

Abstract
Decreased autophagy has been reported to contribute to the progression of cardiac hypertrophy. Our previous research has demonstrated that endophilin A2 (EndoA2) attenuates H2O2-induced cardiomyocyte apoptosis by strengthening autophagy. However, the role of EndoA2 in the regulation of autophagy in cardiac hypertrophy is unknown. In this study, we tested the hypothesis that EndoA2 suppresses cardiac hypertrophy induced by isoproterenol (ISO) by activating autophagy. In vivo, we established a cardiac hypertrophy model by subcutaneous injection of ISO and used intramyocardial delivery of adenovirus vector harboring EndoA2 cDNA (Ad-EndoA2) to overexpress EndoA2. The cardiac hypertrophic response and autophagy level were measured. EndoA2 overexpression suppressed pathological cardiac hypertrophy and enhanced autophagy in rat hearts. In addition, the effects of EndoA2 on cardiac hypertrophy and autophagy were observed in cultured neonatal rat cardiomyocytes (NRCMs) with gain- and loss-of-function approaches to regulate EndoA2 expression. The results were consistent with those of the in vivo study. Furthermore, the involvement of EndoA2-mediated autophagy in the attenuation of ISO-induced cardiac hypertrophy was explored by pharmaceutical inhibition of autophagy. Pretreatment with 3-methyladenine (3-MA) clearly diminished the anti-hypertrophic effects of EndoA2 in ISO-treated NRCMs. The results presented here provide the first evidence that EndoA2 is involved in ISO-induced cardiac hypertrophy. The anti-hypertrophic effects of EndoA2 can be partially attributed to its regulation of autophagy.
Keywords: Endophilin A2, isoproterenol, cardiac hypertrophy, autophagy
Introduction
Although cardiac hypertrophy is initially an adaptive process for increased neurohormonal stress and hemodynamic load, sustained hypertrophy will result in a decompensatory stage and eventually progress to heart failure [1]. It is well recognized that left ventricular hypertrophy is the leading cause of heart failure because of chronic myocardial remodeling and cardiac dysfunction [2]. Hence, exploring the molecular mechanism for cardiac hypertrophy will help establish an effective remedy to prevent heart failure.
Although the precise mechanisms of cardiac hypertrophy are complex and remain to be clarified, autophagy is identified as a crucial factor in controlling cardiac hypertrophy [3,4]. Autophagy is a highly conserved process that exists in eukaryotes. The activation of the autophagy response to nutrient deprivation serves as a means of providing energy to sustain vital cellular functions. In addition, autophagy can eliminate misfolded and dysfunctional proteins. Conversely, disruption of autophagy may result in deleterious effects. Emerging evidence suggests that autophagy participates in many forms of heart diseases, including ischemic heart diseases [5,6], hypertensive heart diseases [7], and load-induced heart diseases [8]. For cardiac hypertrophy, studies suggest that basal levels of autophagy help maintain cardiac morphology and function, whereas overactivated autophagy may be maladaptive. Recently, a study showed that decreasing autophagy evoked a hypertrophic response to isoproterenol (ISO) and that increasing autophagy attenuated cardiac hypertrophy [9]. This evidence suggests that the appropriate level of autophagy may be a novel target therapy for cardiac hypertrophy.
Endophilin A2 (EndoA2), which belongs to the endophilin A family, plays a vital role in the regulation of neurological diseases [10], cardiovascular diseases [11-13] and tumors [14]. EndoA2 was originally identified as a fusion partner of the mixed-lineage leukemia gene in human acute leukemia [15]. Primary studies have focused on neuroregulation, tumor formation and metastasis. During the last few years, the involvement of EndoA2 in cardiovascular diseases has attracted increasing attention. Our recent study showed that EndoA2 attenuated angiotensin-II-induced cardiac hypertrophy by inhibiting angiotensin II type 1 receptor trafficking in neonatal rat cardiomyocytes (NRCMs) [16]. Furthermore, we demonstrated that EndoA2 protected H9C2 cardiomyocytes against H2O2-induced apoptosis [17]. Considering the protective effects of EndoA2 on cardiac hypertrophy and the potential modulation of autophagy by EndoA2, we were prompted to explore the relationship between autophagy and the anti-hypertrophic effects of EndoA2. In the present study, we found that activation of autophagy by EndoA2 contributed to the attenuation of cardiac hypertrophy induced by ISO.
Materials and methods
Primary culture of NRCMs
Neonatal Sprague-Dawley rats (1-3 days old) were exposed to hypothermia until they became unresponsive, the rats were then euthanized by cervical dislocation. Primary cultures of NRCMs were performed as we previously described [16]. Briefly, the cells were seeded at a density of 5×106 cells/ml in DMEM supplemented with 10% FBS and 0.1 mM 5-bromodeoxyuridine. The medium was supplemented with ISO (Sigma-Aldrich, USA), and the cardiomyocytes were further incubated for the indicated times. Incubation with 3-methyladenine (3-MA) (Selleck, Shanghai, CN) occurred 2 h before ISO treatment.
