

Abstract
To examine the role of the p53 homolog p73 in brain development, we studied p73-/-, p73+/-, E2F1-/-, and reeler mutant mice. p73 in developing brain is expressed in Cajal-Retzius (CR) cells, the cortical hem, and the choroid plexus. p73-expressing CR cells are lost in p73-/- embryos, although Reelin is faintly expressed in the marginal zone. Ectopic neurons in the p73-/- preplate and cortical hem at embryonic day 12 implicate p73 in the early developmental program of the cortex; however, preplate partition and early cortical plate formation are not disturbed. Postnatal p73-/- mice show a mild hypoplasia of the rostral cortex and a severely disrupted architecture of the posterior telencephalon. In the developing p73-/- hippocampus, the most striking abnormality is the absence of the hippocampal fissure, suggesting a role of p73 in cortical folding. p73+/- mice have a less severe cortical phenotype; they display a dorsal shift of the entorhinal cortex and a reduced size of occipital and posterior temporal areas, which acquire entorhinal-like features such as Reelin-positive cells in layer II. CR cells appear unaffected by heterozygosity. We relate the malformations of the posterior pole in p73 mutant mice to alterations of p73 expression in the cortical hem and suggest that p73 forms part of an early signaling network that controls neocortical and archicortical regionalization. In mice deficient for the transcription factor E2F1, a main activator of the TAp73 (transactivating p73) isoform, we find a defect of the caudal cortical architecture resembling the p73+/- phenotype along with reduced TAp73 protein levels and propose that an E2F1-TAp73 dependent pathway is involved in cortical patterning.
Keywords: cortical hem, hippocampus, entorhinal cortex, E2F1, TAp73, Reelin
Introduction
p73 is a homolog of the tumor-suppressor gene p53 (Kaghad et al., 1997). In the brain, it is expressed in Cajal-Retzius (CR) cells, the ependyma, and the choroid plexus (Yang et al., 2000; Meyer et al., 2002). The most striking brain abnormalities of p73 knockout mice are loss of CR cells, hydrocephalus, and dysgenesis of the dentate gyrus (Yang et al., 2000). Neurons in various p73-/- brain structures undergo extensive postnatal apoptosis (Pozniak et al., 2002).
p73 is a complex protein that is expressed as transactivating (TA) forms that induce growth arrest and apoptosis and as DeltaN (DNp73) isoforms that lack the transactivating N terminus and are able to inhibit TAp73 and p53 transactivation activities (Yang et al., 2000; Grob et al., 2001; Kartasheva et al., 2002). Expression of these isoforms is controlled by alternative promoters (Jost et al., 1997; Yang et al., 2000). DNp73 has antiapoptotic functions (Pozniak et al., 2000) and is expressed in CR cells, whereas TAp73 is present only in a small percentage of p73-positive cells (Yang and McKeon, 2000; Yang et al., 2000), raising the question of whether DNp73 and TAp73 play different roles in cortical development. Additional diversity is added by differentially spliced C-terminal variants, of which p73α is the predominant form in the brain (Yang et al., 2000; Pozniak et al., 2002).
CR cells are early-born neurons in the cortical marginal zone that secrete high levels of Reelin, an extracellular matrix protein necessary for radial migration (D'Arcangelo et al., 1995). It has been proposed that Reelin is required for the process of “preplate partition,” when the preplate is split into marginal zone and subplate by the developing cortical plate (Marin-Padilla, 1998; Tissir and Goffinet, 2003). This step is disrupted in reeler mice, in which undivided preplate derivatives form the “superplate” on top of an inverted cortical plate (Caviness 1976, 1982). Because CR cells are the main source of Reelin in the embryonic cortex, we ask here whether their absence disturbs preplate partition and the subsequent architectonic development of the p73-/- neocortex in a reeler-like manner.
We reported previously that an important subset of CR cells derive from the cortical hem (Meyer et al., 2002). In the p73-deficient mice, we further examine the possible relationships between CR cells, p73 expression, and the cortical hem, which is known as a signaling center important for archicortical and neocortical development (Grove et al., 1998; Grove and Fukuchi-Shimogori, 2003).
In the course of this study, we observed substantial abnormalities of cortical development also in p73+/- mice. In an attempt to correlate alterations of p73 expression with specific brain defects, we compared p73+/- and E2F1 knock-out mice. E2F1 belongs to a family of transcription factors that transactivate cell-cycle regulators and proapoptotic genes (Field et al., 1996); it is one of the main activators of the TAp73 promoter (Irwin et al., 2000; Stiewe and Putzer, 2000; Zaika et al., 2001). We report here that E2F1 homozygous and p73 heterozygous mice present similar defects of cortical patterning and that TAp73 protein levels are significantly reduced in the E2F1-/- brain. Our findings suggest that an E2F1-TAp73-dependent pathway regulates important aspects of cortical development.
Materials and Methods
We examined p73-/- and p73+/- mutant mice and wild-type (WT) littermates (Yang et al., 2000). A total of 63 animals from heterozygous intercrosses (39 prenatal and 26 postnatal animals) were studied at embryonic day 10 (E10), E11, E12, E13, E15, E15.5, and E18, postnatal day 2 (P2), P3, P4, P7, P10, and P12, and adult. Of the 39 embryos, 13 were p73 homozygotes, 15 were heterozygotes, and 11 were WT; genotyping was performed as by Yang et al. (2000). In addition, we studied C57BL/6 mouse brains at various prenatal (E13, E14, E16, and E18) and postnatal ages. The E10-E15.5 embryos were fixed in 4% paraformaldehyde, postfixed in Bouin's fixative, and embedded in paraffin according to standard procedures. Sections were cut serially at 7 μm and processed for immunohistochemistry (IHC) and in situ hybridization (ISH). The E18 and postnatal p73 mutant mice were frozen and stored at -80°C. Seven of these brains were cut into 20-μm-thick serial sections on a cryostat and also stored at -80°C. For IHC, the frozen sections were air dried, fixed in Bouin's fixative, washed, and then processed as described previously (Meyer et al., 2002). The remaining 19 brains were removed from the freezer and quickly immersed in Bouin's fixative. After fixation for 2 d, they were embedded in paraffin, cut at 10 μm, and processed for IHC. ISH was done as described previously (Yang et al., 2000; Meyer et al., 2002). reeler-Orleans (Relnrl-Orl) mice were studied at E16 and P20. In these mice, a C-terminal truncated protein is produced but not secreted and can be detected using antibodies (Abs) G10 and 142 (de Bergeyck et al., 1997, 1998). E2F1-/- mice were obtained from The Jackson Laboratory (Bar Harbor, ME). We studied P1, P2, and adult animals. Reeler and E2F1-/- mice were fixed in Bouin's fixative, embedded in paraffin, and cut at 10 μm.
For IHC, sections were incubated in the primary antibodies overnight in a humid chamber. After rinsing, they were incubated in the corresponding biotinylated secondary antibodies (rabbit anti-mouse IgG or goat anti-rabbit IgG; Dako, Glostrup, Denmark), diluted at 1:200 in Tris-buffered saline (TBS), washed, and incubated in ABC (Dako). Bound peroxidase was revealed using 0.05% 3,3′-diaminobenzidine (Sigma, St. Louis, MO), 0.04% ammonium nickel (II) sulfate, and 0.03% hydrogen peroxide in TBS, pH 7.6. The sections were dehydrated, cleared, and covered with Eukitt (O. Kindler, Freiburg, Germany).
