

Summary
Variable levels of DNA methylation have been reported at tissue-specific differential methylation regions (DMRs) overlapping enhancers, including super-enhancers (SEs) associated with key cell identity genes, but the mechanisms responsible for this intriguing behavior are not well understood. We used allele-specific reporters at the endogenous Sox2 and Mir290 super-enhancers (SE) in embryonic stem cells and found that the allelic DNA methylation state is dynamically switching, resulting in cell-to-cell heterogeneity. Dynamic DNA methylation is driven by the balance between DNA methyltransferases and transcription factor binding on one side, and co-regulated with the Mediator complex recruitment and H3K27ac level changes at regulatory elements on the other side. DNA methylation at the Sox2 and the Mir290 SEs is independently regulated and has distinct consequences on the cellular differentiation state. Dynamic allele-specific DNA methylation at the two SEs was also seen at different stages in preimplantation embryos, revealing that methylation heterogeneity occurs in vivo.
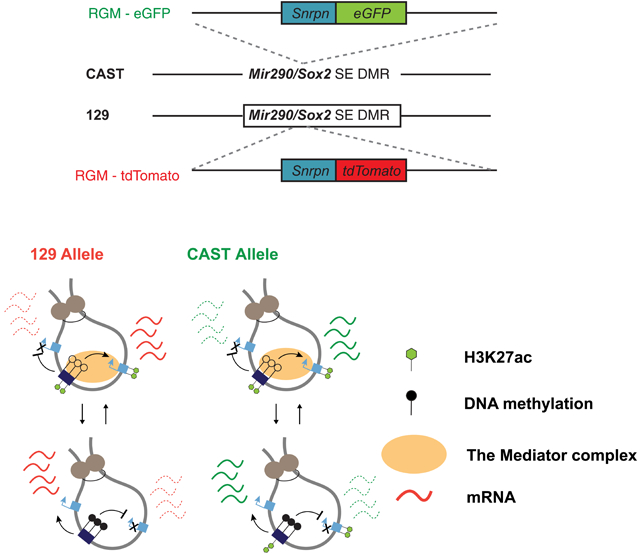
Graphical Abstract
eTOC Blurb
Song et al. used an allelic reporter approach to show super-enhancers allelic DNA methylation dynamics underlies locus-specific heterogeneity, which functionally impacts transcription and cellular states of mouse embryonic stem cells.
Introduction
Tissue-specific differential methylation regions (T-DMRs) have been found to strongly associate with low CpG density and inter-genic enhancers (Ehrlich et al., 2016; Fleischer et al., 2017; Izzi et al., 2016; Jones, 2012; Rinaldi et al., 2016) and the vast majority of cell-type specific DNA methylation changes occur at distal regulatory elements (Luo et al., 2018; Stadler et al., 2011). Whole genome bisulfite sequencing (WGBS) data indicate a low but detectable level of DNA methylation at T-DMRs overlapping active enhancers (Elliott et al., 2015; Heyn et al., 2016; Hon et al., 2013; Jiang et al., 2015; King et al., 2016; Shull et al., 2016; Stadler et al., 2011). Recent single-cell WGBS (scWGBS) data from mouse embryonic stem cells (ESC) and the early mouse embryo suggest that the variable low-to-intermediate DNA methylation levels found at enhancer regions in bulk-cell measurements are largely due to averaging signals across cells with heterogeneous methylation states (Cheow et al., 2015; Guo et al., 2017; Guo et al., 2015; Guo et al., 2013; Rulands et al., 2018; Smallwood et al., 2014). However, due to the static snapshot view of sequencing-based methods, it has been difficult to define the basis, regulation and functional impact of DNA methylation heterogeneity on gene expression and cellular states.
The hierarchy and casual relationship between the regulation of enhancer DNA methylation, active enhancer histone marks, transcription factor (TF) binding and cis-regulated transcription has been challenging to define due to the epigenetic heterogeneity among cells (Jin et al., 2011; King et al., 2016; Zhu et al., 2016). While genome-wide epigenetic profiling provided insights into the relationship between DNA methylation, histone marks, and transcription factors and coactivators binding (King et al., 2016; Roadmap Epigenomics et al., 2015; Wilson and Filipp, 2018), these approaches, even at single-cell level, did not allow resolving fast dynamics of individual epigenetic processes in heterogenous tissues and cell populations. Thus, currently there is no clear understanding of the basis, regulation and functional consequences of DNA methylation heterogeneity.
Our recently developed Reporter of Genome Methylation (RGM) allows tracing of locus-specific DNA methylation based on the on-and-off of a fluorescent signal in single cells in real-time, and has been shown to faithfully reflect the endogenous DNA methylation states at multiple genomic loci (Stelzer et al., 2015; Stelzer et al., 2016). This system allows for robustly tracking locus-specific DNA methylation at enhancer regions and for functionally dissecting the hierarchy of epigenetic events that regulate enhancer activity and cellular states, overcoming the challenges faced by bulk measurements or sequence-based methods. We utilized this system at two pluripotency super-enhancers (SEs), Sox2 and Mir290 SEs in ESCs. Both SEs overlap with ESC-specific DMRs, which display consistently low levels of methylation, indicating potential heterogeneity (Kobayashi et al., 2012; Leung et al., 2014; Rulands et al., 2018; Seisenberger et al., 2012; Stadler et al., 2011). We targeted RGMs to both alleles of the two SEs in F1 129xCasteneous (129xCAST) hybrid ESCs allowing to visualize allele-specific DNA methylation changes. We observed highly dynamic switching between different methylation states on individual alleles resulting in cell-to-cell heterogeneity and were able to distinguish the DNA methylation pathways driving these changes. The RGM system enables isolation of rare and transient populations exclusively based on their locus-specific methylation states, which allowed defining the relationship between dynamic SE DNA methylation changes, the Mediator complex condensation, histone H3K27 acetylation, transcription factor binding, cis-regulated target gene expression, and changes in cellular states. Finally, transgenic methylation reporter mice for both SEs revealed the previously underappreciated epigenetic heterogeneity and dynamics of the pluripotent cells in cleavage embryos, recapitulating and extending the observations in ESCs.
Results
DNA methylation at the Sox2 and Mir290 SE is heterogeneous at the allelic level
Sox2 and Mir290 SEs reside on Chromosome 3 and 7, respectively. Both SEs overlap with T-DMRs which are hypo-methylated in ESCs but become de novo methylated upon differentiation (Stelzer et al., 2015). The T-DMR of the Sox2 SE is located about 100kb upstream of the Sox2 gene whereas the Mir290 SE, consisting of hypo-methylated DMR constituents interspersed by small hyper-methylated regions, is proximal to the Mir290–295 cluster (Figure S1A). WGBS of ESCs indicates that the Sox2 and Mir290 SE DMRs have overall DNA methylation levels higher than that of hypo-methylated promoters of highly expressed genes in ESCs, such as Gapdh and Oct4, but lower than that of imprinting control regions or retroelements, which are monoallelically- and hyper-methylated, respectively (Figure S1B) (Kobayashi et al., 2012; Leung et al., 2014; Seisenberger et al., 2012; Stadler et al., 2011). This low-to-intermediate level of methylation at both SEs in bulk cell WGBS suggests that they are hypermethylated in a small population of cells. Re-analysis of published single cell WGBS (scWGBS) data (Smallwood et al., 2014) revealed that the T-DMRs of both SEs belong to the 5% regions with the most variable DNA methylation level compared to other regions of Chromosome 7 or Chromosome 3 (Figure S1C), further supporting the presence of rare cells with hypermethylated SE DMRs.
Consistent with published scWGBS studies reporting heterogeneity in the wild-type genome (Guo et al., 2017; Guo et al., 2015; Guo et al., 2013; Hu et al., 2016; Singer et al., 2014; Smallwood et al., 2014), we previously observed methylation heterogeneity in ESCs with the endogenous Nanog tagged with eGFP and RGM-tdTomato reporter inserted mono-allelically into the Sox2 or Mir290 SE DMRs (Stelzer et al., 2015). The heterogeneity at these two specific loci was manifested by the bi-modal distribution of RGM activity in Nanog positive (Nanog+) pluripotent cells as seen in FACS (Figure 1A). Sorting cells based on florescence intensity, followed by bisulfite PCR (BS-PCR) and sequencing, validated that RGM methylation strictly correlates with the endogenous methylation in both regions (Figure 1A). Analyzing the Sox2 SE revealed that hyper-methylation occurred on both the targeted and the untargeted alleles in the pluripotent ESC population (Nanog+), indicating that rare allelic methylation exists among cells (Figure S1D). The rare methylated alleles were also detected at the Mir290 SE by high-throughput sequencing of BS-PCR amplicons from the wild-type allele. Figure 1B shows that, comparing to Dnmt3a/b double knockout cells (described later in Figure S3A), we found methylation at the Mir290 SE in non-manipulated wild-type ESCs as well as on the untargeted allele in the Nanog+RGM+ ESCs. These results indicate that SE DNA methylation heterogeneity is created by allele-specific hypermethylation in rare ESC populations independent of RGM targeting. To track DNA methylation heterogeneity on each allele, we targeted the Mir290 and the Sox2 SE independently in 129xCastaneus F1 hybrid ESCs with allele-specific RGM reporters and generated two cell lines, Sox2-129SE-RGM-tdTomato/Sox2-CASTSE-RGM-eGFP (abbreviated below as SOX2-SE-TG) and Mir290-129SE-RGM-tdTomato/Mir290-CASTSE-RGM-eGFP (abbreviated below as MIR290-SE-TG) (Figure 1C, S1E) allowing to visualize the SE locus-specific DNA methylation state at allelic and single cell resolution. These cell lines also enabled dissection of allelic functional output of SE methylation states by distinguishing the two alleles based on the abundance of 129 or CAST allele-specific single nucleotide polymorphisms (SNPs) at both the DNA and the mRNA level.
Figure 1. DNA methylation at the Sox2 and Mir290 SE is heterogeneous at the allelic level.

