

Abstract
Recent studies have shown a strong correlation between air pollution-exposure and detrimental out-comes in the central nervous system, including alterations in blood brain barrier (BBB) integrity, neuroinflammation, and neurodegeneration. However, the mechanisms mediating these pathologies have not yet been fully elucidated. We have previously reported that exposure to traffic-generated air pollution results in increased circulating oxidized low-density lipoprotein (oxLDL), associated with alterations in BBB integrity, in atherosclerotic Apolipoprotein E null (ApoE−/−) mice. Thus, we investigated the role of the lectin-like oxLDL receptor (LOX)-1 in mediating these deleterious effects in ApoE−/− mice exposed to a mixture of gasoline and diesel engine exhaust (MVE: 100 PM μg/m3) for 6h/d, 7d/week, for 30 d by inhalation. Concurrent with exposures, a subset of mice were treated with neutralizing anti-bodies to LOX-1 (LOX-1 Ab) i.p., or IgG (control) i.p., every other day during exposures. Resulting brain microvascular integrity, tight junction (TJ) protein expression, matrix metalloproteinase (MMP)-9/−2 activity, ROS, and markers of cellular adhesion and monocyte/macrophage sequestration were assessed. MVE-exposure resulted in decreased BBB integrity and alterations in microvascular TJ protein expression, associated with increased LOX-1 expression, MMP-9/−2 activities, and lipid peroxidation, each of which was attenuated with LOX-1 Ab treatment. Furthermore, MVE-exposure induced cerebral microvascular ROS and adhesion molecules, expression of which was not normalized through LOX-1 Ab-treatment. Such findings suggest that alterations in brain microvascular structure and integrity observed with MVE-exposure may be mediated, at least in part, via LOX-1 signaling.
Keywords: Air pollution, LOX-1, oxLDL, BBB, MMP-9, inflammation
Introduction
Several recent studies have revealed a clear correlation between exposure to components of air pollution and detrimental central nervous system (CNS)-related outcomes, including exacerbation of cerebrovascular disease, neuroinflammation, and stroke (Block et al., 2013; Brook et al., 2010; Calderón-Garcidueñas et al., 2016; Crichton et al., 2016). While a strong association exists between inhalation exposure to traffic-generated air pollutants and cerebrovascular events such as stroke (Korek et al., 2015; Stafoggia et al., 2014), the pathways involved have not yet been fully elucidated. One possible mechanism that may mediate the effects of air pollution exposure on the CNS is via alterations in blood brain barrier (BBB) integrity and transport (Block et al., 2013, Calderón-Garcidueñas et al., 2008, 2016; Oppenheim et al., 2013). The BBB is a dynamic neurovascular unit consisting of microvascular endothelial cells, pericytes, and the foot-processes of astrocytes, which is responsible for regulating transport between the systemic circulation and the CNS. The microvascular endothelial cells that comprise the BBB are unique in that they are joined via tight junction (TJ) proteins including junctional adhesion molecules, occludin, and claudins (Hawkins & Davis, 2015). Alterations in expression of TJ proteins is associated with increased BBB permeability and dysregulation of transport into the brain parenchyma (Jiao et al., 2011; Oppenheim et al., 2013; Zehendner et al., 2013). The interaction of the extracellular matrix of the basal lamina that surrounds the microvascular endothelium also helps to maintain the integrity of the BBB, as disruption of the extracellular matrix is strongly correlated with altered BBB structure and increased permeability (Fujimoto et al., 2015; Willis et al., 2013). Additionally, alterations in brain microvascular integrity are often associated with increased matrix metalloproteinase (MMP) expression and activity, specifically MMP-9, which is known to degrade components of the extracellular matrix (Abdul-Muneer et al., 2016; Oppenheim et al., 2013; Ren et al., 2015).
One key factor that is reported to be upregulated with exposure to traffic-generated air pollution, in both human exposures and animal research models, is plasma oxidized low-density lipoprotein (oxLDL) (Jacobs et al., 2011; Lund et al., 2009; Zhang et al., 2016). Increased production of oxLDL is associated with endothelial dysfunction and activation of inflammatory signaling pathways (Lubrano & Balzan 2016; Shiraki et al., 2014). The lectin-like low-density lipo-protein oxLDL receptor (LOX-1), a scavenger receptor, is known to be the primary oxLDL receptor present on endothelial cells. LOX-1 has been reported to regulate oxLDL uptake, apoptosis, ligand-mediated signal transduction pathways that regulate expression of proatherogenic molecules such as vascular cellular adhesion molecule (VCAM)-1, intracellular adhesion molecule (ICAM)-1, monocyte chemo-attractant protein (MCP)-1, MMP-9, and also reactive oxygen species (ROS) production via NAD(P)H oxidase activity in the vasculature (Di Pietro et al., 2016; Feng et al., 2014; Lund et al., 2011; Mollace et al., 2015). While the role of LOX-1 signaling has been well characterized in the pathogenesis of atherosclerosis, more recent reports reveal that LOX-1 is also a key player in onset and/or outcome of stroke (Shen et al., 2016). This premise is further evidenced by increased levels of soluble LOX-1 (sLOX-1), which is the soluble form of the (cleaved) LOX-1 membrane receptor found circulating in the plasma (Murase et al., 2000), observed during acute ischemic stroke (Huang et al., 2017; Yokota et al., 2016).
We have previously reported that exposure to vehicle engine exhaust results in elevated plasma oxLDL, increased LOX-1 receptor expression in the systemic vasculature, and BBB disruption associated with increased MMP-9 activity and decreased TJ (claudin-5 and occludin) protein expression in an animal model of atherosclerotic disease, namely the Apolipoprotein (Apo)E−/− mouse (Lund et al., 2011; Oppenheim et al., 2013). However, the mechanisms involved in the observed exposure-mediated alterations in BBB permeability and transport have not yet been elucidated. Additionally, we have shown that plasma sLOX-1 levels are significantly elevated in humans after an acute exposure to diesel exhaust (Lund et al., 2011). Thus, we investigated the hypothesis that exposure to traffic-generated air pollution, namely a mixture of gasoline and diesel engine emissions, results in increased LOX-1 expression and signaling on brain microvascular endothelial cells, which mediates altered BBB integrity and permeability in ApoE−/− mice. The rationale for using the ApoE−/− mouse model in the current study is two-fold: (1) it allows us to investigate the mechanism(s) involved that contribute to pollution-exposure mediated alterations in brain microvascular integrity and BBB integrity reported in our previous studies, using the same animal model (Oppenheim et al., 2013); and (2) when fed a high fat diet, ApoE−/− mice develop atherosclerosis similar in etiology to that observed in much of the human population, including obese children and adolescents (Beauloye et al., 2007; Zhang et al., 1992). As such, this animal model provides a baseline cardiovascular disease state known to contribute to cerebrovascular disease. Since air pollution exposure has been strongly associated with increased incidence (and poorer prognostic outcome) of stroke, in addition to mediating decreased BBB integrity, it is imperative to gain a further understanding of mechanistic pathways involved that may serve as targets for therapeutic intervention, or biomarkers of onset of a clinical occurrence, in susceptible individuals during periods of high air pollution levels.
Materials and methods
Animal inhalation exposure and LOX-1 antibody treatment protocol
Eight-week-old male ApoE−/− mice (Taconic, Hudson, NY) were placed on a high-fat diet (TD88137 Custom Research Diet, Harlan Teklad: 21.2% fat, 1.5g/kg cholesterol) 30 d before exposure. ApoE−/− mice were exposed by whole body inhalation to either 100 μg particulate matter (PM)/m3 mixed vehicle exhaust (MVE: 30 μg PM/m3 gasoline engine exhaust mixed with 70 μg PM/m3 diesel engine exhaust in a pre-exposure chamber upstream of the animal exposure chamber; n = 40) or filtered air (FA, n = 40) 6h/d, 7 d/week, for 30 d, as previously described (Lund et al., 2009, 2011; Oppenheim et al., 2013). These exposure mixtures (gases and PM component size/complexity) have been previously characterized in detail (Lund et al., 2009; Mumaw et al., 2016; Tyler et al., 2016). ApoE−/−mice in each exposure group were randomly assigned to receive either LOX-1 neutralizing antibody (Ab) (anti-mouse LOX-1/SR-E1 Ab; R&D Systems, Minneapolis, MN; n = 20) or control (mouse IgG, n = 20) at 16 mg/ml, 0.1 ml/mouse, i.p., every other day throughout the exposure, as previously described (Lund et al., 2011). Mice were housed in standard shoebox cages within an Association for Assessment and Accreditation of Laboratory Animal Care International-approved rodent housing facility (2 m3 exposure chambers) for the entirety of the study, which maintained constant temperature (20–24 °C) and humidity (between parameters of 30–60% relative humidity). Mice had access to chow and water ad libitum throughout the study period, except during daily exposures when chow was removed. All procedures were approved by the Lovelace Biomedical and Environmental Research Institute’s Animal Care and Use Committee (AAALAC-accredited Assurance #A3083–01; USDA-registered facility #85-R-003) and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Tissue collection
Upon completion of the designated exposure period, animals were sacrificed 14–16 h after their last exposure, and tissues were collected. In an effort to minimize any suffering, mice were anesthetized with Euthasol (390 mg pentobarbital sodium, 50 mg phenytoin sodium/ml; diluted 1:10 and administered at a dose 0.1 ml per 30 g mouse) and euthanized by exsanguination. The brain tissue was carefully dissected from the skull, meninges were removed, split in half at the sagittal suture and were either (1) embedded in OCT (VWR Scientific, West Chester, PA, USA) and frozen on dry ice or (2) immediately snap frozen in liquid nitrogen for molecular assays. All tissues were stored at –80 °C until processed for analysis. For vessel-specific real-time RT-PCR endpoints the vessels were carefully dissected from the cerebrum using a dissecting stereo zoom microscope, with a micrometer to approximate vessel size, as previously described by our laboratory (Oppenheim et al., 2013).
