

Abstract
Recent advances in biotechnology, such as Hi-C, CRISPR/Cas9, and ribosome display, have place nucleoprotein complexes at center stage. Understanding the structural dynamics of these complexes aids in optimizing protocols and interpreting data for these new technologies. The integration of simulation and experiment has helped advance mechanistic understanding of these systems. Coarse-grained simulations, reduced-description models and explicit solvent molecular dynamics simulations yield useful complementary perspectives on nucleoprotein complex structural dynamics. When combined with Hi-C, cryo-EM, and single molecule measurements, these simulations integrate disparate forms of experimental data into a coherent mechanism.
Introduction
Due to the large size of the nucleoprotein complexes (e.g., ~3 million atoms in for the ribosome in explicit solvent) and long time scales involved (~seconds), computational studies must use various approximations, often requiring compromises between force field accuracy and the time scale sampled. One is reminded, however, that simplified models can sometimes be powerful. For example, in the case of the orbits of the planets around the Sun, the Earth and every organism on it are approximated by a single point particle; yet the resulting model produces highly accurate predictions for the Earth’s orbit. As often occurs in molecular dynamics simulations, high levels of detail in the force field are typically attained at the cost of the time scale sampled, and in turn, accuracy of the entropic contribution to the free energy. Similarly, enhanced sampling techniques that produce exhaustive sampling either (1) reduce the level of detail in the force field or (2) simulate portions of the macromolecular complex. Each technique, however, is quite useful on its own right and provides new insight and new perspectives into dynamics that were previously unexplored experimentally. In many cases, the beginning and end states of conformational changes are known from cryo-EM, along with rates determined by single molecule FRET. Molecular simulation provides a powerful way to merge available experimental data into a single, integrated movie, yielding a highly useful perspective unavailable from experimental data alone. In this review, we summarize many of the various computational techniques used to study nucleoprotein complexes. While the review is by no means entirely comprehensive, it serves as a starting point for researchers in the field.
Structural modeling of the ribosome
Early computational studies of the ribosome concentrated on structural modeling of the bacterial ribosome, before higher resolution x-ray crystallography structures were solved [1–6]. RNA homology methods were used to construct predictive models of the E. coli 30S ribosomal subunit and the T. thermophilus 70S ribosome highly consistent with cryo-EM data and used for phasing x-ray crystals [7,8]. After X-ray crystallographic structures of ribosomal subunits were solved, it was possible to perform molecular dynamics simulations of important functional regions [9,10], normal mode calculations [11] and Poisson-Boltzmann analysis on the intact ribosome[12]. In a very interesting use of explicit solvent molecular dynamics simulation, Frank and co-workers performed simulations of the tRNA in solution and selected conformations with very close correlations to their tRNA cryo-EM density inside the ribosome [13].
Computational studies of intact ribosomes: coarse-grained and reduced description simulations
Normal mode analysis and elastic network models have described overall global motions of the ribosome at nucleotide resolution [11,14,15]. These studies reproduced intersubunit rotation, movement of the small subunit head and large-scale motions of the L1 stalk and L7/L12 stalk in bacteria. This technique was also used to perform some of the first automated molecular fitting of cryo-EM reconstructions [16,17]. Further elastic network models have depicted the coupled motions of tRNA and mobile domains of the ribosome [18]. Jernigan and co-workers also examined dynamics of the mRNA channel on the ribosome, a key player in tRNA translocation through the ribosome, suggesting that the entry clamp may assist in base recognition to ensure proper selection of the incoming tRNA [19]. In addition to elastic network models, early electrostatic calculations determined charged regions on the ribosome [12], showing the surface of the ribosome to be mostly negative with scattered positively charged regions corresponding to ribosome proteins.
Reduced-model molecular dynamics simulations have explored larger regions of configurational space and longer time scales. Trylska and co-workers performed coarse-grained molecular dynamics of the entire ribosome on a half a microsecond time scale, accounting for large-scale motions by implementing a Morse potential function, observing intersubunit rotation and stalk motion[20]**. Here, large-scale collective motions such as the rotation of the subunits were produced. In addition, the anti-correlated movement of the ribosomal stalks positioned on the opposite sides of the large subunit was observed. Studies performed by Trylska et al. also enabled the investigation of large-scale conformational changes coupled with electrostatic effects.
All-atom reduced model simulations based on a structure-based potential have enabled systematic studies of spontaneous large-scale conformational changes with atomistic detail [21]. Here, reversible excursions of tRNAs into and out of ribosomal binding sites were observed, consistent with recent single molecule FRET experiments. These simulations produced sampling on the order of ~200 milliseconds for the ribosomal complex in atomistic detail.
In studies of intersubunit rotation of the entire ribosome, Whitford and co-workers, by accounting for molecular flexibility, demonstrated that rotation likely involves asynchronous movement of the bridges, where the transition state is associated with partial rotation and the full displacement of bridge B8 [22].
Noller and co-workers have performed a very interesting computational study on ribosome dynamics, where they use translation-libration-screw (TLS) constraints to refine atomistic models based on x-ray diffraction data [23]. Here, they examine the mobilities of the peptidyl transferase center, the Shine-Dalgarno helix, as well as the 30S rotation relative to the 50S subunit. This was followed by an innovative study placing the head swivel movement in the context of nearly every structural study of translocation, showing that a two-hinge mechanism enables head-swivel [24].
In the first molecular simulations of the eukaryotic ribosome, experimentally determined SHAPE chemical probing data was integrated into a structure-based force field to determine conformational hotspots during translocation [25,26].
Computational studies of intact ribosomes: explicit solvent molecular dynamics simulations
Explicit solvent molecular dynamics simulations of the entire ribosome have examined large-scale conformational changes of the ribosomal complex. One of the more tractable conformational changes is movement (accommodation) of tRNA from its partially bound state (A/T) to its fully bound state (A/A) that occurs during tRNA selection. Targeted molecular dynamics simulations correctly predicted the regions of the large subunit that interact with the tRNA during this conformational change (accommodation corridor) [27]. These simulations are consistent with snoRNA deletion studies in yeast, producing ribosomes with unmodified nucleotides in the accommodation corridor[28,29]. The resulting ribosomes displayed fidelity phenotypes including defects in −1 frameshifting, peptide bond formation and misreading [29]. Mutational studies by the same group demonstrated that mutations of the predicted three-dimensional gate (A2556, 2492 and 2483) are viable for normal ribosomes, but dramatically effect growth when treated by antimicrobials [30]. The Dontsova lab also performed mutations on the same three nucleotides [31,32], finding that two of the nucleotides impair peptide release while the third is lethal and acts to block peptide bond formation, presumably by impeding tRNA accommodation into the fully bound (A/A) conformation. Simulation molecular simulation studies delineated the corridor (hybrid corridor) that facilitates movement of tRNAs from the classical state (A/A, P/P) to the hybrid state (A/P, P/E) [33] [34]. Molecular dynamics simulations of the intact ribosome have also examined dynamics of the L1 stalk and the ribosome exit tunnel in the large subunit of the ribosome [35–37]. Explicit solvent simulations of the 70S ribosome have connected measured kinetic rates of accommodation, intersubunit and head swivel motions to free energy barriers [38] [39]*. These microsecond simulations of the ribosome (3.2 million atoms) measure the diffusion of tRNA inside the ribosome, intersubunit rotation and head swivel, producing an estimate of the pre-factor in the Ahrenius equation for this process. The first simulations of ribosome-EF-P interactions, combined with cryo-EM, showed that EF-P modification and absence of EF-P protein result in the peptidyl-tRNA moving away from the A-site tRNA, generating a geometry that is incompatible with peptide bond formation [40]. A similar study of ErmBL-stalled ribosomes also examined movement in the peptidyl transferase center [41]. The studies nicely integrated simulations of the peptidyl transferase center with simulations of the full ribosome.