The following primary antibodies were used: rabbit polyclonal anti-p73 antibody (1:150) (Kaghad et al., 1997), mouse monoclonal anti-p73 Ab-2 (1:150) (NeoMarkers, Fremont, CA), mouse monoclonal anti-Reelin antibodies G10 and 142 (1:400) (de Bergeyck et al., 1998), rabbit polyclonal antibody against calretinin (1:2500) (Swant, Bellinzona, Switzerland), and mouse monoclonal antibody against GFAP (1: 250) (NeoMarkers).
Immunoblotting. For total protein extraction, the posterior telencephalons of E2F1-/- and wild-type brains at P2 were homogenized with lysis buffer (62.5 mm Tris-HCl, 1% SDS, and 10% glycerol, pH 6.8). Protein quantification was done by bicinchoninic acid assay (Smith et al., 1985). Five percent mercaptoethanol and 0.001% bromophenol blue were then added, and samples were boiled at 95°C for 5 min. Equal amounts (40 pg) of each sample were electrophoresed on 10% SDS-PAGE and transferred to Hybond-P membranes (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Membranes were preincubated with 5% blotting-grade blocker nonfat dry milk (Bio-Rad Laboratories, Hercules, CA) in TBS with 0.1% Tween 20 (TBS-T) at room temperature for 1 hr and washed in TBS-T. Membranes were incubated with a rabbit polyclonal anti-p73 antibody raised against amino acids 1-15 of human p73 (p73 Ab-5; NeoMarkers) and with a rabbit polyclonal anti-p73 that detects amino acids 427-636 (Kaghad et al., 1997). Membrane incubations with both antibodies (1:500 in TBS-T with 5% BSA) were performed overnight at 4°C. Antibody labeling was developed by incubation with an HRP-conjugated goat anti-rabbit (1:20,000) and visualized with the ECL plus Western Blotting Detection System kit (Amersham Biosciences). Membranes were washed in TBS-T and reprobed with monoclonal mouse anti-tubulin antibody (T 9026; Sigma) (1:100,000 in TBS-T with 5% BSA) for 1 hr, followed by incubation with an HRP-conjugated goat anti-mouse secondary antibody (1:10,000). Specific bands were visualized with the ECL kit, scanned with the GS-800 Calibrated Densitometer, and analyzed with the image analysis program Quantity One (Bio-Rad). For immunosignal quantification, band intensities were normalized to basal values obtained in wild-type brains. Data were obtained in optic density per square millimeter and calculated as the percentage of immunostaining values in E2F1 knock-out brains relative to control brains.
Results
Two distinct brain phenotypes in p73-/- and p73+/- mutant mice
On the basis of cytoarchitecture and Reelin expression, we distinguished two main p73 phenotypes (Fig. 1). p73-/- mice showed architectonic abnormalities of practically all structures of the limbic telencephalon, including hippocampal formation, amygdala, retrosplenial, and entorhinal cortex (Fig. 1C, D). In p73+/- mice, the alterations were more subtle and affected mainly the entorhinal cortex, the posterior cortical nucleus of the amygdala, and the caudal neocortex (Fig. 1B). In both p73-/- and p73+/- mice, structures at the posterior pole of the telencephalon were more severely affected than rostral ones. The severe size reduction of the caudal cortex in the p73-/- brain is evident in the parasagittal section of Figure 2B. In p73+/- mice, a similar reduction of the posterior pole was first recognized at E18, when the ventricular/subventricular zone extended farther posteriorly in the WT compared with the p73+/- brain (Fig. 2C-F).
Figure 1.
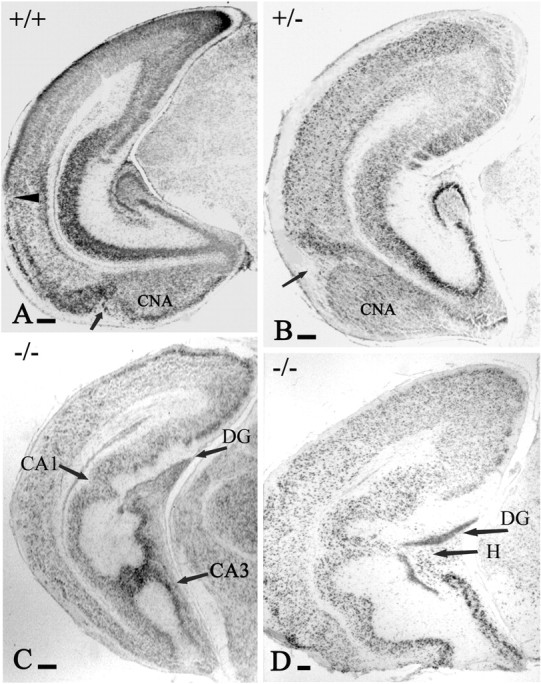
The p73+/- and p73-/- brain phenotypes. A, P3 WT mice. B, P3 +/- mice. C, P3 -/- mice. D, P12 -/- mice, Nissl stain. B, The p73+/- brain displays abnormal shape and cytoarchitecture of caudal cortical areas and posterior cortical nucleus of the amygdala (CNA). A, B, Arrows point to the periamigdaloid fissure, which is shifted dorsally; the arrowhead in A points to the rhinal fissure, which is unrecognizable in B. C, D, The variability in the malformations of hippocampal architecture in p73-/- mice. All of the sections are at comparable levels. DG, Dentate gyrus; H, hilus. Scale bars, 200 μm.
Figure 2.
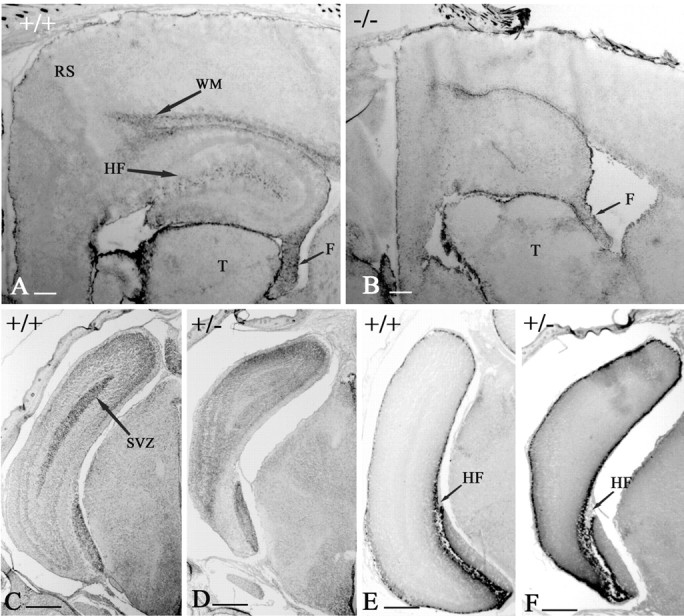
The posterior pole of the telencephalon. A, P7 WT mice. B, P7 -/- mice, parasagittal sections immunostained with GFAP. Cortical areas at the posterior pole are reduced in size in the p73-/- mouse. The white matter (WM) area is also smaller, and the hippocampal fissure (HF) is missing. F, Fimbria; RS, retrosplenial cortex; T, thalamus. C-F, The posterior pole of p73 heterozygous (D, F) and WT (C, E) mice at E18 in coronal Nissl (C, D) and Reelin (E, F) stained sections. Sections are at the same level to show the size reduction of the caudal telencephalon and the dorsal shift of entorhinal cortex. The proliferative subventricular zone (SVZ) is still visible in C (WT) but not in D (p73+/-). E, F, The number and distribution of Reelin-IR CR cells in the hippocampal fissure and in the cortical marginal zone are similar in WT and p73+/- mice. Scale bars, 200 μm.
The postnatal p73-/- telencephalon
p73-/- brains showed the following distinctive features: absence of Cajal-Retzius cells, generalized cortical hypoplasia and caudal dysplasia, severe hippocampal malformations, and abnormal clusters of Reelin-positive stellate cells in layer II of the entorhinal cortex.
