

Abstract
Cytoplasmic deposition of α-synuclein aggregates is a common pathological feature of many neurodegenerative diseases. Strong evidence for the causative role of α-synuclein in these disorders is provided by genetic linkage between this gene and familial Parkinson's disease and by neurodegeneration in transgenic animals that overexpress this protein. In particular, it has been hypothesized that the accumulation of nonfibrillar oligomers of α-synuclein, which serve as intermediates for fibrillar inclusion body formation, causes neurodegeneration. However, little is known about how cells handle potentially toxic protein aggregates. Here we demonstrate that cells are capable of clearing preformed α-synuclein aggregates via the lysosomal degradation pathway. Consequently, blocking this pathway causes the accumulation of the aggregates in non-neuronal cells, differentiated neuroblastoma cells, and primary cortical neurons. This aggregate clearance occurs in an aggregation stage-specific manner; oligomeric intermediates are susceptible to clearance, whereas mature fibrillar inclusion bodies are not. Neutralization of the acidic compartments leads to the accumulation of α-synuclein aggregates and exacerbates α-synuclein toxicity in postmitotic neuronal cells, suggesting that the accumulation of oligomeric intermediates may be an important event leading to α-synuclein-mediated cell death. These results suggest that enhancing lysosomal function may be a potential therapeutic strategy to halt or even prevent the pathogenesis of Parkinson's disease and other Lewy body diseases.
Keywords: α-synuclein, protein aggregation, lysosome, Parkinson's disease, Lewy body, neurodegeneration
Introduction
Deposition of filamentous α-synuclein (α-syn) in the neuronal or glial cytoplasm is a common pathological feature of many neurological diseases, such as Parkinson's disease (PD), dementia with Lewy bodies, multiple system atrophy, and neuronal degeneration with brain iron accumulation type 1 (Trojanowski et al., 1998; Goedert, 2001). All of the genetic variations in human α-syn gene that are causative to the early-onset familial parkinsonism increase the probability to form aggregates (Polymeropoulos et al., 1997; Kruger et al., 1998; Conway et al., 2000; Singleton et al., 2003). Animal models developed in mice and flies have shown that overexpression of α-syn in neurons can cause neuronal loss, along with α-syn aggregation that leads to the formation of both filamentous and granular aggregates (Feany and Bender, 2000; Masliah et al., 2000; Giasson et al., 2002; M. Lee et al., 2002), further supporting the hypothesis that abnormal accumulation of α-syn aggregates may play a critical role in the pathogenesis of neurodegenerative diseases.
Biochemical studies have shown that α-syn can form amyloid-like fibrils with a cross-β-sheet conformation (Serpell et al., 2000). These fibrils have a similar morphology to that found in Lewy bodies (LBs) (Spillantini et al., 1998), suggesting that the mechanism of cell-free fibrillation may reflect the actual pathogenic process in vivo. Fibrillation of α-syn initiates with the dimerization of partially folded monomers (Uversky et al., 2001; Krishnan et al., 2003), followed by the formation of β-sheet-rich nonfibrillar oligomeric intermediates, also known as protofibrils, with several distinct morphologies (Volles and Lansbury, 2003). More recently, we have developed a cell culture model in which overexpression of human α-syn leads to the formation of LB-like inclusion bodies that consist mainly of fibrillar aggregates. In these cells we have demonstrated that nonfibrillar spherical oligomers act as intermediates in the formation of fibrillar inclusion bodies and that the oligomer-to-fibril transition requires microtubule-dependent deposition of oligomers in the pericentriolar region (Lee and Lee, 2002). The mechanism underlying the deleterious effects of α-syn aggregates remains to be elucidated. Several studies have suggested that the oligomeric intermediates are the cause of cellular dysfunction and cell death (Gosavi et al., 2002; Kayed et al., 2003; Volles and Lansbury, 2003), whereas others have pointed to the direct role of fibrillar inclusion bodies in neurodegeneration (for review, see Giasson and Lee, 2003).
The extent of aggregate accumulation likely is determined by a dynamic equilibrium between the production and clearance of aggregates, and this process may, in turn, be critical for cell viability. In contrast to the rapid progress in our understanding of the α-syn aggregation process and its regulation, little is known about the breakdown of preformed aggregates. Previously, we have shown that α-syn aggregation can be promoted by treating cells with the common pesticide rotenone and that the cells can clear the preformed aggregates after removal of the rotenone from the culture medium (H.-J. Lee et al., 2002b). Here we further characterize this aggregate-clearing activity in both non-neuronal and neuronal cells and show that only the oligomeric intermediates, but not the fibrillar inclusion bodies, can be cleared via the lysosomal degradation pathway. We also demonstrate that lysosomal failure leads to the accumulation of α-syn aggregates and concomitant cell death, suggesting that the accumulation of oligomeric intermediates might be the key component of α-syn-mediated cytotoxicity.
Materials and Methods
Materials. Bafilomycin A1 (Baf), E64, cathepsin I, β-lactone, and epoxomicin (Epox) were obtained from Calbiochem (San Diego, CA). Retinoic acid (RA), 3-methyladenine (3-MA), and protease inhibitor mixture were purchased from Sigma (St. Louis, MO). Monoclonal antibodies for α-syn, LB509 and Syn-1, were purchased from Zymed Laboratories (South San Francisco, CA) and BD Biosciences (San Diego, CA), respectively. Polyclonal anti-α-syn 7071 was a gift from Dr. Lansbury (Harvard Medical School, Boston, MA). Monoclonal antibody for lysosome-associated membrane protein 2 (LAMP 2; H4B4) was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA). All of the fluorescently labeled secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Gold-conjugated anti-mouse IgG antibody was obtained from Ted Pella (Redding, CA).