Computational studies of functional regions of the ribosome:
Another class of ribosome simulations focuses on localized regions of the ribosome that are critical for protein synthesis. Quantum mechanical studies of the peptidyl transferase center have explored reaction mechanisms [42,43]. Warshel an co-workers examined the mechanism of GTP hydrolysis by EF-Tu during tRNA selection, concluding that the critical residue H84 contributes to an allosteric effect [44]. Wieden and co-workers have performed extensive studies on the mechanism of EF-Tu, including one very interesting investigation that combines molecular dynamics simulations with rapid kinetics studies [45]. Aqvist and co-workers have also examined the mechanism of the peptidyl transferase reaction, combing an empirical valence bond description with molecular dynamics simulations, concluding that an acid-base catalysis method is unlikely, favoring a proton-shuttling mechanism that does include the P-site adenine O2’ oxygen [42,46]. Aqvist and co-workers have also applied quantum chemical methods to termination [47].
Short time scale simulations of the decoding center complexed with mRNA and tRNA anticodon stem loops have examined the stability of the decoding center hydrogen bond network for a wide range of cognate, near-cognate and non-cognate mRNA-tRNA combinations [48], suggesting that ribosomal RNA had a stabilizing effect on cognate codon-anticodon interactions and a destabilizing effect on certain non-cognate combinations. Aqvist and co-workers performed a similar study with longer aggregate sampling, estimating binding free energies [49]. They applied a similar method to study stop-codon recognition [46] and studied the interaction between the L7/L12 stalk, showing that complementary charge-based interaction between L12-CTD and IF2 is important for fast subunit association [50]. Aqvist and Green studied the mechanism of peptide release, finding that the 2’ OH of the P-site substrate is critical for orienting the nucleophile in a hydrogen-bonding network productive for catalysis [51].
Several studies investigated the dynamics of the decoding center helix [52–55]. Explicit solvent simulations of the decoding center complexed with antibiotics examined the stability of the drug-ribosome interactions [53]. Exhaustive sampling simulations (replica exchange molecular dynamics simulations) show a high degree of convergence after one microsecond in the absence of antibiotics and 15 microseconds the presence of gentamicin. These studies show the decoding center bases (A1492 and A1493) flipping in and out of helix 44 in absence of drug [52,53], while in the presence, the equilibrium shifts towards the flipped out state with time scales consistent with 2-aminopurine fluorescence studies of the decoding helix. The studies produced the first energy landscapes of the ribosome, and suggest that an entropy shuttling mechanism may move the drug from local minimum to local minimum until the binding site is reached. Brooks and co-workers performed studies suggesting that flipping of the decoding bases can distinguish between cognate and near-cognate tRNAs [51].
Trylska and co-workers performed explicit solvent molecular dynamics simulations of decoding introducing various mutations, finding differences in the internal mobility of the A-site, as well as in ion and water density distributions inside the binding cleft, between the prokaryotic and mutated RNA [54]. They also examined the effect of A2058G on clindamycin activity[56]. In an important study, Trylska and co-workers used Brownian dynamics simulations to examine antibiotic binding to the 30S subunit, showing that electrostatic steering is not the sole factor directing the aminoglycosidic antibiotic toward the binding site on the 30S ribosomal subunit. The simulations explain why paromomycin overstabilizes the 70S ribosome complex and precludes its dissociation into 30S and 50S subunits. The study unveiled other binding clefts in ribosomal RNA and explained the physical mechanisms of aminoglycoside diffusion and binding to the ribosomal RNA [57]*. A number of additional studies of localized ribosomal regions have focused on drug-ribosome interactions [53–55,58].
Sponer and co-workers have explicit solvent simulations of three-way junctions present in the ribosome, demonstrating that the junctions contribute to functional fluctuations of the ribosome, including 5S rRNA, A-site finger, and L7/L12 stalk RNA [59]*. Sponer, Frank and co-workers performed studies of the A-site finger [60]. Several groups have used molecular simulations to study dynamics and peptide folding within the exit tunnel of the large subunit of the ribosome [61–66]. O’Brien an co-workers identified a new source of mechanical force acting on the ribosome by combining experimental measurements of changes in nascent-chain extension in the exit tunnel in conjunction with all-atom and coarse-grained simulations, observing that the route of force transmission is shown to be through the nascent polypeptide’s backbone, not through the wall of the ribosome’s exit tunnel [67].
Nucleosomes, di-nucleosomes, tri-nucleosomes, and tetra-nucleosomes
A wide range of single and multiple nucleosome simulations have been performed, both at atomistic and coarse-grained levels of detail. Simulations of single nucleosomes have studied the flexibility of DNA and histone tails in the context of nucleosomes[68]. In the context of a comprehensive multiscale study, multimicrosecond explicit solvent molecular dynamics simulations of dinucleosome, including histone tails were performed with three different state-of-the-art force fields and validated by experimental NMR measurements. Takada and co-workers have examined the effect of histone acetylation on trinucleosome dynamics [69]. Coarse-grained simulations of unrolling have been studied by Langowski and co-workers [70]. The SIRAH force field extension for protein-DNA complexes has been used to perform coarse-grained simulations of tetranucleosomes to study DNA dynamics within the context of this system[71]. Many coarse-grained models of DNA itself have been described previously[72].