Even in the most severe cases, rostral cortical areas were relatively mildly affected (Fig. 3A, B). Lamination and cytoarchitecture were basically preserved, although the cortex was narrower and the white matter and corpus callosum were reduced in size. In contrast, the posterior pole of the hemisphere, including primary and secondary visual areas, the auditory association cortex, and the retrosplenial cortex, was profoundly altered (Fig. 3C). In most cases, these areas had lost the normal lamination pattern and displayed large irregularly arranged cells, many of which were Reelin immunoreactive (IR) (Fig. 3D).
Figure 3.
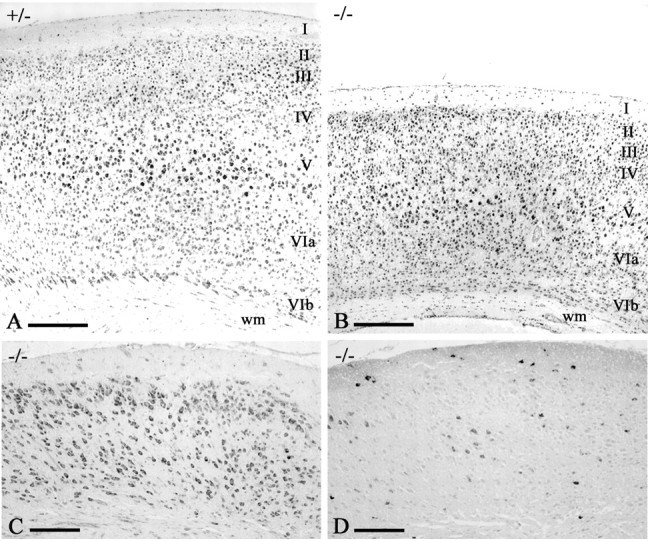
The cortical phenotype of p73-/- mice. A, B, P12 mice, Nissl stain. The frontal (motor) cortex of p73+/- mice (A) displays normal cytoarchitecture. In the p73-/- motor cortex (B), basic lamination and cytoarchitecture are preserved, but the cortex is smaller and white matter (wm) is reduced. C, D, p73-/- (same case as in B), stained with Nissl (C) and Reelin (D). In occipital cortex, lamination is lost and cell density is reduced. Irregular clusters of Reelin-IR neurons are present in the profoundly altered cortex. Scale bars: A, B, 200 μm; C, D, 100 μm.
The hippocampus presented a complex set of anatomical abnormalities in addition to the absence of the infrapyramidal dentate blade reported by Yang et al. (2000). The hippocampal fissure, which normally separates CA1 and CA2 from the dentate gyrus, was missing. The pyramidal cell layer of CA1 and CA2 was deeply folded, and abnormal cell bridges extended between CA1 and CA3 (Fig. 1C) or CA1 and the hilus (Fig. 1D).
p73 in the preplate
In the early preplate (E10-E11), expression of p73 in the WT telencephalon was sparse and mostly close to the roof plate (data not shown). Intense p73 positivity appeared at E12-E12.5 throughout the cortical hem and in CR cells (Meyer et al., 2002). At later embryonic stages, p73 disappeared from the hem and was confined to CR cells and the choroid plexus. ISH, using probes directed against exon 3 (under the control of the first promoter and specific for TAp73 expression) and for exon 3bis (controlled by the second promoter and specific for DNp73 expression), revealed a differential distribution of the respective transcripts. In E12 WT brains, transcripts of exon 3 were restricted to the cortical hem (Fig. 4A, arrowhead), whereas exon 3bis was expressed in the cortical hem and in CR cells all along the cortical wall (Fig. 4D). In E12 p73-/- mice, exon 3 transcripts were still present in the cortical hem (Fig. 4B, arrowhead), but exon 3bis transcripts were absent (data not shown), indicating that there was no functional TAp73 protein to activate the DNp73 promoter. These findings demonstrate a difference in the expression of p73 in the hem and in CR cells and show that CR cell survival depends on DNp73 expression because they are absent in the p73-/- brain.
Figure 4.

The preplate stage. A, B, D, E12 mice, ISH using specific probes for exon 3 (TAp73) in WT (A), exon 3 in p73-/- (B), and exon 3bis (DNp73) in WT (D) mice. TAp73 transcripts are expressed in the cortical hem of WT and p73-/- mice, whereas DNp73 transcripts are present in the WT cortical hem and in CR cells of the cortical preplate. C, E, Nissl-stained sections of a control brain at corresponding levels; C for A and B and E for D. The cortical hem is indicated by arrowheads. F, G, E12 mice, calretinin IHC, horizontal sections. F, WT mice. G, p73-/- embryo. The mutant preplate presents increased numbers of calretinin-IR pioneer neurons compared with the WT preplate. In addition, there are ectopic calretinin-IR neurons in the cortical hem (arrowheads) that are absent in the WT embryo. Arrows indicate the orientation; R, rostral; L, lateral; A, amygdala; E, eye; GE, ganglionic eminence; T, thalamus. Scale bars: A-E, 200 μm; F, G, 100 μm.
The earliest morphological differences between WT and p73-/- brains were detected at E12. In the mutant brains, ectopic calretinin-IR cells appeared in the hem and the posteromedial preplate (Fig. 4G), whereas there were no such cells in WT mice of this age (Fig. 4F). Calretinin-IR pioneer cells in the lateral preplate and basal forebrain were also more numerous in knock-outs than in WT mice (Fig. 4F, G), indicating that the loss of p73 gene products changed the developmental program of the cortical neuroepithelium at the preplate stage.
The development of the cortical plate in the absence of CR cells
We asked whether the absence of CR cells in the E12 p73-/- preplate disrupts preplate partition and early cortical plate development, leading to a reeler-like phenotype. At E15, in WT and p73+/- embryos, CR cells were intensely IR for Reelin (Fig. 5A), p73 (Fig. 5C), and calretinin (Fig. 5E). Cells with these characteristics were not found in the p73-/- marginal zone, which contained only few faintly Reelin-IR cells (Fig. 5B).
Figure 5.
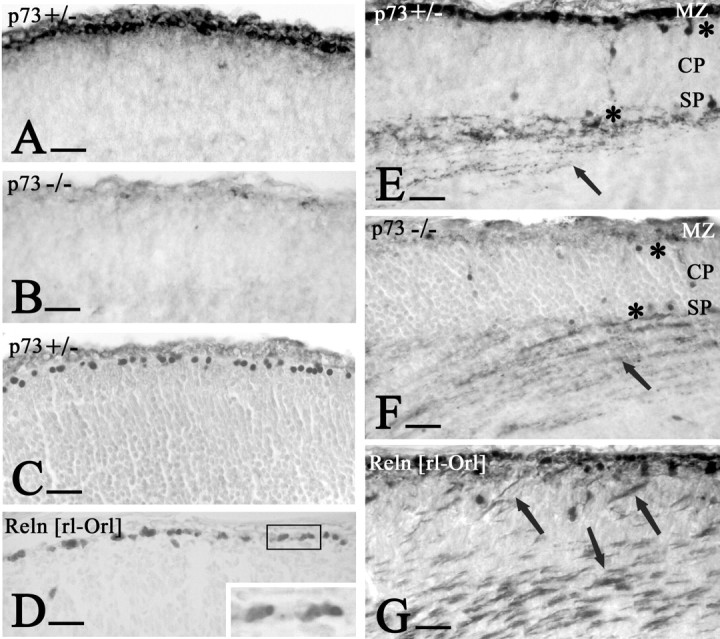
Early development of the cortical plate. A, E15p73+/- mice. B, E15p73-/- mice, Reelin. A, CR cells form a dense cell layer below the pial surface. B, Only few faintly Reelin-positive cells are in the marginal zone (MZ). C, E15 p73+/- mice, high expression of p73 in CR cells. E, E15p73+/- mice. F,p73-/- mice, calretinin. E, CR cells at this stage are calretinin positive and are absent in the p73 knock-out brain. Asterisks mark calretinin-IR pioneer cells above and below the cortical plate (CP), and arrows point to thalamocortical fibers terminating in the subplate (SP); both elements are unchanged in the p73-/- brain. D, G, E16 Relnrl-Orl mice. D, Reelin and p73 are coexpressed in CR cells. Inset, p73 in the nucleus and Reelin in the cytoplasm of the CR cells. G, Calretinin stains superplate elements and aberrant thalamocortical fibers (arrows) traversing the abnormal CP. Compare with F, in which CP and thalamocortical fibers have normal position. Scale bars, 25 μm.
