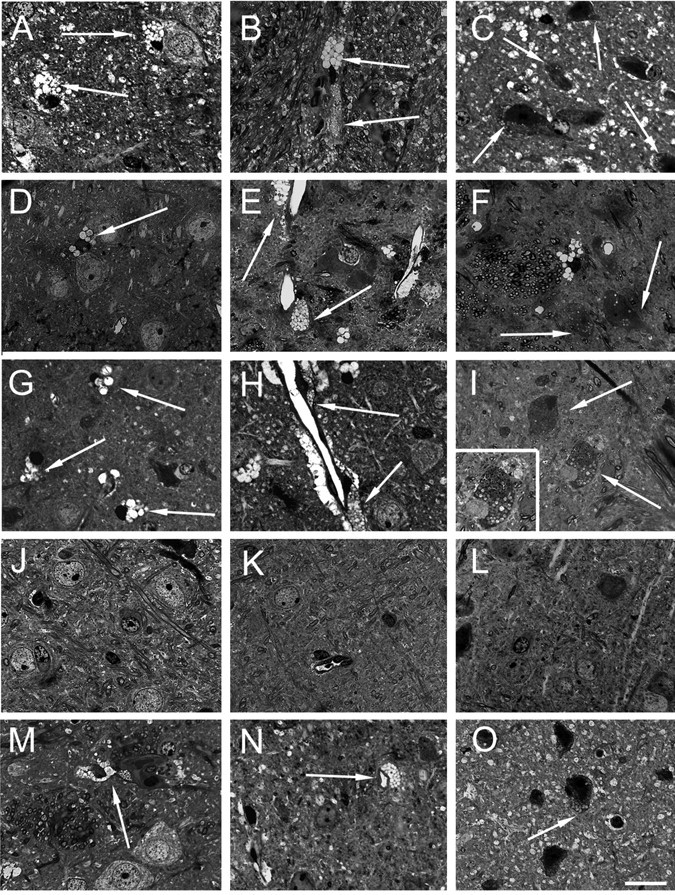
Disappearance or reduction of vacuolation in MPSIIIB mouse brain cells. Semithin sections stained with toluidine blue from brain slices of 6-week-old (A-C), 3-month-old (D-F), and 8-month-old (G-I) untreated MPSIIIB mice and from mouse 1 treated with AAV2-NaGlu vectors (J-L) and mouse 16 treated with AAV5-NaGlu vectors (M-O) are shown. Figure 4 indicates NaGlu activity in slices adjacent to those used for histopathology in these treated mice. Bright fine vacuoles in microglia (A) and larger and darker vacuoles in perivascular cells (B) were observed in the striatum of a 6-week-old untreated MPSIIIB mouse. Few neurons or astrocytes showed fine clear vacuoles in the parietal cortex (C). Large clear vacuoles were visible in microglia in the parietal cortex (D) and in microglia and perivascular cells in the putamen (E) of a 3-month-old untreated MPSIIIB mouse. Neurons or astrocytes with fine clear inclusions are visible in the putamen (F). Vacuoles in cortical microglia (G) and distension in perivascular cells near neurons or astrocytes with normal morphology (H) were observed in the putamen of an 8-month-old untreated mouse. Severely affected cells with abundant polymorphic vacuoles (I) as well as cells with picnotic nuclei (inset in I) were present at several other locations in the putamen of this mouse. Lysosomal distension was not observed in microglia (J), perivascular cells (K), or neurons or astrocytes (J-L) in the parietal cortex of the injected hemisphere of treated mouse 1 (slice 6 from the injected hemisphere). In contrast, residual lesions were observed in microglia (M) and perivascular cells (M, N) in the putamen of mouse 16 (slice 8 from the injected hemisphere). Vacuoles were also observed in neurons or astrocytes in the parietal cortex of the noninjected hemisphere (slice 8) of this mouse. Scale bar, 25 μm.
We examined treated animals killed at 12 (n = 1), 22 (n = 3), or 38 (n = 2) weeks of age for the presence of lesions. Semithin sections were prepared from 1-mm-thick coronal brain slices. Figure 4 indicates enzyme activity in slices adjacent to those in which histology was performed.