Adenoviral vectors. Construction of adeno/α-syn was described previously (H.-J. Lee et al., 2002b). For the construction of adenoviral vectors that contain myc and his-tagged α-syn variants, we first inserted PCR-amplified α-syn cDNAs into the EcoRV-XbaI site of pCDNA3.1-MycHis (Invitrogen, Carlsbad, CA). Coding regions for α-syn variants with the tag then were amplified via PCR and inserted into the transfer vector, pDNR-CMV (BD Biosciences). The sequences of entire coding regions of wild-type, A53T, and A30P variants were confirmed. Recombinant adenoviral DNAs were generated in vitro by the Adeno-X Expression System 2 (BD Biosciences).
Cell culture and α-syn expression. Cells were maintained as described previously (Lee and Lee, 2002). For expression of α-syn in COS-7 cells, 80% confluent cells on 100 mm tissue culture dishes were infected with adenoviral vector containing human α-syn cDNA (adeno/α-syn) (H.-J. Lee et al., 2002b) at a multiplicity of infection (m.o.i.) of 50. After 90 min of incubation fresh medium was added, and the cells were incubated at 37°C until the next day. Cells then were split to ∼40% confluency and maintained for the indicated period. For induction of α-syn aggregation, rotenone was added at a concentration of 100 nm on the same day that the infected cells were split. For washout of rotenone, rotenone-containing medium was replaced with fresh rotenone-free medium. SH-SY5Y cells were split to ∼10% confluency and the next day were induced to differentiate by the addition of 50 μm all-trans RA. The cells were fed with fresh medium containing RA every other day. On day 5 of differentiation the cells were infected with adeno/α-syn at a m.o.i. of 100. Unlike COS-7 cells, infected SH-SY5Y cells were maintained without splitting.
Primary cortical neurons were obtained from embryonic day 16 (E16) embryos of pregnant Sprague Dawley rats (Price and Brewer, 2001). Briefly, embryonic brains were placed in a sterile culture dish containing HBSS (Invitrogen), and cerebral cortices were dissected and collected in a centrifuge tube. The cortical tissues were incubated with the papain solution (HBSS containing 10 U/ml papain, 0.2 mg/ml cysteine, 0.5 mm EDTA, 1 mm CaCl2, and 0.003N NaOH), followed by incubation with the trypsin inhibitor solution (MEM with Earl's salts containing 5% fetal bovine serum, 2.5 mg/ml bovine serum albumin, and 2.5 mg/ml trypsin inhibitor). Then the tissues were dissociated mechanically by pipetting in the culture medium and were centrifuged briefly to collect the cells. Cells were resuspended, plated on either poly-d-lysine/laminin-coated plastic dishes or poly-d-lysine/Matrigel-coated (1:100; BD Biosciences) glass coverslips, and cultured in Neurobasal medium (Invitrogen) with 2% B-27 supplement (Invitrogen) and 0.5 mm Glutamax-1 (Invitrogen). Culture medium was changed every 3 d.
Cell extraction and fractionation. For simple detergent extraction the cells were washed twice with ice-cold PBS before the addition of cold extraction buffer (PBS containing 1% Triton X-100 and protease inhibitor mixture). Cells were scraped, resuspended by pipetting, and put on ice for 10 min. Then the extracts were centrifuged at 16,000 × g for 10 min to separate Triton-soluble (supernatant) and Triton-insoluble (pellet) fractions. The amount of protein was measured with a BCA protein assay kit (Pierce, Rockford, IL). For separation of α-syn oligomers and mature inclusion bodies the procedure was performed as described previously (Lee and Lee, 2002). Briefly, the cells were washed with PBS before the addition of buffer T [containing (in mm): 20 Tris, pH 7.4, 25 KCl, 5 MgCl2 plus 0.25 m sucrose, 1% Triton X-100, protease inhibitor mixture]. After buffer T was removed gently from dishes, the remaining Triton-insoluble material was collected in buffer N (0.1 m Na2CO3, pH 11.5, protease inhibitor mixture) and centrifuged at 80 × g for 10 min. The pellet containing mature inclusion bodies was dissolved in 1× Laemmli sample buffer (SB), and oligomers in the supernatant were centrifuged further at 16,000 × g for 10 min and dissolved in 1× SB.
Western blotting. Western blot analysis was performed according to the procedure described previously (Lee and Lee, 2002).
Immunofluorescence. The cell staining and confocal microscopy were performed as described previously (Lee and Lee, 2002).
Electron microscopy. COS-7 cells were infected with adeno/α-syn at a m.o.i. of 100 and incubated for 4 d. The sections were prepared and incubated with LB509 antibody, followed by incubation with 10 nm gold-conjugated goat anti-mouse IgG antibody as described in Gosavi et al. (2002).
Cell viability assay. SH-SY5Y cells were split in 96-well plates at a density of 1 × 105/well and differentiated for 5 d with 50 μm all-trans RA. Differentiated SH-SY5Y cells were infected with either adeno/α-syn or empty control vector at a m.o.i. of 100. The medium was changed the next day, and the reagents were treated on day 2 after infection. Cell viability was quantified via the MTS reduction assay (Promega, Madison, WI), or Luminescence ATP detection assay (PerkinElmer Life Sciences, Boston, MA), according to the manufacturers' instructions. For trypan blue exclusion assay the cells were trypsinized, resuspended in medium, and mixed with trypan blue dye solution (Sigma) before being counted with a hemacytometer. All assays were performed three times or more.
Results
In our previous study we have shown the progressive maturation of α-syn aggregates from small spherical oligomers to large fibrillar inclusion bodies in a tissue culture model (Lee and Lee, 2002). This process can be promoted by addition of the known mitochondrial inhibitor rotenone, and the preformed aggregates can be cleared with removal of the rotenone (H.-J. Lee et al., 2002b). In the present study we have investigated the susceptibility of different aggregate species to this aggregate-clearing activity, the mechanism of the clearance, and the effect of this mechanism on cell viability. COS-7 cells expressing human α-syn were treated with rotenone for 48, 56, 64, and 72 hr, followed by washout and incubation in rotenone-free medium for 24 or 48 hr. The duration of rotenone treatment correlated positively with the level of aggregates (Fig. 1A). These aggregates disappeared with removal of the rotenone, but the efficiency of aggregate clearance decreased with longer periods of rotenone treatment. Densitometric analysis of α-syn aggregates showed that the level of aggregates induced for 48 and 56 hr dropped by ∼90% after 2 d of washout, but those induced for 72 hr dropped by only 45% after the same washout period (Fig. 1A, bottom panel). One interpretation of this finding is that α-syn aggregates acquire resistance to the clearance mechanism as they evolve into more mature forms.