Coarse-grained simulations of chromatin: nucleosome arrays and intact gene loci
Schlick and co-workers have developed a coarse-grained model for polynucleosome systems over the past 15 years, that captures the physics of nucleosome-nucleosome interactions and sterics, electrostatics, flexible DNA and histone tails, consistent with many forms of experimental data [73–76]. Because the model has much more detail than full chromosome bead polymer models, but less detail than atomistic models, the model is referred to as mesoscale. The model includes different coarse-grained representations for linker DNA, flexible histone tails, linker histone, and nucleosomes [74–79]. The mesoscale model [80–86] has reproduced properties related to internucleosome distances, entry/exit angles, sedimentation coefficients, diffusion contacts, fiber curvature, force versus extension measurements, and cross linking data [73,74,84,87–89]. More recently tools related to electrostatics, Langevin dynamics, Monte Carle and enhanced sampling have been added [82,84,90–92] [87,93,94]. Recently, the mesoscale model was used to study HOXC, producing the first folded gene structure that was defined from first principles (nucleosome positions, epigenetic marks, linker histones) [95]. A second mesoscale model uses a globular histone core and flexible histone tails described by one particle per each amino acid, taking into account their net charge. DNA wrapped around the histone core is approximated at the level of two base pairs represented by one bead (bases and sugar) plus four beads of charged phosphate groups. Simulations of this model reproduced experimental results and the structure of the nucleosome-nucleosome contacts [96].
Intact gene loci: explicit solvent simulations
Recently, the first explicit solvent molecular dynamics simulations of an intact gene locus were performed (GATA4), consisting of 427 nucleosomes and 83 kb of DNA, requiring approximately one billion atoms [97]. Here coarse-grained models of GATA4 from Schlick and co-workers [97,98] were remodeled to construct an all-atom model. This was solvated with water and ion excess ions, minimized and run in molecular dynamics mode with particle mesh Ewald using the GENESIS molecular dynamics code. Scaling past 500,000 processor cores was achieved.
Coarse-grained simulations of intact chromosomes and genomes
The advent of high resolution chromatin capture technology (Hi-C) has produced intense interest in 3-D modeling of chromosomes. By crosslinking distal regions of chromosomes and sequencing them at high resolution, Hi-C allows one to map interactions between chromosome regions, enabling approximate 3-D reconstructions of chromosomes. A working model of chromatin architecture resembling a fractal globule has been proposed [99,100]. Early Hi-C studies demonstrated that chromosomes tend to be organized into large-scale (~5 Mb) A/B compartments and smaller topologically associating domains (TADs) within the compartments (0.3–0.8 Mb). While the TADs entail modular, locally isolated interactions, the compartments form a checker board pattern in the contact map, depicting longer-range interactions that segregate active, A/T rich regions from less active, G/C rich regions [99,100]. The A/B compartments can be extracted by Eigen-vector analysis (i.e., principal component analysis). Interestingly, in the X chromosome in females, one of two X chromosomes is inactivated, whereby a long non-coding RNA (Xist RNA) effectively coats the chromosome, ultimately resulting in a massive reorganization of the chromosome architecture: A/B compartments are replaced by a different set of compartments (S1/S2) [101–103]. Here, the interactions are segregated according to their interactions with the Xist RNA. That is, during X chromosome inactivation, the inactive X chromosome is partitioned into Xist-rich S1 and Xist-poor S2 compartments, which are eventually merged into a single compartment by the SMC protein, SMCHD1[101–103].
Using a very coarse-grained (typically 1 bead > 50 kB) homopolymer model, one can use Monte Carlo [100,104–108] or Langevin dynamics simulations [109–115] to mimic the structure and dynamics of chromatin. These and other simulations often display raindrop shaped features analogous to the topologically associating domains (TADs) characteristic of Hi-C maps [112–116]. More recently, knowledge-based potential terms directly incorporate Hi-C and other data to obtain contact maps quite similar to experimentally measured Hi-C maps [107,109–111,113–115]. Regarding Langevin dynamics, Onuchic and Wolynes have constructed a quasi-equilibrium homopolymer model (MiChroM) including interchromosomal interactions, loop extrusion factors (e.g., CTCF and cohesion), and a maximum entropy term to incorporate interactions observed in Hi-C experiments [114,115]. Their quasi-equilibrium energy landscape studies were used to demonstrate that the confinement of crowding effectively slows diffusion, consistent with experimentally observed diffusion, viscoelasticity and spatial confinement. The model is highly extensible, allowing the authors to construct an excellent model incorporating ChIP-seq data, enabling the study of a variety of epigenetics marks [113].
Innovative coarse-grained methods
Several interesting coarse-grained methods have been developed recently that can be applied to interesting aspects of nucleoprotein complex mechanism. Wu and co-workers developed a Monte Carlo simulation algorithm which incorporates both molecular factors including conformational changes of cellular adhesion molecules and cellular factors including fluctuations of plasma membranes to approach the physical process of adhesion [117]. A highly effective method that combines molecular simulation with SAXS and hydroxyl-radical probing experiments has been developed by Yang and co-workers [118]. Zheng and co-workers developed an interpolated elastic network model (iENM) protocol to construct a transition pathway for large macromolecular complexes such as SNARE [119]. Several other methods are described in a comprehensive review by Kolinski and co-workers [120].
Molecular simulations of the CRISPR-Cas9 and related complexes.
In terms of biotechnology, one of the most exciting areas of molecular simulations of nucleoprotein complexes is the CRISPR-Cas9 system used in gene editing. McCammon and co-workers have performed a number of pioneering studies, incorporating the latest experimental data to shed light on the mechanism of CRISPR-Cas9 [121,122]. Using Gaussian-accelerated molecular dynamics, they reveal the conformational dynamics of Cas9 during activation toward catalysis and find the conformational transition of Cas9 from its apo form to the RNA-bound form. Interesting coarse-grained studies of CRISPR mechanism have been recently performed by Zheng [123].
Single molecule and other in vitro studies
Single molecule studies often reveal the dynamics of a system, connecting structural studies to computer simulations. As a result, a comprehensive picture of mechanism is constructed that integrates multiple experimental perspectives of the same phenomena. For ribosomes, nucleosomes and chromatin, single molecule experiments have played key roles in advancing mechanism.