Animal models, echocardiography and morphometric measurements
SD rats (male, weighing 180-200 g, aged between 8-10 weeks) were obtained from Guangzhou University of Chinese Medicine (Guangzhou, China). All animals were handled according to the EC Directive 86/609/EEC. Animal study protocols were reviewed and approved by the Ethics Committee of Guangzhou Medical University. The SD rats used in this study were randomly divided into the following five groups: (1) sham, (2) adenovirus vector harboring EndoA2 cDNA (Ad-EndoA2), (3) ISO, (4) ISO+Ad-lacZ, and (5) ISO+Ad-EndoA2. The number of animals was 8-10 per group. A total of 100 μL Ad-EndoA2 or Ad-lacZ (109 particles, Shanghai Sunbio Biomedical Technology, Shanghai, CN) was randomly injected into 5 sites along the anterior and posterior left ventricular wall by using a curved 25-gauge needle. Cardiac hypertrophy was induced by subcutaneous injection of ISO (1.5 mg/kg/d) for 7 consecutive days. Rats in the sham group and Ad-EndoA2 group received normal saline (NS). Transthoracic echocardiography was performed with a VisualSonics ultrasound system (Vevo 2100, VisualSonics Inc., Ontario, Canada) equipped with a 30 MHz imaging transducer on day 7 before sacrificing the mice. After that, the rats were sacrificed. The hearts were rapidly removed, and the left ventricle was carefully trimmed. For morphometric measurements, histological cross sections (5 μm thick) of the hearts were stained with hematoxylin-eosin (HE) or Masson’s trichrome.
Transfection with EndoA2 siRNA or Ad-EndoA2
The siRNA duplexes against the rat EndoA2 gene were transiently transfected as described previously [12]. The siRNA and antisense sequences were 5’r (GGU GCU CUA UAA ACA CUA A) dTdT3’ and 5’r (UUA GUGUUU AUA GAG CAC C) dTdT3’, respectively (Qiagen, Valencia, CA, USA). Briefly, siRNA was diluted in high-glucose DMEM without serum (the final concentration was 20 nM), and then Hiperfect Transfect Reagent (Qiagen, Valencia, CA, USA) was added to the diluted siRNA. The complexes were added dropwise onto the cells. The negative siRNA was used as a negative control. The cells were used for studies 48 h later.
The EndoA2 adenovirus was constructed by Shanghai Sunbio Biomedical Technology (Shanghai, CN). The transfection method was performed as we described previously [16,17]. On the day of infection, the cells were 50%-60% confluent; then, the cells were infected with Ad-EndoA2 or Ad-lacZ at a multiplicity of infection (MOI) of 50. Ad-lacZ was utilized as a negative control. The cells were used for studies 48 h later.
RNA isolation and quantitative RT-PCR (qPCR)
Total RNA was extracted from cultured NRCMs or rat heart tissues with Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Rat-specific primers for atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), β-myosin heavy chain (β-MHC), fibronectin, collagen I and GAPDH (listed in Table S1) were synthesized by Invitrogen. One microgram of total RNA was reverse transcribed into first-strand cDNA using a one-step RT kit (Takara Biotechnology, Dalian, CN). The mRNA expression levels were determined using a SYBR-Green Quantitative PCR Kit (Takara Biotechnology, Dalian, CN) on an ABI StepOne real-time PCR system (Thermo Fisher Scientific Inc., Carlsbad, CA). All PCRs were performed in duplicate. GAPDH served as an endogenous control. The fold change of the mRNA expression was calculated using the 2-ΔΔCT method.
Western blot analysis
Western blot analyses were performed as described previously [12]. Briefly, proteins were separated by SDS-PAGE and transferred to PVDF membranes. After blocking with 5% nonfat milk at room temperature for 1 h, the membranes were incubated with primary antibodies against LC3, P62 and Beclin-1 (Cell Signaling, Boston, MA) overnight at 4°C. Then, the membranes were incubated with the corresponding secondary antibodies (Cell Signaling, Boston, MA) for 1 h at room temperature. GAPDH (Boster Biological Technology, Wuhan, CN) was used as a loading control. The target bands were developed using a chemiluminescence system and then visualized by exposure to Kodak X-ray film. The density of each target band was analyzed by ImageJ software.
Measurement of cell surface area
NRCMs plated on coverslips were fixed in 4% paraformaldehyde for 15 min at room temperature, followed by treatment with 1% Triton-100 in PBS for 10 min. Following overnight incubation with α-actinin (Boster Biological Technology, Wuhan, CN), the cells were incubated with the corresponding secondary antibodies (Cell Signaling, Boston, MA) and the nuclei were stained with DAPI solution (Beyotime, Nanjing, CN). Cells were examined under a fluorescence microscope. Cell surface areas were analyzed with NIH ImageJ software. One hundred cells from randomly selected fields were measured for each group.
mRFP-GFP-LC3 adenovirus transfection and confocal microscopy
The mRFP-GFP-LC3 adenovirus were purchased from HanBioInc (Shanghai, China). NRCMs were infected with adenovirus at 50 MOI as described previously [17]. After transfection for 24 h, the cells were washed and fixed with 4% paraformaldehyde. Confocal images were collected with a Nikon A1 laser scanning confocal microscope (Nikon America Inc., Melville, NY) under uniform settings. The number of GFP and mRFP dots was determined by manual counting of fluorescent puncta from at least 5 different myocyte preparations with a 60X objective. At least 40 cells were scored in each experiment. The number of dots/cell was obtained by dividing the total number of dots by the number of nuclei in each microscopic field.
Statistical analysis
All data were analyzed with the statistical software GraphPad Prism5.0 and are expressed as the means ± SEM. Differences were analyzed by unpaired two-tailed Student’s t-tests or one-way analysis of variance (ANOVA) followed by Bonferroni multiple comparison post hoc tests. P<0.05 was considered statistically significant.