Figure 1.

Clearance of α-synuclein oligomeric intermediates. COS-7 cells expressing α-syn were treated with 100 nm rotenone for the indicated times and then incubated in fresh medium without rotenone for 1 or 2 d. A, Cells were extracted with PBS/1% Triton X-100, and the Triton-insoluble fractions were analyzed by Western blotting. The bottom panel shows densitometric analysis of the Western data. The relative density is obtained by calculating the percentage of remaining aggregates after the rotenone washout. B, Oligomeric intermediates (S) and mature fibrillar inclusion bodies (P) were separated before (-) and 1 d after (WO) the rotenone washout and were analyzed by Western blotting. C, Quantitation of the number of mature inclusion bodies. Before and after the rotenone washout the cells were fixed and labeled for α-syn by immunofluorescence. Cells with mature juxtanuclear inclusion bodies were counted in six different randomly selected areas, and the number of these cells was divided by the number of nuclei to obtain the percentage.
To test this hypothesis, we analyzed separately the susceptibility of oligomeric intermediates and mature fibrillar inclusion bodies to the clearance mechanism after the rotenone washout. These aggregates can be separated and quantified via a simple centrifugation procedure; complete separation of these species was confirmed previously with electron microscopy (EM) (Lee and Lee, 2002). At 1 d after removal of the rotenone the level of oligomeric intermediates was significantly lower, whereas that of mature inclusion bodies remained the same (Fig. 1B). The resistance of large inclusions to clearance was confirmed in a microscopic analysis, which showed little change in the number of mature inclusion bodies during the rotenone washout (Fig. 1C). These results suggest that preformed α-syn aggregates can be cleared from the cytoplasm and that early-stage oligomeric intermediates are more susceptible to clearance than mature fibrillar inclusion bodies.
To elucidate the mechanism underlying α-syn aggregate clearance, we investigated the effects of various inhibitors of proteasomes and lysosomes. COS-7 cells expressing α-syn were treated with rotenone for 56 hr; then the medium was replaced with fresh medium lacking rotenone but with various chemicals and incubated for 20 hr. Treatment of the cells with Baf, a specific inhibitor of vacuolar H+-ATPase that disrupts the pH gradient in acidic organelles including lysosomes (Bowman et al., 1988), abolished the aggregate-clearing activity in a dose-dependent manner, causing a net increase in the level of α-syn aggregates (Fig. 2A). In support of these data, E64, an irreversible cysteine-protease inhibitor that affects a subset of lysosomal proteases, also showed a dose-dependent inhibition of aggregate-clearing activity, although to a lesser extent. These results suggest that the lysosomal degradation pathway may be important for the clearance of α-syn aggregates. On the other hand, although the proteasome inhibitors β-lactone (LC) and Epox partially inhibited the aggregate clearance, their effects plateaued at a level that was much less than that of Baf (Fig. 2A). This result suggests that proteasomes contribute only partially to aggregate clearance and that the majority of aggregates might not be susceptible to proteasome-mediated degradation. Because Baf also is known to inhibit macroautophagy by inhibiting autophagosome-lysosome fusion (Kim and Klionsky, 2000), we examined the effect of 3-MA, an inhibitor of the early sequestration stage of macroautophagy (Seglen and Gordon, 1982). Unlike Baf, 3-MA showed little effect on the clearance of α-syn aggregates (Fig. 2B). A macroautophagy stimulator, rapamycin (Blommaart et al., 1995), did not show a significant effect on aggregate clearance either (data not shown). It is unlikely that the effects of these reagents on the levels of α-syn aggregates are attributable to the changes in monomer levels, because none of these reagents showed significant effect on monomer levels within the time scale of the experiment (Fig. 2A,B; TX-s). Using the fractionation procedure that separates small oligomers and mature inclusion bodies, we confirmed that oligomeric intermediates are the targets of Baf-sensitive clearance (Fig. 2C).
Figure 2.
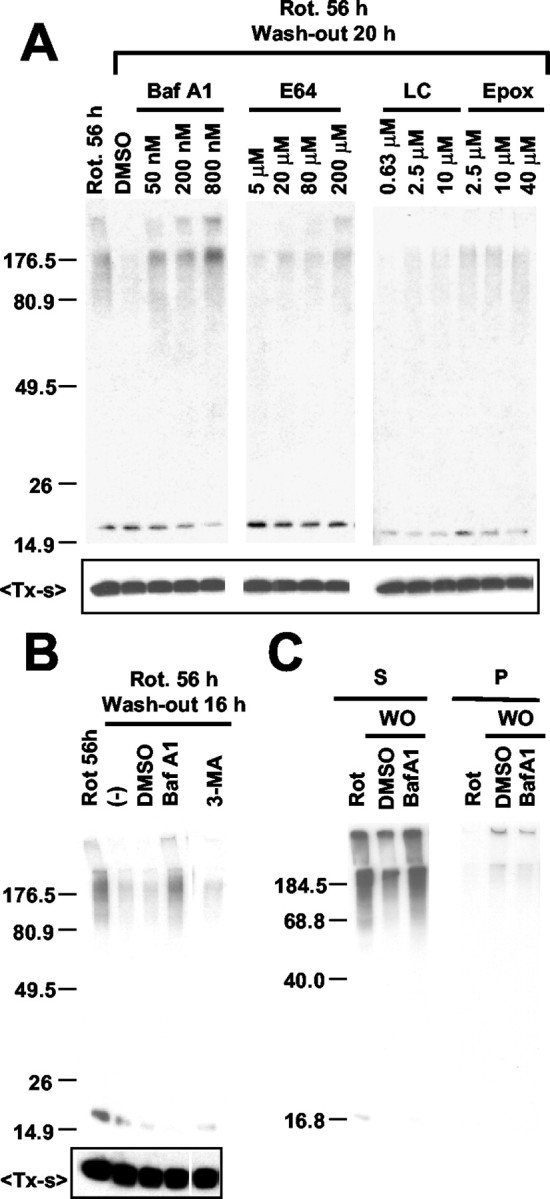
The mechanism of α-syn aggregate clearance. COS-7 cells expressing α-syn were treated with 100 nm rotenone for 56 hr, followed by rotenone washout. A, Lysosomal and proteasomal inhibitors were added at the indicated concentrations during the rotenone washout. B, During the rotenone washout COS-7 cells were treated with Baf (200 nm) or 3-MA (10 mm); (-) indicates no-treatment control. DMSO was treated as a vehicle control for Baf. C, After 16 hr of the rotenone washout (WO) in the presence of DMSO or Baf, oligomeric aggregates (S) and mature inclusion bodies (P) were separated and analyzed by Western blotting. In A and B, the top panels show Triton-insoluble aggregates, and the bottom panels show monomers in the Triton-soluble fractions (TX-s).