(A) Left, DNA methylation heterogeneity at both the Sox2 and the Mir290 SE in v6.5-Nanog-eGFP ESC where the RGM-tdTomato reporter was mono-allelically targeted. Right, BS-PCR followed by sequencing of the Sox2 SE in different populations of the bimodal distribution. (B) Average methylation percentage and standard errors were quantified from high-throughput sequencing of BS-PCR amplicons of the Mir290 SE wild-type alleles in Dnmt3a/b double knockout ESCs, in Nanog+RGM+ ESCs and in untargeted wild-type ESCs. BS-PCRs were amplified allele-specifically as illustrated from potential epigenetic states indicated above. (C) Targeting strategy for generating SOX2-SE-TG and MIR290-SE-TG ESCs using CRISPR/Cas9 and targeting vectors. Methylation tracks from (Stadler et al., 2011) were used as the genome reference with blue bars highlighting the DMRs of the two SEs. Red tracks, 129 allele; green tracks, CAST allele. (D) FACS analysis of CASTx129 F1 ESC clones targeted with allele-specific RGMs at either the Mir290 or the Sox2 SE. (E) Allele-specific BS-PCR of the SEs with RGM (Snprn-tdTomato or Snprn-eGFP) in single PCR amplicons followed by Sanger sequencing in sorted cells from both SOX2-SE-TG and MIR290-SE-TG. See also Figure S1.
The initial FACS analysis detected a small fraction of single-positive (T+G−, T−G+) as well as double-negative (T−G−) cells in both cell lines, though the majority of cells were double-positive (T+G+) (Figure 1D), consistent with the heterogeneity reported in scWGBS data by others (Figure S1C) and in our BS-PCR analysis on both targeted and wild-type alleles (Figure 1A, B, S1D). To confirm that the RGM reporter activity faithfully reflected the allele-specific endogenous DNA methylation state, we sorted the four populations and performed allele-specific BS-PCR followed by Sanger sequencing of the DMRs upstream of the reporters. Figure 1E shows that the reporter activities on both alleles were consistent with the DNA methylation levels of the genomic SE regions and the inserted RGMs in all sorted populations. Quantitative pyro-sequencing further confirmed that T+G+ and T−G− populations represent two extreme methylation states of the intrinsic epigenetic heterogeneity at both SEs (Figure S1F). As expected, both unmethylated alleles in sorted T+G+ cells from both cell lines gained methylation synchronously upon retinoic acid (RA) induced differentiation. This confirms that the RGM-targeted SEs undergo the predicted methylation changes when exiting pluripotency (Figure S1G).
Dynamic allele-specific SE DNA methylation is regulated by de novo methylation and passive demethylation during cell proliferation.
To gain insights into the origin of DNA methylation heterogeneity, we FACS sorted equal numbers of the four populations from both reporter cell lines and monitored the RGM activity upon passaging in serum + LIF medium (Figure 2A). Figures 2B (serum + LIF) and Figure S2A show that the SE DNA methylation states in the four sorted populations were not stable but highly dynamic with each allele independently switching the RGM on-and-off over the course of only a few days. This indicates that the observed SE DNA methylation heterogeneity is a result of fast dynamic and reversible switching of allelic DNA methylation states. When sorted cells were passaged and cultured in “2i” (GSKi and MAPKi) medium, the kinetics of the transitions between different methylation states was significantly altered with slowed de novo methylation for both SEs and an initial acceleration of demethylation at the Mir290 SE (Figure 2B, C and Figure S2B). Demethylation of T−G− population of SOX2-SE-TG in “2i”, however, is slower over long term than that in serum + LIF, possibly due to impaired cell division as shown below. The observed DNA methylation difference between “2i” and serum + LIF is consistent with the extensive global demethylation induced in “2i” by down-regulation of de novo and maintenance methyltransferases (Choi et al., 2017; Leitch et al., 2013; Sim et al., 2017; von Meyenn et al., 2016; Yagi et al., 2017).
Figure 2. SE DNA methylation heterogeneity is created by dynamic switching of methylation states.

(A) Experiment setup for monitoring SE DNA methylation dynamics. Yellow cells: T+G+; grey cells: T−G−; red cells: T+G−; green cells: T−G+. (B) FACS analyses on the dynamics of T+G−, T−G+, T+G+ and T−G− populations 4 days post-sorting for both MIR290-SE-TG in serum + LIF or “2i” medium. (C) Quantifications of the dynamics of 4 sorted populations from MIR290-SE-TG in percentages change over time when cultured in the serum + LIF or the “2i” medium after sorting. See also Figure S2.
Demethylation in “2i” suggests that changes in DNA methyltransferase (Dnmt) activities modulate the observed dynamics. To determine the main de novo methyltransferase driver for SE methylation, we compared RGM activities in Dnmt3a or Dnmt3b single-knockout and Dnmt3a/3b double-knockout cells (DKO) (Figure S3A). Although the number of RGM negative cells was reduced in Dnmt3a or Dnmt3b single-knockout cells, cells with methylated SEs were eliminated only in the absence of both de novo methyltransferases in DKO cells preventing any de novo methylation (Figure 3A, S3B). The hypomethylation of both SEs was further confirmed by pyro-sequencing in DKO ESCs as well as in cells induced to differentiate by RA (Figure S3C). These results suggest that both DNMT3A and DNMT3B have redundant functions and independently contribute to de novo methylation of SE DMRs.
Figure 3. The dynamics of SE DNA methylation is driven by de novo methylation and passive demethylation during cell proliferation.

(A) Elimination of the population with methylated SEs in Dnmt3a and Dnmt3b double knockout (DKO) v6.5-Nanog-eGFP ESCs with the RGM-tdTomato reporter targeted mono-allelically at either the Sox2 or the Mir290 SE. (B) Demethylation of sorted T+G−, T−G+ and T−G− cells from SOX2-SE-TG (and (C) MIR290-SE-TG) cells with and without thymidine block. (D) Expected changes in the T+G+ cell percentage for each demethylation mechanism upon CRIPSR/Cas9-mediated gene disruptions. Changes in the percentage of T+G+ cells indicate the rate of demethylation on the 129SE-RGM-tdTomato allele. (E) Relative changes in the T+G+ percentage upon transfecting sgRNAs against enzymes involved in DNA demethylation, as compared to cells transfected with the same vector without sgRNA (sgControl). (F) A model for the origin of locus-specific DNA methylation heterogeneity. See also Figure S3.
DNA demethylation can occur either passively in rapidly dividing cells, caused by inhibition of DNMT1 or by active removal of the methyl group mediated by Tet enzymes and base excision repair (BER) pathways (Wu and Zhang, 2017). To assess whether demethylation of the SEs involved active or passive mechanisms, we analyzed whether DNA demethylation would be affected in cells upon delaying cell cycle progression using thymidine block. In all three populations carrying at least one methylated allele, the kinetics of demethylation upon thymidine block was significantly decreased upon 3 days in culture (Figure 3B, C). This suggests that cell proliferation-driven passive demethylation is responsible for SE demethylation. To confirm this observation genetically, we transfected 129SE-RGM-tdTomato T−G+ cells with Cas9 and sgRNAs against genes encoding the maintenance enzymes DNMT1/UHRF1, which upon downregulation would lead to genome-wide passive dilution of methylation. In addition, we used sgRNAs against enzymes implicated in mediating active demethylation (Tets/Tdg/Aid). Figure 3D shows the predicted outcomes of 129SE-RGM-tdTomato allele demethylation (changes of the fraction of T+G+ cells) after disruption of these genes. When Dnmt1 or Uhrf1 were disrupted, the 129SE-RGM-tdTomato allele became demethylated in a substantial fraction of cells (Figure 3E). In contrast, transduction of sgRNAs against Tet enzymes, Aid, or Tdg had no substantial effect indicating that active demethylation is not significantly involved in SE demethylation. To confirm that the lack of methylation changes upon disruption of Tets, Aid, or Tdg was not due to inefficient Cas9-sgRNA transfection, we further compared the demethylation kinetics of the 129SE-RGM-tdTomato allele in single clones harboring homozygous Tdg and Aid frame-shift mutations (Figure S3D) with that of wild-type cells, and observed no difference (Figure S3E). In addition, DNA methylation levels, as quantified by pyro-sequencing, did not reveal a significant difference among Tet1, 2 and 3 single knockout, Tet1, 2 double knockout, Tet1, 2, 3 triple knockout ESCs, and the isogenic wild-type cells (Dawlaty et al., 2014; Dawlaty et al., 2013; Dawlaty et al., 2011a) (Figure S3C). Given the rapid proliferation of ESCs, our data are consistent with the notion that locus-specific DNA methylation at both SEs is subjected to intrinsically dynamic changes at the allelic level in each cell due to unsynchronized cell division and passive DNA demethylation, which leads to heterogeneous SE methylation at a snapshot sampling time (t1, …, t4, Figure 3F, top). The steady-state of such dynamic heterogeneity reflects a balance between de novo methylation dependent on both DNMT3A and DNMT3B and passive demethylation during rapid cell proliferation (Figure 3F, bottom).
Transcription factor binding at SEs promotes demethylation and inhibits de novo methylation
To explore additional regulators of SE DNA methylation dynamics besides DNMTs activities and cell division, we investigated the impact of transcription factor (TF) binding on the transition between DNA methylation states. Some TFs can serve as readers of DNA methylation or inducing changes to DNA methylation states upon binding to target sequences (Feldmann et al., 2013; Maurano et al., 2015; Yin et al., 2017; Zhu et al., 2016). The Sox2 SE harbors multiple enrichment sites for the master TFs OCT4 and NANOG in ESCs (Hnisz et al., 2013) (Figure 4A, top). We deleted enrichment sites for the two TFs (peak 1 for NANOG and 2 for both NANOG and OCT4) at the Sox2 SE DMR on either the 129SE-RGM-tdTomato or the CASTSE-RGM-eGFP allele using sgRNAs against allele-specific SNPs (Figure 4A, bottom) and generated ESC clones harboring allele-specific peak deletions (ΔPeak 1-CAST, ΔPeak 2-CAST and ΔPeak 2–129 clones, Figure S4A). We sorted the T−G− and T+G+ populations from these clones and monitored the re-establishment of allelic heterogeneity across deletion genotypes (Figure 4B). The fraction of T+G− or T−G+ cells transitioning from T+G+ or T−G− cells were quantified as allelic de novo methylation rates or demethylation rates, respectively (Figure 4C). We found that both the 129SERGM-tdTomato and the CASTSE-RGM-eGFP allele exhibited a faster de novo methylation rate after deletion of its TF enrichment sites as compared to the intact wild-type allele (Figure 4D, top), indicating higher susceptibility to de novo methylation upon loss of TF binding. Similarly, the allele that had its TF enrichment site deleted showed a slower demethylation rate than the wild-type allele, indicating less resistance to maintenance methylation upon loss of TF binding (Figure 4D, bottom). To confirm that the observed RGM activity changes correspond to changes in DNA methylation, we performed BS-PCR followed by Sanger sequencing on sorted cells from ΔPeak 1-CAST and ΔPeak 2–129 clones. This analysis confirmed that the methylation status of the endogenous SE region was consistent with that of the Snrpn promoter as well as RGM activities at allelic resolution after genetic manipulation (Figure S4B). The TF binding effect on methylation dynamics was not only seen in cloned cells but also in sorted T+G+ cell population transfected with allele-specific sgRNAs against TF enrichment sites (Figure 4E). Consistent with the single-cell clone analyses, the allele with TF enrichment site deletion showed a faster de novo methylation rates than the wild-type allele that was not targeted by the sgRNAs (Figure 4F).