Results
EndoA2 attenuated ISO-induced cardiac structural and functional changes in SD rats
ISO, a nonselective β-adrenergic receptor (β-AR) agonist, has been widely used as a stimulus for cardiac hypertrophy [18,19]. In this study, primary NRCMs were treated with different concentrations of ISO for 24 h, and then the expression of EndoA2 was determined by western blot analysis. As shown in Figure S1A, EndoA2 protein levels were significantly decreased by 0.5 μM ISO and 1 μM ISO compared with the control. Furthermore, NRCMs were incubated with 1 μM ISO as the times indicated. As shown in Figure S1B, the EndoA2 protein levels were decreased after 3 h. According to the results of previous study [9,19], we treated NRCMs with 1 μM ISO for 24 h to induce cardiac hypertrophy. For the in vivo studies, SD rats received a subcutaneous injection of ISO (1.5 mg/kg/d) for 7 days. A significant downregulation of EndoA2 expression was observed after ISO treatment in the heart tissues of SD rats (Figure S1C). We speculated that EndoA2 might play a protective role in cardiac hypertrophy.
Ad-EndoA2 was transduced into the rat left ventricle by intramyocardial injection. Ad-lacZ was utilized as a negative control. As shown in Figure 1, the delivery of Ad-EndoA2 clearly ameliorated ISO-induced hypertrophic changes and impaired systolic function, as revealed by echocardiography (Figure 1A and Table S2), the heart weight to body weight (HW/BW) ratio and the left ventricle weight to body weight (LVW/BW) ratio (Figure 1B), the mRNA levels of hypertrophic biomarkers (Figure 1C) and HE staining (Figure 1D, 1E). Taken together, our results show that EndoA2 is a negative regulator of cardiac hypertrophy in vivo.
Figure 1.
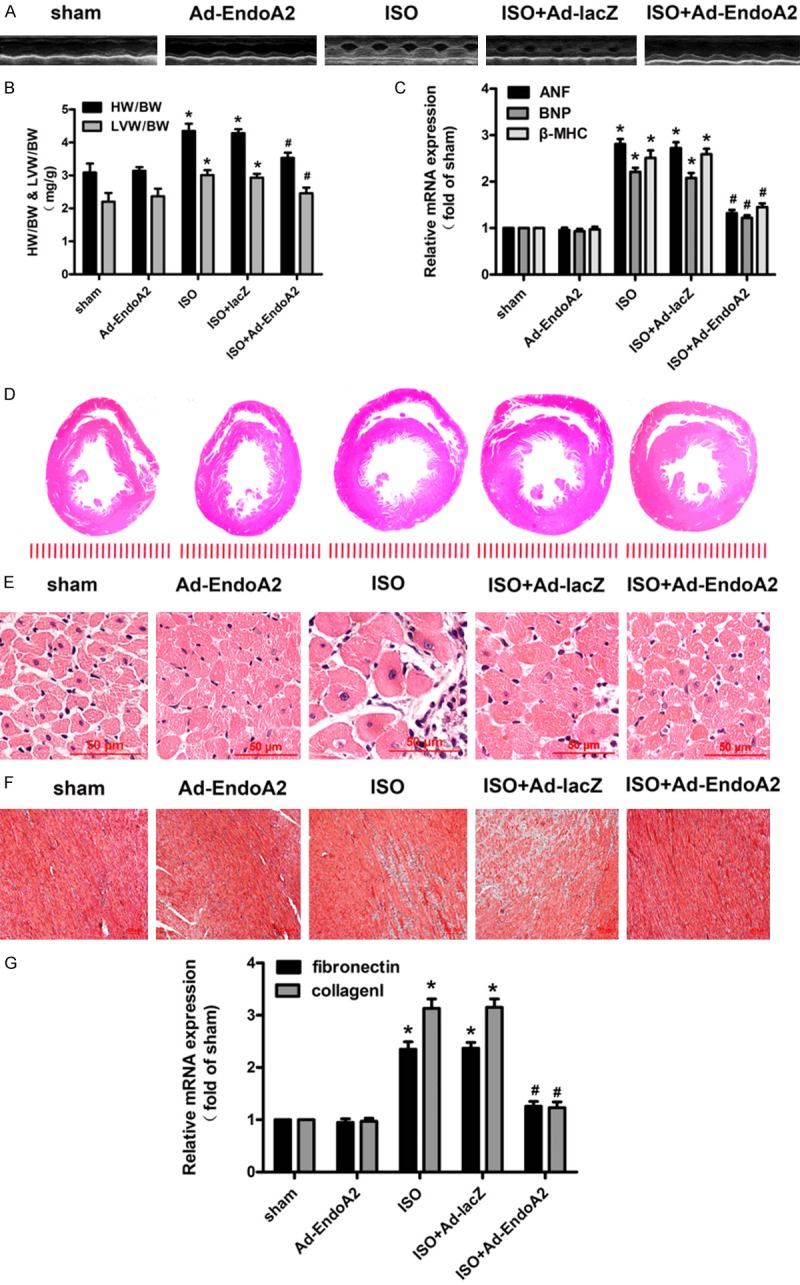
EndoA2 attenuated ISO-induced cardiac structural and functional changes in SD rats. SD rats were subjected to intramyocardial injection of Ad-lacZ or Ad-EndoA2. The animals were further subjected to subcutaneous injection of ISO (1.5 mg/kg/d) or normal saline (NS) for 7 days. The sham rats received NS. The Ad-EndoA2 rats were subjected to intramyocardial injection of Ad-EndoA2 and then received NS. Pathological changes in the hearts were observed by echocardiography (A); the heart weight to body weight (HW/BW) ratio and left ventricular weight to body weight (LVW/BW) ratio (B); the mRNA levels of hypertrophic biomarkers including atrial natriuretic factor (ANF), brain natriuretic polypeptide (BNP) andβ-myosin heavy chain (β-MHC) (C); and hematoxylin-eosin (HE)-stained cross sections of the left ventricle (D, E). Myocardial fibrosis was determined by Masson’s trichrome staining (F) and the mRNA levels of fibrotic biomarkers including fibronectin and collagen I (G). The results showed that EndoA2 attenuated ISO-induced cardiac hypertrophy and fibrosis. n=6, *P<0.05 vs. sham group, #P<0.05 vs. ISO group.