Double immunofluorescence
Brain sections (10 μm, midsagittal cut/plane) were prepared for either LOX-1, occludin, claudin-5, MMP-9, or 4-hydroxynonenal (4-HNE) by double immunofluorescence, as previously described by our laboratory (Lund et al., 2011; Oppenheim et al., 2013). Briefly, sections were blocked in 10% goat serum for 30 min at RT, rinsed in
PBS, and incubated with 150 μl per section of mixed anti-rabbit LOX-1 (1:1000 dilution; Abcam, Cambridge, MA, USA), or occludin (1:500; Abcam, Cambridge, MA, USA), or claudin-5 (1:500; Abcam, Cambridge, MA, USA), or MMP-9 (1:1000; Abcam, Cambridge, MA, USA), or 4-HNE (1:1000; Abcam, Cambridge, MA, USA), and anti-sheep vWF (1:1000 dilution; Abcam, Cambridge, MA, USA), in order to determine endothelial cell-specific co-localization, for 1 h at RT. Slides were then rinsed three times with PBS and incubated with 150 μl per section of a mixture of secondary antibodies Alexa Fluor 488 (anti-rabbit) and Alexa Flour 555 (anti-sheep) (1:500 dilution; ThermoFisher Scientific, Richardson, TX, USA) and Hoechst nuclear stain in the dark for 1 h at RT. Slides were then rinsed three times in PBS, and coverslipped with Aqueous Gel Mount (Sigma, Darmstadt, Germany). Slides were imaged by fluorescent microscopy at 10 ×, 40 ×, and 100 × using the appropriate excitation/emission filters, digitally recorded, and analyzed by image densitometry using Image J software (NIH). Double immunofluorescence was quantified by merging Alexa 488 (fluorescein isothiocyanate) and Alexa 555 (RFP) signals into red-green-blue (RGB) images. Colocalization was determined by quantifying total fluorescence of over-layed signals from a minimum of three slides, two sections each, three regions from each section (n = 3–4 per group). Only vessels less than 100 μm in size from cerebral sections were utilized for fluorescence analysis.
BBB permeability
Changes in BBB permeability were assessed using the fluorescent tracer, sodium fluorescein (Na-F) in a subset of mice on the study. A subset of the ApoE−/− mice exposed to either FA (n = 6 LOX-1 Ab-treated, n = 6 IgG treated) or MVE (n = 6 LOX-1 Ab-treated, n = 6 IgG treated) were injected i.p. with 100 μl of 2% Na-F in 1× PBS 30 min prior to the end of their final exposure on day 30. For this procedure, the chambers were opened briefly by technicians wearing the appropriate personal protective equipment, and animals were quickly injected with Na-F i.p., put back in the cages and then chambers were closed to complete exposures. Mice were anesthetized 1 h post exposure and transcardially perfused with sterile saline until colorless perfusion was visualized. The brains were isolated, and the meninges, cerebellum, and brain stem were gently dissected away. The brains were weighed, homogenized and analyzed for Na-F fluorescence, as previously described (Oppenheim et al., 2013). Data are expressed as the amount of tracer per gram of tissue.
In situ zymography
Matrix metalloproteinase activity was analyzed on serial brain sections (10 μm thick), using 150 μl of 10 μg/ml dye-quenched (DQ)-Gelatin (EnzChek, Molecular Probes, Invitrogen, Carlsbad, CA, USA) and 1 μg/ml DAPI (nuclei stain; Invitrogen, Carlsbad, CA, USA) in 1% UltraPure™ low melting point agarose (Invitrogen, Carlsbad, CA, USA) cover-slipped, chilled for 5 min at 4 °C, and then incubated for 6h in a dark, humid chamber at 37 °C, as previously described by our laboratory (Lund et al., 2009, 2011; Oppenheim et al., 2013). Slides were analyzed using fluorescent microscopy and densitometry was calculated using white/black images and quantified using Image J software (NIH, Bethesda, MD, USA; performed on six sections per sample, three regions per section, six samples per group). Background fluorescence (fluorescence present in total image outside of the vessel) was subtracted from each section before statistical comparison between groups.
Dihydroethidium (DHE) staining
To visualize ROS levels in the brains of study animals, 10 μm unfixed, frozen sections (midsagittal cut/plane) were processed through DHE staining using 150 μl of 10 μM DHE, slides were then cover-slipped and incubated at 37 °C for 45 min, as previously described by our laboratory (Lund et al., 2009, 2011; Oppenheim et al., 2013). Ethidium staining was visualized by fluorescent microscopy at 40 × on an EVOS FL-inverted microscope (ThermoFisher, Scientific, Richardson, TX, USA), digitally recorded, and analyzed by image densitometry (color images converted to white/black) using Image J software, from minimum of three slides, two sections each, three regions from each section (n = 3–4 per group). A subset of slides (two sections per slide, n = 3 per treatment/exposure group) were pretreated with flavoenzyme inhibitor, diphenyleneiodonium (DPI, 0.1 mM, Sigma, St Louis, MO, USA) in dark, moist chamber at 37 °C for 30 min prior to DHE staining.
Real-time reverse transcriptase polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cerebral vessels that were dissected from the cerebrum, using a zoom stereoscope (45×), as previously described by our laboratory (Oppenheim et al., 2013). RNA was isolated using an RNAEasy Mini kit (Qiagen, Valencia, CA, USA), per kit instructions. cDNA was synthesized using an iScript cDNA Synthesis kit (Biorad, Hercules, CA, USA; Cat. #170–8891). Real-time PCR analyzes of LOX-1, MCP-1, MMP-9, VCAM-1, ICAM-1, or GAPDH (house-keeping gene) were conducted using specific primers (Table 1) and SYBR Green detection (SSo Advanced Universal SYBR Green Supermix; Biorad, Hercules, CA, USA; Cat #172–5271), following manufacturer’s protocol. Real-time PCR analysis was run on a Biorad CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). ΔΔCT values calculated using CFX Manager™ Software (Bio-Rad, Hercules, CA, USA) and normalized (to GAPDH) on triplicate samples, as previously described by our laboratory (Lund et al., 2009, 2011). Results are expressed as normalized gene expression as a percentage of GAPDH controls.
Table 1.
Primer sets utilized for real time qPCR.
| Gene/primer | Accession # | Sequence (5’–3”) |
|---|---|---|
| Mouse LOX-1 forward | NM_138648 | CACAAGACTGGCTCTGGCATA |
| Mouse LOX-1 reverse | NM_138648 | GCAGGTCTGCCGGIIIIII |
| Mouse MCP-1 forward | NM_011333.3 | ATTGGGATCATCTTGCTGGT |
| Mouse MCP-1 reverse | NM_011333.3 | CCTGCTGTTCACAGTTGCC |
| Mouse MMP-9 forward | NM_013599.4 | TTGGTTTCTGCCCTAGTGAGAGA |
| Mouse MMP-9 reverse | NM_013599.4 | AAAGATGAACGGGAACACACAGG |
| Mouse VCAM-1 forward | NM_011693.3 | ACTTTCTATTTCACTCACACCAGCC |
| Mouse VCAM-1 reverse | NM_011693.3 | ATCTTCACAGGCATTTCAAGTCTCT |
| Mouse ICAM-1 forward | NM_010493.3 | CCATAAAACTCAAGGGACAAGCC |
| Mouse ICAM-1 reverse | NM_010493.3 | TACCATTCTGTTCAAAAGCAGCA |
| Mouse GAPDH forward | NM_001289726.1 | CATGGCCTTCCGTGTTCCTA |
| Mouse GAPDH reverse | NM_001289726.1 | GCGGCAGTCAGATCCA |
LOX-1: lectin-like oxidized low density lipoprotein (LDL) receptor; MCP-1: monocyte chemoattractant protein-1; MMP-9: matrix metalloproteinase-9; VCAM-1: vascular cellular adhesion molecule-1; ICAM-1: intracellular adhesion molecule-1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase (house-keeping gene).
Statistical analysis
Data are expressed as mean ± SEM. Two-way analysis of variance with a post hoc Holm-Sidak test was used for analysis of multiple treatment and exposure groups. Statistical analyzes were conducted in SigmaPlot v12 (Systat Software, Inc., San Jose, CA, USA) software. A p ≤ 0.050 was considered statistically significant.
Results
MVE-exposure mediated LOX-1 expression in cerebral microvascular endothelial cells is normalized by LOX-1 Ab-treatment in ApoE−/− mice
We have previously reported that exposure to traffic-generated air pollution results in increased circulating oxLDL and vascular endothelial cell LOX-1 expression by day 7 of exposure, which is normalized in ApoE−/− mice with LOX-1 Ab treatment (Lund et al., 2011). In the current study, compared to FA controls (Figure. 1(A–C)) and FA + LOX-1 Ab (Figure 1(D–F)), we observed a more than two-fold increase in cerebral microvascular endothelial LOX-1 levels in MVE-exposed ApoE−/− mice (+IgG) (Figure 1(G–I)) at both the tissue level (Figure 1(M)) and transcript level (Figure 1(N)), which was attenuated through LOX-1 Ab-treatment (Figure 1(J–L)). There is also a slight decrease in endothelial microvascular LOX-1 levels observed between FA + IgG vs. FA + LOX-1 Ab animal groups (Figure 1(C vs. F), respectively), though not statistical. These findings are not unexpected considering oxLDL expression mediates LOX-1 expression in the vasculature, and baseline oxLDL levels are increased in ApoE−/− mice when placed on a high-fat diet (Kato et al., 2009). Therefore, these results suggest that inhalation MVE-exposure results in increased LOX-1 expression in the endothelial cells of the cerebral microvasculature. Figure 1(M) shows representative (co-localized) endothelial LOX-1 expression across study groups; whereas Figure 1(N) is cerebral microvessels LOX-1 mRNA levels, as assessed by real-time qPCR.