Figure 5F shows that, despite the absence of the CR cells, the p73-/- preplate split into marginal zone and subplate. Calretinin stains pioneer cells above and below the WT cortical plate (Meyer et al., 1998, 2000); these cells were in their correct position in the p73+/- and p73-/- mice (Fig. 5E, F). Early thalamocortical fibers were also calretinin IR and terminated in their correct target layer, the subplate (Fig. 5E, F) (Allendoerfer and Shatz, 1994); in contrast, in reeler embryos they traversed the cortical plate to reach the superplate (Fig. 5G). The difference between reeler and p73-/- mice is important insofar as reeler CR cells express p73 but no functional Reelin (Fig. 5D), which leads to a disruption of preplate partition. In p73-/- mice, very low amounts of Reelin in the marginal zone were able to initiate preplate partition and cortical plate migration; the absence of p73 seemed to play no role in these events.
The absence of the hippocampal fissure in the p73-/- brain
We compared the development of the hippocampal fissure in E15 and E16 WT, p73-/-, and reeler embryos. In WT mice, the prospective fissure was marked by an indentation of the pial surface together with an approximately triangular collection of CR cells that coexpressed calretinin, Reelin, and p73 (Fig. 6A-C). The dentate primordium was just ventral to the incipient fissure and characterized by calretinin (Fig. 6A) and proliferating cell nuclear antigen immunoreactivity (data not shown). In p73-/- mice, CR cells were missing, and the pial surface did not invaginate (Fig. 6D, E). In contrast, in E16 reeler mice, p73/Reelin-IR CR cells were collecting in the fissure, which showed normal folding (Fig. 6F). In fact, the development of the hippocampal fissure is not disturbed in reeler mice.
Figure 6.
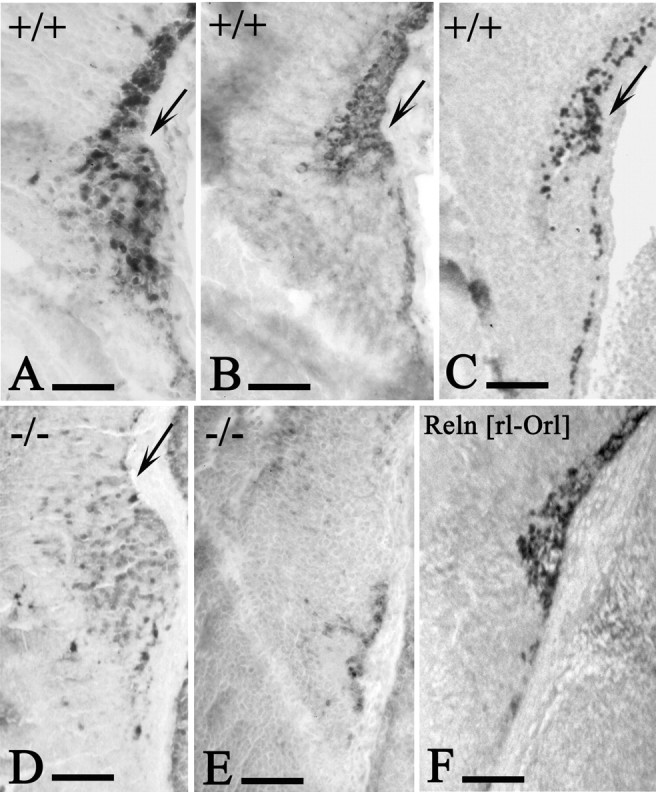
Early development of the hippocampal fissure. A-C, E15 WT mice. D, E, E15 p73-/- mice. F, E16 Relnrl-Orl mice. CR cells express calretinin (A), Reelin (B), and p73 (C) and occupy the fissure (arrows), which begins to fold. In E15 p73-/- embryos (D, calretinin; E, Reelin), CR cells are absent and the fissure does not fold. F, Reelin. In E16 Relnrl-Orl mice, the hippocampal fissure develops normally. Scale bars, 100 μm.
Examination of the postnatal hippocampus provided additional clues to understand the fissure defect. In the early postnatal WT and p73+/- mice, a band of GFAP-IR astrocytes extended all along the pial surface into the hippocampal fissure (Fig. 7E), whereas the underlying molecular layers of dentate gyrus and Ammon's horn were densely populated by CR cells (Fig. 7A, C). In p73-/- mice, the hippocampal fissure was reduced to a small indentation of the pial surface that did not extend farther into the brain (Fig. 7F). Subpial astrocytes were restricted to the brain surface, and CR cells were missing (Fig. 7B, D). In consequence, the molecular layers of the dentate and CA1/subiculum were fused instead of being separated by the fissure elements. At approximately P10 in the WT mice, GFAP-IR astrocytes invaded all strata of Ammon's horn, and the radial glia scaffold was established in the dentate (Fig. 7G). In contrast, astrocytes were sparse and dispersed in p73-/- mice, and the radial glia scaffold of the dentate gyrus was unrecognizable (Fig. 7H). Also, calretinin-positive cells of the hilus had an abnormal position (Fig. 7C, D), indicating that p73 deficiency severely alters the architecture of all components of the hippocampal formation.
Figure 7.
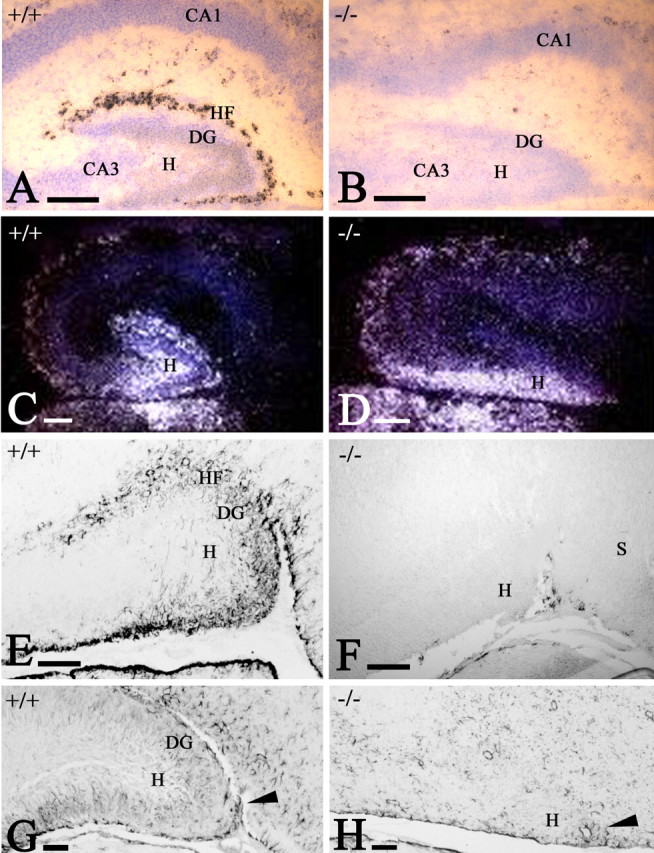
The postnatal hippocampal fissure (HF). A, B, Reelin ISH in WT (A) and p73-/- (B) mice at P2. B, CR cells are absent, and the molecular layers of dentate gyrus (DG) and CA1/CA2 are fused. C, D, Calretinin ISH in WT (C) and p73-/- (D) mice at P2. In the mutant, calretinin-positive hilus (H) cells have abnormal distribution. E, F, GFAP IHC in WT (E) and p73-/- (F) mice at P2. GFAP stains subpial astrocytes in the HF and the incipient radial glia scaffold in the dentate gyrus. In the mutant, astrocytes are found only along the medial brain surface. G, H, GFAP in WT (G) and p73-/- (H) mice at P12. The radial glia scaffold is fully established in the WT dentate; in the mutant, astrocytes are sparse and lack radial orientation. Arrowheads mark the entrance of the fissure, which fails to fold in the mutant. S, Subiculum. Scale bars: A, B, 200 μm; C-H, 100 μm.