Figure 4. Transcription factor binding at SEs promotes demethylation and inhibits de novo methylation.

(A) Top, schematic representation of TF enrichment sites (based on the ENCODE ChIP-seq data; Accession: ENCSR779CZG (NANOG, pink track, peak 1 and 2) and ENCSR392DGA (OCT4, blue track, peak 2) relative to the RGM targeted site (orange). Bottom, allele-specific deletions of individual peaks after overlapping NANOG (N) and OCT4 (O) ChIP tracks. Red: 129SE-RGM-tdTomato allele, green: CASTSE-RGM-eGFP allele. Scissors illustrate sgRNA targeting sites. (B) Experimental setup using cells with different allelic TF enrichment site deletions in assessing the impacts of TF binding on SE methylation dynamics. (C) Top, T+G+ cells were sorted from the genotyped single-cell clones with allelic TF enrichment site deletions. Bottom, T−G− cells were sorted from the same clones. (D) Quantification of allele-specific de novo methylation rates (top panels, T+G− or T−G+ cells derived from T+G+ cells) and demethylation rates (bottom panels, T+G− or T−G+ cells derived from T−G− cells) of the respective ESC clones compared to that of an unmodified parental wild-type clone (dotted line level). (E) Bulk T+G+ cells were sorted from the SOX2-SE-TG cell line, and transfected with allele-specific sgRNA pairs to delete TF enrichment sites or with empty vectors. (F) Quantification of allele-specific de novo methylation rates of the bulk cells transfected with different sgRNAs. See also Figure S4.
DNA methylation decreases MED1 association with SEs, enhancer-promoter H3K27ac, and in-cis transcription of the target genes
We investigated whether the rapid changes in SE DNA methylation would dynamically affect target gene transcription. Promoter DNA methylation has long been associated with stable silencing of gene expression (Deaton and Bird, 2011; Dor and Cedar, 2018; Schubeler, 2015; Smith and Meissner, 2013), in comparison, enhancer methylation’s role in transcription is less well characterized. The Mediator complex has been shown to be dynamically involved in phase-separated condensates concentrating at SEs for transcription of key cell-identity genes (Sabari et al., 2018). Since SE DNA methylation is dynamically changing, we investigated whether different allelic methylation states affect association of MED1 condensates with the Mir290 SE. We performed DNA FISH at the Mir290 SE locus and MED1 immunostaining on sorted cell populations. Figure 5A and Figure S5A show that MED1 was not enriched at the methylated Mir290 SE as T−G− cell populations did not have DNA FISH foci that overlapped with MED1 enrichment as compared to cells in which at least one Mir290 SE was unmethylated. Since the Mediator complex interacts with both the SE and the promoter (Whyte et al., 2013), a loss of MED1 enrichment upon SE DNA methylation may affect promoter activity as well. We therefore performed H3K27ac ChIP-seq as a proxy epigenetic mark defining active enhancers and promoters on four sorted populations from both reporter cell lines. H3K27ac was significantly reduced at both methylated SE regions, as measured by total (Figure 5B, C Sox2 SE and Mir290 SE boxes, Figure S5B enhancer panels) as well as allele-specific H3K27ac enrichment (Figure S5C enhancer panels). As expected, a decrease in H3K27ac was also observed at promoters residing on the same chromosome with the methylated SE (Figure 5B, C Sox2 and Mir290 boxes, and Figure S5B, S5C promoter panels), but not at adjacent regions (Figure S5B adjacent regions panels). This demonstrates that SE methylation affects the promoter H3K27ac level, likely through a loss of enhancer-promoter communication.
Figure 5. DNA methylation decreases MED1 association at SE, enhancer-promoter H3K27ac, and in-cis transcription of the target genes.

(A) Averaged DNA FISH (Magenta, Mir290 SE) and co-immuno-fluorescence staining (Green, MED1) signal in the nuclei of MIR290-SE-TG cells sorted based on allelic methylation states. Random spots were selected in the same image away from the DNA FISH spots. (B) Peak calling from H3K27ac ChIP-seq of 4 sorted populations from MIR290-SE-TG. Mir290 SE and Mir290–295 cluster are boxed in blue. (C) Peak calling from H3K27ac ChIP-seq of 4 sorted populations from SOX2-SE-TG. Sox2 SE and Sox2 gene are boxed in blue. (D) Allele-specific expression of Mir290–295 pri-miRNA (top) and Sox2 mRNA (bottom) in 3 sorted populations, with VIC-TaqMan probe detecting the 129SE-RGM-tdTomato allele, and FAM-TaqMan probe detecting the CASTSE-RGM-eGFP allele in both SE cases. Independently targeted clones for each SE were used as biological replica. Data are represented as mean ± SD. (E) Fold-change of total Mir290–295 pri-miRNA (left) and total Sox2 mRNA (right) from the 4 sorted populations normalized to that of the T+G− population. Independently targeted clones for each SE were used as biological replica. Data are represented as mean ± SD. (F) Quantification of Mir290–295 expression on sorted SOX2-SE-TG cells compare to Sox2 expression (left), and quantification of Sox2 expression on sorted MIR290-SE-TG cells compare to Mir290–295 expression (right). Data are represented as mean ± SD. See also Figure S5.
To test whether synchronized H3K27ac changes upon transient DNA methylation at enhancers and promoters affects in-cis target gene expression, we performed allele-specific qRT-PCR on the four sorted cell populations from both reporter cell lines. As shown in Figure 5D, methylation of either allele of the SEs resulted in decreased target gene expression on the same chromosome. However, the Sox2 SE and the Mir290 SE have different effects on the total expression level of their respective target genes. The suppressive effect of transient DNA methylation was independent and additive when either Mir290 SE allele was methylated (Figure 5E, left). In contrast, total Sox2 expression only significantly decreased when both Sox2 SE alleles were methylated (Figure 5E, right), and in single-positive cells only single-molecule RNA FISH (smFISH) could detect a slight decrease of Sox2 transcripts (Figure S5D, E), indicating a compensating mechanism on total Sox2 transcripts when one SE allele is methylated. Notably, DNA methylation at two SEs exclusively anti-correlated with their respective in-cis target genes, and little difference is seen in Mir290–295 expression if cells were sorted based on the methylation state at the Sox2 SE locus, and vice versa (Figure 5F). This indicates that the DNA methylation state of the two SEs switches independently of each other.
To determine whether SE methylation has a causal role in suppressing enhancer-promoter H3K27ac and transcription, we transfected Cas9-sgRNAs targeting Dnmt1 and Uhrf1 and removed DNA methylation in sorted T−G− MIR290-SE-TG cells to induce rapid passive demethylation (Figure 6A). Figure 6B shows that cells deficient for Dnmt1 or Uhrf1 displayed significantly faster demethylation resulting in a higher proportion of T+G+ cells as compared to the control. Both acetylation of H3K27 at the SE (Figure 6C) and Mir290–295 expression (Figure 6D) were significantly increased upon Dnmt1/Uhrf1 disruptions, as measured by ChIP-qPCR and qRT-PCR from the same cultures, respectively. This suggests that change in DNA methylation directly regulates SE function and transcription in-cis.
Figure 6. DNA methylation directly suppresses SE activity and affects ES cell state.

(A) Experimental setup for assessing the causal role of SE DNA methylation suppresses H3K27ac. FACS (DNA methylation), RT-qPCR (Mir290–295) and ChIP-qPCR (H3K27ac) were co-assessed from the same pool of cells from each sample. (B) Loss of DNA methylation in MIR290-SE-TG T−G− cells 8 days post Dnmt1 and Uhrf1 sgRNA transfection as compared to controls. (C) H3K27ac ChIP-qPCR at the Mir290 SE from the experimental groups in (B), respectively. Data are represented as mean ± SD. (D). Mir290–295 pri-miRNA level from the experimental groups in (B). Data are represented as mean ± SD. (E) Summary of the dynamic regulation and functional impact of allelic SE methylation. (F) Colony formation assays in “2i” starting from 100 sorted cells. Data are represented as mean ± SD. (G) Growth curves measured by AlamarBlue Cell Viability Reagent. Data are represented as mean ± SD. (H) Principal component analysis of the top 5% highly variable genes from different populations of SOX2-SE-TG (Labeled as S. red: T+G−, green, T−G+, black: T−G−, yellow: T+G+) and MIR290-SE-TG (Labeled as M. color code same as S). See also Figure S5, S6, S7.
Since correlating abundance in RNA allele-specific SNPs with allele-specific RGM activities allows distinguishing direct targets regulated in-cis by the SE methylation status versus expression changes caused by secondary effects, we searched additional genomic targets on the same chromosomes that are directly regulated by SE methylation by allele-specific RNA-seq analysis on sorted populations. We quantified allele-specific expression of genes on Chromosome 3 (for MIR290-SE-TG) and Chromosome 7 (for SOX2-SE-TG) in single positive cells and calculated the ratio between expressions from the allele with an unmethylated SE over that of the other allele with a methylated SE. We plotted this ratio of each gene calculated in T−G+ cells as the X-axis value and the ratio calculated in T+G− cells as the Y-axis value (Figure S6A). As expected, Mir290–295 and Sox2 both appeared in the upper right corner as they were in-cis directly suppressed by allelic SE methylation. Surprisingly, two antisense transcripts relative to Sox2 and Mir290–295, Ecm1 and AU018091, respectively, were oppositely regulated by allele-specific Sox2 or Mir290 SE methylation: SE hyper-methylation strongly correlated with up-regulations of both anti-sense transcripts, whereas SE hypo-methylation correlated with inhibition (Figure S6B, C). This result shows that direct transcriptional targets of SE methylation are highly specific with possibly opposite effects on some cis-regulated genes. Though the detailed mechanism of such regulation remains to be elucidated, Ecm1 was upregulated in Sox2 SE deletion cells (Gagnon et al., 2014).