Figure 1.
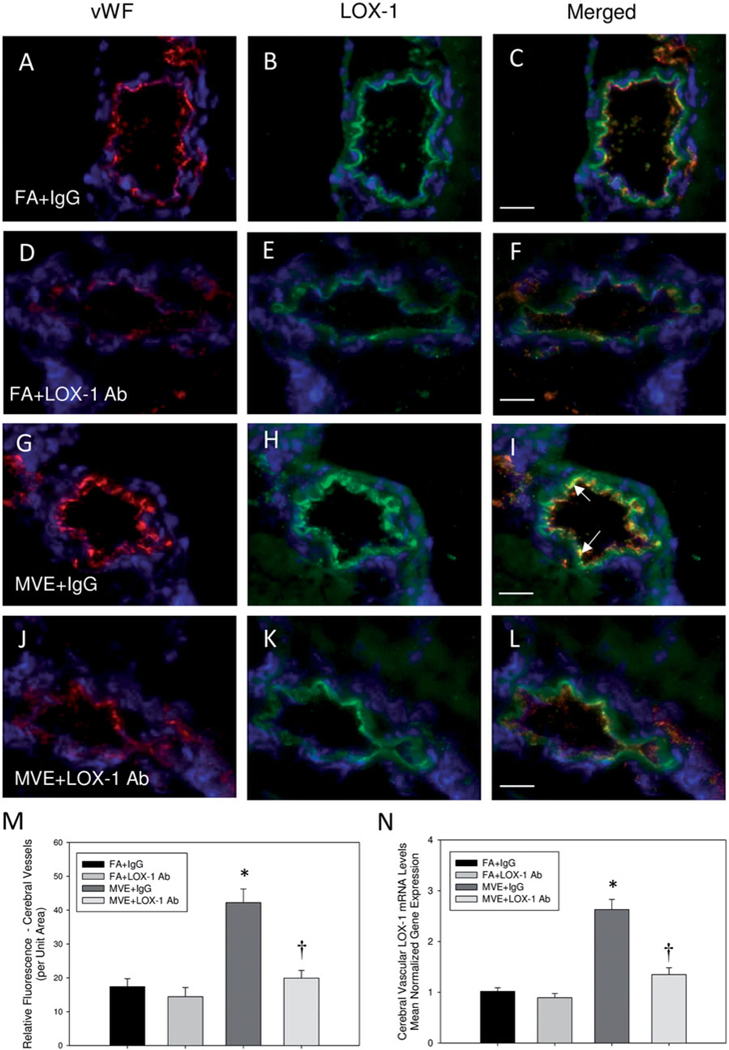
LOX-1 Ab treatment normalized MVE-induced LOX-1 expression in cerebral microvessels of ApoE−/− mice. Double immunofluorescence of LOX-1 (green fluorescence) and endothelial-cell specific vonWillebrand factor (vWF) (red fluorescence) in cerebral microvessels (frontal cortex) from ApoE−/− mice exposed to either (A-C) FA + IgG; (D-F) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (G-I) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or (J-L) MVE + LOX-1 Ab for 6 hr/d, 7 d/wk, for 30 d; blue fluorescence = Hoechst nuclear stain. Colocalization was determined by quantifying total fluorescence of overlayed signals from minimum of three slides, two sections each, three regions from each section (n = 5–7 per group), which is represented by the graph shown in panel M. Graph presented in panel N shows mRNA quantification of LOX-1 expression in cerebral microvessels, as quantified by real-time RT-PCR. Arrows indicate increased expression of endothelial-cell specific LOX-1 expression. Scale bar =10 μm; 100× magnification. Control slides with no primary anti-body were also done (not shown) to confirm specific binding. *p < 0.050 compared to FA + IgG control. †p < 0.050 compared to MVE + IgG, as determined by ANOVA.
MVE-mediated BBB disruption, associated with increased MMP-9 expression, is attenuated with LOX-1 ab treatment in ApoE−/− mice
To assess alterations in BBB transport and permeability resulting from LOX-1 signaling induced by MVE-exposure, a subset of mice from each group on the study were injected with the molecular fluorescent tracer, sodium fluorescein (Na-F), on the final day of the study exposures. As Na-F is a molecule with an MW of 376.3 Da, it would typically have limited transport from the blood across an intact BBB under homeostatic conditions, thus the presence of FITC fluorescence in the cerebral parenchyma indicates altered BBB integrity and increased permeability. In agreement with our previous studies, we observe that MVE-exposure results in greater than a 2-fold increase in FITC fluorescence in the cerebrum of ApoE−/− mice, as compared to FA controls (Figure 2), suggesting MVE-exposure mediated increase in BBB permeability. Furthermore, brains from LOX-1 Ab-treated MVE-exposed mice showed a significant decrease in fluorescence compared to MVE (+IgG) exposed animals (Figure 2).
Figure 2.
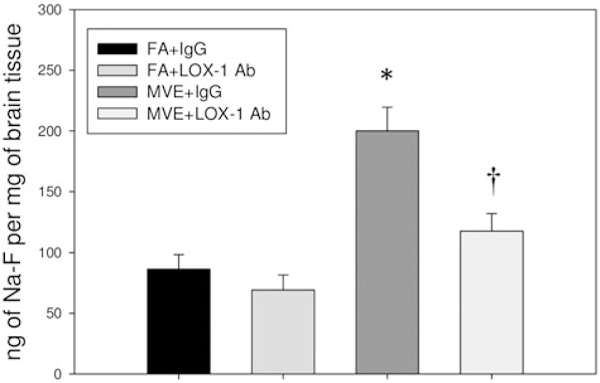
LOX-1 Ab treatment attenuates MVE-exposure induced BBB permeability. Changes in BBB permeability were assessed using the fluorescent tracer, sodium fluorescein (Na-F). ApoE−/− mice were exposed by inhalation to either mixed vehicular engine exhaust (MVE: 100μg PM/m3)+ LOX-1 Ab (16mg/ml, 0.1 ml/mouse, every other day throughout 30 d exposure); MVE + IgG (16 mg/ ml, 0.1 ml/mouse); filtered air (FA) + LOX-1 Ab; or FA + IgG, and a subset of mice (n = 6 per group) were injected i.p. with 100 μl of 2% Na-F-PBS 30min prior to the end of their final exposure on day 30. Graph shows total fluorescence in the cerebrum, as measured by fluorometry. Data are expressed as the amount of tracer per gram of tissue. *p < 0.050 compared to FA + IgG control; †p < 0.050 compared to MVE + IgG, as determined by ANOVA.
Lectin-like oxLDL receptor signaling is known to mediate induction of MMP-9 activity in vascular endothelial cells, and increased MMP-9 activity in the BBB has been shown to decrease BBB integrity via increased TJ protein degradation (Mahajan et al., 2012; Ren et al., 2015). We have previously reported that MMP-9 expression and activity are upregulated in the cerebral microvasculature of MVE-exposed ApoE−/− mice (Oppenheim et al., 2013), which was confirmed in the current studies (Figure 3). To determine whether LOX-1 signaling mediates MMP expression and/or activity in the cerebral microvasculature, we analyzed transcript levels of MMP-9 and in situ zymography for gelatinase (MMP-2 and −9) activity. LOX-1 Ab-treatment resulted in decreased MMP-9 activity levels in MVE exposed ApoE−/− cerebral microvessels (Figure 3(D,E)), compared to MVE + IgG animals (Figure 3(C)); however, these levels were still elevated in comparison to FA-exposed animals (Figure 3(A,B)). Interestingly, a more significant decrease was observed in cerebral microvascular MMP-9 mRNA transcript levels from the MVE + LOX-1 Ab-treated group, in comparison to gelatinase activity levels; however, overall trends in transcript expression were similar to those observed for activity with in situ zymography analysis, as shown in Figure 3(E vs. F). The difference between MMP activities vs. transcript levels may be due, in part, to the fact that in situ zymography cannot differentiate MMP-2 vs. MMP-9 activity, whereas RT-qPCR analysis is specific for MMP-9 expression; suggesting a possible role for MMP-2 activity and/or alterations in tissue inhibitor of matrix metalloproteinase (TIMP1/2) expression in MVE-exposure mediated BBB disruption.
Figure 3.