Regarding larger regions of chromatin, a number of interesting studies have produced new insight into the compaction of chromatin architecture within a rigorous, in vitro setting. Nordenskiold and co-workers recently performed single molecule studies of a 165 kb mimic of chromatin, reconstituting chromatin from T4 phage DNA (165 kb) and recombinant human histone octamers. In this exciting study, the authors probed the compaction of chromatin as a function of magnesium and spermine ions using single molecule fluorescence microscopy and dynamic light scattering, identifying ~250–400 nm condensates, comparable to the compaction observed in vivo. In addition, the authors performed transmission electron microscopy (TEM) and atomic force microscopy to clearly identify single nucleosomes, beads-on-a-string formations and aggregrated nucleosomes [124]. Fierz and co-workers use single molecule FRET to measure the time scales of nucleosome-nucleosome stacking, revealing that even tightly packed nucleosomes undergo reshuffling of configurations, exhibiting a hierarchy of dynamics of micro- to milliseconds [125]. Specifically, they examine an array of twelve nucleosomes organized into three tetranucleosome units, where Alexa 568 donors and Alexa 647 acceptors are placed on the DNA of neighboring nucleosomes, directly measuring nucleosome-nucleosome stacking. Discrete tetranucleosome units exchange nucleosome stacking partners (register interconversion) on the order of hundreds of microseconds. Distorted and open configurations are also sampled. They examine compaction caused by HP1a, a factor that cross bridges H3K9me3-modified nucleosomes. When HP1α is added, causing even more compaction, significant dynamics persist. Dekker and co-workers used flow through microfluidic chambers to dynamically image the process of DNA loop extrusion by yeast condensing, extruding ~10kb of DNA at 1.5 kb/s using ATP hydrolysis[126]. Bulk FRET studies of show that the chromatin architectural factors, linker histone (H1) and MeCP2 (Rett Syndrome) produce compaction by trapping the nucleosomes in more tightly wrapped states[127]. In combination with restriction enzyme accessibility studies for 17-mer nucleosome arrays, the study suggests that linker histone and MeCP2 may create higher-order chromatin structure susceptible to remodeling by ISWI. In small angle X-ray scattering studies (SAXS), Luger and co-workers show that nucleosomes with extra-nucleosomal DNA engage additional binding sites in MeCP2, resulting in a compact higher-order complex[128]. Time resolved SAXS experiments by Pollack and co-workers examined salt-induced disassembly of nucleosome particles, demonstrating that histone protein dimers are released sequentially, with the first H2A-H2B dimer released only after the DNA has formed a teardrop-shaped conformer[129]. Lastly, a large number of single molecule studies of nucleosomes have been performed, gaining insight into nucleosome wrapping, sliding, and dynamics, among other aspects of nucleosome function[130–133].
Conclusions: Outstanding issues and future outlook:
A wide variety of computational techniques have been used to capture the dynamics and mechanisms of nucleoprotein complexes, ranging from the highly detailed explicit solvent molecular dynamics simulations of nucleosomes and ribosomes to coarse-grained polymer bead models used to visualize Hi-C data for entire chromosomes. While the ribosome represents one of the most well-studied biophysical systems, where workers are computing free energies of transitions, chromosomes represent the next frontier, where emerging fundamental questions are arising. The vast array of diverse techniques provides one with a myriad of perspectives, whether it be global motions on long time scales or conformational changes of single residues that are crucial for biological function. Overall, computational methods allow one to synthesize a wide range of different experiments into a more coherent mechanism of the nucleoprotein complex. In one sense, movies generated by molecular simulation produce a more realistic view of macromolecular function because they include the constant background of small and large thermal fluctuations that give rise to conformational changes, painting a vivid picture of, for example, the stochastic Brownian motion required for ribosome conformational changes [134]. In contrast, movies accompanying X-ray crystallography or cryo-EM studies can sometimes be over simplified, showing (i) tractor-beam like behavior of one protein being mysteriously guided in a bee line to its target on a macromolecular complex, (ii) linear morphing between states with no fluctuations whatsoever, or (iii) flashing between two single snapshots. Inexpensive simulators, such as SMOG and others, provide a convenient method to visualize stochastic conformational changes [135].
Figure 1.
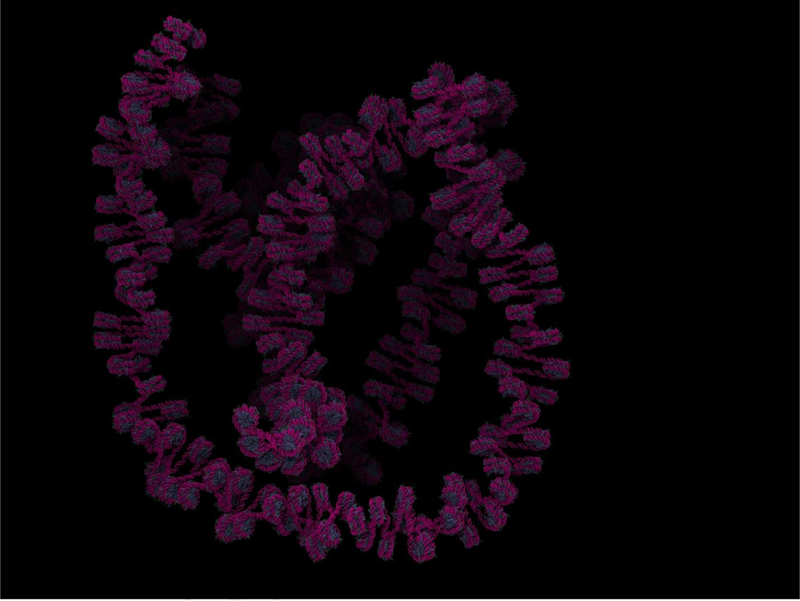
Explicit solvent simulations of the GATA4 gene locus, including 427 nucleosomes, 83 kb and 1 billion atoms. Magenta, DNA; blue, proteins (histone octamers).