Lesions had disappeared in enzyme-positive regions in three mice. In the injected hemisphere of mouse 1 (AAV2-NaGlu; age, 12 weeks) (Fig. 8L) and mouse 3 (AAV2-NaGlu; age, 22 weeks) (Fig. 8J,K), regions adjacent to enzyme-positive slices were completely devoid of lesion and showed normal histology. However, lesions persisted in the noninjected hemisphere, where NaGlu activity was low or undetectable (≤0.1 U per milligram of protein). In mouse 21 (AAV5-NaGlu; age, 22 weeks), in which enzyme activity was detected at physiological or high levels in the entire brain, lesions had disappeared and histology was normal throughout the injected and noninjected hemisphere (data not shown). Histology was also normal in slice 6 of mouse 23 (data not shown). Because untreated animals showed lesions in the entire brain before the age of vector injection, these results indicated that NaGlu delivery presumably reversed pre-existing lesions and prevented the development of pathology.
Persisting lysosomal distensions were observed in three mice in regions in which enzyme activity was detected at physiological or supraphysiological levels (Fig. 8M-O). Mouse 16 (AAV5-NaGlu; age, 22 weeks), in which NaGlu activity was detected in the entire brain, showed normal histology in slices 2, 4, and 6 of the injected hemisphere, whereas lesions persisted in perivascular cells and microglia in slices 8 and 10. Few neurons or astrocytes showed large clear vacuoles in the olfactory bulb and in slices 4, 6, 8, and 10 of the noninjected hemisphere. Purkinje cells contained vacuoles. However, lesions were less severe than in age-matched untreated MPSIIIB mice (6 months of age). Mouse 9 (AAV2-NaGlu; age, 38 weeks) showed normal histology in slice 6 of the injected hemisphere, but vacuoles persisted in neurons, glia, and perivascular cells in slices 4 and 8 as well as in slice 8 of the noninjected hemisphere, although NaGlu activity was presumably present at these locations. Similar pictures were seen in slices 4, 6, and 8 of the injected hemisphere of mouse 11 (AAV2-NaGlu; age, 38 weeks). However, persisting lesions in treated mice killed at 38 weeks of age were less severe than in age-matched untreated controls (8 months of age). These results showed that NaGlu delivery improved but did not clear all lysosomal storage lesions in these mice.
Discussion
We show that AAV2 or AAV5 vector-mediated gene transfer in the brain has the capacity to treat neuropathology in the entire brain of the MPSIIIB mouse. Our study raises a number of issues that must be addressed to assess the clinical relevance of our results.
NaGlu delivery to the brain
A striking observation was the induction of very high NaGlu activity levels in the brains of treated mice principally, but not only, in regions surrounding the vector injection site. Fu et al. (2002) reported similar findings, although to a lesser extent. Comparable treatment with AAV2 or AAV5 vectors in MPSI mice did not result in comparable high enzyme activities, although gene expression was similarly controlled by the mPGK promoter (Desmaris et al., 2004). Confocal microscopy on cryosections of overexpressing brain tissue showed that NaGlu accumulated in intracellular organelles. Only a small fraction of the enzyme was correctly targeted to lysosomes in this region, as shown by colabeling with anti-NaGlu and anti-Lamp1 antibodies. Cells in which NaGlu accumulated were neurons, a previously known target cell of AAV5 vectors. The absence of NaGlu overexpression in astrocytes and microglia indicates either that AAV5 vectors did not penetrate these cells or that the PGK promoter was not active in these cells. Biochemical, histological, and behavioral improvements in animals overexpressing NaGlu in the brain suggest that accumulation was well tolerated by neurons. However, long-term consequences were not addressed in this study.
NaGlu synthesis was sustained over 38 weeks in the treated mouse brain. Activity levels and the number of positive mice did not decrease with time. Conversely, activity drastically increased with time in AAV5-NaGlu vector-treated mice. Thus, PGK promoter activity was sustained in target neurons. These cells persisted and continuously produced active enzyme over several months.