The involvement of the lysosomal pathway in α-syn aggregate clearance was demonstrated further by microscopic examination. COS-7 cells expressing α-syn were treated with rotenone for 56 hr, followed by incubation in rotenone-free medium for 20 hr. Immunofluorescence labeling of these cells showed that α-syn aggregates occasionally are surrounded by LAMP 2, a protein that is localized in the limiting membranes of the late endosomes and lysosomes (Fig. 3A, arrows). Immuno-EM study also showed the localization of α-syn-positive aggregates in vacuolar structures (Fig. 3B). These results further strengthen the conclusion that spherical α-syn oligomers are cleared from the cytoplasm via the lysosomal degradation pathway.
Figure 3.

Localization of α-syn aggregates in the lysosomes. A, COS-7 cells expressing α-syn were treated with 100 nm rotenone for 56 hr, followed by incubation in rotenone-free medium. Cells were fixed and labeled with antibodies for α-syn (red) and LAMP 2 (green). Arrows indicate α-syn aggregates captured within LAMP 2-positive compartments. Nuclei were stained with Hoechst 33258 (blue). B, EM analysis of COS-7 cells with α-syn aggregates. α-Syn aggregates are visualized by 10 nm gold-conjugated secondary antibody. Boxed area is magnified on the right. Scale bars: left, 0.4 μm; right, 0.2 μm. Arrowheads indicate immunogold particles that label spherical and amorphous aggregates in a vacuolar structure. The amorphous aggregate may represent ongoing degradation. N, Nucleus; C, centrosome.
To determine whether postmitotic neuronal cells also can clear α-syn aggregates, we characterized α-syn aggregation in SH-SY5Y human neuroblastoma cells. With RA treatment these cells differentiate into postmitotic neuron-like cells with extended neurites and neuronal marker proteins. At different stages of the differentiation the cells were infected with adeno/α-syn and incubated for 3 d (Fig. 4A). The cells were lysed in the presence of 1% Triton X-100, and the Triton-soluble and Triton-insoluble fractions were subjected to Western blotting by the use of anti-α-synuclein antibody. The Triton-soluble and Triton-insoluble fractions are defined in this study as the supernatant and the pellet of 16,000 × g centrifugation, respectively. Naive SH-SY5Y cells overexpressing α-synuclein did not produce a detectable level of aggregates, whereas the same level of expression induced aggregation in differentiated SH-SY5Y cells; the level of aggregation increased with a longer period of differentiation (Fig. 4B).
Figure 4.
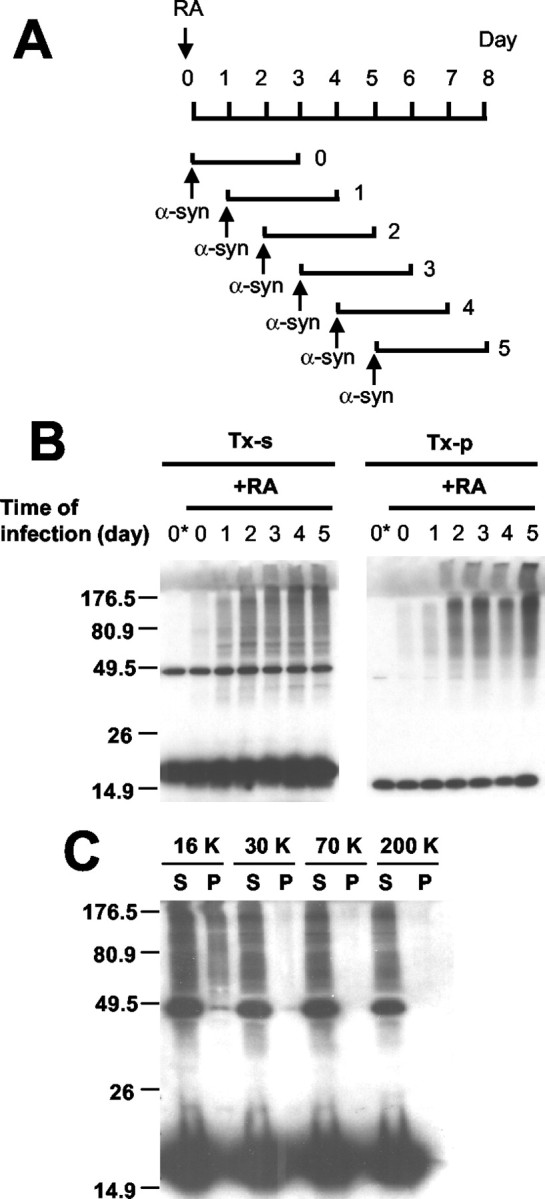
α-Synuclein aggregation in differentiated human neuroblastoma cells. A, Schematic presentation of differentiation and α-synuclein expression. α-Synuclein is expressed for 3 d in cells at different stages of differentiation. B, Western analysis of α-synuclein aggregation. The first lane (0*) of each panel shows the naive SH-SY5Y cells infected and processed in the same way. Note that the level of α-synuclein aggregation increases with a longer period of differentiation in both Triton-soluble (TX-s) and Triton-insoluble (TX-p) fractions. C, Sedimentation analysis of α-syn aggregates. Detergent extract of differentiated SH-SY5Y cells overexpressing α-syn was subjected to a sequential differential centrifugation. Cells were treated with 20 nm Baf before the extraction to enrich the aggregates (see Fig. 5). Numbers at the top indicate the centrifugal forces in gravity (g). S and P indicate the supernatant and the pellet of each centrifugation, respectively.