Our results suggest that DNA methylation at both SEs fluctuates independently and dynamically, altering Mediator complex condensates at the SE and allelic H3K27ac at enhancers and promoters in-cis and ultimately leading to heterogeneous allelic transcription of the target genes (Figure 6E).
Sox2 and Mir290 SE methylation heterogeneities have different biological impacts on ESC state
Culture in “2i” medium has been shown to only allow naïve pluripotent cells to proliferate (Nichols and Smith, 2009). Long-term culture of MIR290-SE-TG and SOX2-SE-TG cells in “2i” after passaging from serum + LIF media, though favoring T+G+ population, decreased but did not abolish heterogeneity completely (Figure S6D). The persistence of all four populations in both reporter cell lines indicates that DNA methylation at both SEs have different degrees of heterogeneity in different culture conditions. Both Sox2 and Mir290–295 are highly expressed in ESCs (Calabrese et al., 2007; Hnisz et al., 2013; Jaenisch and Young, 2008; Thomson et al., 2011), raising the possibility that allelic transcriptional heterogeneity caused by SE methylation heterogeneity may lead to co-existing heterogeneous cellular states of ESCs. In “2i” media, SOX2-SE-TG T−G− cells exhibited significantly impaired colony-forming ability and proliferation (Figure 6F, G). However, under the same condition, the heterogeneous DNA methylation at the Mir290 SE did not lead to any obvious changes of ESCs, despite the slight colony formation disadvantage of MIR290-SE-TG T−G− cells (Figure 6F, G). We further explored the functional differences among populations in vivo by injecting sorted cells to form teratomas. Surprisingly, despite the significant growth disadvantage of SOX2-SE-TG T−G− population, they were able to contribute to all three germ layers in teratoma formation assays with no obvious contribution bias towards any germ layer compared to SOX2-SE-TG T+G+, MIR290-SE-TG T+G+ and T−G− cells (Figure S6E). This indicates that ESCs with biallelic methylation at the Sox2 SE are still pluripotent. However, when examined at the molecular level, these cells were distinct from other populations in principal component analysis on difference in the 5% most highly variably expressed genes (Figure 6H) and 17,000 uniquely distinct H3K27ac enrichment peaks in ChIP-seq (Figure S6F). GO analysis on RNA-seq revealed that the SOX2-SE-TG T−G− population preferentially expressed genes in differentiation-related pathways as compared to the MIR290-SE-TG T−G− population (Figure S7A). The epigenetic and transcriptional differences of SOX2-SE-TG T−G− cells indicate that these cells down-regulate Sox2 expression and are prone to differentiate but not as yet committed to a certain fate. Our results are consistent with the notion that pluripotent ESC are heterogeneous as reflected by the dynamic allelic DNA methylation of key pluripotency super-enhancers.
DNA methylation is dynamic at both SEs in blastocysts, while exhibiting spatial-temporal differences in pre-implantation embryos
In vivo, both Sox2 and Mir290–295 are expressed in preimplantation embryos. As reported previously Sox2 expression increases between the morula and the blastocyst stage (Mistri et al., 2018) and Mir290–295 expression significantly upregulates at the 4-cell stage (Medeiros et al., 2011). To investigate changes in DNA methylation of the two SEs at single cell and allelic resolution, we generated transgenic mice homozygous for the 129SE-RGM-tdTomato allele or the CASTSE-RGM-eGFP allele and obtained 2–4 cell embryos carrying one 129SE-RGM-tdTomato allele and one CASTSE-RGM-eGFP allele by mating animals homozygous for RGM-eGFP or RGM-tdTomato (Figure 7A). The two SEs gained allelic DNA methylation heterogeneity at different times: reporter activity became apparent as early as the 4-cell stage for the Mir290 SE but only at the morula stage for the Sox2 SE (Figure 7B). At the blastocyst stage, Sox expression was restricted to the inner cell mass (ICM) whereas the Mir290–295 displayed broad expression in both ICM and trophectoderm (TE) (Nichols and Smith, 2009; Paikari et al., 2017; Wicklow et al., 2014). Heterogeneous SE DNA methylation was consistent with the established spatial expression pattern of the two genes in blastocysts (Figure 7C). We further investigated whether the observed methylation heterogeneity was due to dynamic allelic methylation state switching in vivo. We sorted the four populations from SOX2-SE-TG and MIR290-SE-TG ESCs, injected each population into 8-cell stage wild-type CD1-IGS host embryos and cultured embryos for 2 days to monitor de novo methylation or demethylation at single cell resolution (Figure S7B). A long-term membrane bound dye (Cy5) was used to track the injected cells (Figures 7D and Figure S7C). Figure 7D (SOX2-SE-TG cells) and Figure S7C (MIR290-SE-TG cells) show that at the blastocyst stage, injected T−G− cells demethylated the SE as they turned on the RGMs on either or both alleles and became single positive or T+G+ cells (T−G− columns, white arrows). Demethylation also was observed in injected single positive cells as the originally methylated allele at injection became unmethylated and cells became T+G+ (T+G− and T−G+ columns, white arrows). Similarly, dynamic de novo methylation was observed in vivo, as injected T+G+ cells shut down RGM activities on either or both alleles (T+G+ columns, yellow arrows) and single positive cells became T−G− cells (TG+ and T−G+ columns, yellow arrows).
Figure 7. DNA methylation is dynamic at both SEs in blastocysts, while exhibiting spatial-temporal differences in pre-implantation embryos.

(A) Mating scheme for generating SOX2-SE-TG and MIR290-SE-TG mice and heterozygous pre-implantation embryos genetically carry 129SE-RGM-tdTomato and CASTSE-RGM-eGFP at the Sox2 or the Mir290 SE for imaging analyses. (B) live 4–8 cell (MIR290-SE-TG) and morula stage (SOX2-SE-TG) embryos. (C) Live E3.5-E4.5 blastocysts of SOX2-SE-TG and MIR290-SE-TG in 10x low magnification, 40x high magnification, and 3D projections (left to right in each group). Red: tdTomato, green: eGFP, blue: Hoechst 33342. (D) Tracking Sox2 SE DNA methylation dynamics in vivo. Columns are sorted and injected populations and rows are different imaging channels. Red: RGM-tdTomato; Green: RGM-eGFP; Cy5: Qtracker™ 705 was used to label and track injected cells. White arrows indicate demethylation, and yellow arrows de novo methylation, at 2 days post-injection compare to 5hr post-injection. Channels were adjusted for brightness and contrast for optimal visibility. See also Figure S7.
In summary, our data indicate that dynamic DNA methylation exists at active SEs in early preimplantation embryos creating locus-specific epigenetic heterogeneity, recapitulating and extending our observations in ESCs in-vitro.
Discussion
The importance of DNA methylation regulation at cis-regulatory elements is increasingly recognized as many developmental- and disease-associated DMRs overlap with these regions (Schultz et al., 2015; Weigel et al., 2016; Ziller et al., 2013). Locus-specific DNA methylation heterogeneity across cells has been shown by recent scWGBS as a potential explanation for the variable low-to-intermediate levels of methylation at active enhancers in bulk measurements. The present work was based on an experimental paradigm that overcomes some of the limitations of single-cell sequencing approaches using an allele-specific reporter system. This allowed us to address questions that were not resolved by previously used sequencing-based methods. (i) Our study shows that in ESCs the methylation state of the two alleles of the Sox2 and Mir290 SEs change dynamically and independently of each other. (ii) We demonstrate that the dynamic change of SE DNA methylation is driven by the balance between three DNMTs and cell proliferation, with transcription factor binding promoting the hypomethylated state. (iii) We show that DNA methylation dynamically regulates target genes in-cis and inhibits formation of Mediator complex condensates at the SE as well as enhancer-promoter H3K27 acetylation. (iv) Allelic variation of SE DNA methylation, reflecting the epigenetic heterogeneity of ESCs, can originate from cells of different transcriptional landscapes and proliferative potentials as for the Sox2 SE or of developmentally identical states as for the Mir290 SE. (v) Finally, we show that dynamic DNA methylation is not only seen in cultured ESCs but also in preimplantation embryos.
Allele-specific RGM reporters targeted to the endogenous Sox2 and the Mir290 SEs allowed us to trace DNA methylation both in vitro and in vivo. Detailed analyses showed that the low levels of DNA methylation of the Sox2 and the Mir290 SE are due to the presence of a small fraction of cells with hypermethylated SE alleles. The methylation heterogeneity in these cells results from highly dynamic and reversible switching between allelic DNA methylation states. Because the RGM reporter allowed isolation of cells with defined allele-specific SE DNA methylation states, we were able to demonstrate that dynamic changes in SE DNA methylation are tightly anti-correlated in-cis with enhancer-promoter H3K27ac levels. This is likely due to disruption of enhancer-promoter interactions consistent with the Mediator complex condensates showing decreased association at the methylated Mir290 SE. The Mediator complex and its unit MED1 have been shown previously to form condensates with liquid-like properties, which allows dynamic interactions with transcription factors and the transcription apparatus (Cho et al., 2018; Sabari et al., 2018). Our study shows that DNA methylation can affect these transcriptional condensates. Given the dynamic state switching of allelic SE DNA methylation as well as the dynamic nature of MED1 condensate formation, it is highly likely that one process mediates the other. We also show that SE DNA methylation can have opposing effects on transcription of different genes located on the same chromosome: the direct target genes Sox2 and Mir290–295 were repressed while the antisense genes Ecm1 and AU018091 were activated by SE methylation. By removing DNA methylation at the Mir290 SE through Dnmt1/Uhrf1 deletion we showed that changes in SE DNA methylation is a dynamic process actively regulating its transcriptional activity. By enabling sorting for a particular epigenetic state and combined with allelic expression analyses, we demonstrate that dynamic DNA methylation serves as an epigenetic basis for allelic heterogeneity in gene expression and that dynamic DNA methylation at SEs is a likely mechanism for dynamic random monoallelic transcription seen in mammalian cells (Deng et al., 2014; Reinius and Sandberg, 2015). However, it warrants further exploration to establish the causal link between allelic epigenetic and transcriptional heterogeneity in vivo.