Masson’s trichrome staining results showed a dramatically elevated collagen in the hearts of the ISO group compared with the sham group (Figure 1F). Intramyocardial Ad-EndoA2 delivery reduced the collagen volume fraction. Furthermore, the qPCR results of fibrotic biomarkers showed that EndoA2 overexpression inhibited the increased mRNA levels of fibronectin and collagen I induced by ISO treatment (Figure 1G).
EndoA2 attenuated hypertrophic responses induced by ISO in NRCMs
To further explore the role of EndoA2 in ISO-induced cardiac hypertrophy, NRCMs were transfected with Ad-EndoA2 or siRNA to overexpress or knockdown EndoA2 expression, respectively. As shown in Figure 2A, 2B, EndoA2 overexpression remarkably suppressed the increase in ISO-induced cell surface area. Conversely, EndoA2 siRNA knockdown further increased the ISO-induced cell surface area. Furthermore, we employed qPCR to determine the effects of EndoA2 on cardiac hypertrophy. As shown in Figure 2C, EndoA2 overexpression attenuated the effects of ISO on the mRNA levels of ANF, BNP and β-MHC. In contrast, EndoA2 siRNA knockdown enhanced the effects of ISO on these genes. These data reveal that EndoA2 is also a negative regulator of cardiac hypertrophy in vitro.
Figure 2.
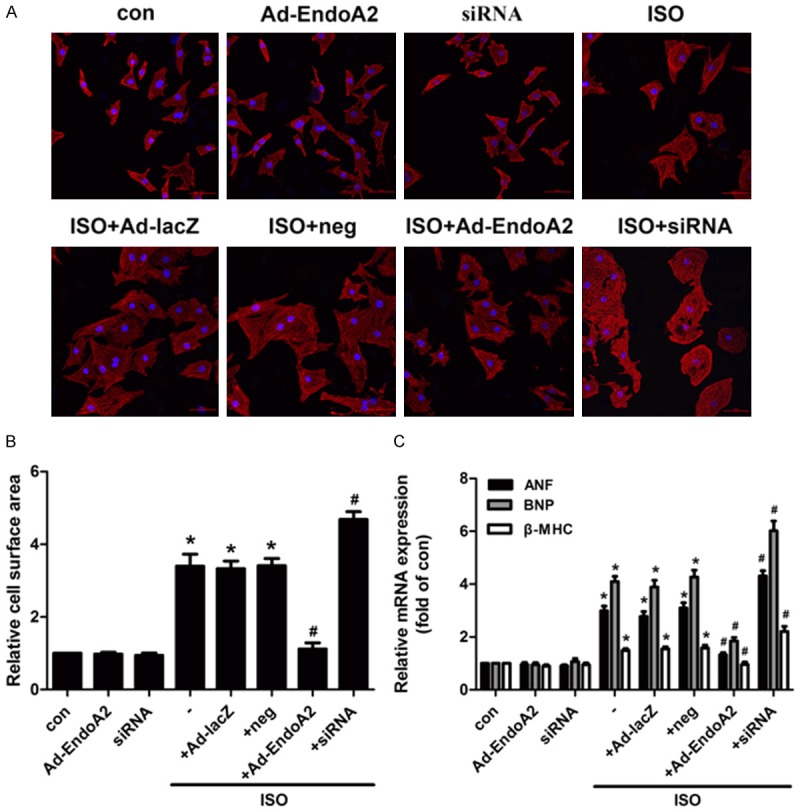
EndoA2 attenuated hypertrophic responses induced by ISO in neonatal rat cardiomyocytes (NRCMs). A, B. NRCMs were transfected with Ad-EndoA2 or siRNA followed by incubation with ISO (1 μM for 24 h). Morphological changes were observed by staining with α-actinin. ISO increased cell surface area, whereas EndoA2 overexpression decreased the ISO-induced cell surface area, and EndoA2 siRNA knockdown further enhanced the effects of ISO on the cell surface area. n=6, *P<0.05 vs. control group, #P<0.05 vs. ISO group. C. The mRNA expression levels were determined by qPCR. Statistical analyses showed that ISO increased the mRNA expression levels of ANF, BNP and β-MHC, whereas EndoA2 overexpression significantly decreased the ISO-induced increases in ANF, BNP and β-MHC mRNA expression levels in NRCMs, EndoA2 siRNA knockdown further enhanced the effects of ISO on the mRNA expression levels of ANF, BNP and β-MHC. n=6, *P<0.05 vs. control group, #P<0.05 vs. ISO group.
EndoA2 promoted autophagy in rat hearts and NRCMs
Autophagy is a regulated, highly conserved, and functionally complex process that participates in basal cellular homeostasis, and dysfunction of autophagy is closely associated with various heart diseases [20]. Our previous study revealed that EndoA2 inhibited H2O2-induced H9C2 cardiomyocyte apoptosis by strengthening autophagy [17], which suggests that EndoA2 plays a critical role in controlling autophagosome formation and the degradation process. Accordingly, we further explored the possible participation of EndoA2 in regulating autophagy in cardiac hypertrophy.