Aggregation of α-syn in differentiated SH-SY5Y has some characteristics that are distinct from those in COS-7 cells. Unlike COS-7 cells in which most aggregates are found in the Triton-insoluble fraction, we found that differentiated SH-SY5Y cells produced Triton-soluble and Triton-insoluble aggregates equally well (Fig. 4B). The Triton-soluble fraction was centrifuged sequentially at increasing centrifugal forces for 1 hr, and we found that the Triton-soluble aggregates remained in the supernatant even at 200,000 × g (Fig. 4C). This indicates that differentiated SH-SY5Y cells produce stable small oligomers in addition to larger oligomers that sediment at 16,000 × g. Rotenone had relatively little effect on the level of α-syn aggregates in differentiated SH-SY5Y cells, resulting in less than a twofold increase (data not shown), indicating subtle cell type-specific differences in the regulation of α-syn aggregation. However, microscopic characterization demonstrated that differentiated SH-SY5Y cells produce both spherical oligomers and mature inclusion bodies that have structural and compositional properties similar to those produced in COS-7 cells (Supplementary Fig. 1; available at www.jneurosci.org). Thus despite some differences the overall mechanism of α-syn aggregation in differentiated SH-SY5Y cells appears to be similar to that in COS-7 cells.
To determine whether α-syn aggregates also can be removed via the lysosomal pathway in differentiated SH-SY5Y cells, we treated cells overexpressing α-syn with lysosomal inhibitors. Treatment with Baf led to the accumulation of both the Triton-soluble and Triton-insoluble aggregates in a dose- and time-dependent manner, whereas little change was observed in the levels of monomer (Fig. 5A,B). On the other hand, Baf treatment did not cause the accumulation of α-syn aggregates in undifferentiated SH-SY5Y cells (Fig. 5C), in which aggregate formation was not detected (Fig. 4). This suggests that Baf affects the clearance of preformed aggregates rather than their de novo formation; if production were altered, Baf treatment would have increased the aggregate levels in undifferentiated cells. Cathepsin I, an inhibitor of lysosomal cathepsin proteases, also increased the level of α-syn aggregates, confirming the role of the lysosomal pathway. In contrast, neither aggregate nor monomer levels changed after the treatment with macroautophagy inhibitor 3-MA. These results suggest that the non-macroautophagic lysosomal degradation pathway might be involved in α-syn aggregate clearance in postmitotic neuronal cells. This is supported further by the finding that oligomeric aggregates often colocalize with LAMP 2 in differentiated SH-SY5Y cells treated with a lysosomal protease inhibitor (Fig. 6).
Figure 5.

Lysosomal inhibitors stabilize α-syn aggregates in differentiated SH-SY5Y cells. A, Differentiated SH-SY5Y cells overexpressing α-syn were treated with Baf, cathepsin inhibitor I, or 3-MA for 24 hr at the indicated concentrations. Triton-soluble (TX-s) and Triton-insoluble (TX-p) fractions were obtained from cell extracts and analyzed by Western blotting. B, Progressive accumulation of aggregates in differentiated SH-SY5Y after treatment with 20 nm Baf. C, Baf treatment does not lead to aggregate accumulation in undifferentiated SH-SY5Y cells.
Figure 6.

Overlap between α-syn punctate pattern and LAMP 2 in differentiated SH-SY5Y cells. Cells overexpressing α-syn were treated with 4 μm cathepsin inhibitor I for 24 hr and labeled with antibodies for α-syn (red) and LAMP 2 (green). Arrows indicate granular α-syn stains that overlap with LAMP 2-positive compartments. Nuclei were stained with Hoechst 33258 (blue).
We have shown previously that overexpression of α-syn in COS-7 cells leads to a reduction in cell viability and that cell death correlates with the formation of oligomeric intermediates (Gosavi et al., 2002). Differentiated SH-SY5Y cells overexpressing α-syn also showed reduced cell viability as compared with cells transduced with empty control vector, indicating that overproduction of α-syn has a toxic effect (Fig. 7A, filled square vs filled circle). To study the effect of Baf on α-syn-induced cell toxicity, we transduced the cells with either empty vector or adeno/α-syn and then treated them with Baf on day 2 after infection. Cell viability was measured by the MTS reduction assay. Treatment with Baf increased the toxic effect of α-syn even further, resulting in a greater rate of cell death as compared with DMSO-treated α-syn-expressing cells (Fig. 7A, filled vs open circle). The effect of Baf on α-syn-overexpressing cells became apparent on day 4, which is 2 d after the treatment, whereas Baf treatment of cells transduced with empty control vector did not cause significant cell death during the same period (Fig. 7A, open square). This suggests that Baf itself did not contribute directly to cell death until day 4, and that it did so by potentiating the toxic effects of α-syn. Enhancement of α-syn cytotoxicity in Baf-treated cells was confirmed by two additional assays: live cell counting (Fig. 7B) and ATP measurement (Fig. 7C). These results indicate that stabilization of α-syn oligomers by inhibition of the lysosomal pathway exacerbates α-syn toxicity.
Figure 7.