While Sox2 and Mir290 SE methylation affect target gene expression similarly, we detected some differences on cellular growth and differentiation. Cells with biallelically methylated Sox2 SE revealed impaired growth and upregulation of differentiation-related pathways (Figure 6F, G, Figure S7A). In contrast, Mir290 SE methylation had little effects on cell state. We identified additional differences of how DNA methylation suppresses activity of the two SEs. Mir290–295 expression was independently suppressed by methylation at either Mir290 SE DMR allele consistent with the observation that individual DMR constituents have independent activities (Suzuki et al., 2017). In contrast, monoallelic Sox2 SE methylation did not significantly affect the overall Sox2 expression, suggesting additional regulatory mechanisms.
The experimental platform described here allows rapid tracing and isolating rare cell populations based on their transient methylation signatures at specific loci and thus can provide mechanistic insights into the nature of enhancer DNA methylation in heterogeneous cell populations both in vivo and in vitro in real-time, which is difficult in sequencing-based approaches. Furthermore, this system enables manipulation of different molecular components to define interactions and hierarchies between layers of epigenetic regulation in dynamic systems with rapid changes. Our study provides a path towards the mechanistic understanding of dynamic T-DMR regulation in heterogeneous tissues and complex biological processes, such as development and diseases (Heyn and Esteller, 2012; Robertson, 2005).
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rudolf Jaenisch (jaenisch@wi.mit.edu).
EXPERIMENTAL MODEL AND JECT DETAILS
ESC cell lines
ESC cell culture and proliferation assays
All cells were cultured at 37°C with 5% CO2. 129xCAST or v6.5 mouse male ESCs were cultured on irradiated mouse embryonic fibroblasts (MEFs) with standard ESCs medium: (500 ml) DMEM supplemented with 10% FBS (Hyclone), 10 mg recombinant leukemia inhibitory factor (LIF), 0.1 mM beta-mercaptoethanol (Sigma-Aldrich), penicillin/streptomycin, 1 mM L-glutamine, and 1% nonessential amino acids (all from Invitrogen). For experiments in 2i culture conditions, ESCs were cultured on gelatin-coated plates with N2B27 + 2i + LIF medium containing: (500 ml), 240 ml DMEM/F12 (Invitrogen; 11320), 240 ml Neurobasal media (Invitrogen; 21103), 5 ml N2 supplement (Invitrogen; 17502048), 10 ml B27 supplement (Invitrogen; 17504044), 10 mg recombinant LIF, 0.1 mM beta-mercaptoethanol (Sigma-Aldrich), penicillin/streptomycin, 1 mM L-glutamine, and 1% nonessential amino acids (all from Invitrogen), 50 mg/ml BSA (Sigma), PD0325901 (Stemgent, 1 mM), and CHIR99021 (Stemgent, 3 mM). For measuring cell proliferation, AlamarBlue Cell Viability Reagent (Bio-Rad, BUF012A) was added to cell culture and incubated at 37°C with 5% CO2 and emission at 590nm was monitored every 50hrs. At each sampling time point, relative changes in cell numbers were compared to 0hr after sorting.
Generating biallelically targeted reporter cell lines
To generate SOX2-SE-TG and MIR290-SE-TG reporter cell lines, targeting vectors (Mir290-SE-RGM-tdTomato, Mir290-SE-RGM-eGFP, Sox2-SE-RGM-tdTomato, Sox2-SE-RGM-eGFP), and CRISPR/Cas9 were transfected into ESCs using Xfect ESC Transfection Reagent (Clontech, Cat#631320), according to the provider’s protocol. Forty-eight hours following transfection, cells were selected for puromycin resistance (Sigma Aldrich, Cat#P7255) and plated on MEF feeder plates. Single colonies were further analyzed for proper and single integration by Southern blot and Junction PCR analysis. PGK-Puromycin resistance cassette were looped out by overexpression of Cre recombinase (pTurbo-Cre, GenBank accession number AF334827) and followed by Southern blot validation.
ESCs with CRISPR-Cas9-mediated deletions
Tet-enzyme single-, double- and triple knockouts were generated and described previously (Dawlaty et al., 2014; Dawlaty et al., 2013; Dawlaty et al., 2011a). sgRNA sequences are cloned into px330-BFP vector under U6 promoter. px330-BFP-sgRNA vectors were transfected into pre-plated ESC cells using Xfect ESC Transfection Reagent, according to the provider’s protocol. For analysis in populations, cells were sorted for BFP 48 hours post-transfection and cultured on MEF feeder plates. For single clone analysis, cells were genotyped using Southern blot or TA cloning of PCR products of CRISPR targeting site from each allele followed by sequencing. For Dnmt3a and Dnmt3b, Aid and Tdg single knockouts, single clones with frame-shifting indels were selected for further analysis; for TF binding site deletions, single clones have allele-specific entire peak site deletions were selected for further analysis. For TF enrichment site deletion experiments, sgRNA pairs for generating deletion are transfected as following: Δpeak 1-CAST: sgTFBS-Sox2-SE-1(CAST) and sgTFBS-Sox2-SE-2(CAST); Δpeak 2-CAST: sgTFBS-Sox2-SE-2(CAST) and sgTFBS-Sox2-SE-3(CAST); Δpeak 2–129: sgTFBS-Sox2-SE-2(129) and sgTFBS-Sox2-SE-3(Both); Δ Peak 1+2-CAST: sgTFBS-Sox2-SE-1(CAST) and sgTFBS-Sox2-SE-3(CAST). All sgRNA sequences are listed in Table S1.
Animals
Blastocyst injections and generation of reporter mice
Blastocyst injections were performed using (C57BL/6xDBA) B6D2F1 (Charles River) or CD1 (Charles River) host embryos. In brief, 6–7-week old B6D2F1 females were hormone primed by an intraperitoneal (i.p.) injection of pregnant mare serum gonadotropin (PMS, EMD Millipore) followed 46 hr later by an injection of human chorionic gonadotropin (hCG, VWR). Embryos were harvested at the morula stage and cultured in a CO2 incubator overnight. To obtain tetraploid (4n) blastocysts, electrofusion was performed at approximately 44–47 h post hCG using a BEX LF-301 cell fusion device (Protech International Inc., Boerne, TX). On the day of the injection, groups of embryos were placed in drops of M2 medium using a 16-um diameter injection pipet (CytoSpring). Approximately ten cells were injected into the blastocoel cavity of each embryo using a Piezo micromanipulator (Prime Tech). Approximately 20 blastocysts were subsequently transferred to each recipient female; the day of injection was considered as 2.5 days postcoitum (DPC). Male chimera mice were mated to C57BL/6 females and the ones that gave birth to agouti pups (F1) have germ-line transmitted CASTX129 ESC. Mice were handled in accordance with institutional guidelines and approved by the Committee on Animal Care (CAC) and Department of Comparative Medicine (DCM) of Massachusetts Institute of Technology.
Mouse mating scheme and genotyping
All mouse F1 mice heterozygous for either the SE-RGM-tdTomato (abbreviated as SOX2-SE-T0 or MIR290-SE-T0) or the SE-RGM-eGFP allele (abbreviated as SOX2-SE-G0 or MIR290-SE-G0) were obtained by mating germ-line transmitted chimeras to C57BL/6 females. F2 mice homozygous for either SE-RGM-tdTomato (abbreviated as SOX2-SE-TT or MIR290-SE-TT) or the SE-RGM-eGFP allele (abbreviated as SOX2-SE-GG or MIR290-SE-GG) were generated by inbreeding (SOX2-SE-T0 x SOX2-SE-T0, SOX2-SE-G0 x SOX2-SE-G0, MIR290-SE-T0 x MIR290-SE-T0, MIR290-SE-G0 x MIR290-SE-G0). Mice are genotyped by PCR the 5’junction of the SE RGM: SOX2-SEF (or MIR290-SE-F) with tdTomato-R for the RGM-tdTomato allele, SOX2-SE-F(or SOX2-SE-F) with eGFP-R for the RGM-eGFP allele, and SOX2-SE-F (or MIR290-SE-F) with SOX2-SE-R (or MIR290-SE-R) for the wild-type allele (Table S5).
Confocal imaging of live pre-implantation embryos
2-cell embryos were obtained from mating SOX2-SE-TT or MIR290-SE-TT females hormone primed step-wise with PMS and hCG to SOX2-SE-GG or MIR290-SE-GG males, respectively, or the opposite mating strategy (SOX2-SE-GG or MIR290-SE-GG females to SOX2-SE-TT or MIR290-SE-TT males, respectively). 2-cell embryos were flushed out from the oval-duct by M2 media with BSA (CytoSpring # m2113) 48hrs post mating. The embryos were then cultured in 25–50ul KSOM media droplets (CytoSpring # KO102) covered by mineral oil in a 37 5% CO2 incubator. Embryos will become blastocysts at E3.5. For monitoring methylation dynamics in vivo, ESCs were cultured in serum + LIF, pre-plated and sorted based on RGM activity before injection. 2–3 cells were injected into 8-cell stage CD1 host embryos and cultured in M2 media with BSA at 37°C in 5% CO2. Images were taken by a Zeiss LSM 710 Laser Scanning Confocal microscope. Images were taken using either 10x or 40x water lenses and saved in LSM format. Channels for eGFP (excitation 488nm), tdTomato (excitation 594nm), Cy5 (excitation 633nm), and Hoechst 33342 (excitation 405nm) were merged into image composites.
Teratoma formation assays and H&E staining
0.5–1 million sorted ESCs in serum + LIF media were 1:1 mixed with Matrigel and injected subcutaneously into the femur on both sides of the NSG mice. Tumors were taken when reaching 1cm in diameter and mice euthanized. Mice were handled in accordance with institutional guidelines and approved by the Committee on Animal Care (CAC) and Department of Comparative Medicine (DCM) of Massachusetts Institute of Technology. Tissues were dissected and fixed in 10% formalin overnight. Tissues were embedded in paraffin, sectioned, and stained for H&E.
METHOD DETAILS
Southern blots
Genomic DNA (10–15 mg) was digested with appropriate restriction enzymes overnight. Subsequently, genomic DNA was separated on a 0.8% agarose gel, transferred to a nylon membrane (Amersham) and hybridized with 32P probe labeled by Prime-It II Random Primer Labeling Kit (Agilent Technologies, Cat#300385).