To determine the autophagy level, we assessed the expression of autophagosome markers, including LC3, Beclin-1 and P62. In the heart tissues of SD rats, adenovirus-mediated EndoA2 gene transfer resulted in elevated autophagy levels, as revealed by the increase in the levels of LC3-II, Beclin-1 and the ratio of LC3-II/LC3-I, and by the decrease in the levels of P62 compared with the ISO group (Figures 3A, 3B and S2). Furthermore, in cultured NRCMs, transfection of Ad-EndoA2 led to enhanced Beclin-1 and LC3-II formation and decreased P62 expression compared with ISO treatment (Figures 3C and S2). In contrast, downregulation of endogenous EndoA2 further suppressed autophagy, as indicated by accumulation of P62 accompanied by decreased Beclin-1 and LC3-II compared with ISO treatment (Figures 3D and S2).
Figure 3.
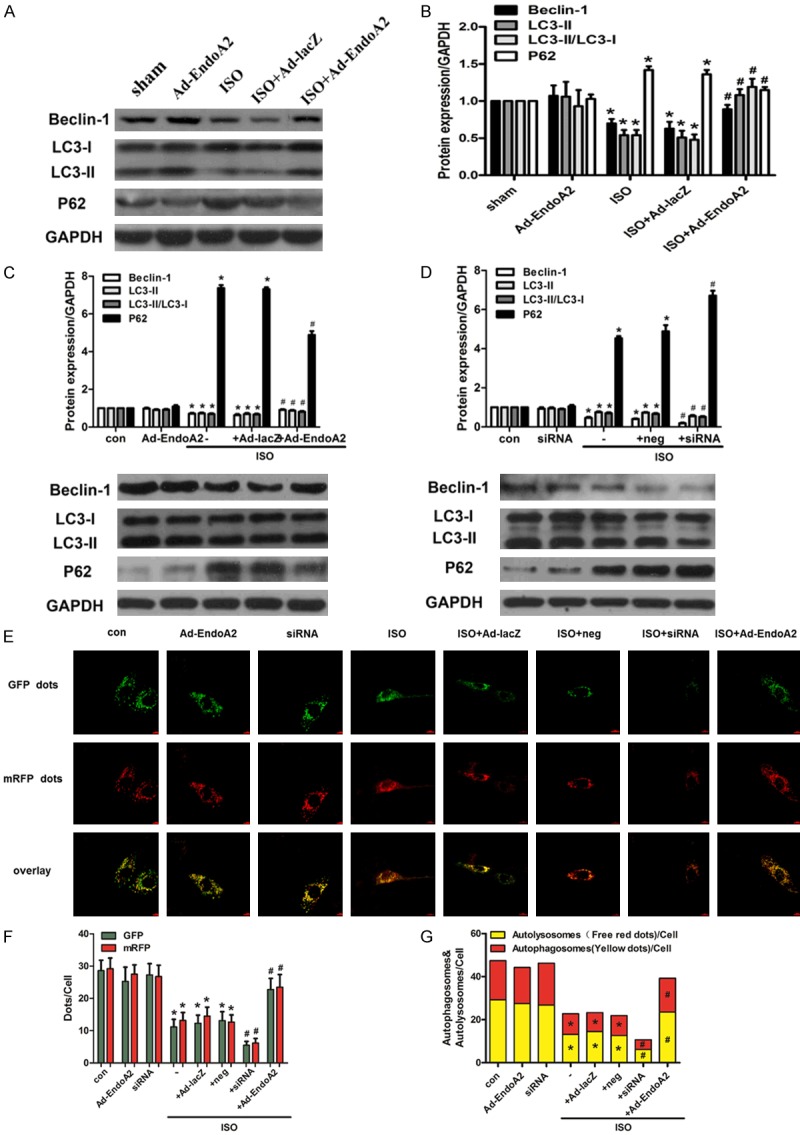
EndoA2 promoted autophagy in rat hearts and NRCMs. A, B. Protein levels of Beclin-1, LC3 and P62 were measured in rat hearts treated with ISO (1.5 mg/kg/d) for 7 days after adenovirus-mediated gene delivery of EndoA2 or lacZ. Densitometric analyses showed that the Ad-EndoA2 plus ISO-treatment group increased the expression levels of LC3-II and Beclin-1 and the LC3-II/LC3-I ratio and decreased the expression levels of P62 compared with the ISO group. n=6, *P<0.05 vs. sham group, #P<0.05 vs. ISO group. C, D. NRCMs were transfected with Ad-EndoA2 or EndoA2 siRNA to overexpress or knockdown EndoA2 expression, followed by incubation with ISO (1 μM for 24 h). The protein levels of Beclin-1, LC3 and P62 were determined by western blot analysis. Densitometric analyses showed that EndoA2 overexpression strengthened autophagy by increasing the expression levels of LC3-II and Beclin-1 and the LC3-II/LC3-I ratio and by decreasing the expression levels of P62 compared with ISO treatment alone, while EndoA2 siRNA knockdown further attenuated autophagy. n=6, *P<0.05 vs. control group, #P<0.05 vs. ISO group. E-G. NRCMs were transfected with the mRFP-GFP-LC3 adenovirus and then treated with ISO. The autophagosomes are represented by yellow puncta and autolysosomes are represented by red puncta in the merged images. The results represent the means from 5 independent experiments. EndoA2 overexpression strengthened the autophagosome formation and autophagic flux, while EndoA2 siRNA knockdown attenuated it. n=5, *P<0.05 vs. control group, #P<0.05 vs. ISO group.