Lysosomal inhibition exacerbates α-syn-induced cytotoxicity. A, Differentiated SH-SY5Y cells were infected with empty control vector (dotted lines) or adeno/α-syn (solid lines) and were treated with DMSO (filled symbols) or 10 nm Baf (open symbols) on day 2 after infection. Cell viability was measured on days 3, 4, and 5 after infection, using the MTS reduction assay. To obtain relative viability, we used data from empty vector-infected cells on day 3 as a reference. B, Trypan blue exclusion assay. Cells were treated with 10 nm Baf on day 2 after infection, and live cell numbers were obtained on days 3 (white bars) and 4 (gray bars). C, ATP measurement. Cells were treated with 10 nm Baf, and ATP levels were measured on day 4. A, B, n = 3; C, n = 5. Error bars represent SEM. Statistical significance was assessed by one-way ANOVA and is indicated with *p < 0.01 and #p < 0.05.
Having demonstrated the effect of the lysosomal defect on the stabilization of α-syn aggregates in cells transiently overexpressing α-syn, we then asked whether the lysosomal defect has the same effect on endogenous neuronal α-syn. To investigate this, we studied the effect of Baf on α-syn aggregate levels in rat embryonic cortical neurons, which express high levels of endogenous α-syn. Neurons (11 d in vitro, DIV 11) were treated with increasing amounts of Baf, and high-molecular-weight α-syn aggregates were visualized by Western blotting. After Baf treatment the neurons accumulated a 62 kDa oligomeric α-syn in both Triton-soluble and Triton-insoluble fractions (Fig. 8A). Because the 62 kDa oligomer was identified in denaturing polyacrylamide gel, this oligomer could be a stable unit of much larger complex rather than an independent oligomer on its own. This finding confirms that lysosomal defect leads to the accumulation of an oligomeric form of α-syn generated from the endogenous protein in cortical neurons. Furthermore, when the neurons were treated with a mixture of protease inhibitors that block the activities of most of lysosomal proteolytic enzymes, the punctate pattern of α-syn immunoreactivity appeared in a time-dependent manner and overlapped with the LAMP 2-positive compartments (Fig. 8B), suggesting that the oligomers that fail to be degraded accumulate in the lysosomes.
Figure 8.
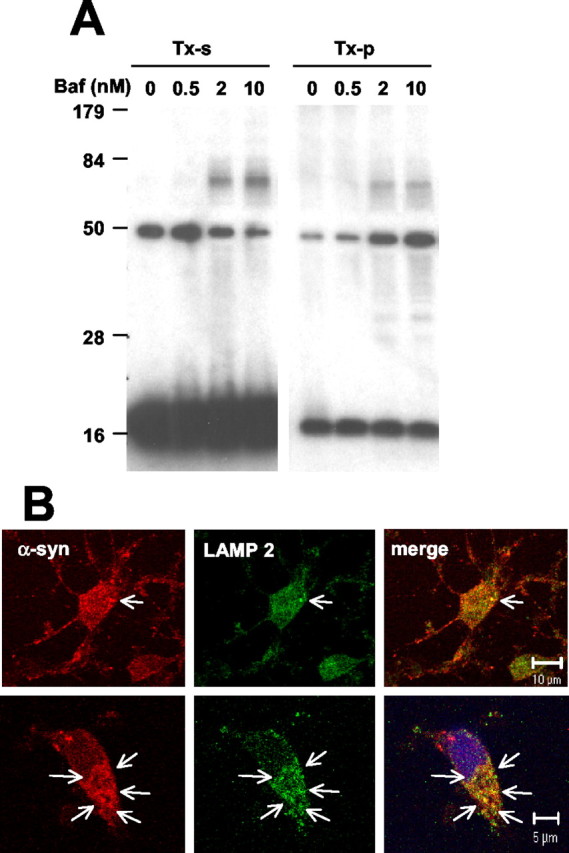
Accumulation of α-syn oligomers in rat cortical neurons. A, Neurons (DIV 11) were treated with Baf at the indicated concentrations for 24 hr. Endogenous α-syn in the Triton-soluble (TX-s) and Triton-insoluble (TX-p) fractions was analyzed by Western blotting. B, DIV 11 cortical neurons were treated with the mixture of serine, cysteine, and aspartic protease inhibitors for 2 hr (top images) and 4 hr (bottom images) and fluorescently labeled for α-syn (red) and LAMP 2 (green). Arrows indicate the colocalization between α-syn and LAMP 2. Nuclei were stained with Hoechst 33258 (blue).
Discussion
In the present study we confirmed that cells, including those of neuronal origin, are capable of clearing preformed α-syn aggregates. More importantly, the following findings point to the conclusion that only the oligomeric intermediates, but not the mature fibrillar inclusion bodies, are susceptible to the aggregate-clearing mechanism. First, α-syn aggregates become resistant to breakdown as the aggregates advance to more mature states (i.e., fibrillar inclusion bodies). Second, aggregate breakdown is observed only in the oligomer-enriched fraction, not in the mature inclusion body fraction. Third, the number of mature inclusion bodies does not decrease during the period in which oligomers undergo an extensive breakdown. Inhibition of this clearance mechanism causes rapid accumulation of aggregates in both differentiated human neuroblastoma cells and rat embryonic cortical neurons. These results suggest that aggregate clearance is a major determinant of the cellular α-syn aggregate load, thus demonstrating that the extent of accumulation of α-syn aggregates is determined by a dynamic equilibrium between production and removal of the aggregates.
Our study shows that the lysosomal degradation pathway is mostly responsible for the clearance of α-syn oligomers. This is supported by the findings that neutralization of acidic compartments and inhibition of lysosomal proteases block the oligomer clearance and that α-syn oligomers are present in LAMP 2-positive vacuolar structures. Although the proteasomal pathway does appear to contribute to aggregate clearance, it does so to a much lesser extent than the lysosomal pathway. This is consistent with the finding that oligomeric aggregates are mostly ubiquitin-negative and rarely colocalize with proteasomes (Lee and Lee, 2002) (see also Supplementary Fig. 1). Whether there are different populations of aggregates that are cleared distinctly by either the lysosomal or the proteasomal pathway is an important issue that needs to be addressed.