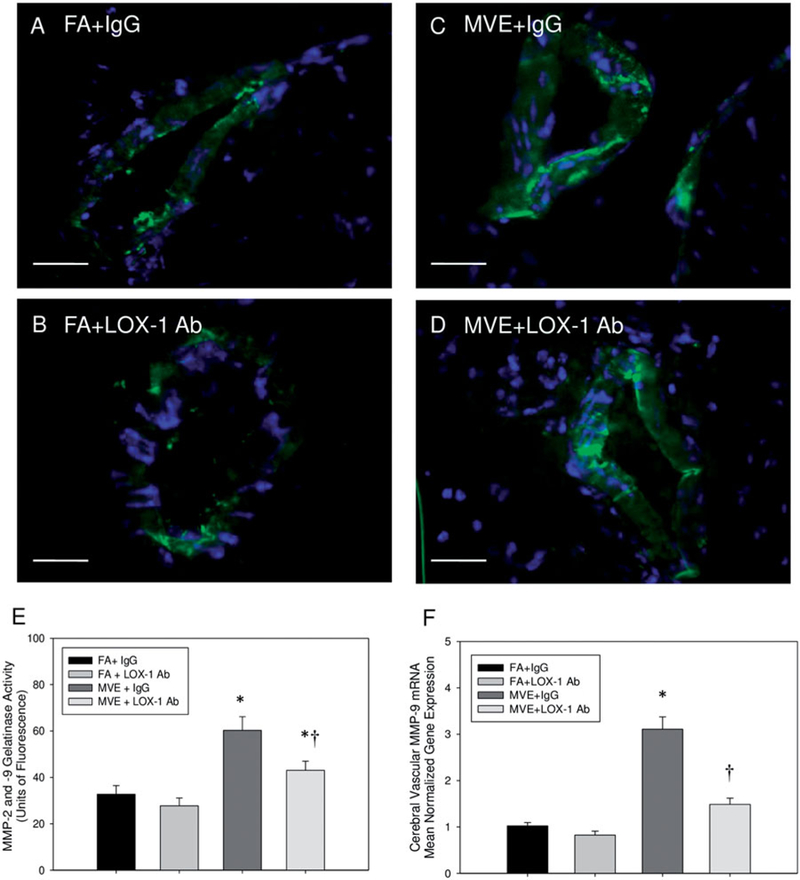
Gelatinase (MMP-2 and −9) activity and MMP-9 expression are decreased in cerebral microvessels of LOX-1-treated ApoE−/− mice exposed to MVE. MMP-9 (and −2) activity, as shown by in situ zymography, in the (frontal cortex) cerebral microvasculature in ApoE−/− mice exposed (A) FA + IgG; (B) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (C) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or MVE + LOX-1 Ab for 6 h/d, 7 d/ week, for 30 d. Gelatinase activity, quantified as units of fluorescence per unit area, is shown in (E) compared to MMP-9 mRNA transcript levels (F). Scale bar = 10 μm; arrows indicate increased areas of MMP activity (green fluorescence); blue fluorescence = Hoechst nuclear stain. Background fluorescence (fluorescence present in total image outside of the vessel) was subtracted from each section before statistical comparison between groups. n = 5–7 per group, two slides (two sections each) per sample, 8–12 vessels on each section, throughout the cerebrum, were used for analysis. *p < 0.050 compared to FA + IgG control; †p<0.050 compared to MVE + IgG, as determined by ANOVA.
LOX-1 Ab treatment attenuates MVE-exposure mediated decreases in BBB TJ proteins and oxidative stress
As increased BBB permeability is often associated with decreased expression of TJ proteins, we analyzed the endothelial expression of claudin-5 and occludin in the cerebral microvasculature of our study animals via double-immunofluorescence/co-localization. In agreement with our previous studies, compared to FA-exposed mice + IgG (Figures 4(A–C) and 5(A-C)) and FA + LOX-1 Ab mice (Figures 4(D–F) and 5(D–F)), MVE-exposure results in decreased expression of both claudin-5 (Figure 4: ~37% reduction) and occludin (Figure 5: ~51% reduction) in cerebral micro-vessels of ApoE−/− mice + IgG (Figures 4(G–I) and 5(G-I), respectively). And while LOX-1 Ab-treatment prevented the significant reduction in TJ protein expression mediated by MVE-exposure (Figures 4(J–L) and 5(J–L)), levels were not completely normalized compared to FA (+IgG or + LOX-1 Ab) control animals. Interestingly, LOX Ab-treatment appears to increase TJ protein expression of both claudin-5 and occludin in the cerebral microvasculature of ApoE−/− mice, albeit not statistically. This may be due to the overall decreased baseline integrity of the BBB observed in the ApoE−/− mouse model, due to the universal knockout down of ApoE−/−, which is known to contribute to BBB integrity. Relative fluorescence, as quantified by total co-localized fluorescence per unit area, is shown in Figures 4(M) and 5(M). Such findings suggest that LOX-1 signaling is involved in altering brain microvascular integrity, which can lead to BBB disruption and deregulated transport from the blood into the brain parenchyma.
Figure 4.
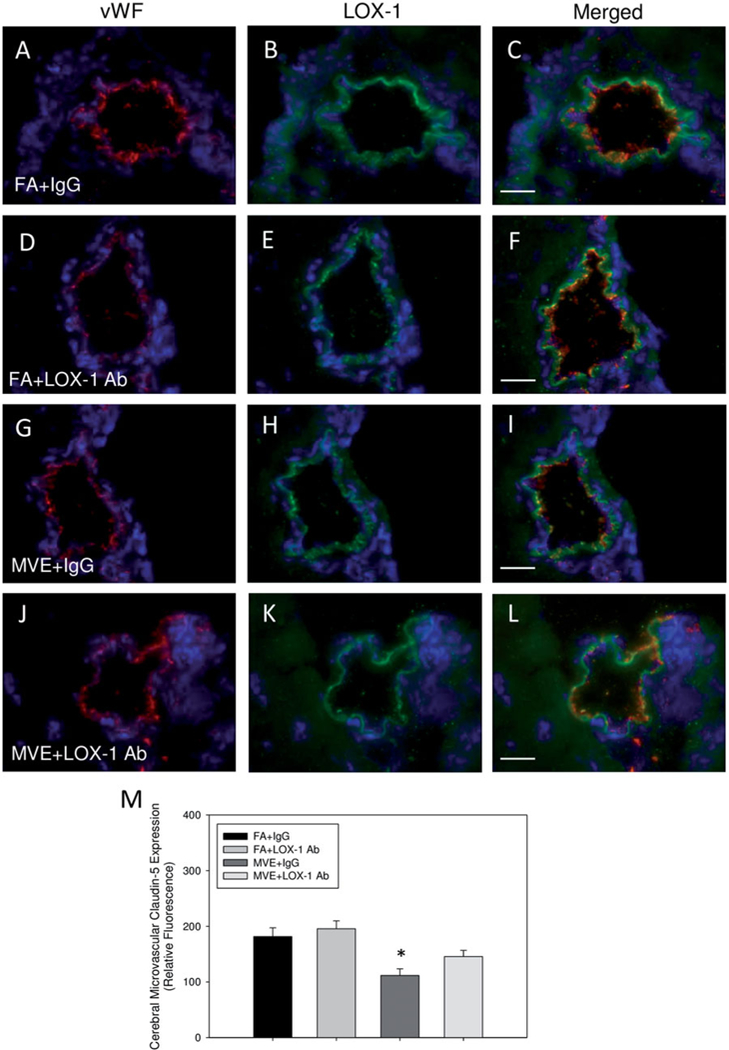
Expression of TJ protein claudin-5 in the cerebral microvasculature of ApoE−/− mice exposed to mixed vehicular emissions or filtered air. Double immune-fluorescence of claudin-5 (green fluorescence) and endothelial-cell specific vonWillebrand factor (vWF) (red fluorescence) in cerebral microvessels (frontal cortex) from ApoE−/− mice exposed to either (A-C) FA + IgG; (D-F) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (G-I) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or (J-L) MVE + LOX-1 Ab for 6 h/d, 7 d/week, for 30 d; blue fluorescence = Hoechst nuclear stain. Colocalization was determined by quantifying total fluorescence of overlayed signals from a minimum of three slides, two sections each, three regions from each section (n = 5–7 per group), which is represented by the graph shown in panel M. Scale bar =10 μm; 100 × magnification. Control slides with no primary antibody were also done (not shown) to confirm specific binding. *p < 0.050 compared to FA + IgG control, as determined by ANOVA.
Figure 5.
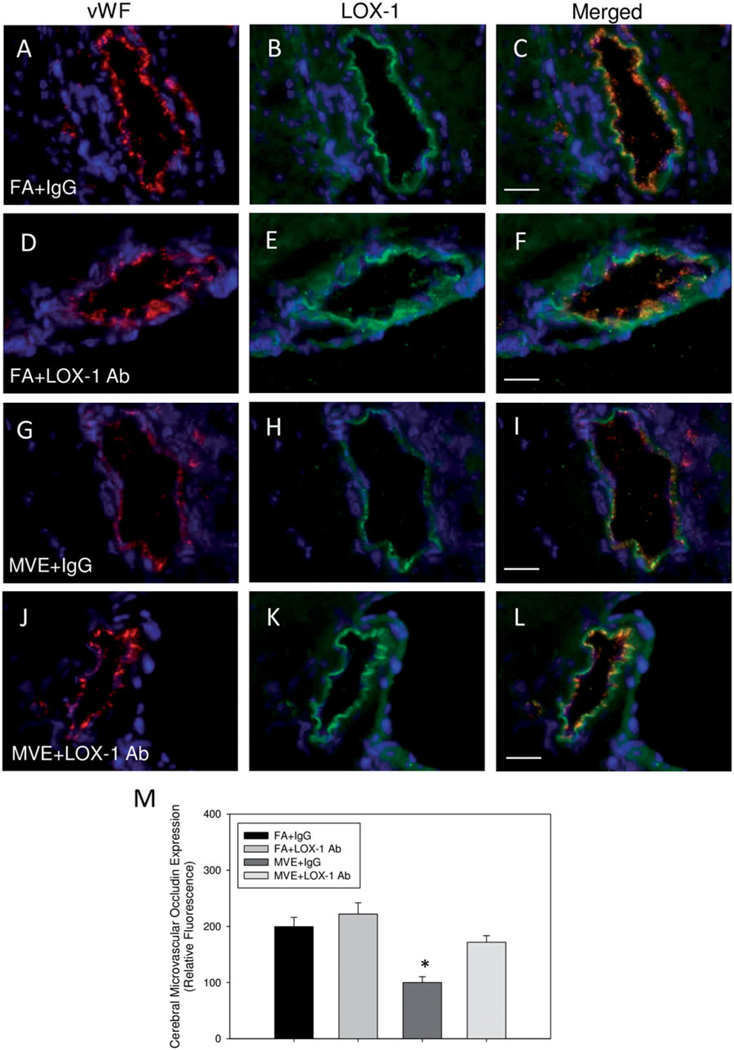
Expression of TJ protein, occludin, in the cerebral microvasculature of ApoE−/− mice exposed to mixed vehicular emissions or filtered air. Double immunofluorescence of occludin (green fluorescence) and endothelial-cell specific vonWillebrand factor (vWF) (red fluorescence) in cerebral microvessels (frontal cortex) from ApoE−/− mice exposed to either (A-C) FA + IgG; (D-F) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (G-I) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or (J-L) MVE + LOX-1 Ab for 6h/d, 7 d/week, for 30 d; blue fluorescence = Hoechst nuclear stain. Colocalization was determined by quantifying total fluorescence of overlayed signals from a minimum of three slides, two sections each, three regions from each section (n = 5–7 per group), which is represented by the graph shown in panel M. Scale bar =10 μm; 100 × magnification. Control slides with no primary antibody were also done (not shown) to confirm specific binding. *p < 0.050 compared to FA + IgG control, as determined by ANOVA.