Flow cytometry
To assess the proportion of eGFP and tdTomato in the established reporter cell lines, a single-cell suspension was filtered and assessed on the BD Aria or FACSCanto II. Compensation was achieved by using cells with either tdTomato or eGFP fluorescence. Fsc files were analyzed by FlowJo.
Bisulfite conversion-PCR (BS-PCR) and pyro-sequencing
Bisulfite conversion of genomic DNA, nested PCR, and sequencing was established as described previously (Stelzer et al., 2015). Pyro-seq of all bisulfite converted genomic DNA samples were performed with PyroMark Q48 Autoprep (QIAGEN) according to the manufacturer’s instructions. Primers used for BS-PCR and pyro-sequencing are listed in Table S2.
Retinoic acid differentiation
ESCs carrying the reporter for both Mir290 and Sox2 SE regions were sorted for Nanog-eGFP positive and RGM-tdTomato positive and plated on gelatin-coated plates in ESC medium (+LIF). The next day, cells were washed with PBS, re-suspended in basal N2B27 medium (2i medium without LIF, insulin, and the two inhibitors), and supplemented with 0.25uM retinoic acid (RA, Sigma Aldrich, Cat#R2625–50MG). Medium was replaced every other day.
Double thymidine block
10–20k cells/per well were plated onto 12-well plates after sorting with media containing 2.5mM thymidine for 12hrs. Blocking was released by washing twice with PBS and culturing in serum + LIF mouse ES media for 9hrs. Cells were then again blocked with 2.5mM thymidine for 14hrs and FACS analyses were done 6hrs post release.
QUANTIFICATION AND STATISTICAL ANALYSIS
qRT-PCR and TaqMan assays
Total mRNA was extracted from ESCs using Direct-zol RNA Miniprep (Zymo Research, Cat#R2050) after pre-plating for elimination of MEF feeders, treated with DNase A defined amount of mRNA reverse-transcribed into cDNA using SuperScript III First-Strand Synthesis SuperMix (Life Technologies, Cat#18080400) using random hexamers. Total expression of transcripts were quantified by qRT-PCR using Fast SYBR Green Master Mix (Life Technologies), and allele-specific transcripts are quantified by TaqMan Assay customized probes (Sigma, Table S3) targeting Sox2 and Mir290–295 pri-mRNA SNPs. Tukey’s multiple comparison (****P<0.0001, **P<0.01, *P<0.05). Both qRT-PCR and TaqMan assays used at least 2 independently targeted clones as biological replica. The probes and context sequences are listed in Table S3.
ChIP-qPCR
ChIP was done on 2–5 million cells of each same-culture-sorted population from both reporter cell lines as described previously (Lesch et al., 2013), 2ug of anti-H3K27ac antibody (abcam ab4729) was used for precipitation. Eluted DNA was quantified using real-time qPCR with Fast SYBR Green Master Mix. Each ChIP-qPCR was repeated 3 times. Enrichment was calculated using as percentage of input. Statistical differences between samples are calculated with two-way ANOVA (alpha = 0.05), followed by Tukey’s multiple comparison (****P<0.0001, **P<0.01). Primers used for detecting positive and negative control sequences, and SE targets are listed in Table S4.
H3K27ac ChIP-Seq and analysis
ChIP samples of 4 same-culture-sorted populations from SOX2-SE-TG and MIR290-SE-TG, respectively, were validated for positive and negative targets using qPCR. Libraries of Input-ChIP pairs were prepared with Accel-NGS 2S PCR-Free Library Kit (Cat#20096) and sequenced using Illumina HiSeq 2500. Raw reads were aligned to the reference genome mm10 using BWA using default parameters. Peak calling was done using MACS2. Peak intensities at SE, promoter and in-between regions are quantified and compared using bamCompare – deepTools 3.0.2 with FPKM from 10bp genomic bins of each sample. SNPs specific to 129 or CAST genomes at SE and promoters were counted from mapped raw reads and SNPs covered by more than 3 reads are accepted for quantification. Coordinates for analysis (mm10): Mir290-SE: chr7:3198900–3202780, Mir290-promoter: chr7:3215340–3221110; Sox2-SE: chr3:34752523–34766449, Sox2-promoter chr3:34649995–34652460.
RNA-Seq and analysis
For each reporter cell line, 2 independently targeted clones are independently sorted twice, generating 2 biological replica x 2 experimental replica = 4 replica in total. Stranded mRNA libraries were prepared using KAPA HyperPrep (SOX2-SE-TG) and TrueSeq Stranded PolyA prep (MIR290-SE-TG). mRNA libraries were sequenced on Illumina HiSeq 2500. Allele-specific RNA expression was quantified with a custom pipeline. In short, raw fastq files are aligned to a consensus genome using STAR (v.2.5.3.a). The reference transcriptome includes the Mir290–295 pri-miRNA or Sox2 and the RGMs on pseudo-chromosomes. After alignment SNPsplit (v0.3.2) splits the reads into four files based on single nucleotide variations (SNV). The reads were either allele specific (for CAST or S129), unassigned (if there are no SNVs present) or conflicting (if the SNVs in the read are from both alleles). The split read files were quantified using RSEM (v1.2.31) separately for each sample. Raw counts were then normalized to library using DESeq2 (v1.18.1) for each split. To obtain sample-level quantifications raw counts were summed over the splits before normalization. Differential expression analysis (DEA) was performed using DESeq2 (v1.18.1) at the level of samples. Samples were corrected for genetic clone and batch effect. GO analyses were performed using PANTHER. All expressed genes in the respective cell lines were used as the reference backgrounds. All P-values were controlled for false discovery rate (Benjamin-Hochberg procedure).
RNA smFISH and image analyses
Cells were fixed for 15 min with 4% PFA at room temperature and subsequently permeabilized in 70% EtOH overnight. Custom designed smFISH probes for Sox2 labeled with Quasar 670 (Stellaris® DesignReady FISH Probes, Cat# VSMF-3075–5-BS) were incubated with the samples for 16 hours at 30°C in hybridization buffer (100 mg/mL dextran sulfate, 25% formamide, 2X SSC, 1 mg/mL E.coli tRNA, 1 mM vanadyl ribonucleoside complex, 0.25 mg/mL BSA). Samples were washed twice for 30 min at 30°C with wash buffer (25% formamide, 2X SSC) containing DAPI (1 μg/mL, Sigma D9542). All solutions were prepared with RNAse-free water. Finally, the sections were mounted using ProlongGold (Life Technologies, P36930) and imaged two days later. Mounted samples were imaged on a Nikon Ti-Eclipse epifluorescence microscope equipped with an Andor iXON Ultra 888 EMCCD camera, using a 100X /1.45 Plan Apo Lambda oil objective (Nikon) and dedicated, custom-made fluorescence filter sets (Nikon). z-stacks with a distance of 0.3 μm between planes were collected. The number of Sox2 (mRNA) signals per cell was quantified using home-made MATLAB scripts.
DNA FISH, Med1 IF and average image analyses
DNA FISH of the Mir290 SE and IF of MED1 were done as previously described (Sabari et al., 2018). For analysis of RNA/DNA FISH with immunofluorescence, custom Python scripts were written to process and analyze 3D image data gathered in FISH and IF channels. Nuclear stains were blurred with a Gaussian filter (sigma = 2.0), maximally projected in the z plane, and clustered into 2 clusters (nuclei and background) by K-means. FISH foci were either manually called with ImageJ or automatically called using the scipy ndimage package. For automatic detection, an intensity threshold (mean + 3*standard deviation) was applied to the FISH channel. The ndimage find_objects function was then used to call contiguous FISH foci in 3D. These FISH foci were then filtered by various criteria, including size (minimum 100 voxels), circularity of a max z-projection (circularity = 4*areaperimeter2; 0.7), and being present in a nucleus (determined by nuclear mask described above). For manual calling, FISH foci were identified in maximum z-projections of the FISH channel, and the x and y coordinates were used as reference points to guide the automatic detection described above. The FISH foci were then centered in a 3D-box (length size l = 3.0 μm). The IF signal centered at FISH foci for each FISH and IF pair are then combined and an average intensity projection is calculated, providing averaged data for IF signal intensity within a l × l square centered at FISH foci. As a control, this same process was carried out for IF signal centered at an equal number of randomly selected nuclear positions. These average intensity projections were then used to generate 2D contour maps of the signal intensity. Contour plots are generated using the matplotlib python package. For the contour plots, the intensity-color ranges presented were customized across a linear range of colors (n! = 15). For the FISH channel, black to magenta was used. For the IF channel, we used chroma.js (an online color generator) to generate colors across 15 bins, with the key transition colors chosen as black, blueviolet, medium-blue, lime. This was done to ensure that the reader’s eye could more readily detect the contrast in signal. The generated colormap was employed to 15 evenly spaced intensity bins for all IF plots. The averaged IF centered at FISH or at randomly selected nuclear locations are plotted using the same color scale, set to include the minimum and maximum signal from each plot.
High-throughput sequencing of bisulfite PCR
PCR amplicons were sonicated using Covarius into 150–200bp range. NEBNext® Ultra™ DNA Library Prep Kit for Illumina and NEBNext® Multiplex Oligos for Illumina® were used to construct libraries according to manufacturer’s protocol. Single barcoded library was prepared from sonicated bisulfite PCR amplicon fragments of the Mir290 SE wildtype-allele using NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB #E7370S) and NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1, NEB #E7335S). Libraries were sequenced with 40bp single reads, adapter trimmed, aligned and analyzed with Bismark v0.21.0 (bismark - nondirectional). CpGs with >1000 coverage were counted to generate average percentage of methylation. Methylation percentage and its standard error were estimated as described in (Smallwood et al., 2014), and number of methylated counts was assumed to be a binomial random variable.
DATA AND SOFTWARE AVAILABILITY
Description: http://dx.doi.org/10.17632/6vbc6htfnf.1
The raw confocal, gel and film images and original fsc files have been deposited at Mendeley Data
Description: Raw and processed high-throughput sequencing data have been deposited at NCBI Gene Expression Omnibus under ID code GSE132416 (subseries: GSE132376 for H3K27ac ChIP-seq, GSE132404 for BS-seq, and GSE132414 for RNA-seq).