To further confirm the changes in cardiomyocyte autophagy, immunofluorescence assay was conducted. We transfected cardiomyocytes with an adenovirus harboring tandem fluorescent mRFP-GFP-LC3 to measure the autophagy flux rate. Remarkably, cardiomyocytes stimulated with ISO presented diminished LC3 aggregation. In the presence of ISO, EndoA2 overexpression strengthened autophagosome formation and autophagic flux by enhancing the conversion of autophagosomes to autophagolysosomes, as indicated by the significant increase in yellow puncta (indicating autophagosomes) and free red puncta (indicating autophagolysosomes) (Figure 3E-G). On the other hand, EndoA2 siRNA knockdown attenuated autophagosome formation and autophagic flux. These evidences convergently demonstrate that EndoA2 promotes autophagy in rat hearts and NRCMs.
EndoA2 overexpression-induced autophagy accounted for the protective effects of EndoA2 in cardiac hypertrophy induced by ISO
It has been suggested that the activation of autophagy plays a beneficial role in the prevention of cardiac hypertrophy. To investigate the involvement of autophagy in EndoA2-mediated regression of cardiac hypertrophy, NRCMs were treated with 3-MA, a widely used autophagy inhibitor. As shown in Figure 4, inhibition of autophagy by pretreatment with 3-MA clearly diminished the anti-hypertrophic effects of EndoA2 in ISO-treated NRCMs, as evidenced by the increased cell surface area (Figure 4A, 4B) and elevated mRNA levels of hypertrophic biomarkers (Figure 4C) compared with the ISO+Ad-EndoA2 group. These data reveal that upregulation of autophagy is at least partially responsible for the effects of EndoA2 in the regression of cardiac hypertrophy.
Figure 4.
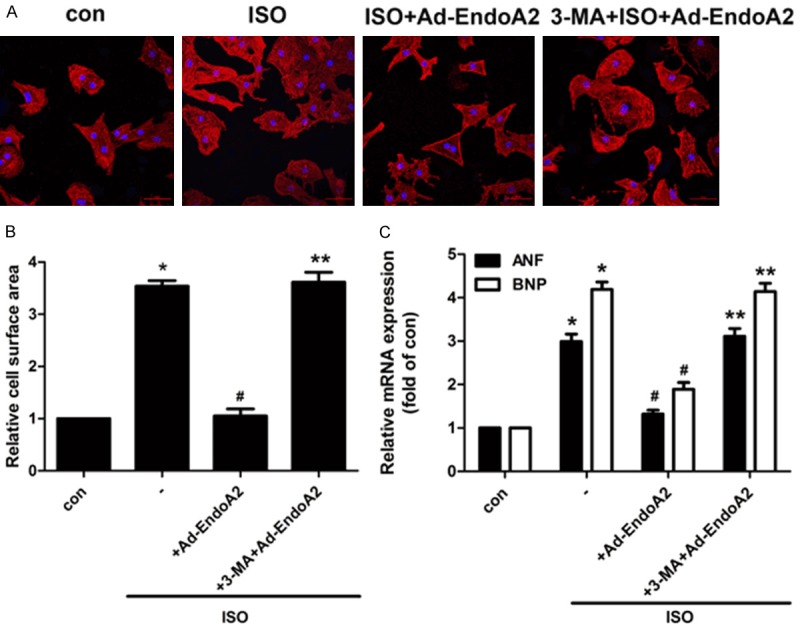
EndoA2 overexpression-induced autophagy accounted for the protective effects of EndoA2 on cardiac hypertrophy induced by ISO. A-C. NRCMs were treated with the autophagy inhibitor 3-MA (1 mM, 24 h). Immunofluorescence and qPCR were employed to determine the hypertrophic response in NRCMs. Representative images of cell surface area and the densitometric analyses of qPCR showed that pretreatment with 3-MA abrogated the protective effects of EndoA2 on ISO-induced cardiac hypertrophy, as evidenced by increased cell surface area and mRNA expression levels of ANF and BNP compared with the ISO+Ad-EndoA2 group. n=6, *P<0.05 vs. control group, #P<0.05 vs. ISO group, **P<0.05 vs. ISO+Ad-EndoA2 group.
Discussion
To date, investigations into EndoA2 have mainly focused on neuroregulation [10], tumor formation and metastasis [14]. In recent years, the roles of EndoA2 in cardiovascular diseases have been increasingly recognized [11-13]. In this study, we identified EndoA2 as a novel molecule with anti-hypertrophic properties both in vivo and in vitro. We found that the expression of EndoA2 was markedly downregulated in response to ISO treatment both in vivo and in vitro. EndoA2 overexpression ameliorated, whereas EndoA2 siRNA knockdown promoted, cardiac hypertrophy induced by ISO. Furthermore, EndoA2 overexpression resulted in activation of autophagy, an effect that might be critical for the anti-hypertrophic effects of EndoA2, but knockdown of EndoA2 significantly suppressed autophagy.