To analyze the role of LOX-1 signaling in MVE-induced oxidative stress in the cerebral microvasculature of ApoE−/− mice, which is also associated with BBB disruption, we used dihydroethidium (DHE) staining as an indicator of super-oxide production and endothelial-specific 4-hydroxynonenal (4-HNE) immunofluorescence as a marker of lipid peroxidation. In comparison to that observed in FA + IgG animals (Figures 6(A) and 7(A–C)) and FA + LOX-1 Ab (Figures 6(B) and 7(D–F)), MVE-exposure induced a statistical increase in both cerebral microvascular ROS (~2.2-fold) and lipid peroxidation (~3.7-fold) levels (Figures 6(C) and 7(G–I)) in MVE + IgG ApoE−/− mice, as determined by ANOVA. While LOX-1 Ab-treatment did not effectively reduce ROS (DHE) levels in the cerebral microvasculature (Figure 6(D, E)), 4-HNE levels were attenuated in the MVE + LOX-1 Ab treatment group (Figure 7(J–M)), compared to those levels observed in the MVE + IgG group. In a subset of slides processed through DHE staining, sections were pretreated with the broad-spectrum flavoenzyme inhibitor, diphenyleneiodonium (DPI), in an effort to determine whether MVE-mediated LOX-1 activation is driving production of ROS via NADPH oxidase signaling. Interestingly, while DPI-treatment did decrease ROS (as indicated by DHE) in cerebral microvessels of MVE-exposed ApoE−/− mice (~40%), ROS levels were still significantly increased compared to FA control animals (Figure S1(A,B) vs. (C,D)). Based on these observations, it appears NADPH oxidase is not the only source of ROS production in the cerebral microvasculature of ApoE−/− mice exposed to MVE. However, LOX-1 receptor signaling does promote NADPH oxidase-mediated ROS production in the cerebral vasculature of MVE-exposed animals (Figure S1(C)), as evidenced by the significant reduction in DHE in the LOX-1- Ab treated animals (Figure S1(D)).
Figure 6.
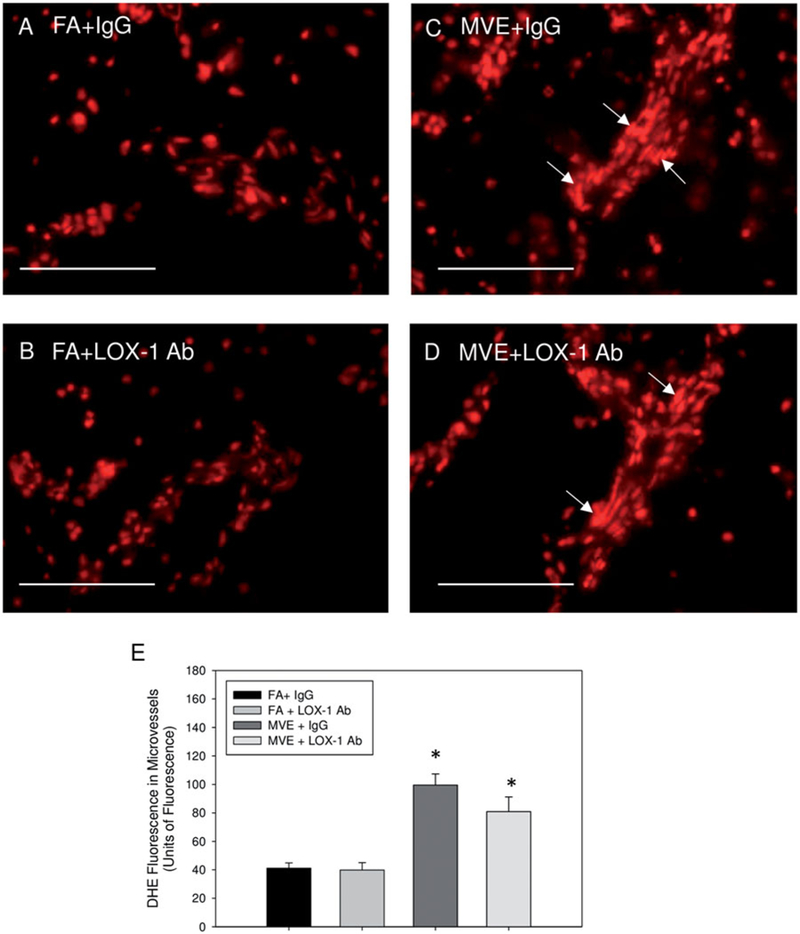
LOX-1 Ab-treatment does not mitigate MVE-exposure mediated induction of ROS in the cerebral microvessels of ApoE−/− mice. Reactive oxygen species fluorescence staining of the cerebral microvessels from brains of ApoE−/− mice exposed to mixed vehicle emissions or filtered air. Representative dihydroethidium (DHE, red) fluorescence of ApoE−/− mice exposed to either (A) FA + IgG; (B) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (C) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or MVE + LOX-1 Ab for 6h/d, 7 d/week, for 30 d. Scale bar =100 μm. Arrows indicate areas of increased DHE staining. E = graphical quantification of total fluorescence per unit area. n = 5–7 per group, three slides (six section) per animal, two sites (areas) each, were used for analysis. *p < 0.050 compared to FA + IgG control.
Figure 7.
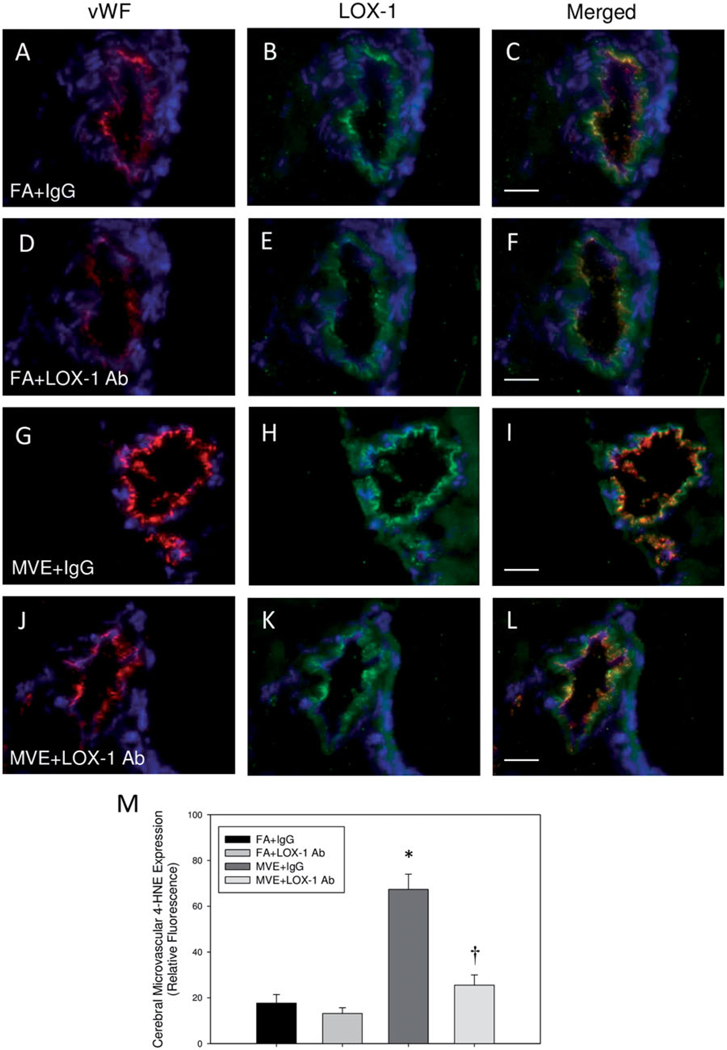
MVE-exposure induces lipid peroxidation in the cerebral microvasculature of ApoE−/− mice, which is attenuated with LOX-1 Ab treatment. Double immunofluorescence of 4-hydroxynonenol (4-HNE), a marker of lipid peroxidation (green fluorescence) and endothelial-cell specific vonWillebrand factor (vWF) (red fluorescence) in cerebral microvessels (frontal cortex) from ApoE−/− mice exposed to either (A-C) FA + IgG; (D-F) FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); (G-I) 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or (J-L) MVE + LOX-1 Ab for 6h/d, 7 d/week, for 30 d; blue fluorescence = Hoechst nuclear stain. Colocalization was determined by quantifying total fluorescence of overlayed signals from a minimum of three slides, two sections each, three regions from each section (n = 5–7 per group), which is represented by the graph shown in panel M. Arrows indicate increased 4-HNE expression in the cerebral microvasculature. Scale bar =10 μm; 100 × magnification. Control slides with no primary antibody were also done (not shown) to confirm specific binding. *p < 0.050 compared to FA + IgG control, †p < 0.050 compared to MVE + IgG, as determined by ANOVA.