Acknowledgements
The author is grateful to NIH NIGMS GM072686, NSF and LANL LDRD for the support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- 1.Brimacombe R, Atmadja J, Stiege W, Schuler D: A detailed model of the three-dimensional structure of Escherichia coli 16 S ribosomal RNA in situ in the 30 S subunit. Journal of molecular biology 1988, 199:115–136. [DOI] [PubMed] [Google Scholar]
- 2.Malhotra A, Tan RK, Harvey SC: Prediction of the three-dimensional structure of Escherichia coli 30S ribosomal subunit: a molecular mechanics approach. Proc Natl Acad Sci U S A 1990, 87:1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Easterwood TR, Major F, Malhotra A, Harvey SC: Orientations of transfer RNA in the ribosomal A and P sites. Nucleic Acids Res 1994, 22:3779–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malhotra A, Harvey SC: A quantitative model of the Escherichia coli 16 S RNA in the 30 S ribosomal subunit. J Mol Biol 1994, 240:308–340. [DOI] [PubMed] [Google Scholar]
- 5.VanLoock MS, Agrawal RK, Gabashvili IS, Qi L, Frank J, Harvey SC: Movement of the decoding region of the 16 S ribosomal RNA accompanies tRNA translocation. J Mol Biol 2000, 304:507–515. [DOI] [PubMed] [Google Scholar]
- 6.Fink DL, Chen RO, Noller HF, Altman RB: Computational methods for defining the allowed conformational space of 16S rRNA based on chemical footprinting data. RNA 1996, 2:851–866. [PMC free article] [PubMed] [Google Scholar]
- 7.Tung CS, Joseph S, Sanbonmatsu KY: All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat Struct Biol 2002, 9:750–755. [DOI] [PubMed] [Google Scholar]
- 8.Tung CS, Sanbonmatsu KY: Atomic model of the Thermus thermophilus 70S ribosome developed in silico. Biophys J 2004, 87:2714–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Ma B, Shapiro B: Binding interactions between the core central domain of 16S rRNA and the ribosomal protein S15 determined by molecular dynamics simulations. NUCLEIC ACIDS RESEARCH 2003, 31:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanbonmatsu KY, Joseph S: Understanding discrimination by the ribosome: stability testing and groove measurement of codon-anticodon pairs. J Mol Biol 2003, 328:33–47. [DOI] [PubMed] [Google Scholar]
- 11.Chacon P, Tama F, Wriggers W: Mega-Dalton biomolecular motion captured from electron microscopy reconstructions. J Mol Biol 2003, 326:485–492. [DOI] [PubMed] [Google Scholar]
- 12.Baker N, Sept D, Joseph S, Holst M, McCammon J: Electrostatics of nanosystems: Application to microtubules and the ribosome. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA 2001, 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Frank J: Transfer RNA in the hybrid P/E state: correlating molecular dynamics simulations with cryo-EM data. Proceedings of the National Academy of Sciences of the United States of America 2007, 104:16540–16545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tama F, Valle M, Frank J, Brooks CL 3rd: Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc Natl Acad Sci U S A 2003, 100:9319–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Rader AJ, Bahar I, Jernigan RL: Global ribosome motions revealed with elastic network model. J Struct Biol 2004, 147:302–314. [DOI] [PubMed] [Google Scholar]
- 16.Gorba C, Miyashita O, Tama F: Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data. Biophys J 2008, 94:1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitra K, Schaffitzel C, Shaikh T, Tama F, Jenni S, Brooks CL 3rd, Ban N, Frank J: Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature 2005, 438:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurkcuoglu O, Doruker P, Sen TZ, Kloczkowski A, Jernigan RL: The ribosome structure controls and directs mRNA entry, translocation and exit dynamics. Physical biology 2008, 5:046005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmermann MT, Jia K, Jernigan RL: Ribosome Mechanics Informs about Mechanism. J Mol Biol 2016, 428:802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trylska J, Tozzini V, McCammon JA: Exploring global motions and correlations in the ribosome. Biophys J 2005, 89:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusupova G, Jenner L, Rees B, Moras D, Yusupov M: Structural basis for messenger RNA movement on the ribosome. Nature 2006, 444:391–394. [DOI] [PubMed] [Google Scholar]
- 22.Levi M, Whitford PC: Dissecting the energetics of subunit rotation in the ribosome. Journal of Physical Chemistry B In Press 2019.This important study of the intersubunit bridges of the ribosome demonstrated that intersubunit rotation likely involves asynchronous movement of the bridges.
- 23.Korostelev A, Noller HF: Analysis of structural dynamics in the ribosome by TLS crystallographic refinement. Journal of molecular biology 2007, 373:1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohan S, Donohue JP, Noller HF: Molecular mechanics of 30S subunit head rotation. Proc Natl Acad Sci U S A 2014, 111:13325–13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25.Gulay SP, Bista S, Varshney A, Kirmizialtin S, Sanbonmatsu KY, Dinman JD: Tracking fluctuation hotspots on the yeast ribosome through the elongation cycle. Nucleic Acids Res 2017, 45:4958–4971.First molecular simulation of the eukaryotic ribosome, to our knowledge. This new methodology tunes the force field potential to mobility data from each residue from SHAPE chemical probing.
- 26.Kirmizialtin S, Hennelly SP, Schug A, Onuchic JN, Sanbonmatsu KY: Integrating molecular dynamics simulations with chemical probing experiments using SHAPE-FIT. Methods Enzymol 2015, 553:215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben-Shem A, Jenner L, Yusupova G, Yusupov M: Crystal structure of the eukaryotic ribosome. Science 2010, 330:1203–1209. [DOI] [PubMed] [Google Scholar]
- 28.Baxter-Roshek JL, Petrov AN, Dinman JD: Optimization of ribosome structure and function by rRNA base modification. PLoS One 2007, 2:e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meskauskas A, Dinman JD: Ribosomal protein L3: gatekeeper to the A site. Mol Cell 2007, 25:877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakauskaite R, Dinman JD: Mutations of highly conserved bases in the peptidyltransferase center induce compensatory rearrangements in yeast ribosomes. RNA 2011, 17:855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burakovsky DE, Sergiev PV, Steblyanko MA, Konevega AL, Bogdanov AA, Dontsova OA: The structure of helix 89 of 23S rRNA is important for peptidyl transferase function of Escherichia coli ribosome. FEBS letters 2011, 585:3073–3078. [DOI] [PubMed] [Google Scholar]
- 32.Burakovsky DE, Sergiev PV, Steblyanko MA, Kubarenko AV, Konevega AL, Bogdanov AA, Rodnina MV, Dontsova OA: Mutations at the accommodation gate of the ribosome impair RF2-dependent translation termination. RNA 2010, 16:1848–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitford PC, Sanbonmatsu KY: Simulating movement of tRNA through the ribosome during hybrid-state formation. J Chem Phys 2013, 139:121919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen K, Whitford PC: Capturing Transition States for tRNA Hybrid-State Formation in the Ribosome. J Phys Chem B 2016, 120:8768–8775. [DOI] [PubMed] [Google Scholar]
- 35.Trabuco LG, Schreiner E, Eargle J, Cornish P, Ha T, Luthey-Schulten Z, Schulten K: The role of L1 stalk-tRNA interaction in the ribosome elongation cycle. Journal of molecular biology 2010, 402:741–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trabuco LG, Harrison CB, Schreiner E, Schulten K: Recognition of the regulatory nascent chain TnaC by the ribosome. Structure 2010, 18:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gumbart J, Trabuco LG, Schreiner E, Villa E, Schulten K: Regulation of the protein-conducting channel by a bound ribosome. Structure 2009, 17:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenner LB, Demeshkina N, Yusupova G, Yusupov M: Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 2010, 17:555–560. [DOI] [PubMed] [Google Scholar]
- *39.Whitford PC, Blanchard SC, Cate JH, Sanbonmatsu KY: Connecting the kinetics and energy landscape of tRNA translocation on the ribosome. PLoS Comput Biol 2013, 9:e1003003.Microsecond sampling of the ribosome in explicit solvent allowed the calculation of diffusive motions of the ribosome. Diffusion coefficients were used to construct a table connecting experimentally measured kinetic rates to free energy barrier heights.