The p73+/- telencephalon
In p73+/- mice, the cortex appeared mostly normal, except for the most caudal regions, and, most strikingly, the entorhinal cortex. The large projection cells of entorhinal layer II express Reelin (Drakew et al., 1998; Perez-Garcia et al., 2001) and define this area. In p73+/- mice, the entorhinal cortex was shifted into a more dorsal position, concurrent with an enlargement of the posterior cortical nucleus of the amygdala (Fig. 8A-C). The caudal neocortex (primary and secondary visual cortex and auditory association area) was significantly reduced in size, and the cytoarchitecture appeared disorganized. Furthermore, a band of large Reelin-IR neurons extended from the displaced entorhinal layer II into the altered neocortical territory (Fig. 8 A-D), suggesting a transformation of lateral and dorsal neocortex into an entorhinal-like cortex. The presence of ectopic Reelin-IR cells and fibers in the white matter of the posterior pole suggested abnormal axonal projections. Early postnatally, the molecular layer of the altered cortex was populated by numerous CR cells (Fig. 8 E, F), which were intensely IR for Reelin (Fig. 8 E) and p73 (Fig. 8 F). At P12, most CR cells had disappeared (Fig. 8 B), as had their counterparts in WT brains.
Figure 8.
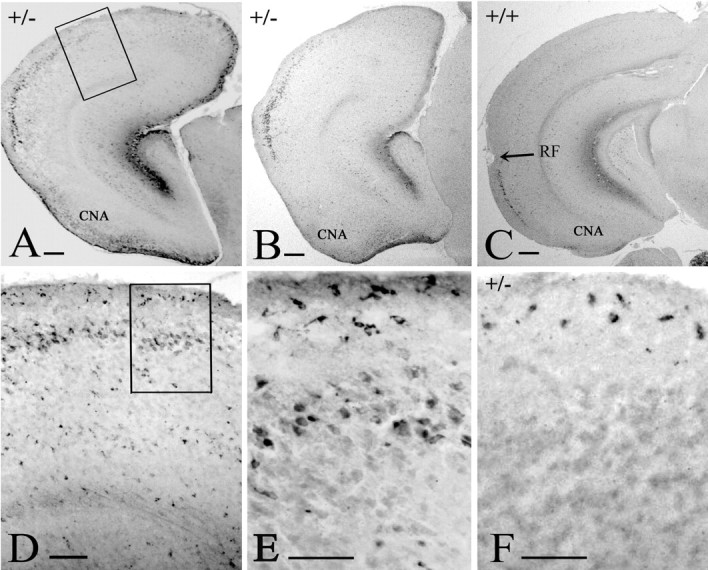
Architecture of the p73+/- telencephalon. Reelin in P3 p73+/- (A), P12 p73+/- (B), and P12 WT (C) brains. Reelin-IR cells in layer II mark the entorhinal cortex in the WT brain. In p73+/- brains, they are dorsally displaced and expanded, whereas the neocortex is reduced in size. The posterior cortical nucleus of the amygdala (CNA) is hyperplastic. The rhinal fissure (RF) is not visible in the p73+/- brains. D, E, Higher magnification of the boxed areas in A and D, respectively, to show ectopic Reelin-IR cells in layer II of the abnormal cortex and CR cells in layer I. F, At P3, CR cells in the abnormal p73+/- cortex are intensely p73 IR. Scale bars: A-C, 200 μm; D, 100 μm; E, F, 50 μm.
The E2F1-/- brain closely resembles the p73+/- phenotype
The transcription factor E2F1 is one of the main activators of TAp73 (Irwin et al., 2000; Stiewe and Putzer, 2000). In early postnatal E2F1-/- mice, we detected a brain phenotype similar to that of p73+/- mice, characterized by an increase of the posterior cortical nucleus of the amygdala, a dorsal shift of the entorhinal cortex, and a reduction and entorhinal-like transformation of the caudal neocortex, along with the presence of Reelin-positive cells in layer II of the altered territory (Fig. 9B-D). The rhinal fissure was unrecognizable. Calretinin immunostaining revealed that the laminar distribution of calretinin-IR interneurons in the auditory and visual areas of E2F1-/- animals was clearly distinct from the WT pattern (Fig. 9A, B), indicating an alteration of the normal cortical architecture. The white matter of the mutant posterior pole was reduced in size and populated by increased numbers of calretinin- and Reelin-IR interstitial neurons. These anatomical abnormalities developed in the presence of CR cells, which were indistinguishable from those of WT mice. In particular, p73 immunoreactivity in CR cells was apparently unchanged (data not shown) and did not reflect the minor reduction of DNp73 levels observed in the E2F1-/- brains (see below).
Figure 9.
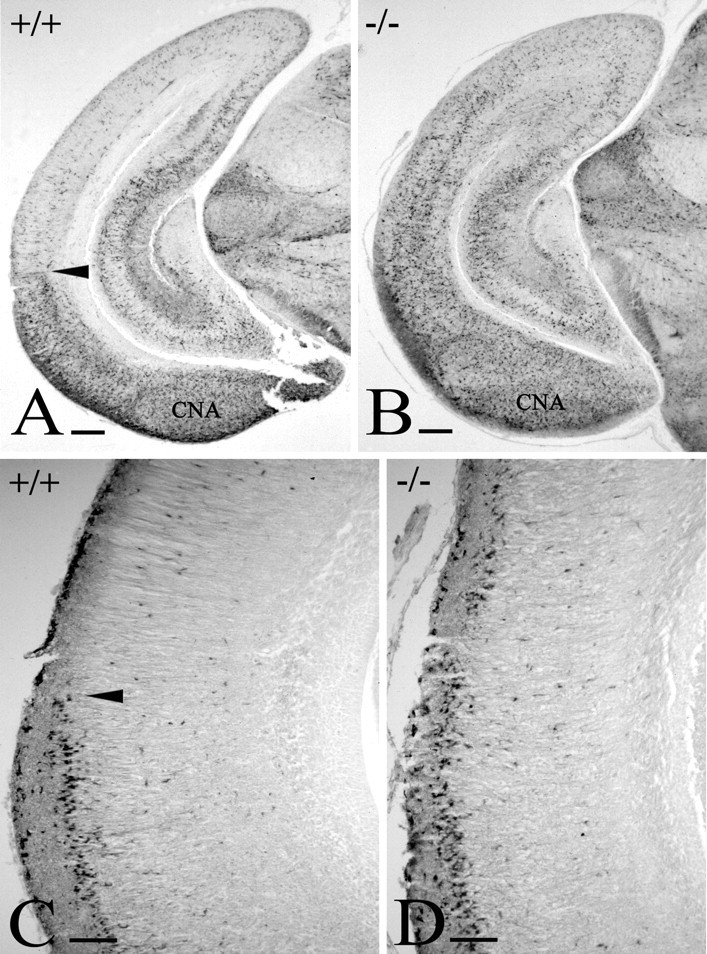
The E2F1-/- cortex. Calretinin immunostaining in WT (A) and E2F1-/- (B) cortex at P1 shows that the caudal neocortex is transformed and acquires entorhinal-like features. Calretinin-IR interneurons are distributed differently in WT and E2F1-/- cortex, and the posterior cortical nucleus of the amygdala (CNA) is slightly larger in the mutant. C, In WT, Reelin-IR cells in layer II stop at the rhinal fissure (arrowheads in A and C). D, in the mutant, they extend dorsally into the altered neocortex. Scale bars: A, B, 200 μm; C, D, 100 μm.