LOX-1 mediates expression of VCAM-1 and MCP-1, but not ICAM-1 in the cerebral vasculature of MVE-exposed ApoE−/− mice
In an effort to investigate other proatherogenic signaling pathways, known to contribute to atherosclerotic plaque growth and/or other stroke-related vascular events, we analyzed alterations in MVE and LOX-1- mediated mRNA transcript expression of VCAM-1, ICAM-1, and MCP-1. Our results show that MVE-exposure increases ICAM-1, VCAM-1, and MCP-1 in the cerebral vasculature (Figure 8(A–C), respectively), which is mediated in part via the LOX-1 receptor, suggesting MVE-exposure stimulates increased leukocyte recruitment and adhesion to endothelial cells in the cerebral vasculature. Interestingly, while we did observe increases in both ICAM-1 and MCP-1 mRNA in the cerebral vasculature of MVE-exposed ApoE−/− mice (Figure 8(C)), LOX-1 Ab treatment did not significantly attenuate the increase in expression. Such results suggest that both ICAM-1 and MCP-1 expression are likely mediated by other signaling pathways, in addition to LOX-1, following MVE-exposure.
Figure 8.
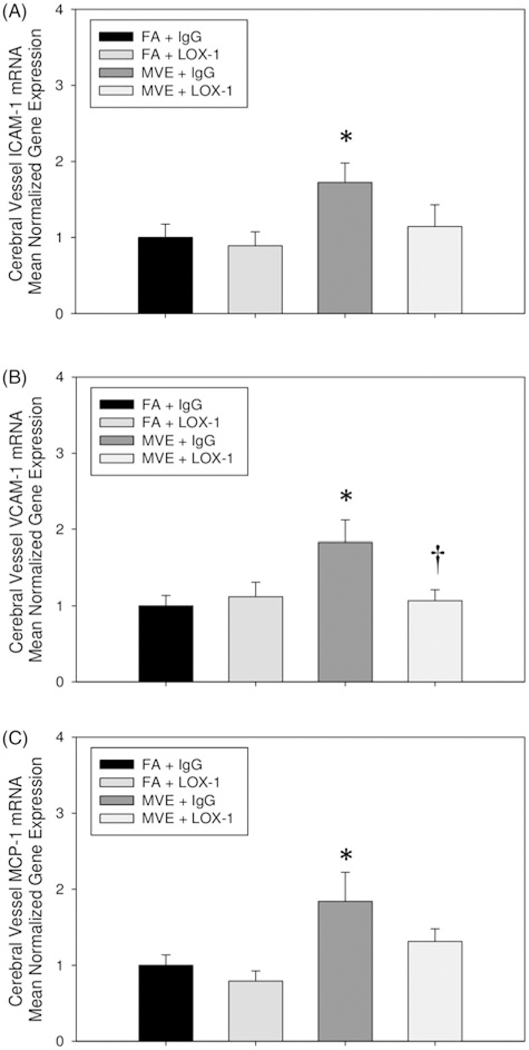
MVE-exposure mediates increased expression of adhesion and cytokine molecules, which is mediated via LOX-1 receptor signaling. RT-qPCR analysis of in the cerebral microvessels of (A) intracellular adhesion molecule (ICAM)-1; (B) vascular cell adhesion molecule (VCAM)-1; and (C) monocyte chemoattractant protein (MCP)-1 in ApoE−/− mice exposed to either filtered air (FA)+ IgG; FA + LOX-1 Ab (16 mg/ml, 0.1 ml/mouse, every other day throughout exposure); 100 PM μg/m3 of mixed vehicular emission (MVE) + IgG; or MVE + LOX-1 Ab for 6 h/d, 7 d/week, for 30 d. *p < 0.050 compared to FA control, †p < 0.050 compared to MVE + IgG, as determined by ANOVA.
Discussion
There are an increasing number of reports in the literature that show a strong association between exposure to air pollution and detrimental outcomes in the CNS, which include both neuroinflammatory and neurodegenerative pathologies (Calderón-Garcidueňas et al., 2008, 2016; Stafoggia et al., 2014). Additionally, recent reports detail exposure to environmental air pollution as a leading risk factor for stroke and/or mortality from stroke (Block et al., 2013; Crichton et al., 2016; Feigin et al., 2016; Scheers et al., 2015; Vidale et al., 2010). Thus, it is imperative to investigate the path-ways by which this environmental pollutant source may be contributing to these detrimental effects in the CNS. We, as well as others, have previously reported that exposure to air pollution results in neurovascular dysfunction, specifically disruption of BBB integrity and increased permeability (Calderón-Garcidueňas et al., 2008, 2016; Oppenheim et al., 2013). These previous studies report that exposure-mediated alterations in brain microvascular integrity were associated with decreased TJ protein expression and inflammation; however, the signaling pathways involved have not yet been fully elucidated. Thus, we investigated the hypothesis that exposure to traffic-generated air pollution results in altered cerebral microvascular integrity and transport, via an LOX-1 signaling pathway, in ApoE−/− mice. Utilizing this particular animal model presents limitations, as the Apo E protein is involved in the structural integrity and transport across the BBB (Bell et al., 2012; Nishitsuji et al., 2011). Nevertheless, using this model also provides benefits to this study, as it allows us to utilize a model with a baseline atherosclerotic pathology present in the vasculature that is consistent with that observed in the majority of the human population, including obese children and adolescents (Beauloye et al., 2007). Additionally, since our previous studies on MVE- mediated alterations in BBB transport and integrity were completed with the ApoE−/− mouse model, utilizing the same model for the current study allows for investigation into mechanistic pathways and also a more comprehensive understanding of what may occur when multiple “insults” and/or pathologies are present during periods of exposure to high levels of air pollution. Importantly, we utilized the same physiologically relevant route of exposure and concentrations (100 PM μg/m3) of MVE as those in our previously reported study (Oppenheim et al., 2013). While higher than that which would likely be experienced in a daily environmental exposure scenario, the concentration of 100 PM μg/m3 of MVE used in the current studies are comparable to theoretical “high exposure” environmental scenarios (e.g. close proximity to congested roadway “plumes”), occupational exposure scenarios, and/or those experienced in highly populated urban settings (Occupational Health Regulations: Exposure Limits for Particulates. Dieselnet, 2006; World Health Organization (WHO) Ambient Air Pollution Database, 2016).
The LOX-1 scavenger receptor is the primary receptor for oxLDL on endothelial cells (Sawamura et al., 1997), which typically has low basal expression. However, LOX-1 expression is significantly upregulated in the vasculature during pathophysiologic conditions and/or when circulating oxLDL is elevated (Li & Mehta, 2000; Mehta et al., 2006). The LOX-1 receptor can undergo proteolytic cleavage of the extracellular domain, resulting in a circulating soluble fraction sLOX-1 (Murase et al., 2000). Thus, sLOX-1 levels can be a useful diagnostic biomarker of cardiovascular disease (Hayashida et al., 2005). A potential role for LOX-1 signaling in the pathogenesis of stroke is only beginning to emerge; however, sLOX-1 has been reported to be elevated during acute ischemic stroke in humans and/or animal models (Shen et al., 2016; Yokota et al., 2016). We have previously reported that exposure to traffic-generated pollutants results in increased circulating oxLDL, aortic LOX-1 expression, and elevated plasma sLOX-1 in both animal models and human exposure scenarios (Lund et al., 2011). Therefore, LOX-1 signaling may be a key mechanistic pathway mediating the effects of air pollution exposure on the cerebral vasculature contributing to and/or exacerbating stroke outcomes. Thus, we investigated the role of MVE- mediated induction of LOX-1 signaling in the cerebral microvessels using LOX-1 antibody treatment throughout the exposures. Our data show that LOX-1 Ab treatment resulted in reduced endothelial LOX-1 expression in the cerebral microvessels of ApoE−/− mice; however, LOX-1 mRNA levels remained elevated in the MVE + LOX-1 Ab, compared to control (FA + IgG) expression, though differences observed are not significant. The lack of complete “normalization” of LOX-1 mRNA levels in the cerebral microvasculature is likely due to the premise that LOX-1 is a multi-ligand scavenger receptor whose expression is known to be upregulated via ligand interactions. The ligands and other factors known to upregulate LOX-1 expression include proinflammatory cytokine expression and/or vaso-active peptides, such as endothelin (ET)-1 (Xu et al., 2013). While the current study did not assess these specific endpoints, it has previously been reported that exposure to traffic-generated air pollutants increases ET-1 signaling path-ways in both animal models and human exposures (Lund et al., 2009; Shen et al., 2016). In agreement with our previous findings in the systemic vasculature (Lund et al., 2011), we observed a decrease in oxidative stress in the cerebral microvasculature, assessed by 4-HNE staining, in the LOX-1 Ab-treated ApoE−/− mice exposed to MVE. Interestingly, we did not observe an attenuation of ROS (DHE) levels in the cerebral microvasculature with LOX-1 Ab treatment. As DHE fluorescence is indicative of superoxide production, these collective findings suggest that multiple types of ROS/ radicals (e.g. hydrogen peroxide, hydroxyl radicals, lipid peroxyl radicals, etc.) are likely involved in promoting the overall increase in oxidative stress observed in the cerebral microvasculature of MVE-exposed animals. However, we did measure a decrease in LOX-1-mediated ROS levels through pretreatment with DPI (NOX-inhibitor), suggesting LOX-1 driven NAPDH oxidase activity is responsible for the production of at least some ROS observed in the cerebral microvasculature resulting from MVE-exposure.