Global NaGlu delivery in the brains of MPSIIIB mice was remarkable and is in contrast to a previous report in which AAV2 vectors encoding NaGlu were used (Fu et al., 2002). Quantitative PCR analysis of vector genomes in serial brain slices showed that vector dispersion in tissues surrounding the injection site accounted for NaGlu expression in 30% to >80% of the injected hemisphere. AAV5-NaGlu vectors were also detected at restricted regions of the noninjected hemisphere in few mice. More efficient dispersion of AAV5 than AAV2 vectors in brain tissues (Davidson et al., 2000) resulted in wider delivery of the enzyme. However, vector dispersion did not account for the detection of NaGlu in 80-100% of the brain, as observed in 2 of 15 AAV2-NaGlu-treated and 12 of 15 AAV5-NaGlu-treated mice. NaGlu activity in brain slices devoid of AAV vector genome demonstrates that the protein itself spread in brain tissues. The extent of enzyme release beyond vector-positive regions was proportional to gene transfer and expression levels. Thus, in addition to more efficient dispersion, more efficient gene transfer and expression with AAV5-NaGlu than AAV2-NaGlu vectors also contributed to wider delivery of the enzyme. Long-distance NaGlu delivery may be processed via axonal transport, as suggested for another lysosomal hydrolase (Passini et al., 2002), although direct evidence for this mechanism requires additional investigation. Capture of NaGlu by microglia, astrocytes, and neurons was demonstrated by the disappearance of lysosomal storage lesions in areas devoid of vector genomes. We did not determine the extent to which capture occurred through the mannose-6-phosphate or other internalization pathways. However, disease correction indicates that captured enzyme was adequately targeted to lysosomes. Comparison with our previous observations in MPSI mice that underwent a similar treatment (Desmaris et al., 2004) suggests that the entire process of enzyme release, transport, and capture was less efficient for NaGlu than for α-l-iduronidase. Additional investigations will determine which step is altered.
Disease correction
Lysosomal enzyme defects are usually associated with compensatory changes in the activity of other lysosomal enzymes (Li et al., 1999). We show that the correction of NaGlu deficiency tended to normalize the activity level of several lysosomal enzymes, providing evidence for a global improvement of lysosomal functions. NaGlu overexpression was not deleterious to lysosomal functions, because improvement was equally observed in brain regions containing physiological or supraphysiological NaGlu activity levels.
MPSIIIB mice accumulate GM2 gangliosides (and, more markedly, GM3 gangliosides) in brain cells (Li et al., 1999). GM2 and GM3 accumulation in lysosomes and various cell membranes presumably contributes to neuropathology (Walkley et al., 2000). We show that accumulation occurred early in life and was presumably present in animals treated at 6 weeks of age. Gangliosides were measured in the brain of a subpopulation of AAV2-NaGlu vector-treated animals, in which NaGlu activities were representative of the entire population. NaGlu delivery induced a rapid reduction in GM2 and GM3 levels, which became equivalent to those of normal mice at 12 weeks of age. Reduction was observed in the injected hemisphere, where NaGlu activity levels were supraphysiological, indicating that NaGlu overexpression was compatible with the reduction in ganglioside overload. Reduction was also observed in extracts of the noninjected hemisphere with low or undetectable NaGlu activity.
Vacuolations characterizing lysosomal storage were observed in MPSIIIB mouse brain cells at 6 weeks of age and became more prominent with age, as described previously (Li et al., 1999). We examined the presence of pathology at multiple locations in the brains of six treated mice. Analyzed fragments were taken from brain slices in which mean NaGlu activity could be extrapolated from measurements made in adjacent slices. A complete disappearance of pathology or minimal lesions was observed in areas in which NaGlu was present at physiological or supraphysiological levels, whereas pathology was as severe as that seen in age-matched untreated MPSIIIB mice in regions containing low or no NaGlu activity. This observation is in contrast to our previous findings in MPSI mice, where histology was normalized in regions in which enzyme activity was below detection threshold (Desmaris et al., 2004). Thus, despite higher expression levels, NaGlu did not spread as efficiently as α-l-iduronidase in brain tissues.
A puzzling observation, also in contrast to our previous results in MPSI mice, was the persistence or the reappearance of ganglioside overload and vacuolation in some mice when high NaGlu activity was detected in tissue extracts in these regions. The observation suggests either that cells did not capture enzyme efficiently or that enzyme capture did not rescue altered functions. Microglia activation and astrocytosis are hallmarks and possible determinants of pathophysiology in MPSIIIB (Li et al., 2002; Ohmi et al., 2003). A plausible hypothesis is that abnormal HS would trigger a specific inflammatory process that would further become self-induced, either by altering enzyme uptake and HS degradation or by perpetuating pathology notwithstanding enzyme delivery and HS degradation. An important issue in that respect would be the assessment of microglia activation and astrocytosis at locations where pathology reversed or persisted.