- *40.Huter P, Arenz S, Bock LV, Graf M, Frister JO, Heuer A, Peil L, Starosta AL, Wohlgemuth I, Peske F, et al. : Structural Basis for Polyproline-Mediated Ribosome Stalling and Rescue by the Translation Elongation Factor EF-P. Mol Cell 2017, 68:515–527 e516.The first molecular dynamics simulation of EF-P ribosome interactions.
- 41.Arenz S, Bock LV, Graf M, Innis CA, Beckmann R, Grubmuller H, Vaiana AC, Wilson DN: A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat Commun 2016, 7:12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trobro S, Aqvist J: Mechanism of peptide bond synthesis on the ribosome. Proceedings of the National Academy of Sciences of the United States of America 2005, 102:12395–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *43.Sharma PK, Xiang Y, Kato M, Warshel A: What are the roles of substrate-assisted catalysis and proximity effects in peptide bond formation by the ribosome? Biochemistry 2005, 44:11307–11314. *Warshel and co-workers found solvation entropy is significant and while the 2’-hydroxyl group of tRNA A76 accelerates the reaction, it does not directly catalyze.
- 44.Adamczyk AJ, Warshel A: Converting structural information into an allosteric-energy-based picture for elongation factor Tu activation by the ribosome. Proceedings of the National Academy of Sciences of the United States of America 2011, 108:9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wieden HJ, Mercier E, Gray J, Steed B, Yawney D: A combined molecular dynamics and rapid kinetics approach to identify conserved three-dimensional communication networks in elongation factor Tu. Biophysical journal 2010, 99:3735–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallin G, Aqvist J: The transition state for peptide bond formation reveals the ribosome as a water trap. Proceedings of the National Academy of Sciences of the United States of America 2010, 107:1888–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trobro S, Aqvist J: Mechanism of the translation termination reaction on the ribosome. Biochemistry 2009, 48:11296–11303. [DOI] [PubMed] [Google Scholar]
- 48.Becker T, Bhushan S, Jarasch A, Armache JP, Funes S, Jossinet F, Gumbart J, Mielke T, Berninghausen O, Schulten K, et al. : Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 2009, 326:1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Almlof M, Ander M, Aqvist J: Energetics of codon-anticodon recognition on the small ribosomal subunit. Biochemistry 2007, 46:200–209. [DOI] [PubMed] [Google Scholar]
- 50.Ge X, Mandava CS, Lind C, Aqvist J, Sanyal S: Complementary charge-based interaction between the ribosomal-stalk protein L7/12 and IF2 is the key to rapid subunit association. Proc Natl Acad Sci U S A 2018, 115:4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw JJ, Trobro S, He SL, Aqvist J, Green R: A Role for the 2’ OH of peptidyl-tRNA substrate in peptide release on the ribosome revealed through RF-mediated rescue. Chem Biol 2012, 19:983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trakhanov S, Yusupov M, Shirokov V, Garber M, Mitschler A, Ruff M, Thierry JC, Moras D: Preliminary X-ray investigation of 70 S ribosome crystals from Thermus thermophilus. J Mol Biol 1989, 209:327–328. [DOI] [PubMed] [Google Scholar]
- 53.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF: Crystal structure of the ribosome at 5.5 A resolution. Science 2001, 292:883–896. [DOI] [PubMed] [Google Scholar]
- 54.Romanowska J, Setny P, Trylska J: Molecular dynamics study of the ribosomal A-site. J Phys Chem B 2008, 112:15227–15243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaiana AC, Westhof E, Auffinger P: A molecular dynamics simulation study of an aminoglycoside/A-site RNA complex: conformational and hydration patterns. Biochimie 2006, 88:1061–1073. [DOI] [PubMed] [Google Scholar]
- 56.Kulczycka-Mierzejewska K, Sadlej J, Trylska J: Molecular dynamics simulations suggest why the A2058G mutation in 23S RNA results in bacterial resistance against clindamycin. J Mol Model 2018, 24:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **57.Dlugosz M, Trylska J: Aminoglycoside association pathways with the 30S ribosomal subunit. J Phys Chem B 2009, 113:7322–7330.This innovative study probed the pathways of drug binding to the ribosome using Brownian dynamics simulations of aminoglycosides binding to the ribosome.
- 58.Ge X, Roux B: Absolute binding free energy calculations of sparsomycin analogs to the bacterial ribosome. J Phys Chem B 2010, 114:9525–9539. [DOI] [PubMed] [Google Scholar]
- 59.Besseova I, Reblova K, Leontis NB, Sponer J: Molecular dynamics simulations suggest that RNA three-way junctions can act as flexible RNA structural elements in the ribosome. Nucleic Acids Res 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reblova K, Razga F, Li W, Gao H, Frank J, Sponer J: Dynamics of the base of ribosomal A-site finger revealed by molecular dynamics simulations and Cryo-EM. Nucleic acids research 2010, 38:1325–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirmizialtin S, Ganesan V, Makarov DE: Translocation of a beta-hairpin-forming peptide through a cylindrical tunnel. The Journal of chemical physics 2004, 121:10268–10277. [DOI] [PubMed] [Google Scholar]
- 62.Ziv G, Haran G, Thirumalai D: Ribosome exit tunnel can entropically stabilize alpha-helices. Proceedings of the National Academy of Sciences of the United States of America 2005, 102:18956–18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez LM Contreras, Martinez-Veracoechea FJ, Pohkarel P, Stroock AD, Escobedo FA, DeLisa MP: Protein translocation through a tunnel induces changes in folding kinetics: a lattice model study. Biotechnology and bioengineering 2006, 94:105–117. [DOI] [PubMed] [Google Scholar]
- 64.Ishida H, Hayward S: Path of nascent polypeptide in exit tunnel revealed by molecular dynamics simulation of ribosome. Biophysical journal 2008, 95:5962–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrone PM, Snow CD, Lucent D, Pande VS: Side-chain recognition and gating in the ribosome exit tunnel. Proceedings of the National Academy of Sciences of the United States of America 2008, 105:16549–16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucent D, Snow CD, Aitken CE, Pande VS: Non-bulk-like solvent behavior in the ribosome exit tunnel. PLoS computational biology 2010, 6:e1000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fritch B, Kosolapov A, Hudson P, Nissley DA, Woodcock HL, Deutsch C, O’Brien EP: Origins of the Mechanochemical Coupling of Peptide Bond Formation to Protein Synthesis. J Am Chem Soc 2018, 140:5077–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bishop TC: Molecular dynamics simulations of a nucleosome and free DNA. J Biomol Struct Dyn 2005, 22:673–686. [DOI] [PubMed] [Google Scholar]
- 69.Chang L, Takada S: Histone acetylation dependent energy landscapes in tri-nucleosome revealed by residue-resolved molecular simulations. Sci Rep 2016, 6:34441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wocjan T, Klenin K, Langowski J: Brownian dynamics simulation of DNA unrolling from the nucleosome. J Phys Chem B 2009, 113:2639–2646. [DOI] [PubMed] [Google Scholar]
- 71.Brandner A, Schuller A, Melo F, Pantano S: Exploring DNA dynamics within oligonucleosomes with coarse-grained simulations: SIRAH force field extension for protein-DNA complexes. Biochem Biophys Res Commun 2018, 498:319–326. [DOI] [PubMed] [Google Scholar]
- 72.Dans PD, Walther J, Gomez H, Orozco M: Multiscale simulation of DNA. Curr Opin Struct Biol 2016, 37:29–45. [DOI] [PubMed] [Google Scholar]
- 73.Collepardo-Guevara R, Schlick T: Insights into the chromatin fiber structure by in vitro and in silico single-molecule stretching experiments. Biochem. Soc. Trans 2013, 41:494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ozer G, Luque A, Schlick T: The chromatin fiber: Multiscale problems and approaches. Curr. Opin. Struct. Biol 2015, 31:124–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bascon G, Schlick T: Chromatin mesoscale modeling 2017:123–148. [Google Scholar]
- *76.Bascom G, Kim T, Schlick T: Kilobase pair chromatin fiber contacts promoted by living-system-like dna linker length distributions and nucleosome depletion. J. Phys. Chem. B Special Issue: K. Schulten Memorial 2017, 121:3882–3894.This mesoscale model of chromatin is one of the few models available at the nucleosome level of detail capable of folding entire gene loci.