Although the mechanism of α-syn degradation is far from clearly understood, both the proteasome and lysosome systems appear to play a role in normal metabolism of this protein (Bennett et al., 1999; Paxinou et al., 2001; Tofaris et al., 2001; Liu et al., 2003; Webb et al., 2003). Because protein aggregation is sensitive to the expression level of protein, the accumulation of α-syn aggregates in response to various inhibitors of these degradation systems actually might be the consequence of simply elevating the cytoplasmic α-syn concentration. However, in our study the accumulation of α-syn oligomers occurs rapidly, within a matter of hours, whereas the estimated half-life of α-syn is as long as several days (Okochi et al., 2000; Paxinou et al., 2001). In fact, within the time scale of our experiments we did not detect significant changes in the monomer levels. Thus it is unlikely that changes in the monomer levels play a significant role in the observed effects of the lysosomal inhibitors on the aggregate accumulation.
One crucial question is how the aggregates are delivered to the lysosomes for destruction. Although little is known specifically about the delivery of protein aggregates to the lysosomes, a process called autophagy has been considered a major pathway for targeting various cytoplasmic substrates destined for degradation in lysosomes. Autophagy has been linked with some programmed cell death (Xue et al., 1999; Inbal et al., 2002) and is hyperactivated both in a mouse model of neurodegeneration (Yue et al., 2002) and in human neurodegenerative diseases, such as PD (Anglade et al., 1997) and Alzheimer's disease (Nixon et al., 2000). In culture the expression of mutant huntingtin in striatal neurons (Kegel et al., 2000; Petersen et al., 2001) and exposure of dopaminergic neurons to methamphetamine (Larsen et al., 2002) caused autophagy-associated degeneration. Furthermore, overexpression of the A53T mutant form of α-syn stimulated autophagic cell death in PC12 cells (Stefanis et al., 2001). There are distinct autophagic mechanisms: macroautophagy, microautophagy, and chaperone-mediated autophagy. Delivery of bulk cytoplasmic “garbage” to lysosomes is mediated by macroautophagy. This process is initiated by enclosing obsolete organelles and proteins with double membrane structures called autophagosomes, which then are fused with the lysosomes to form autophagic vacuoles (Kim and Klionsky, 2000). Microautophagy, on the other hand, occurs when lysosomes invaginate their own membranes, resulting in direct uptake of parts of the cytoplasm (Dunn, 1994). Another mechanism called chaperone-mediated autophagy involves direct import of cytosolic proteins into the lysosomes (Chiang et al., 1989).
To assess the role of autophagy in α-syn aggregate clearance, we investigated the effects of macroautophagy inhibition and stimulation and found that neither inhibition nor stimulation showed a significant effect. Consistent with this finding, visualization of autophagic vesicles with a fluorescent dye, monodansylcadaverine (Biederbick et al., 1995), showed no colocalization between α-syn aggregates and these vesicles (data not shown). Although macroautophagy does not appear to play an important role in the breakdown of α-syn aggregates, other autophagic mechanisms might be involved and thus are worth further investigation. Alternatively, it is also possible that the endosomal pathway, which is another mechanism of substrate delivery for lysosomal degradation, may play a role in the clearance of α-syn aggregates. Interestingly, we have shown recently that α-syn aggregation can initiate and develop in membranes (H.-J. Lee et al., 2002a). These membrane-associated aggregates thus may be targeted to the lysosomes via the endosomal pathway.
We have identified several distinct oligomeric species both in COS-7 cells (Lee and Lee, 2002) and in differentiated SH-SY5Y cells (S.-J. Lee, unpublished observations); their sizes range widely from dimer to multimers. COS-7 and differentiated SH-SY5Y cells show distinct patterns of size distribution at steady state: COS-7 cells mostly have multimers large enough to sediment at 16,000 × g, whereas more than one-half of the SH-SY5Y aggregates are found in the supernatant of a 200,000 × g spin. The multimers found in the 16,000 × g pellet are spherically shaped, with sizes of 15-60 nm in diameter (Lee and Lee, 2002), much larger than the spherical protofibrils (2-8 nm) produced from purified recombinant α-syn by Lansbury and colleagues (Ding et al., 2002). Some smaller multimers, on the other hand, show sedimentation properties similar to the in vitro protofibrils (Y. S. Kim and S.-J. Lee, unpublished observations). Except for the relative sizes, there is no structural information available at the moment for distinguishing these aggregate species between themselves and from the in vitro protofibrils. Therefore, we call all of the nonfibrillar aggregates collectively “oligomers” in this report. In cells overexpressing α-syn, these nonfibrillar oligomers appear before the formation of the juxtanuclear inclusion bodies that contain α-syn fibrils, and both the fibrillation and the inclusion body formation are blocked by disrupting the transport of these oligomers (Lee and Lee, 2002). These results support the conclusion that some of these oligomers, if not all, are the precursors of the inclusion bodies and the intermediates of the cellular fibrillation process.
In the differentiated SH-SY5Y model we show that the accumulation of oligomeric intermediates correlates with α-syn-mediated cytotoxicity. Some studies have raised the possibility that α-syn oligomers act directly as proteotoxins (Volles et al., 2001; Kayed et al., 2003). Alternatively, it is also plausible that the accumulation of these oligomers might overburden the cellular machinery that is involved in the transport and clearance of these aggregates. This abnormally high burden not only might disturb normal cell physiology but also might render cells highly sensitive to additional stresses. However, it needs to be noted that the accumulation of oligomers would lead to the formation of mature inclusion bodies, and there is a view that inclusion bodies are participating directly in cellular dysfunction and demise (Giasson and Lee, 2003). Both in cells and in test tubes the α-syn fibrillation process appears to involve multiple rounds of conversion from one intermediate form to more advanced forms. How this process occurs in a cellular context and how the cells respond to these aggregates on their accumulation are critical questions that remain to be addressed. Solving these questions should advance our understanding of the mechanism that underlies α-syn cytotoxicity and, eventually, of the pathogenesis of α-synucleinopathies.