Untreated MPSIIIB mice appeared hyperactive in the open field and, more convincingly, during a 24 hr recording of their movements in home cages. This observation is in contrast to previous findings by Li et al. (1999) showing a decreased activity of MPSIIIB mice in the open field. This discrepancy may result from a higher number of backcrossing to the C57BL/6 background in our colony, differences in mouse age and sex, and/or differences in apparatus and methodology. Using the elevated plus-maze, we noticed a reduced propensity for anxiety in MPSIIIB mice compared with normal controls. This feature may reflect impaired behavioral inhibition (Avale et al., 2004). Hyperactivity and impaired behavioral inhibition are relevant to clinical manifestations in affected children. They were not observed in treated animals. In AAV5-NaGlu vector-treated mice, normal activity in home cages at 18 weeks of age was associated with high and widespread NaGlu activity in the brain at 22 or 38 weeks of age. AAV2-NaGlu vector-treated mice showing normal behavior in the elevated plus-maze at 18 and 34 weeks of age also showed high NaGlu activity at 38 weeks of age, although the enzyme was primarily located in the injected hemisphere. Consistent with previous reports, this observation suggests that behavioral improvement in the elevated plus-maze may not require enzyme delivery to brain hemispheres (Graeff et al., 1993; File and Gonzalez, 1996).
Conclusion
A treatment for MPSIIIB is highly desirable. Enzyme replacement therapy faces challenging issues in this disease (Yu et al., 2000). The present report provides evidence that a single injection of AAV vectors coding for NaGlu has the potential to completely alleviate the disease in the entire brain of the MPSIIIB mouse. However, biochemical, histological, and behavioral improvements were obtained through the expression of highly supraphysiological levels of the enzyme. Although deleterious effects linked to NaGlu overexpression were not demonstrated, it is likely that similar conditions would raise serious concerns in humans. A control of the gene expression level aimed at delivering minimal effective amounts of the missing enzyme would therefore be desirable. Data from some animals suggest suboptimal release capture or transport of the enzyme, presumably synthesized in mouse neurons. This limitation may be reminiscent of that encountered when NaGlu was produced in CHO cells (Zhao and Neufeld, 2000; Weber et al., 2001) and certainly requires improvement. However, it is remarkable in this context that NaGlu produced at a single site by AAV vector-transduced neurons could spread and reverse pathology in the entire brain of some MPSIIIB mice.
Footnotes
This work was supported by grants from Vaincre les Maladies Lysosomales, the Groupement d'Intérêt Scientifique Maladies Rares, and the Association Française contre la Myopathie. We thank C. Bronnert for excellent technical assistance. We are grateful to Prof. E. Neufeld for the generous gift of the MPSIIIB mice and the anti-NaGlu antibody. AAV vector stocks were generously prepared by the vector core of the Gene Vector Production Network (Nantes, France).
Correspondence should be addressed to Dr. Jean Michel Heard, Unité Rétrovirus et Transfert Génétique, Département Neuroscience, Institut Pasteur, 28 rue du Roux, 75015 Paris, France. E-mail: jmheard@pasteur.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410229-11$15.00/0
A.C. and N.D. contributed equally to this work.
References
- Adra CN, Boer PH, McBurney M (1987) Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene 60: 65-74. [DOI] [PubMed] [Google Scholar]
- Avale ME, Falzone TL, Gelman DM, Low MJ, Grandy DK, Rubinstein M (2004) The dopamine D4 receptor is essential for hyperactivity and impaired behavioral inhibition in a mouse model of attention deficit/hyperactivity disorder. Mol Psychiatry 9: 718-726. [DOI] [PubMed] [Google Scholar]
- Bhaumik M, Muller VJ, Rozaklis T, Johnson L, Dobrenis K, Bhattacharyya R, Wurzelmann S, Finamore P, Hopwood JJ, Walkley SU, Stanley P (1999) A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 9: 1389-1396. [DOI] [PubMed] [Google Scholar]
- Bosch A, Heard JM (2003) Gene therapy for mucopolysaccharidosis. Int Rev Neurobiol 55: 271-296. [DOI] [PubMed] [Google Scholar]