MMP-9 is an enzyme that is known to degrade several components of the extracellular matrix in the basal lamina surrounding cerebral vessels. Increased MMP-9 levels have been associated with BBB disruption, increased stroke infarct size and/or severity, and also occurrence of hemorrhagic transformation in stroke (Castellanos et al., 2003; Horstmann et al., 2003; Montaner et al., 2001; Rodriguez-Yanez et al., 2006; Romanic et al., 1998; Sotgiu et al., 2006). We have previously reported that MVE-exposure increases MMP-9 expression and activity in the vasculature, which was (at least in part) mediated by increased ET-1 signaling (Lund et al., 2009). MMP-9 expression in vascular endothelial cells is also known to be upregulated through multiple other pathways, including Angiotensin (Ang)-II and LOX-1 signaling (Guo et al., 2015; Mehta et al., 2006). While LOX-1 Ab treatment resulted in a significant reduction of MMP-9 mRNA expression and activity in the microvasculature of MVE-exposed ApoE−/− mice, these levels were not normalized to those observed in the control animals. Interestingly, we observed a more pronounced attenuation in MMP-9 mRNA expression vs. gelatinase (MMP-2 and −9) activity with LOX-1 Ab-treatment in MVE-exposed cerebral micro-vessels. These findings suggest MMP-2 may also be upregulated with MVE-exposure (thus the higher activity level), which is not directly mediated by LOX-1 signaling. Additionally, there may be exposure-mediated alterations in expression of tissue inhibitors of matrix metalloproteinases (TIMP)-1 or −2, and subsequent inhibition of MMP-2 or −9 activity. When considering factors that mediate MMP-9 expression and/or activity, and that we have previously reported at least some of these factors are upregulated with traffic-generated pollutant-exposure, it is likely that there are multiple signaling pathways involved in MMP-9 expression and activity in these tissues. Additional studies, with appropriate inhibitors, are currently underway in our laboratory to further elucidate the signaling pathways involved in MVE-induced MMP-9 expression and activity in the cerebral vasculature.
Importantly, increased MMP-9 activity has been reported to mediate BBB disruption, via alterations in TJ protein expression, in both human and animal model exposure scenarios (Barr et al., 2010; Bauer et al., 2010). We have previously reported that MVE-exposure results in BBB disruption associated with decreased TJ protein expression in ApoE−/− mice (Oppenheim et al., 2013), which has also been observed in the brains of children living in urban areas with high levels of air pollution (Mexico City) (Calderon-Garciduenas et al., 2016). In the current study, our results show that LOX-1 mediated signaling is correlated to decreased microvascular TJ protein expression and BBB integrity in ApoE−/− mice. These findings may, at least in part, account for the poorer prognostic outcome associated with stroke occurrence on days with elevated levels of ambient air pollution, as increased MMP-9 activity and BBB disruption are associated with hemorrhagic transformation and/or complications during ischemic stroke (Jickling et al., 2014; Ljungman & Mittleman, 2014; Turner & Sharp, 2016). Additionally, neurovascular BBB dysfunction is also associated with neurodegeneration and dementia-related disorders, such as Alzheimer’s disease (Nelson et al., 2016), occurrence of which is reported to be associated with exposure to high levels of ambient and/or traffic-generated air pollution in both children and adults [rev. in Xu et al. (2016)]. Thus, it is imperative to gain a more in-depth understanding of the pathways involved, as well as the components and/or mixtures of air pollution that drive these detrimental responses, in order to address both environmental regulatory issues and therapeutic options for exposure-related effects in the CNS.
In agreement with previously published reports of air pollution exposure-mediated induction of VCAM-1, ICAM-1, and MCP-1, both in vivo and in vitro (Bind et al., 2012; Shaw et al., 2011), we observe an MVE-mediated increase in these proatherogenic signaling factors in the cerebral micro-vasculature of ApoE−/− mice. Adhesions molecules, such as ICAM-1 and VCAM-1, are known to play a role in inflammation, activation of endothelial cells, and transendothelial migration of leukocytes. Expression of ICAM-1 and VCAM-1 are reported to be elevated during the pathogenesis of acute stroke, as well as the progression of ischemic injury following stroke (Frijns et al., 1997; Supanc et al., 2011). Additionally, the infiltration of leukocytes during stroke, sequestered via signaling molecules such as MCP-1, can result in increased tissue injury during an ischemic stroke (Losy & Zaremba, 2001). Thus, while the upregulation of these factors may not directly lead to the onset of a stroke, they may exacerbate the effects of air pollution on the neurovascular unit leading to exaggerated injury and/or poorer prognostic outcome. Interestingly, while LOX-1 signaling appears to mediate VCAM-1 expression in the cerebral vasculature of ApoE−/− mice, LOX-1 Ab-treatment did not significantly attenuate MVE-induced ICAM-1 or MCP-1 expression, although there is a notable reduction. Since both vascular endothelial ICAM-1 and MCP-1 expression are mediated through several different signaling pathways, including proinflammatory cytokines [e.g. interleukin (IL)-1p] and vasoactive peptides, it is likely there are multiple pathways involved in the induction of these proatherogenic factors in the cerebral vasculature from our exposure groups. While this study did not directly quantify the expression of these proinflammatory cytokines, we have previously shown that MVE exposure significantly increases cerebral IL-1p expression (Oppenheim et al., 2013), while others have reported an association between air pollution exposure and vascular endothelial cell cytokine expression (Schisler et al., 2015).
It is important to address the limitations of the current study. As previously mentioned, the use of the ApoE−/− mouse model, while beneficial for providing baseline atherosclerotic pathology that is similar in etiology to that observed in humans, still provides limitations in that: (1) there will be a higher baseline level of BBB disruption, due to the absence of ApoE protein expression; (2) there are extremely high levels of cholesterol present in these animals when fed a high fat diet that can contribute to systemic inflammation and/or metabolic abnormalities; and (3) the endpoints of this study represent only a subchronic exposure scenario and thus we cannot directly extrapolate these findings to acute (hourly, etc.) exposure scenarios. Future studies with different chemical components and/or concentrations of traffic-generated air pollutants, acute vs. chronic end-points, as well as those using healthy or “wildtype” models, are necessary to further elucidate diet- vs. exposure- mediated effects of MVE in the neurovascular unit.
Conclusions
In conclusion, the findings of this study provide insight into a novel LOX-1-mediated signaling pathway involved in traffic-generated pollution-mediated alterations in BBB integrity, which is associated with increased MMP-9 activity and resulting decreased TJ protein expression in the cerebral microvasculature of ApoE−/− mice on a high-fat diet. These findings suggest that LOX-1 signaling likely mediates some of the detrimental outcomes associated with air pollution exposure in the neurovascular unit and/or CNS. Furthermore, we observe MVE-exposure results in increased oxidative stress and lipid peroxidation in the cerebral micro-vasculature, which is associated with increased signaling/ sequestration of monocytes/macrophage, as shown through increased MCP-1, VCAM-1, and ICAM-1 expression. Expression of several of these factors was attenuated, though not completely normalized, with LOX-1 Ab-treatment during MVE exposure. The lack of normalization observed in these study endpoints, with LOX-1 Ab-treatment, suggests that there are likely other key mechanistic pathways involved in the MVE-mediated responses in the cerebral microvasculature, for which further mechanistic studies are necessary to determine. Collectively, findings from this study have identified at least one mechanistic signaling pathway involved in air pollution-mediated alterations in BBB integrity, namely the LOX-1 receptor.
Supplementary Material
Acknowledgements
We would like to thank the Chemistry and Inhalation Exposure group, in the Environmental Respiratory Health Program, at Lovelace Biomedical and Environmental Research Institute for the characterization and monitoring of the animal exposures.
Funding
This work was supported by National Institute of Environmental Health Sciences (NIEHS) at National Institute of Health [5R00ES016586 to A.K.L.] and University of North Texas Research Initiation Grant (RIG) [GA93601 to A.K.L] and Environmental Protection Agency Center Grant RD-83479601 [Project 3 to A.K.L., coInvestigator and Project 2 to J.D.M., coPI] for the animal exposures. However, the authors declare no financial gains to these entities associated with this publication.