- 77.Schlick T, Hayes J, Grigoryev SA: Toward convergence of experimental studies and theoretical modeling of the chromatin fiber. J. Biol. Chem 2012, 287:5183–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grigoryev S, Bascom G, Schubert M, Woodcock C, Schlick T: Hierarchical looping of zigzag nucleosome chains in metaphase chromosomes 2016:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rao SSP, Huang S-c, Hilaire BGS, Engreitz JM, Perez EM, Kieffer-Kwon K-r, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. : Cohesin loss eliminates all loop domains, leading to links among superenhancers and downregulation of nearby genes. Cell 2017, 171:305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun J, Zhang Q, Schlick T: Electrostatic mechanism of nucleosomal array folding revealed by computer simulation 2005:8180–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arya G, Schlick T: Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model 2006:16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T: Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions 2009:13317–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Collepardo-Guevara R, Schlick T: Chromatin fiber polymorphism triggered by variations of DNA linker lengths 2014:8061–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luque A, Collepardo-Guevara R, Grigoryev S, Schlick T: Dynamic condensation of linker histone C -terminal domain regulates chromatin structure. Nucl. Acids Res 2014, 42:7553–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Collepardo-Guevara R, Portella G, Vendruscolo M, Frenkel D, Schlick T, Orozco M: Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: A multiscale computational study. J. Amer. Chem. Soc 2015, 137:10205–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luque A, Ozer G, Schlick T: Correlation among DNA linker length, linker histone concentration, and histone tails in chromatin. Biophys. J 2016, 110:2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arya G, Schlick T: Efficient global biopolymer sampling with end-transfer configurational bias Monte Carlo. J. Chem. Phys 2007, 106:044107. [DOI] [PubMed] [Google Scholar]
- 88.Arya G, Schlick T: A tale of tails: H ow histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A 2009, 113:4045–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Collepardo-Guevara R, Schlick T: The effect of linker histone’s nucleosome binding affinity on chromatin unfolding mechanisms. Biophys. J 2011, 101:1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carruthers LM, Bednar J, Woodcock CL, Hansen J: Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochem 1998, 37:14776–14787. [DOI] [PubMed] [Google Scholar]
- 91.Butler PJG, Thomas JO: Changes in chromatin folding in solution. J. Mol. Biol 1980, 140:505–529. [DOI] [PubMed] [Google Scholar]
- 92.Scheffer MP, Eltsov M, Bednar J, Frangakis AS: Nucleosomes stacked with aligned dyad axes are found in native compact chromatin in vitro. J. Struc. Biol 2012, 178:207–214. [DOI] [PubMed] [Google Scholar]
- 93.Beard D, Schlick T: Modeling salt-mediated electrostatics of macromolecules: T he algorithm DiSCO ( D iscrete C harge S urface C harge O ptimization) and its application to the nucleosome. Biopolymers 2001, 58:106–115. [DOI] [PubMed] [Google Scholar]
- 94.Beard D, Schlick T: Inertial stochastic dynamics: I. long-timestep methods for langevin dynamics. J. Chem. Phys 2000, 112:7313–7322. [Google Scholar]
- **95.Bascom Gavin D., Myers Christopher G., and Tamar Schlick: Mesoscale Modeling Reveals Formation of an Epigenetically Driven HOXC Gene Hub. Proc Natl Acad Sci U S A 2019, in press.A mesoscale model of chromatin was used to study HOXC, producing the first folded gene structure that was defined from first principles (nucleosome positions, epigenetic marks, linker histones).
- 96.Fan Y, Korolev N, Lyubartsev AP, Nordenskiold L: An advanced coarse-grained nucleosome core particle model for computer simulations of nucleosome-nucleosome interactions under varying ionic conditions. PLoS One 2013, 8:e54228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jung J, Nishima W, Daniels M, Bascom G, Kobayashi C, Adedoyin A, Wall M, , Lappala A, Fischer W, Tung C-T, Schlick T, Sugita Y, Sanbonmatsu KY: Scaling Molecular Dynamics Beyond 100,000 Processor Cores for Large-Scale Biophysical Simulations. Journal of Computational Chemistry 2019, In press.* Molecular dynamics simulations of the GATA4 gene locus in explicit solvent include 427 nucleosomes, 83 kb of DNA and 1 billion atoms, representing the largest biomolecular simulation to date.