- Brument N, Morenweiser R, Blouin V, Toublanc E, Raimbaud I, Chérel Y, Folliot S, Gaden F, Boulanger P, Kroner-Lux G, Moullier P, Rolling F, Salvetti A (2002) A versatile and scalable two-step ion-exchange chromatography process for the purification of recombinant adeno-associated virus serotypes-2 and -5. Mol Ther 6: 678-686. [DOI] [PubMed] [Google Scholar]
- Chiorini JA, Kim F, Yang L, Kotin RM (1999) Cloning and characterization of adeno-associated virus type 5. J Virol 73: 1309-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Dersken TA, Zabner J, Ghodsi A, Chiorini JA (2000) Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA 97: 3428-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmaris N, Verot L, Puech JP, Caillaud C, Vanier MT, Heard JM (2004) Prevention of neuropathology in the mouse model of Hurler syndrome by AAV vector-mediated gene therapy. Ann Neurol 56: 68-76. [DOI] [PubMed] [Google Scholar]
- File SE, Gonzalez LE (1996) Anxiolytic effects in the plus-maze of 5-HT1A-receptor ligands in dorsal raphe and ventral hippocampus. Pharmacol Biochem Behav 54: 123-128. [DOI] [PubMed] [Google Scholar]
- Fu H, Samulski RJ, McCown TJ, Picornell YJ, Fletcher D, Muenzer J (2002) Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIb mouse model by adeno-associated virus-mediated gene delivery. Mol Ther 5: 42-49. [DOI] [PubMed] [Google Scholar]
- Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H (1996) Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet 5: 711-725. [DOI] [PubMed] [Google Scholar]
- Gehler J, Cantz M, Tolksdorf M, Spranger J, Gilbert E, Drube H (1974) Mucopolysaccharidosis. VII. β-glucuronidase deficiency. Humangenetik 23: 149-158. [DOI] [PubMed] [Google Scholar]
- Gondre-Lewis MC, McGlynn R, Walkley SU (2003) Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr Biol 13: 1324-1329. [DOI] [PubMed] [Google Scholar]
- Goodwin E, Rottman F (1992) The 3′ flanking sequence of the bovine growth hormone gene contains novel elements required for efficient and accurate polyadenylation. J Biol Chem 267: 16330-16334. [PubMed] [Google Scholar]
- Graeff FG, Silveira MC, Nogueira RL, Audi EA, Oliveira RM (1993) Role of the amygdala and periaqueductal gray in anxiety and panic. Behav Brain Res 58: 123-131. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kern A, Rittner K, Kleinschmidt J (1998) Novel tools for production and purification of recombinant adeno-associated virus vectors. Hum Gene Ther 9: 2745-2760. [DOI] [PubMed] [Google Scholar]
- Haust MD, Gordon BA (1986) Ultrastructural and biochemical aspects of the Sanfilippo syndrome type III. Connect Tissue Res 15: 57-64. [DOI] [PubMed] [Google Scholar]
- Hogg S (1996) A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol Biochem Behav 54: 21-30. [DOI] [PubMed] [Google Scholar]
- Huang JQ, Trasler JM, Igdoura S, Michaud J, Hanai N, Gravel RA (1997) Apoptotic cell death in mouse models of GM2 gangliosidosis and observation on human Tay-Sachs and Sandhoff diseases. Hum Mol Genet 6: 1879-1885. [DOI] [PubMed] [Google Scholar]
- Inui K, Yutaka T, Okada S, Yabuuchi H, Wenger DA, Desnick RJ (1985) Hexosaminidase A activity in skin fibroblasts from various types of GM2 gangliosidosis using a fluorogenic sulphated substrate. J Inherit Metab Dis 8: 149-150. [DOI] [PubMed] [Google Scholar]
- Kyrklund T (1987) Two procedures to remove polar contaminants from a crude brain lipid extract by using prepacked reverse-phase columns. Lipids 22: 274-277. [DOI] [PubMed] [Google Scholar]
- Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, Neufeld EF (1999) Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding α-N-acetylglucosaminidase. Proc Natl Acad Sci USA 96: 14505-14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Zhoa HZ, Neufeld EF, Cai Y, Gomez-Pinilla S (2002) Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J Neurosci Res 69: 30-38. [DOI] [PubMed] [Google Scholar]
- Marsh J, Fensom AH (1985) 4-Methylumbelliferyl α-N-acetylglucosaminidase activity for diagnosis of Sanfilippo B disease. Clin Genet 27: 258-262. [DOI] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel K, Ryazantsev S, Li HH, Neufeld EF (2003) Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA 100: 1902-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Lee EB, Heuer GG, Wolfe JH (2002) Distribution of a lysosomal enzyme in the adult brain by axonal transport and by cells of the rostral migratory stream. J Neurosci 22: 6437-6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelled D, Lloyd-Evans E, Riebeling C, Jeyakumar M, Platt FM, Futerman AH (2003) Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J Biol Chem 278: 29496-29501. [DOI] [PubMed] [Google Scholar]