Although this malformation closely resembled the p73+/- phenotype, the dorsalization of the entorhinal cortex and the reduction of the neocortical territory were less pronounced.
TAp73 expression is reduced in the E2F1-/- brain
Immunoblot analysis of E2F1-deficient and WT brains at P2 revealed a reduction of TAp73 expression in the mutant (Fig. 10). In WT brains probed with an N-terminal antibody (Fig. 10A), we observed a large band of 82 kDa corresponding to TAp73α and a smaller band of 72 kDa corresponding to TAp73β (Yang and McKeon, 2000). In the E2F1-/- brains, we observed a 50% reduction of the 82 kDa band and a 20% reduction of the 72 kDa band. Immunoblotting using the C-terminal antibody (this antibody consistently stains DNp73-expressing CR cells) revealed a single band of ∼66 kDa (Fig. 10C), which is in accordance with the predicted molecular weight of DNp73α (Yang and McKeon, 2000). This band showed only a 7% reduction in the E2F1 mutant brains. Using the N-terminal antibody, we observed an additional band of higher molecular weight in the E2F1-/- brains (Fig. 10A), which could not be attributed to the known p73 isoforms. Our results show that the loss of function of E2F1 leads to a significant reduction of TAp73 protein levels and a minor decrease of DNp73 levels in the brain.
Figure 10.

Expression of p73 in E2F1 knock-out mice. A, C, Immunoblot analysis of p73 in cortical tissue extracts probed with N-terminal (A) and C-terminal (C) antibodies. Note the important reduction of TAp73 levels at 82 kDa (TAp73α) and at 72 kDa (probably representing TAp73β) in mutant mice compared with the controls. After incubation with a C-terminal antibody, a DNp73 isoform at 66 kDa shows only a slight reduction in the E2F1 mutant (C). B, D, α-Tubulin controls to test the amount of protein analyzed.
Discussion
Our study of p73-deficient brains points to extensive activities of p73 in cell proliferation, cell survival, and cortical patterning. The dramatic defects of p73 deficiency are difficult to reconcile with its rather selective expression in the brain. We propose that p73 may play a role in the signaling network of the cortical hem, which regulates neocortical and archicortical regionalization and the generation of CR cells. We discuss the possible role of p73 in CR cells, the differential expression of the main products of the p73 gene (TAp73 and DNp73) in early brain development, and the involvement of an E2F1-TAp73-dependent signaling pathway in the regionalization of the posterior telencephalon.
Cajal-Retzius cells in cortical development
CR cells clearly depend on p73 and do not develop in the p73-/- brain. CR cells secrete Reelin, which activates a signaling pathway that leads to phosphorylation of the adapter protein Dab1 in cortical plate cells (Howell et al., 1999). In reeler mice, cortical layers are inverted and radial architecture is disturbed (Lambert de Rouvroit and Goffinet, 1998). The absence of CR cells as the main source of Reelin would be expected to produce reeler-like defects of cortical migration and lamination. However, lack of CR cells in the p73 mutant is compatible with quite normal lamination and cytoarchitecture, perhaps because Reelin is faintly expressed at the critical stages for preplate (Meyer et al., 2002) and cortical plate formation, respectively. Later-appearing Reelin-positive interneurons (Alcántara et al., 1998; Pesold et al., 1999; Meyer et al., 2002) may also be able to compensate for the loss of CR cells.
The intense Reelin signal in CR cells is thus redundant for lamination and architecture of the mouse cortex. However, to judge from the prominence of CR cells in the human brain (Meyer and Goffinet, 1998; Meyer et al., 1999, 2002), they gain in importance during evolution and may be involved in other aspects of cortical development, such as size increase and folding of the cortical surface. These aspects are not manifest in the lissencephalic mouse cortex but may be relevant for the formation of the gyrated primate cortex. The loss of the hippocampal fissure in the p73-/- mice supports a possible relationship between p73 expression in CR cells and cortical folding.
Although p73-deficient mice do not have reeler-like malformations, cortical development is abnormal, and the ectopic cells in the preplate reflect an early alteration of the developmental program. The cortical hypoplasia points to a generalized defect of cell proliferation in the ventricular zone attributable to the loss of p73 and indicates that the activities of the p73 gene in brain development are wider than expected from the restricted expression in CR cells.
The p73-/- hippocampus
The embryonic p73-/- hippocampus has three distinctive features: absence of CR cells, lack of the hippocampal fissure, and dysgenesis of the dentate gyrus. Hippocampal CR cells and Reelin are essential for the establishment of the entorhinal-hippocampal pathways (Del Rio et al., 1997; Super et al., 1998; Ceranik et al., 2000), and the connectivity of the p73-/- hippocampus is probably profoundly disturbed. We show that CR cells may also be critical for the folding of the hippocampal fissure. Because the fissure develops normally in reeler mice, folding does not depend on Reelin; however, CR cells may play a role in this process via p73 or other proteins, e.g., the chemokine receptor CXCR4 (Stumm et al., 2003; Tissir et al., 2004), whose ligand, SDF1 (stromal cell-derived factor 1), is highly expressed in the meninges and in cells investing the hippocampal fissure (Lu et al., 2002). Meningeal cells strongly influence dentate development, and their destruction induces a loss of the infrapyramidal blade (Hartmann et al., 1992) similar to the p73-/- dentate defect.
On the other hand, it has been proposed that subpial astrocytes in the fissure form a transient boundary between Ammon's horn and the dentate gyrus that allows the establishment of an independent radial glia scaffold in the dentate subgranular zone (Sievers et al., 1992). The radial glia scaffold is essential for the migration of dentate granule cells and defective in reeler and scrambler mice (Forster et al., 2002; Frotscher et al., 2003; Weiss et al., 2003). We show that it is also reduced and disorganized in p73-/- mice. Importantly, astrocytes in the subgranular zone are also progenitors of late-born granule cells (Seri et al., 2001). Loss or reduction of this precursor population may lead to a decrease in adult neurogenesis, which has been correlated with learning and memory (Kempermann et al., 1997; Gould et al., 1999). In summary, CR cells, meninges, subpial astrocytes, and dentate radial glia are closely interconnected in hippocampal morphogenesis, and p73 is deeply involved in this process.