- 98.Bascom GD, Sanbonmatsu KY, Schlick T: Mesoscale Modeling Reveals Hierarchical Looping of Chromatin Fibers Near Gene Regulatory Elements. The Journal of Physical Chemistry B 2016, 120:8642–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lajoie BR, Dekker J, Kaplan N: The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods (San Diego, Calif.) 2015, 72:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lieberman-Aiden E, Berkum NLV, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. : Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang C-Y, Jégu T, Chu H-P, Oh HJ, Lee JT: SMCHD1 Merges Chromosome Compartments and Assists Formation of Super-Structures on the Inactive X. Cell 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jegu T, Aeby E, Lee JT: The X chromosome in space. Nat Rev Genet 2017, 18:377–389. [DOI] [PubMed] [Google Scholar]
- 103.Froberg JE, Pinter SF, Kriz AJ, Jegu T, Lee JT: Megadomains and superloops form dynamically but are dispensable for X-chromosome inactivation and gene escape. Nat Commun 2018, 9:5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bon M, Marenduzzo D, Cook PR: Modeling a self-avoiding chromatin loop: relation to the packing problem, action-at-a-distance, and nuclear context. Structure 2006, 14:197–204. [DOI] [PubMed] [Google Scholar]
- 105.Marenduzzo D, Micheletti C, Cook PR: Entropy-driven genome organization. Biophys J 2006, 90:3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cook PR, Marenduzzo D: Entropic organization of interphase chromosomes. J Cell Biol 2009, 186:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sorensen AL, Vigouroux C, Buendia B, et al. : Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol 2017, 18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giorgetti L, Galupa R, Nora EP, Piolot T, Lam F, Dekker J, Tiana G, Heard E: Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 2014, 157:950–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fudenberg G, Getz G, Meyerson M, Mirny LA: High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature Biotech 2011, 29:1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Leonid A Mirny: Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 2016, 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, et al. : Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lappala A, Terentjev EM: “Raindrop” Coalescence of Polymer Chains during Coil–Globule Transition. Macromolecules 2013, 46:1239–1247. [Google Scholar]
- *113.Di Pierro M, Cheng RR, Aiden E Lieberman, Wolynes PG, Onuchic JN: De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc Natl Acad Sci U S A 2017, 114:12126–12131.Onuchic and co-workers extend their model to include ChIP-seq data, enabling the study of a variety of epigenetics marks.
- **114.Di Pierro M, Potoyan DA, Wolynes PG, Onuchic JN: Anomalous diffusion, spatial coherence, and viscoelasticity from the energy landscape of human chromosomes. Proc Natl Acad Sci U S A 2018, 115:7753–7758.Onuchic and co-workers use quasi-equilibrium energy landscape studies to demonstrate that confinement effectively slows diffusion, consistent with experimentally observed diffusion, viscoelasticity and spatial confinement.
- **115.Di Pierro M, Zhang B, Aiden EL, Wolynes PG, Onuchic JN: Transferable model for chromosome architecture. Proc Natl Acad Sci U S A 2016, 113:12168–12173.Onuchic and co-workers constructed a Langevin dynamics quasi-equilibrium homopolymer model (MiChroM) including interchromosomal interactions, loop extrusion factors (e.g., CTCF and cohesion). The model uses the powerful maximum entropy method, including a maximum entropy term to incorporate interactions observed in Hi-C experiments by minimizing the differences between observed and modeled structures.
- 116.Shi G, Liu L, Hyeon C, Thirumalai D: Interphase human chromosome exhibits out of equilibrium glassy dynamics. Nat Commun 2018, 9:3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen J, Wu Y: Understanding the Functional Roles of Multiple Extracellular Domains in Cell Adhesion Molecules with a Coarse-Grained Model. J Mol Biol 2017, 429:1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang W, Ravikumar KM, Parisien M, Yang S: Theoretical modeling of multiprotein complexes by iSPOT: Integration of small-angle X-ray scattering, hydroxyl radical footprinting, and computational docking. J Struct Biol 2016, 196:340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zheng W: Probing the structural dynamics of the SNARE recycling machine based on coarse-grained modeling. Proteins 2016, 84:1055–1066. [DOI] [PubMed] [Google Scholar]
- 120.Kmiecik S, Kouza M, Badaczewska-Dawid AE, Kloczkowski A, Kolinski A: Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int J Mol Sci 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Palermo G, Ricci CG, Fernando A, Basak R, Jinek M, Rivalta I, Batista VS, McCammon JA: Protospacer Adjacent Motif-Induced Allostery Activates CRISPR-Cas9. J Am Chem Soc 2017, 139:16028–16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Palermo G, Miao Y, Walker RC, Jinek M, McCammon JA: CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc Natl Acad Sci U S A 2017, 114:7260–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zheng W: Probing the structural dynamics of the CRISPR-Cas9 RNA-guided DNA-cleavage system by coarse-grained modeling. Proteins 2017, 85:342–353. [DOI] [PubMed] [Google Scholar]
- **124.Zinchenko A, Berezhnoy NV, Wang S, Rosencrans WM, Korolev N, van der Maarel JRC, Nordenskiold L: Single-molecule compaction of megabase-long chromatin molecules by multivalent cations. Nucleic Acids Res 2018, 46:635–649.This ground breaking study measures and visualizes compaction of chromatin due to cations.
- *125.Kilic S, Felekyan S, Doroshenko O, Boichenko I, Dimura M, Vardanyan H, Bryan LC, Arya G, Seidel CAM, Fierz B: Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1alpha. Nat Commun 2018, 9:235.Single molecule FRET experiments show that highly compact chromatin exhibits shuffling of nucleosomes between stacking partners, along with a distorted state and an open state.
- 126.Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, Dekker C: Real-time imaging of DNA loop extrusion by condensin. Science 2018, 360:102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Riedmann C, Fondufe-Mittendorf YN: Comparative analysis of linker histone H1, MeCP2, and HMGD1 on nucleosome stability and target site accessibility. Sci Rep 2016, 6:33186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang C, van der Woerd MJ, Muthurajan UM, Hansen JC, Luger K: Biophysical analysis and small-angle X-ray scattering-derived structures of MeCP2-nucleosome complexes. Nucleic Acids Res 2011, 39:4122–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen Y, Tokuda JM, Topping T, Meisburger SP, Pabit SA, Gloss LM, Pollack L: Asymmetric unwrapping of nucleosomal DNA propagates asymmetric opening and dissociation of the histone core. Proc Natl Acad Sci U S A 2017, 114:334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Killian JL, Li M, Sheinin MY, Wang MD: Recent advances in single molecule studies of nucleosomes. Curr Opin Struct Biol 2012, 22:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Leuba SH, Bustamante C, van Holde K, Zlatanova J: Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys J 1998, 74:2830–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ngo TT, Yoo J, Dai Q, Zhang Q, He C, Aksimentiev A, Ha T: Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat Commun 2016, 7:10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bowman GD: Mechanisms of ATP-dependent nucleosome sliding. Curr Opin Struct Biol 2010, 20:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *134.Whitford PC, Geggier P, Altman RB, Blanchard SC, Onuchic JN, Sanbonmatsu KY: Accommodation of aminoacyl-tRNA into the ribosome involves reversible excursions along multiple pathways. RNA 2010, 16:1196–1204.The study simulated spontaneous large-scale conformational changes of the ribosome (accommodation events). Reversible accommodation events were consistent with single molecule FRET experiments.
- *135.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN: SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res 2010, 38:W657–661.SMOG is a user-friendly tool for reduced-model, inexpensive simulations of conformational changes, allowing one to upload structures, download GROMACS set up files and also aid in performing simulations.