- Peters SP, Coyle P, Glew RH (1976) Differentiation of β-glucocerebrosidase from β-glucosidase in human tissues using sodium taurocholate. Arch Biochem Biophys 175: 569-582. [DOI] [PubMed] [Google Scholar]
- Picard L, Simmons G, Power CA, Meyer A, Weiss RA, Clapham PR (1997) Multiple extracellular domains of CCR-5 contribute to HIV-1 entry and fusion. J Virol 71: 5003-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss B, von Bohlen und Halbach O (2003) Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res 313: 139-157. [DOI] [PubMed] [Google Scholar]
- Saifer A, Perle G (1974) Automated determination of serum hexosaminidase A by pH inactivation for detection of Tay-Sachs disease heterozygotes. Clin Chem 20: 538-543. [PubMed] [Google Scholar]
- Salafsky IS, Nadler HL (1973) A fluorometric assay of α-glucosidase and its application in the study of Pompe's disease. J Lab Clin Med 81: 450-454. [PubMed] [Google Scholar]
- Salvetti A, Orève S, Chadeuf G, Favre D, Champion-Arnaud P, David-Ameline J, Moullier P (1998) Factors influencing recombinant adeno-associated virus production. Hum Gene Ther 9: 695-706. [DOI] [PubMed] [Google Scholar]
- Samulski RJ, Chang LS, Shenk T (1987) A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J Virol 61: 3096-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinus-Blanc C (1996) Animal spatial cognition: behavioral and neural approaches. Singapore: World Scientific.
- Turnbull J, Powell A, Guimond S (2001) Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol 11: 75-81. [DOI] [PubMed] [Google Scholar]
- Ullrich K, Basner R, Gieselmann V, Von Figura K (1979) Recognition of human urine α-N-acetylglucosaminidase by rat hepatocytes. Involvement of receptors specific for galactose, mannose 6-phosphate and mannose. Biochem J 180: 413-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoof F, Hers HG (1968) The abnormalities of lysosomal enzymes in mucopolysaccharidoses. Eur J Biochem 7: 34-44. [DOI] [PubMed] [Google Scholar]
- Vasar E, Peuranen E, Oopik T, Harro J, Mannisto PT (1993) Ondansetron, an antagonist of 5-HT3 receptors, antagonizes the anti-exploratory effect of caerulein, an agonist of CCK receptors, in the elevated plus-maze. Psychopharmacology (Berl) 110: 213-218. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Haskins ME, Shull R (1988) Alterations in neuron morphology in mucopolysaccharidosis type I. Acta Neuropathol 75: 611-620. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Baker HJ, Rattazzi MC, Haskins ME, Wu JY (1991) Neuroaxonal dystrophy in neuronal storage disorders: evidence for major GABAergic neuron involvement. J Neurol Sci 104: 1-8. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Zervas M, Wiseman S (2000) Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb Cortex 10: 1028-1037. [DOI] [PubMed] [Google Scholar]
- Weber B, Hopwood JJ, Yogalingam G (2001) Expression and characterization of human recombinant α-N-acetylglucosaminidase. Protein Expr Purif 21: 251-259. [DOI] [PubMed] [Google Scholar]
- Wood S (1975) A sensitive fluorometric assay for α-KL-fucosidase. Clin Chim Acta 58: 251-256. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ (1998) Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72: 2243-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EP (1992) Prenatal diagnosis of Hurler disease by analysis of α-iduronidase in chorionic villi. J Inherit Metab Dis 15: 224-230. [DOI] [PubMed] [Google Scholar]
- Yu WH, Zhao KW, Ryazantsev S, Rozengurt N, Neufeld EF (2000) Short-term enzyme replacement in the murine model of Sanfilippo syndrome type B. Mol Genet Metab 71: 206-214. [DOI] [PubMed] [Google Scholar]
- Zhao HG, Li HH, Schidthen A, Neufeld EF (1996) The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci USA 93: 6101-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao KW, Neufeld EF (2000) Purification and characterization of recombinant human α-N-acetylglucosaminidase secreted by Chinese hamster ovary cells. Protein Expr Purif 19: 202-211. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R (2001) Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci USA 98: 1982-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N (1999) Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973-985. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Donello JE, Trono D, Hope TJ (1999) Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol 73: 2886-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]