Accumulating evidence demonstrates that EndoA2 plays an important role in cardiovascular diseases. Our previous study revealed that EndoA2 attenuated angiotensin-II-induced cardiac hypertrophy in NRCMs by regulating angiotensin II type 1 receptor intracellular trafficking [16], which suggests a fundamental role of EndoA2 in cardiac hypertrophy. However, whether EndoA2 regulates other forms of cardiac hypertrophy remains unknown. Stimulation of the β-AR by ISO is a well-established model of cardiac hypertrophy both in vivo and in vitro [21,22]. In this study, we demonstrated that the expression of EndoA2 significantly declined in response to hypertrophic stimuli in rat hearts and NRCMs. Therefore, we speculated that EndoA2 overexpression suppresses ISO-induced cardiac hypertrophy. We constructed an animal cardiac hypertrophy model consistent with the method in a previous study [9]. Our results demonstrated that intramyocardial injection of Ad-EndoA2 significantly ameliorated ISO-induced cardiac hypertrophy and repressed cardiac dysfunction, as evidenced by the results of echocardiography, HW/BW ratio and LVW/BW ratio, mRNA levels of hypertrophic biomarkers and HE staining. ISO induces cardiac hypertrophy as well as cardiac dysfunction through several mechanisms, including oxidative stress [23], an increase in collagen deposition [24], and intracellular calcium overload [25]. Then, we measured collagen deposition in rat hearts. Masson’s trichrome staining results showed dramatically elevated collagen in the hearts of the ISO group and intramyocardial Ad-EndoA2 delivery inhibited collagen deposition. The qPCR results of fibrotic biomarkers were consistent with the results of Masson’s trichrome staining. Additionally, EndoA2 overexpression attenuated the cardiomyocyte hypertrophy induced by ISO, whereas gene silencing of EndoA2 aggravated cardiomyocyte hypertrophy in NRCMs. Thus, the present study provides new evidence that EndoA2 is involved in ISO-induced cardiac hypertrophy and cardiac dysfunction.
The role of autophagy in cardiac hypertrophy remains debatable. Some evidence supports that the upregulation of autophagy is detrimental in cardiac hypertrophy. Under severe pressure, amplified cardiac growth is induced in Beclin-1 transgenic mice [26]. Inhibition of Beclin-1 by 3-MA or siRNA ameliorates cardiac hypertrophy [27]. Other studies argue that upregulation of autophagy plays a beneficial role in cardiac hypertrophy due to misfolded protein degeneration and energy support. Cardiac hypertrophy is characterized by increased cell size, enhanced protein synthesis, reorganization of the cytoskeleton, and aggregation of misfolded proteins and damaged organelles. In fact, autophagy, which can clear misfolded proteins and damaged organelles, declines in the progression of cardiac hypertrophy. Cardiac-specific deficiency of Atg5 quickly progresses to cardiac hypertrophy and dysfunction after transverse aortic constriction [28]. Macrophage migration inhibitory factor protects against cardiac hypertrophy via an mTOR activated autophagy signaling pathway [29]. Therefore, proper activation of autophagy antagonizes cardiac hypertrophy. We preliminarily discovered that EndoA2 attenuated H2O2-induced H9C2 cardiomyocyte apoptosis by strengthening autophagy [17], indicating the relationship between EndoA2 and autophagy. Then, we measured autophagy indicators in this study. Beclin-1 and the conversion of LC3-I to LC3-II are specific indicators of autophagy activation. P62, a selective substrate for autophagy, and the inhibition of autophagy will block its degradation. We showed that autophagy was impaired after ISO treatment, as evident in the downregulation of LC3-II and Beclin-1, the decrease in the LC3-II/LC3-I ratio and the upregulation of P62. EndoA2 overexpression activated autophagy, whereas EndoA2 siRNA knockdown further inactivated autophagy. Moreover, immunofluorescence results showed that EndoA2 overexpression strengthened autophagosome formation and autophagic flux, whereas EndoA2 siRNA further attenuated it compared with ISO treatment group. 3-MA, a potent inhibitor of autophagy, reversed the anti-hypertrophic effects of EndoA2, which suggests the involvement of autophagy in the anti-hypertrophic effects of EndoA2.
In summary, our study reveals that autophagy activation by EndoA2 contributes to the attenuation of ISO-induced cardiac hypertrophy. This finding will help us to better understand the potential roles of EndoA2 in cardiovascular diseases. In this regard, as a negative regulator of cardiac hypertrophy, EndoA2 may represent a novel therapeutic target for preventing heart failure.
Acknowledgements
This work was funded by National Natural Science Foundation of China (No. 81803531 to Dr. Y Liu, No. 81872869 to Dr. J Luo) and Natural Science Foundation of Guangdong Province (No. 2018A030310173 to Dr. Y Liu).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Yan G, Zhu N, Huang S, Yi B, Shang X, Chen M, Wang N, Zhang GX, Talarico JA, Tilley DG, Gao E, Sun J. Orphan nuclear receptor nur77 inhibits cardiac hypertrophic response to beta-adrenergic stimulation. Mol Cell Biol. 2015;35:3312–3323. doi: 10.1128/MCB.00229-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo J, Song W, Yang G, Xu H, Chen K. Compound danshen (salvia miltiorrhiza) dripping pill for coronary heart disease: an overview of systematic reviews. Am J Chin Med. 2015;43:25–43. doi: 10.1142/S0192415X15500020. [DOI] [PubMed] [Google Scholar]