Supplementary Material
Table S1. Table S1. sgRNAs for targeting and knockout, Related to the STAR Methods
Table S2. Primers for bisulfite PCR and pyro-sequencing, Related to the STAR Methods
Table S3. Primers for qRT-PCR, Related to the STAR Methods
Table S4. Primers for ChIP-qPCR, Related to the STAR Methods
Table S5. Primers for reporter ESC line, KO cell line, and mouse genotyping, Related to the STAR Methods
Highlights.
Allele-specific reporters revealed dynamic DNA methylation of Sox2 and miR290 SEs
DNMTs and transcription factor binding regulate methylation dynamics
SE DNA methylation directly regulates transcription in-cis
Dynamic DNA methylation is co-regulated with MED1 recruitment and H3K27ac level
Acknowledgements
We thank George Bell, Prathapan Thiru and Bingbing Yuan for their help in ChIP-seq analysis and BS-PCR sequencing analysis; Ruth Flannery and Dina Rooney for their help with animal husbandry, injections of the ESCs and harvesting pre-implantation embryos; Dongdong Fu for sectioning and processing of teratoma samples. We would like to thank Tom Volkert, Sumeet Gupta, Kevin Truong, Amanda Chilaka, and Jennifer Love of the Whitehead Genome Technology Core for their help in ChIP-seq; Wendy Salmon of the W.M. Keck Microscopy Facility for help with confocal microscopy; Glenn Paradis, Patti Wisniewski, Patrick Autissier, Michael Jennings, Michele Griffin, Mervelina Saturno-Condon, Hanna Aharonov, and Eleanor Kincaid of the Whitehead Institute and MIT flow cytometry facilities for their help with cell sorting. We thank Dr. Roderick Bronson and Kathleen Cormier at the KI Swanson Biotechnology Center Histology Core for teratoma sample consultation. We thank Raaji Alagappan, Tenzin Lungjangwa, and Carrie Garrett-Engele for their technical support. We thank Alicia V. Zamudio from Young Lab, Jian Shu, Shawn Liu, Haiting Ma, Emile Wogram, and all of the members of the Jaenisch lab for helpful discussions. Y.S. was supported by HFSP long-term fellowship, ISF grant no. 1610/18 and is the incumbent of the Louis and Ida Rich Career Development Chair. RJ was supported by NIH grants HD 045022, R37-CA084198 and 1U19AI131135-01
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
R.J. is a cofounder of Fate Therapeutics, Fulcrum Therapeutics and Omega Therapeutics and an advisor to Dewpoint Therapeutics. R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics and Dewpoint Therapeutics.
References
- Calabrese JM, Seila AC, Yeo GW, and Sharp PA (2007). RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 104, 18097–18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheow LF, Quake SR, Burkholder WF, and Messerschmidt DM (2015). Multiplexed locus-specific analysis of DNA methylation in single cells. Nature Protocols 10, 619–631. [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Huebner AJ, Clement K, Walsh RM, Savol A, Lin K, Gu H, Di Stefano B, Brumbaugh J, Kim SY, et al. (2017). Prolonged Mek½ suppression impairs the developmental potential of embryonic stem cells. Nature 548, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, Powell BE, Cheng AW, Faull KF, Lyko F, et al. (2014). Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev Cell 29, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, et al. (2013). Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 24, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. (2011). Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A (2011). CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, and Sandberg R (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- Dor Y, and Cedar H (2018). Principles of DNA methylation and their implications for biology and medicine. Lancet 392, 777–786. [DOI] [PubMed] [Google Scholar]
- Ehrlich KC, Paterson HL, Lacey M, and Ehrlich M (2016). DNA Hypomethylation in Intragenic and Intergenic Enhancer Chromatin of Muscle-Specific Genes Usually Correlates with their Expression. Yale J Biol Med 89, 441–455. [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Hong CB, Xing XY, Zhou X, Li DF, Coarfa C, Bell RJA, Maire CL, Ligon KL, Sigaroudinia M, et al. (2015). Intermediate DNA methylation is a conserved signature of genome regulation. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann A, Ivanek R, Murr R, Gaidatzis D, Burger L, and Schubeler D (2013). Transcription Factor Occupancy Can Mediate Active Turnover of DNA Methylation at Regulatory Regions. Plos Genet 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer T, Tekpli X, Mathelier A, Wang SX, Nebdal D, Dhakal HP, Sahlberg KK, Schlichting E, Borresen-Dale AL, Borgen E, et al. (2017). DNA methylation at enhancers identifies distinct breast cancer lineages. Nature Communications 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, and Schier AF (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. Plos One 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Li L, Li J, Wu X, Hu B, Zhu P, Wen L, and Tang F (2017). Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27, 967–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Zhu P, Guo F, Li X, Wu X, Fan X, Wen L, and Tang F (2015). Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc 10, 645–659. [DOI] [PubMed] [Google Scholar]
- Guo H, Zhu P, Wu X, Li X, Wen L, and Tang F (2013). Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23, 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, and Esteller M (2012). DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13, 679–692. [DOI] [PubMed] [Google Scholar]
- Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, et al. (2016). Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 17, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, and Ren B (2013). Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nature Genetics 45, 1198–U1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Huang K, An Q, Du G, Hu G, Xue J, Zhu X, Wang CY, Xue Z, and Fan G (2016). Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi B, Pistoni M, Cludts K, Akkor P, Lambrechts D, Verfaillie C, Verhamme P, Freson K, and Hoylaerts MF (2016). Allele-specific DNA methylation reinforces PEAR1 enhancer activity. Blood 128, 1003–1012. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, and Young R (2008). Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Jones MJ, Chen E, Neumann SM, Fraser HB, Miller GE, and Kobor MS (2015). Discordance of DNA methylation variance between two accessible human tissues. Sci Rep 5, 8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Li Y, and Robertson KD (2011). DNA methylation: superior or ordinate in the epigenetic hierarchy? Genes Cancer 2, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics 13, 484–492. [DOI] [PubMed] [Google Scholar]
- King AD, Huang K, Rubbi L, Liu S, Wang CY, Wang YS, Pellegrini M, and Fan GP (2016). Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep 17, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. (2012). Contribution of Intragenic DNA Methylation in Mouse Gametic DNA Methylomes to Establish Oocyte-Specific Heritable Marks. Plos Genet 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, et al. (2013). Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch BJ, Dokshin GA, Young RA, McCarrey JR, and Page DC (2013). A set of genes critical to development is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. P Natl Acad Sci USA 110, 16061–16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Du TT, Wagner U, Xie W, Lee AY, Goyal P, Li YJ, Szulwach KE, Jin P, Lorincz MC, et al. (2014). Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. P Natl Acad Sci USA 111, 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CY, Hajkova P, and Ecker JR (2018). Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, and Stamatoyannopoulos JA (2015). Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep 12, 1184–1195. [DOI] [PubMed] [Google Scholar]
- Medeiros LA, Dennis LM, Gill ME, Houbaviy H, Markoulaki S, Fu D, White AC, Kirak O, Sharp PA, Page DC, et al. (2011). Mir-290–295 deficiency in mice results in partially penetrant embryonic lethality and germ cell defects. Proc Natl Acad Sci U S A 108, 14163–14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistri TK, Arindrarto W, Ng WP, Wang CY, Lim LH, Sun LL, Chambers I, Wohland T, and Robson P (2018). Dynamic changes in Sox2 spatio-temporal expression promote the second cell fate decision through Fgf4/Fgfr2 signaling in preimplantation mouse embryos. Biochem J 475, 1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, and Smith A (2009). Naive and primed pluripotent states. Cell Stem Cell 4, 487–492. [DOI] [PubMed] [Google Scholar]
- Paikari A, C DB, Saw D, and Blelloch R (2017). The eutheria-specific Mir-290 cluster modulates placental growth and maternal-fetal transport. Development 144, 3731–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, and Sandberg R (2015). Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nature reviews Genetics 16, 653–664. [DOI] [PubMed] [Google Scholar]
- Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A, et al. (2016). Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 19, 491–501. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD (2005). DNA methylation and human disease. Nat Rev Genet 6, 597–610. [DOI] [PubMed] [Google Scholar]
- Rulands S, Lee HJ, Clark SJ, Angermueller C, Smallwood SA, Krueger F, Mohammed H, Dean W, Nichols J, Rugg-Gunn P, et al. (2018). Genome-Scale Oscillations in DNA Methylation during Exit from Pluripotency. Cell Syst 7, 63-76 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeler D (2015). Function and information content of DNA methylation. Nature 517, 321–326. [DOI] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, and Reik W (2012). The Dynamics of Genome-wide DNA Methylation Reprogramming in Mouse Primordial Germ Cells. Molecular Cell 48, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull AY, Luo JF, Pei LR, Lee EJ, Liu JM, Choi J, Awan FT, and Shi HD (2016). DNA Hypomethylation within B-Cell Enhancers and Super Enhancers Reveal a Dependency on Immune and Metabolic Mechanisms in Chronic Lymphocytic Leukemia. Blood 128. [Google Scholar]
- Sim YJ, Kim MS, Nayfeh A, Yun YJ, Kim SJ, Park KT, Kim CH, and Kim KS (2017). 2i Maintains a Naive Ground State in ESCs through Two Distinct Epigenetic Mechanisms. Stem Cell Reports 8, 1312–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, and Elowitz MB (2014). Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 55, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, and Kelsey G (2014). Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nature methods 11, 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, and Meissner A (2013). DNA methylation: roles in mammalian development. Nature reviews Genetics 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Stelzer Y, Shivalila CS, Soldner F, Markoulaki S, and Jaenisch R (2015). Tracing dynamic changes of DNA methylation at single-cell resolution. Cell 163, 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y, Wu H, Song YL, Shivalila CS, Markoulaki S, and Jaenisch R (2016). Parent-of-Origin DNA Methylation Dynamics during Mouse Development. Cell Rep 16, 3167–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Young RA, and Sharp PA (2017). Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 168, 1000-1014 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, and Ramanathan S (2011). Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F, Iurlaro M, Habibi E, Liu NQ, Salehzadeh-Yazdi A, Santos F, Petrini E, Milagre I, Yu M, Xie Z, et al. (2016). Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Mol Cell 62, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel C, Veldwijk MR, Oakes CC, Seibold P, Slynko A, Liesenfeld DB, Rabionet M, Hanke SA, Wenz F, Sperk E, et al. (2016). Epigenetic regulation of diacylglycerol kinase alpha promotes radiation-induced fibrosis. Nat Commun 7, 10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklow E, Blij S, Frum T, Hirate Y, Lang RA, Sasaki H, and Ralston A (2014). HIPPO Pathway Members Restrict SOX2 to the Inner Cell Mass Where It Promotes ICM Fates in the Mouse Blastocyst. Plos Genet 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S, and Filipp FV (2018). A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst Biol Appl 4, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nature reviews Genetics 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, Yamamoto T, and Yamada Y (2017). Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature 548, 224–227. [DOI] [PubMed] [Google Scholar]