Lysosomal processing capacity appears to diminish progressively over the lifespan of an animal (Brunk and Brun, 1972; Nakamura et al., 1989), and lysosomal malfunction has been associated with age-related neurodegenerative disorders (Nixon et al., 2000; Bahr and Bendiske, 2002). The link between lysosomal dysfunction and neurodegeneration has been strengthened further by the finding that the inhibition of lysosomal enzymes leads to some of the features of age-related neurodegenerative diseases, such as protein deposition, synaptic loss, and neuronal demise (Takauchi and Miyoshi, 1989; Okada et al., 1994; Bendiske et al., 2002; Felbor et al., 2002). The results presented in the current study link lysosomal disturbance to the accumulation of α-syn oligomers and α-syn-mediated cell death, implicating the importance of the lysosomal pathway in the cellular response to α-syn aggregation. We propose that the lysosomal pathway is a central protective mechanism against α-syn oligomer-mediated toxicity and that stimulation of this pathway may be an effective approach to the prevention and treatment of PD and other related disorders.
Footnotes
This work was supported by the Michael J. Fox Foundation for Parkinson's Research and the Abramson Family Foundation. Monoclonal antibody for LAMP 2 (H4B4) developed by Drs. August and Hildreth was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA. We thank Stephen Lee and Amy Manning-Bog for critically reading this manuscript and Nafisa Ghori for technical assistance with EM.
Correspondence should be addressed to Seung-Jae Lee, The Parkinson's Institute, 1170 Morse Avenue, Sunnyvale, CA 94089. E-mail: slee@thepi.org.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241888-09$15.00/0
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12: 25-31. [PubMed] [Google Scholar]
- Bahr BA, Bendiske J (2002) The neuropathogenic contributions of lysosomal dysfunction. J Neurochem 83: 481-489. [DOI] [PubMed] [Google Scholar]
- Bendiske J, Caba E, Brown QB, Bahr BA (2002) Intracellular deposition, microtubule destabilization, and transport failure: an “early” pathogenic cascade leading to synaptic decline. J Neuropathol Exp Neurol 61: 640-650. [DOI] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM (1999) Degradation of α-synuclein by proteasome. J Biol Chem 274: 33855-33858. [DOI] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66: 3-14. [PubMed] [Google Scholar]
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ (1995) Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 270: 2320-2326. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K (1988) Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA 85: 7972-7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk U, Brun A (1972) The effect of aging on lysosomal permeability in nerve cells of the central nervous system. An enzyme histochemical study in rat. Histochemie 30: 315-324. [DOI] [PubMed] [Google Scholar]
- Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246: 382-385. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee S-J, Rochet JC, Ding TT, Williamson RE, Lansbury Jr PT (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA 97: 571-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding TT, Lee S-J, Rochet JC, Lansbury Jr PT (2002) Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41: 10209-10217. [DOI] [PubMed] [Google Scholar]
- Dunn Jr W (1994) Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol 4: 139-143. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394-398. [DOI] [PubMed] [Google Scholar]
- Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR (2002) Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci USA 99: 7883-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Lee VM (2003) Are ubiquitination pathways central to Parkinson's disease? Cell 114: 1-8. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34: 521-533. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2: 492-501. [DOI] [PubMed] [Google Scholar]
- Gosavi N, Lee H-J, Lee JS, Patel S, Lee S-J (2002) Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem 277: 48984-48992. [DOI] [PubMed] [Google Scholar]
- Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A (2002) DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 157: 455-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486-489. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M (2000) Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20: 7268-7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Klionsky DJ (2000) Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem 69: 303-342. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF (2003) Oxidative dimer formation is the critical rate-limiting step for Parkinson's disease alpha-synuclein fibrillogenesis. Biochemistry 42: 829-837. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18: 106-108. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D (2002) Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci 22: 8951-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Lee S-J (2002) Characterization of cytoplasmic α-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem 277: 48976-48983. [DOI] [PubMed] [Google Scholar]
- Lee H-J, Choi C, Lee S-J (2002a) Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem 277: 671-678. [DOI] [PubMed] [Google Scholar]
- Lee H-J, Shin SY, Choi C, Lee YH, Lee S-J (2002b) Formation and removal of α-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277: 5411-5417. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002) Human α-synuclein harboring familial Parkinson's disease-linked Ala-53→ Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99: 8968-8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ (2003) Endoproteolytic activity of the proteasome. Science 299: 408-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265-1269. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Takeda M, Suzuki H, Morita H, Tada K, Hariguchi S, Nishimura T (1989) Lysosome instability in aged rat brain. Neurosci Lett 97: 215-220. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM (2000) The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res 25: 1161-1172. [DOI] [PubMed] [Google Scholar]
- Okada M, Miyake T, Kitamura T, Kawasaki K, Mizushima Y (1994) Anti-ubiquitin immunoreactivity associates with pyramidal cell death induced by intraventricular infusion of leupeptin in rat hippocampus. Neurosci Res 19: 59-66. [DOI] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C (2000) Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J Biol Chem 275: 390-397. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H (2001) Induction of α-synuclein aggregation by intracellular nitrative insult. J Neurosci 21: 8053-8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D (2001) Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet 10: 1243-1254. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Price PJ, Brewer GJ (2001) Serum-free media for neural cell cultures: adult and embryonic. In: Protocols for neural cell culture, 3rd Ed (Fedoroff S, Richardson A, eds), pp 255-264. Totowa, NJ: Humana.
- Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79: 1889-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci USA 97: 4897-4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95: 6469-6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA (2001) Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21: 9549-9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takauchi S, Miyoshi K (1989) Degeneration of neuronal processes in rats induced by a protease inhibitor, leupeptin. Acta Neuropathol (Berl) 78: 380-387. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG (2001) Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509: 22-26. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM (1998) Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death Differ 5: 832-837. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Lee H-J, Li J, Fink AL, Lee S-J (2001) Stabilization of partially folded conformation during α-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem 276: 43495-43498. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lansbury Jr PT (2003) Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry 42: 7871-7878. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee S-J, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury Jr PT (2001) Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40: 7812-7819. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278: 25009-25013. [DOI] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM (1999) Autophagy is activated by apoptotic signaling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci 14: 180-198. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N (2002) A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35: 921-933. [DOI] [PubMed] [Google Scholar]