The p73+/- brain phenotype
The p73+/- malformation is mainly a patterning defect of the posterior pole. Areal territories are altered, with ventral regions (posterior cortical nucleus of the amygdala and entorhinal cortex) dominating over dorsal ones (posterior temporal and occipital neocortex). The neocortex is reduced and transformed into an entorhinal-like cortex. Furthermore, cell numbers in the caudal p73+/- cortex continue to decrease postnatally by apoptosis (Pozniak et al., 2002). These abnormalities indicate a decisive role of p73 in the areal patterning and neuronal survival of the posterior telencephalon.
p73 heterozygosity affects number and position of the Reelin-positive stellate cells of entorhinal layer II, which are a major origin of the entorhinal-hippocampal pathway (Steward and Scoville, 1976; Witter, 1993). The ectopic Reelin-positive cells in the altered p73+/- neocortex possibly establish aberrant connections. Another important aspect of the p73+/- brain is the size reduction and transformation of visual and secondary auditory areas, which raises the question of whether the sensory pathways are correctly established. Remarkably, p73+/- mice display apparently normal behavior despite their cortical defect. However, it is questionable whether a p73+/--like phenotype would be compatible with normal cognitive functions in the human brain. Extrapolation of the mouse brain defect on the human brain would predict a substantial reduction of primary visual cortex and occipitotemporal association areas, a damage that, combined with the entorhinal malformation, would lead to severe cognitive disabilities and mental retardation.
A comparable dosage-dependent defect has been described for the neurodevelopmental control gene PAX6, homozygous mutation of which is lethal in mice and humans. PAX6 heterozygous individuals present aniridia and behavioral and cognitive defects associated with structural abnormalities of specific cortical areas (Sisodiya et al., 2001; Ellison-Wright et al., 2004).
An E2F1-TAp73 signaling pathway in the cortical hem
We reported previously that CR cells derive from the cortical hem and migrate tangentially into the neocortex (Meyer et al., 2002; Takiguchi-Hayashi et al., 2004). The cortical hem is a posteromedial signaling center that releases signaling molecules such as BMP (bone morphogenetic) and WNT (winglessint) proteins, which control the expression of region-specific transcription factors; disruption of the signaling pathways in the hem results in distorted cortical and hippocampal patterning (Ragsdale and Grove, 2001; Grove and Fukuchi-Shimogori, 2003). Our proposal that early alterations of p73 expression in the hem lead to alterations of the posterior cortical map is consistent with this concept, although the possible molecular mechanisms are as yet unknown.
Does the role of p73 in cortical patterning depend on CR cells? Our findings point to a CR cell-independent mechanism. The severe malformation of the posterior pole in the p73-/- mice cannot be attributed to CR cells because they are lost throughout the cortex, not only in the most affected region. On the other hand, CR cells are apparently unchanged in p73+/- and E2F1-/- mice, both of which present abnormal caudal regionalization. The differential expression of the main p73 products may give a clue for solving this question because CR cells express DNp73 (Yang and McKeon, 2000; Yang et al., 2000), whereas TAp73 transcripts are detected in the cortical hem.
The E2F1-/- mice may help to shed light on the p73 patterning defect. E2F1 is a member of a transcription factor family that functions as a positive regulator of apoptosis and a suppressor of cell-cycle progression (Helin et al., 1992; Kaelin et al., 1992; Shan et al., 1992; Field et al., 1996). E2F1-deficient mice appear to develop normally, although postnatal and adult neurogenesis is impaired in olfactory bulb, dentate gyrus, and cerebellum (Cooper-Kuhn et al., 2002). E2F1 is a major activator of the TAp73 promoter (Irwin et al., 2000; Seelan et al., 2002), which is consistent with the significant decrease of TAp73 protein in the E2F1-/- brain. In contrast, DNp73 seems to be less affected by E2F1 deficiency than TAp73, which correlates with the preserved p73 immunoreactivity in CR cells. E2F1 has other targets in addition to TAp73 (Stanelle et al., 2002), and, in fact, the E2F1-/- phenotype is not completely identical to the p73+/- phenotype. The rather mild defect in E2F1-/- mice and the more severe form in the p73+/- mice point to a dosage-dependent effect of TAp73 in cortical patterning.
Our data in the p73 and E2F1 mice suggest that the malformation of the posterior pole is unrelated to CR cells. The common feature in p73-/-, p73+/-, and E2F1-/- brains is the loss and reduction, respectively, of TAp73 expression, supporting the hypothesis that an E2F1-TAp73 pathway in the cortical hem is important for the regionalization of the posterior pole.
Footnotes
This study was supported by European Union Grant QLG3-CT-2000-00158 (Concorde), Ministerio de Ciencia y Tecnologia Grant BFI 2002-03594, and Gobierno de Canarias PI2002-122. C.G.P.G. is supported by Grant AP2000-1769 from the Ministerio de Educación, Cultura, y Deporte. We thank An dré Goffinet for providing the reeler mice and Reelin antibodies. We also thank Cristina M. Ramirez for help with the immunoblot analysis.
Correspondence should be addressed to Gundela Meyer, Department of Anatomy, Faculty of Medicine, University La Laguna, 38071 La Laguna, Tenerife, Spain. E-mail: gmeyer@ull.es.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249878-10$15.00/0
Retired, 2003.
References
- Alcántara S, Ruiz M, D'Arcangelo G, Ezan F, De Lecea L, Curran T, Sotelo C, Soriano E (1998) Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci 18: 7779-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendoerfer KL, Shatz CJ (1994) The subplate, a transient neocortical structure; its role in the development of connections between thalamus and cortex. Annu Rev Neurosci 17: 185-218. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS (1976) Patterns of cell and fiber distribution in the neocortex of the reeler mutant mouse. J Comp Neurol 170: 435-448. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS (1982) Neocortical histogenesis in normal and reeler mice: a developmental study based upon 3H-thymidine autoradiography. Dev Brain Res 4: 297-326. [DOI] [PubMed] [Google Scholar]
- Ceranik K, Zhao S, Frotscher M (2000) Development of the entorhinohippocampal projection: guidance by Cajal-Retzius cell axons. Ann NY Acad Sci 911: 43-54. [DOI] [PubMed] [Google Scholar]
- Cooper-Kuhn CN, Vroemen M, Brown HY, Thompson MA, Winkler J, Kuhn HG (2002) Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol Cell Neurosci 21: 312-323. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374: 719-723. [DOI] [PubMed] [Google Scholar]
- de Bergeyck V, Nakajima K, Lambert de Rouvroit C, Naerhuyzen B, Goffinet AM, Miyata T, Ogawa M (1997) A truncated Reelin protein is produced but not secreted in the “Orleans” reeler mutation (Reln[rl-Orl]). Brain Res Mol Brain Res 50: 85-90. [DOI] [PubMed] [Google Scholar]
- de Bergeyck V, Naerhuyzen B, Goffinet AM, Lambert de Rouvroit C (1998) A panel of monoclonal antibodies against reelin, the extracellular matrix protein defective in reeler mutant mice. J Neurosci Methods 82: 17-24. [DOI] [PubMed] [Google Scholar]
- Del Rio JA, Heimrich B, Borrell V, Forster E, Drakew A, Alcantara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E (1997) A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature 385: 70-74. [DOI] [PubMed] [Google Scholar]
- Drakew A, Frotscher M, Deller T, Ogawa M, Heimrich B (1998) Developmental distribution of a reeler gene-related antigen in the rat hippocampal formation visualized by CR-50 immunocytochemistry. Neuroscience 82: 1079-1086. [DOI] [PubMed] [Google Scholar]
- Ellison-Wright Z, Heyman I, Frampton I, Rubia K, Chitnis X, Ellison-Wright I, Williams SC, Suckling J, Simmons A, Bullmore E (2004) Heterozygous PAX6 mutation, adult brain structure and fronto-striato-thalamic function in a human family. Eur J Neurosci 19: 1505-1512. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin Jr WG, Livingston DM, Orkin SH, Greenberg ME (1996) E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 85: 549-561. [DOI] [PubMed] [Google Scholar]