- Yin YM, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang GH, and Qian J (2016). Transcription factors as readers and effectors of DNA methylation. Nature Reviews Genetics 17, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Seila AC, Yeo GW, and Sharp PA (2007). RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 104, 18097–18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheow LF, Quake SR, Burkholder WF, and Messerschmidt DM (2015). Multiplexed locus-specific analysis of DNA methylation in single cells. Nature Protocols 10, 619–631. [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Huebner AJ, Clement K, Walsh RM, Savol A, Lin K, Gu H, Di Stefano B, Brumbaugh J, Kim SY, et al. (2017). Prolonged Mek½ suppression impairs the developmental potential of embryonic stem cells. Nature 548, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, Powell BE, Cheng AW, Faull KF, Lyko F, et al. (2014). Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev Cell 29, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, et al. (2013). Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 24, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. (2011a). Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. (2011b). Tet1 Is Dispensable for Maintaining Pluripotency and Its Loss Is Compatible with Embryonic and Postnatal Development. Cell Stem Cell 9, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A (2011). CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, and Sandberg R (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, and Cedar H (2018). Principles of DNA methylation and their implications for biology and medicine. Lancet 392, 777–786. [DOI] [PubMed] [Google Scholar]
- Ehrlich KC, Paterson HL, Lacey M, and Ehrlich M (2016). DNA Hypomethylation in Intragenic and Intergenic Enhancer Chromatin of Muscle-Specific Genes Usually Correlates with their Expression. Yale J Biol Med 89, 441–455. [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Hong CB, Xing XY, Zhou X, Li DF, Coarfa C, Bell RJA, Maire CL, Ligon KL, Sigaroudinia M, et al. (2015). Intermediate DNA methylation is a conserved signature of genome regulation. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann A, Ivanek R, Murr R, Gaidatzis D, Burger L, and Schubeler D (2013). Transcription Factor Occupancy Can Mediate Active Turnover of DNA Methylation at Regulatory Regions. Plos Genet 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer T, Tekpli X, Mathelier A, Wang SX, Nebdal D, Dhakal HP, Sahlberg KK, Schlichting E, Borresen-Dale AL, Borgen E, et al. (2017). DNA methylation at enhancers identifies distinct breast cancer lineages. Nature Communications 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, and Schier AF (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. Plos One 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Li L, Li J, Wu X, Hu B, Zhu P, Wen L, and Tang F (2017). Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27, 967–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Zhu P, Guo F, Li X, Wu X, Fan X, Wen L, and Tang F (2015). Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc 10, 645–659. [DOI] [PubMed] [Google Scholar]
- Guo H, Zhu P, Wu X, Li X, Wen L, and Tang F (2013). Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23, 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, and Esteller M (2012). DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13, 679–692. [DOI] [PubMed] [Google Scholar]
- Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, et al. (2016). Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 17, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, and Ren B (2013). Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nature Genetics 45, 1198–U1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Huang K, An Q, Du G, Hu G, Xue J, Zhu X, Wang CY, Xue Z, and Fan G (2016). Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi B, Pistoni M, Cludts K, Akkor P, Lambrechts D, Verfaillie C, Verhamme P, Freson K, and Hoylaerts MF (2016). Allele-specific DNA methylation reinforces PEAR1 enhancer activity. Blood 128, 1003–1012. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, and Young R (2008). Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Jones MJ, Chen E, Neumann SM, Fraser HB, Miller GE, and Kobor MS (2015). Discordance of DNA methylation variance between two accessible human tissues. Sci Rep 5, 8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Li Y, and Robertson KD (2011). DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics 13, 484–492. [DOI] [PubMed] [Google Scholar]
- King AD, Huang K, Rubbi L, Liu S, Wang CY, Wang YS, Pellegrini M, and Fan GP (2016). Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep 17, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. (2012). Contribution of Intragenic DNA Methylation in Mouse Gametic DNA Methylomes to Establish Oocyte-Specific Heritable Marks. Plos Genet 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, et al. (2013). Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch BJ, Dokshin GA, Young RA, McCarrey JR, and Page DC (2013). A set of genes critical to development is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. P Natl Acad Sci USA 110, 16061–16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Du TT, Wagner U, Xie W, Lee AY, Goyal P, Li YJ, Szulwach KE, Jin P, Lorincz MC, et al. (2014). Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. P Natl Acad Sci USA 111, 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CY, Hajkova P, and Ecker JR (2018). Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, and Stamatoyannopoulos JA (2015). Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep 12, 1184–1195. [DOI] [PubMed] [Google Scholar]
- Medeiros LA, Dennis LM, Gill ME, Houbaviy H, Markoulaki S, Fu D, White AC, Kirak O, Sharp PA, Page DC, et al. (2011). Mir-290–295 deficiency in mice results in partially penetrant embryonic lethality and germ cell defects. Proc Natl Acad Sci U S A 108, 14163–14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, et al. (2005). The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 33, D284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistri TK, Arindrarto W, Ng WP, Wang CY, Lim LH, Sun LL, Chambers I, Wohland T, and Robson P (2018). Dynamic changes in Sox2 spatio-temporal expression promote the second cell fate decision through Fgf4/Fgfr2 signaling in preimplantation mouse embryos. Biochem J 475, 1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, and Smith A (2009). Naive and primed pluripotent states. Cell Stem Cell 4, 487–492. [DOI] [PubMed] [Google Scholar]
- Paikari A, C DB, Saw D, and Blelloch R (2017). The eutheria-specific miR-290 cluster modulates placental growth and maternal-fetal transport. Development 144, 3731–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, and Tyagi S (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nature methods 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, and Sandberg R (2015). Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nature reviews Genetics 16, 653–664. [DOI] [PubMed] [Google Scholar]
- Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A, et al. (2016). Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 19, 491–501. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD (2005). DNA methylation and human disease. Nat Rev Genet 6, 597–610. [DOI] [PubMed] [Google Scholar]
- Rulands S, Lee HJ, Clark SJ, Angermueller C, Smallwood SA, Krueger F, Mohammed H, Dean W, Nichols J, Rugg-Gunn P, et al. (2018). Genome-Scale Oscillations in DNA Methylation during Exit from Pluripotency. Cell Syst 7, 63-76 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeler D (2015). Function and information content of DNA methylation. Nature 517, 321–326. [DOI] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, and Reik W (2012). The Dynamics of Genome-wide DNA Methylation Reprogramming in Mouse Primordial Germ Cells. Molecular Cell 48, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull AY, Luo JF, Pei LR, Lee EJ, Liu JM, Choi J, Awan FT, and Shi HD (2016). DNA Hypomethylation within B-Cell Enhancers and Super Enhancers Reveal a Dependency on Immune and Metabolic Mechanisms in Chronic Lymphocytic Leukemia. Blood 128. [Google Scholar]
- Sim YJ, Kim MS, Nayfeh A, Yun YJ, Kim SJ, Park KT, Kim CH, and Kim KS (2017). 2i Maintains a Naive Ground State in ESCs through Two Distinct Epigenetic Mechanisms. Stem Cell Reports 8, 1312–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, and Elowitz MB (2014). Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 55, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, and Kelsey G (2014). Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nature methods 11, 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, and Meissner A (2013). DNA methylation: roles in mammalian development. Nature reviews Genetics 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Stelzer Y, Shivalila CS, Soldner F, Markoulaki S, and Jaenisch R (2015). Tracing dynamic changes of DNA methylation at single-cell resolution. Cell 163, 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y, Wu H, Song YL, Shivalila CS, Markoulaki S, and Jaenisch R (2016). Parent-of-Origin DNA Methylation Dynamics during Mouse Development. Cell Rep 16, 3167–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Young RA, and Sharp PA (2017). Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 168, 1000-1014 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, and Narechania A (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, and Ramanathan S (2011). Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F, Iurlaro M, Habibi E, Liu NQ, Salehzadeh-Yazdi A, Santos F, Petrini E, Milagre I, Yu M, Xie Z, et al. (2016). Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Molecular cell 62, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel C, Veldwijk MR, Oakes CC, Seibold P, Slynko A, Liesenfeld DB, Rabionet M, Hanke SA, Wenz F, Sperk E, et al. (2016). Epigenetic regulation of diacylglycerol kinase alpha promotes radiation-induced fibrosis. Nat Commun 7, 10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklow E, Blij S, Frum T, Hirate Y, Lang RA, Sasaki H, and Ralston A (2014). HIPPO Pathway Members Restrict SOX2 to the Inner Cell Mass Where It Promotes ICM Fates in the Mouse Blastocyst. Plos Genet 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S, and Filipp FV (2018). A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst Biol Appl 4, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nature reviews Genetics 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, Yamamoto T, and Yamada Y (2017). Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature 548, 224–227. [DOI] [PubMed] [Google Scholar]
- Yin YM, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang GH, and Qian J (2016). Transcription factors as readers and effectors of DNA methylation. Nature Reviews Genetics 17, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Table S1. sgRNAs for targeting and knockout, Related to the STAR Methods
Table S2. Primers for bisulfite PCR and pyro-sequencing, Related to the STAR Methods
Table S3. Primers for qRT-PCR, Related to the STAR Methods
Table S4. Primers for ChIP-qPCR, Related to the STAR Methods
Table S5. Primers for reporter ESC line, KO cell line, and mouse genotyping, Related to the STAR Methods
Data Availability Statement
Description: http://dx.doi.org/10.17632/6vbc6htfnf.1
The raw confocal, gel and film images and original fsc files have been deposited at Mendeley Data
Description: Raw and processed high-throughput sequencing data have been deposited at NCBI Gene Expression Omnibus under ID code GSE132416 (subseries: GSE132376 for H3K27ac ChIP-seq, GSE132404 for BS-seq, and GSE132414 for RNA-seq).