- 3.Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K, Thum T. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue R, Jiang J, Dong B, Tan W, Sun Y, Zhao J, Chen Y, Dong Y, Liu C. DJ-1 activates autophagy in the repression of cardiac hypertrophy. Arch Biochem Biophys. 2017;633:124–132. doi: 10.1016/j.abb.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Qin Y, Zhu X, Liu D, Chen F, Xu S, Zheng D, Zhou Y, Luo J. Increased expression of DRAM1 confers myocardial protection against ischemia via restoring autophagy flux. J Mol Cell Cardiol. 2018;124:70–82. doi: 10.1016/j.yjmcc.2018.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Wu X, He L, Chen F, He X, Cai Y, Zhang G, Yi Q, He M, Luo J. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One. 2014;9:e112891. doi: 10.1371/journal.pone.0112891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang ZV, Rothermel BA, Hill JA. Autophagy in hypertensive heart disease. J Biol Chem. 2010;285:8509–8514. doi: 10.1074/jbc.R109.025023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Sun D, Liu Z, Li M, Hong H, Liu C, Gao S, Li H, Cai Y, Chen S, Li Z, Ye J, Liu P. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl Res. 2016;172:96–112. e116. doi: 10.1016/j.trsl.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Murdoch JD, Rostosky CM, Gowrisankaran S, Arora AS, Soukup SF, Vidal R, Capece V, Freytag S, Fischer A, Verstreken P, Bonn S, Raimundo N, Milosevic I. Endophilin-A deficiency induces the Foxo3a-Fbxo32 network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep. 2016;17:1071–1086. doi: 10.1016/j.celrep.2016.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CZ, Li XY, Du RH, Gao M, Ma MM, Li FY, Huang EW, Sun HS, Wang GL, Guan YY. Endophilin A2 influences volume-regulated chloride current by mediating ClC-3 trafficking in vascular smooth muscle cells. Circ J. 2016;80:2397–2406. doi: 10.1253/circj.CJ-16-0793. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Gao M, Ma MM, Tang YB, Zhou JG, Wang GL, Du YH, Guan YY. Endophilin A2 protects H2O2-induced apoptosis by blockade of Bax translocation in rat basilar artery smooth muscle cells. J Mol Cell Cardiol. 2016;92:122–133. doi: 10.1016/j.yjmcc.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Huang EW, Liu CZ, Liang SJ, Zhang Z, Lv XF, Liu J, Zhou JG, Tang YB, Guan YY. Endophilin-A2-mediated increase in scavenger receptor expression contributes to macrophage-derived foam cell formation. Atherosclerosis. 2016;254:133–141. doi: 10.1016/j.atherosclerosis.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Wu X, Gan B, Yoo Y, Guan JL. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev Cell. 2005;9:185–196. doi: 10.1016/j.devcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 15.So CW, Caldas C, Liu MM, Chen SJ, Huang QH, Gu LJ, Sham MH, Wiedemann LM, Chan LC. EEN encodes for a member of a new family of proteins containing an src homology 3 domain and is the third gene located on chromosome 19p13 that fuses to MLL in human leukemia. Proc Natl Acad Sci U S A. 1997;94:2563–2568. doi: 10.1073/pnas.94.6.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Shen HJ, Wang XQ, Liu HQ, Zheng LY, Luo JD. EndophilinA2 protects against angiotensin II-induced cardiac hypertrophy by inhibiting angiotensin II type 1 receptor trafficking in neonatal rat cardiomyocytes. J Cell Biochem. 2018;119:8290–8303. doi: 10.1002/jcb.26862. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Liu HQ, Xiao JY, Ma KT, Wang XQ, Shen HJ, Luo JD. Autophagy is involved in the protective effect of endophilin A2 on H2O2-induced apoptosis in H9C2 cardiomyocytes. Biochem Biophys Res Commun. 2018;499:299–306. doi: 10.1016/j.bbrc.2018.03.151. [DOI] [PubMed] [Google Scholar]
- 18.Feng XJ, Gao H, Gao S, Li Z, Li H, Lu J, Wang JJ, Huang XY, Liu M, Zou J, Ye JT, Liu PQ. The orphan receptor NOR1 participates in isoprenaline-induced cardiac hypertrophy by regulating PARP-1. Br J Pharmacol. 2015;172:2852–2863. doi: 10.1111/bph.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitsuyama S, Takeshita D, Obata K, Zhang GX, Takaki M. Left ventricular mechanical and energetic changes in long-term isoproterenol-induced hypertrophied hearts of SERCA2a transgenic rats. J Mol Cell Cardiol. 2013;59:95–106. doi: 10.1016/j.yjmcc.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 20.Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Curr Hypertens Rep. 2009;11:406–411. doi: 10.1007/s11906-009-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galindo CL, Skinner MA, Errami M, Olson LD, Watson DA, Li J, McCormick JF, McIver LJ, Kumar NM, Pham TQ, Garner HR. Transcriptional profile of isoproterenol-induced cardiomyopathy and comparison to exercise-induced cardiac hypertrophy and human cardiac failure. BMC Physiol. 2009;9:23. doi: 10.1186/1472-6793-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morisco C, Marrone C, Galeotti J, Shao D, Vatner DE, Vatner SF, Sadoshima J. Endocytosis machinery is required for beta1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc Res. 2008;78:36–44. doi: 10.1093/cvr/cvn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khatua TN, Borkar RM, Mohammed SA, Dinda AK, Srinivas R, Banerjee SK. Novel sulfur metabolites of garlic attenuate cardiac hypertrophy and remodeling through induction of Na(+)/K(+)-ATPase expression. Front Pharmacol. 2017;8:18. doi: 10.3389/fphar.2017.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergaro G, Prud’homme M, Fazal L, Merval R, Passino C, Emdin M, Samuel JL, Cohen Solal A, Delcayre C. Inhibition of galectin-3 pathway prevents isoproterenol-induced left ventricular dysfunction and fibrosis in mice. Hypertension. 2016;67:606–612. doi: 10.1161/HYPERTENSIONAHA.115.06161. [DOI] [PubMed] [Google Scholar]
- 25.Tsai CY, Kuo WW, Shibu MA, Lin YM, Liu CN, Chen YH, Day CH, Shen CY, Viswanadha VP, Huang CY. E2/ER beta inhibit ISO-induced cardiac cellular hypertrophy by suppressing Ca2+-calcineurin signaling. PLoS One. 2017;12:e0184153. doi: 10.1371/journal.pone.0184153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertro phy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, Xiong L, Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One. 2013;8:e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension. 2014;63:490–499. doi: 10.1161/HYPERTENSIONAHA.113.02219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.