- Forster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Muller U, Frotscher M (2002) Reelin disabled 1, and β1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci USA 99: 13178-13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Haas CA, Forster E (2003) Reelin controls granule cell migration in the dentate gyrus by acting on the radial glial scaffold. Cereb Cortex 13: 634-640. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2: 260-265. [DOI] [PubMed] [Google Scholar]
- Grob TJ, Novak U, Maisse C, Barcaroli D, Lüthi AU, Pirnia F, Tobler A (2001) Human ΔNp73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ 8: 1213-1223. [DOI] [PubMed] [Google Scholar]
- Grove EA, Fukuchi-Shimogori T (2003) Generating the cerebral cortical area map. Annu Rev Neurosci 26: 381-410. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S, Limon J, Yip L, Ragsdale CW (1998) The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125: 2315-2325. [DOI] [PubMed] [Google Scholar]
- Hartmann D, Sievers J, Pehlemann WF, Berry M (1992) Destruction of meningeal cells over the medial cerebral hemisphere of newborn hamsters prevents the formation of the infrapyramidal blade of the dentate gyrus. J Comp Neurol 320: 33-61. [DOI] [PubMed] [Google Scholar]
- Helin K, Lees JA, Vidal M, Dyson N, Harlow E, Fattaey A (1992) A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 70: 337-350. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA (1999) Reelin-induced tryosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev 13: 643-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden K, Kaelin WG (2000) Role for the 53 homologue p73 in E2F-1 induced apoptosis. Nature 407: 645-648. [DOI] [PubMed] [Google Scholar]
- Jost CA, Marin MC, Kaelin WJ (1997) p73 is a simian p53-related protein that can induce apoptosis. Nature 389: 191-194. [DOI] [PubMed] [Google Scholar]
- Kaelin Jr WG, Krek W, Sellers WR, DeCaprio JA, Ajchenbaum F, Fuchs CS, Chittenden T, Li Y, Farnhem PJ, Blanar MA (1992) Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell 70: 351-364. [DOI] [PubMed] [Google Scholar]
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D (1997) Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90: 809-819. [DOI] [PubMed] [Google Scholar]
- Kartasheva NN, Contente A, Lenz-Stöppler C, Roth J, Dobbelstein M (2002) p53 induces the expression of its antagonist p73ΔN, establishing an auto-regulatory feedback loop. Oncogene 21: 4715-4727. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386: 493-495. [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM (1998) The reeler mouse as a model of brain development. Adv Anat Embryol Cell Biol 150: 1-108. [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ (2002) Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci USA 99: 7090-7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M (1998) Cajal-Retzius cells and the development of the neocortex. Trends Neurosci 21: 64-71. [DOI] [PubMed] [Google Scholar]
- Meyer G, Goffinet A (1998) Prenatal development of reelin-immunoreactive neurons in the human neocortex. J Comp Neurol 397: 29-40. [PubMed] [Google Scholar]
- Meyer G, Soria JM, Martinez-Galan JR, Martin-Clemente B, Fairen A (1998) Different origins and developmental histories of transient neurons in the marginal zone of the fetal and neonatal rat cortex. J Comp Neurol 397: 493-518. [PubMed] [Google Scholar]
- Meyer G, Goffinet AM, Fairen A (1999) What is a Cajal-Retzius cell? A reassessment of a classical cell type based on recent observations in the developing neocortex. Cereb Cortex 9: 765-775. [DOI] [PubMed] [Google Scholar]
- Meyer G, Schaaps JP, Moreau L, Goffinet AM (2000) Embryonic and early fetal development of the human neocortex. J Neurosci 20: 1858-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Perez-Garcia GC, Abraham H, Caput D (2002) Expression of p73 and Reelin in the developing human cortex. J Neurosci 22: 4973-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia CG, Gonzalez-Delgado FJ, Suarez-Sola ML, Castro-Fuentes R, Martin-Trujillo JM, Ferres-Torres R, Meyer G (2001) Reelinimmunoreactive neurons in the adult vertebrate pallium. J Chem Neuroanat 21: 41-51. [DOI] [PubMed] [Google Scholar]
- Pesold C, Liu WS, Guidotti A, Costa E, Caruncho HJ (1999) Cortical bitufted, horizontal, and Martinotti cells preferentially express and secrete reelin into perineuronal nets, nonsynaptically modulating gene expression. Proc Natl Acad Sci USA 96: 3217-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Radinovik S, Yang A, McKeon F, Kaplan DR, Miller FD (2000) An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 289: 304-306. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD (2002) p73 is required for survival and maintenance of CNS neurons. J Neurosci 22: 9800-9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale CW, Grove EA (2001) Patterning in the mammalian cerebral cortex. Curr Opin Neurobiol 11: 50-58. [DOI] [PubMed] [Google Scholar]
- Seelan RS, Irwin M, van der Stoop P, Qian C, Kaelin Jr WG, Liu W (2002) The human p73 promoter: characterization and identification of functional E2F binding sites. Neoplasia 4: 195-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21: 7153-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Zhu X, Chen PL, Durfee T, Yang Y, Sharp D, Lee WH (1992) Molecular cloning of cellular genes encoding retinoblastoma-associated proteins: identification of a gene with properties of transcription factor E2F. Mol Cell Biol 12: 5620-5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers J, Hartmann D, Pehlemann WF, Berry M (1992) Development of astroglial cells in the proliferative matrices, the granule cell layer, and the hippocampal fissure of the hamster dentate gyrus. J Comp Neurol 320: 1-32. [DOI] [PubMed] [Google Scholar]
- Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, Moore AT, van Heyningen V (2001) PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet 28: 214-216. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85. [DOI] [PubMed] [Google Scholar]
- Stanelle J, Stiewe T, Theseling CC, Peter M, Pützer BM (2002) Gene expression changes in response to E2F1 activation. Nucleic Acids Res 30: 1859-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Scoville SA (1976) Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol 169: 347-370. [DOI] [PubMed] [Google Scholar]
- Stiewe T, Putzer BM (2000) Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 26: 464-469. [DOI] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S (2003) CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci 23: 5123-5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Super H, Martinez A, Del Rio JA, Soriano E (1998) Involvement of distinct pioneer neurons in the formation of layer-specific connections in the hippocampus. J Neurosci 18: 4616-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiguchi-Hayashi K, Sekiguchi M, Ashigaki S, Takamatsu M, Hasegawa H, Suzuki-Migishima R, Yokoyama M, Nakanishi S, Tanabe Y (2004) Generation of reelin-positive marginal zone cells from the caudomedial wall of telencephalic vesicles. J Neurosci 24: 2286-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM (2003) Reelin and brain development. Nature Rev Neurosci 4: 496-505. [DOI] [PubMed] [Google Scholar]
- Tissir F, Wang CE, Goffinet AM (2004) Expression of the chemokine receptor Cxcr4 mRNA during mouse brain development. Brain Res Dev Brain Res 149: 63-71. [DOI] [PubMed] [Google Scholar]
- Weiss KH, Johansen C, Tielsch A, Herz J, Deller T, Frotscher M, Forster E (2003) Malformation of the radial glial scaffold in the dentate gyrus of reeler mice, scrambler mice, and ApoER2/VLDLR-deficient mice. J Comp Neurol 460: 56-65. [DOI] [PubMed] [Google Scholar]
- Witter MP (1993) Organization of the entorhinal-hippocampal system: a review of current anatomical data. Hippocampus 3: 33-44. [PubMed] [Google Scholar]
- Yang A, McKeon F (2000) p63 and p73: p53 mimics, menaces and more. Nat Rev 1: 199-207. [DOI] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkens P, Sharpe A, McKeon F, Caput D (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404: 99-103. [DOI] [PubMed] [Google Scholar]
- Zaika A, Irwin M, Sansome C, Moll UM (2001) Oncogenes induce and activate endogenous p73 protein. J Biol Chem 276: 11310-11316. [DOI] [PubMed] [Google Scholar]