Footnotes
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Abdul-Muneer PM, Pfister BJ, Haorah J, Chandra N. (2016). Role of matrix metalloproteinases in the pathogenesis of traumatic brain injury. Mol Neurobiol. 53:6106–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TL, Latour LL, Lee K-Y, et al. (2010). Blood brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke. 41:e123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AT, Burgers HF, Rabie T, Marti HH. (2010). Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 30:837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauloye V, Zech F, Tran HT, et al. (2007). Determinants of early atherosclerosis in obese children and adolescents. J Clin Endocrinol Metab. 92:3025–32. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 485:512–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind MA, Baccarelli A, Zanobetti A, et al. (2012). Air pollution and markers of coagulation, inflammation, and endothelial function: associations and epigene-environment interactions in an elderly cohort. Epidemiology. 23:332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Elder A, Auten RL, et al. (2013). The outdoor air pollution and brain health workshop. Neurotoxicology. 5:972–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook RD, Rajagopalan S, Pope CA, et al. (2010). Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 121:2331–78. [DOI] [PubMed] [Google Scholar]
- Calderón-Garcidueňas L, Solt AC, Henríquez-Roldán C, et al. (2008). Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood- brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol. 36:289–310. [DOI] [PubMed] [Google Scholar]
- Calderón-Garcidueňas L, Reynoso-Robles R, Vargas-Martínez J, et al. (2016). Prefrontal white matter pathology in air pollution exposed Mexico City young urbanites and their potential impact on neurovascular unit dysfunction and the development of Alzheimer’s disease. Environ Res. 146:404–17. [DOI] [PubMed] [Google Scholar]
- Calderón-Garcidueňas L, Leray E, Heydarpour P, et al. (2016). Air pollution, a rising environmental risk factor for cognition, neuroinflammation and neurodegeneration: The clinical impact on children and beyond. Rev Neurol (Paris). 172:69–80. [DOI] [PubMed] [Google Scholar]
- Castellanos M, Leira R, Serena J, et al. (2003). Plasma metalloprotei- nase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke. 34:40–6. [PubMed] [Google Scholar]
- Crichton S, Barratt B, Spiridou A, et al. (2016). Associations between exhaust and non-exhaust particulate matter and stroke incidence by stroke subtype in South London. Sci Total Environ. 568:278–84. [DOI] [PubMed] [Google Scholar]
- Di Pietro N, Formoso G, Pandolfi A. (2016). Physiology and patho-physiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascul Pharmacol. S1537–1891:30101–4. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ca ZR, Tang Y, et al. (2014). TLR4/NF-KB signaling pathway-mediated and oxLDL-induced up-regulation of LOX-1, MCP-1, and VCAM-1 expressions in human umbilical vein endothelial cells. Genet Mol Res. 3:680–95. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Roth GA, Naghavi M, et al. (2016). Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 1990–2013. Lancet Neurol. 15:913–24. [DOI] [PubMed] [Google Scholar]
- Frijns CJ, Kappelle LJ, van Gijn J, et al. (1997). Soluble adhesion molecules reflect endothelial cell activation in ischemic stroke and in carotid atherosclerosis. Stroke. 28:2214–18. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Shiba M, Kawakita F, et al. (2015). Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg. 124:1693–702. [DOI] [PubMed] [Google Scholar]
- Guo Y-S, Wu Z-G, Yang J-K, Chen X-J. (2015). Impact of losartan and angiotensin II on the expression of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in rat vascular smooth muscle cells. Mol Med Rep. 11:1587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. (2015). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 57:173–85. [DOI] [PubMed] [Google Scholar]
- Hayashida K, Kume N, Murase T, et al. (2005). Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome. Circulation. 112:812–18. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kalb P, Koziol J, et al. (2003). Profiles of matrix metallo-proteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 34:2165–70. [DOI] [PubMed] [Google Scholar]
- Huang W, Li Q, Chen X, et al. (2017). Soluble lectin-like oxidized low-density lipoprotein receptor-1 as a novel biomarker for large-artery atherosclerotic stroke. Int J Neurosci. [Epub ahead of print]. doi: 10.1080/00207454.2016.1272601 [DOI] [PubMed] [Google Scholar]
- Jacobs L, Emmerechts J, Hoylaerts MF, et al. (2011). Traffic air pollution and oxidized LDL. PLoS One. 6:e16200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Wang Z, Liu Y, et al. (2011). Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 44:130–9. [DOI] [PubMed] [Google Scholar]
- Jickling GC, Lui D, Stomova B, et al. (2014). Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 34:185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato R, Mori C, Kitazato K, et al. (2009). Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipo-protein E knockout mice. Arterioscler Thromb Vasc Biol. 29:33–9. [DOI] [PubMed] [Google Scholar]
- Korek MJ, Bellander TD, Lind T, et al. (2015). Traffic-related air pollution exposure and incidence of stroke in four cohorts from Stockholm. J Expo Sci Environ Epidemiol. 25:517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Mehta JL. (2000). Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL an implication of apoptosis of human coronary artery endothelial cells: evidence from use of anti-sense LOX-1 mRNA and chemical inhibitors. Arterioscler Thromb Vasc Biol. 20:1116–22. [DOI] [PubMed] [Google Scholar]
- Ljungman PL, Mittleman MA. (2014). Ambient air pollution and stroke. Stroke. 45:3734–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losy J, Zaremba J. (2001). Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke. 32:2695–6. [DOI] [PubMed] [Google Scholar]
- Lubrano V, Balzan S. (2016). Roles of LOX-1 in microvascular dysfunction. Microvasc Res. 105:132–40. [DOI] [PubMed] [Google Scholar]
- Lund AK, Lucero J, Lucas S, et al. (2009). Vehicular emissions induce vascular MMP-9 expression and activity associated with endothelin- 1 mediated pathways. Arterioscler Thromb Vasc Biol. 29:511–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund AK, Lucero J, Harman M, et al. (2011). The oxidized low-density lipoprotein receptor mediates vascular effects of inhaled vehicle emissions. Am J Respir Crit Care Med. 184:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Reynolds JL, et al. (2012). Suppression of MMP-9 expression in brain microvascular endothelial cells (BMVEC) using a gold nanorod (GNR)-siRNA nanoplex. Immunol Invest. 41:337–55. [DOI] [PubMed] [Google Scholar]
- Mehta JL, Chen J, Hermonat PL, et al. (2006). Lectin-like oxidized LDL receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 69:36–45. [DOI] [PubMed] [Google Scholar]
- Mollace V, Gliozzi M, Musolino V, et al. (2015). Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: role of oxidative stress and LOX-1 receptor expression. Int J Cardiol. 184:152–8. [DOI] [PubMed] [Google Scholar]
- Montaner J, Alvarez-Sabín J, Molina CA, et al. (2001). Matrix metallo-proteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 32:2762–77. [DOI] [PubMed] [Google Scholar]
- Mumaw CL, Levesque S, McGraw C, et al. (2016). Microglial priming through the lung-brain axis: the role of air pollution-induced circulating factors. FASEB J. 30:1880–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase T, Kume N, Kataoka H, et al. (2000). Identification of soluble forms of lectin-like oxidized LDL receptor-1. Arterioscler Thromb Vasc Biol. 20:715–20. [DOI] [PubMed] [Google Scholar]
- Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 1862:887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitsuji K, Hosono T, Nakamura T, et al. (2011). Apolipoprotein E regulates the integrity of tight junctions in an isoforms-dependent manner in an in vitro blood brain barrier model. J Biol Chem. 286:17536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occupational Health Regulations: Exposure Limits for Particulates. Dieselnet, 2006. Available from: https://www.dieselnet.com/standards/us/ohs.php [last accessed May 2017].
- Oppenheim HA, Lucero J, Guyot AC, et al. (2013). Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part Fibre Toxicol. 10:62. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Li N, Wang B, et al. (2015). Limb ischemic preconditioning attenuates blood-brain barrier disruption by inhibiting activity of MMP-9 and occludin degradation after focal cerebral ischemia. Aging Dis. 6:406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Yáñez M, Castellanos M, Blanco M, et al. (2006). New-onset hypertension and inflammatory response/poor outcome in acute ischemic stroke. Neurology. 67:1973–8. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, et al. (1998). Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 29:1020–30. [DOI] [PubMed] [Google Scholar]
- Sawamura T, Kume N, Aoyama T, et al. (1997). An endothelial receptor for oxidized low density lipoprotein. Nature. 386:73–7. [DOI] [PubMed] [Google Scholar]
- Scheers H, Jacobs L, Casas L, et al. (2015). Long-term exposure to particulate matter air pollution is a risk factor for stroke: meta-analytical evidence. Stroke. 46:3058–66. [DOI] [PubMed] [Google Scholar]
- Schisler JC, Ronnebaum SM, Madden M, et al. (2015). Endothelial inflammatory transcriptional responses to an altered plasma expo- some following inhalation of diesel emissions. Inhal Toxicol. 27:272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CA, Robertson S, Miller MR, et al. (2011). Diesel exhaust particulate-exposed macrophages cause marked endothelial cell activation. Am J Respir Cell Mol Biol. 44:840–51. [DOI] [PubMed] [Google Scholar]
- Shen MY, Chen FY, Hsu JF, et al. (2016). Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood. 127:1336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T, Aoyama T, Yokoyama C, et al. (2014). LOX-1 plays an important role in ischemia-induced angiogenesis of limbs. PLoS One. 9:e114542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu S, Zanda B, Marchetti B, et al. (2006). Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 13:505–13. [DOI] [PubMed] [Google Scholar]
- Stafoggia M, Cesaroni G, Peters A, et al. (2014). Long-term exposure to ambient air pollution and incidence of cerebrovascular events: results from 11 European cohorts within the ESCAPE project. Environ Health Perspect. 122:919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supanc V, Bioglav Z, Kes VB, Demarin V. (2011). Role of cell adhesion molecules in acute ischemic stroke. Ann Saudi Med. 31:365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR. (2016). Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci. 10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler CR, Zychowski KE, Sanchez BN, et al. (2016). Surface area- dependence of gas-particle interactions influences pulmonary and neuroinflammatory outcomes. Part Fibre Toxicol. 13:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidale S, Bonanomi A, Guidotti M, et al. (2010). Air pollution positively correlates with daily stroke admission and in hospital mortality: a study in the urban area of Como, Italy. Neurol Sci. 31:179–82. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Ambient Air Pollution Database, 2016. Available from: http://www.who.int/phe/health_topics/out-doorair/databases/cities/en/ [last accessed May 2017].
- World Health Organization (WHO) Ambient (outdoor) air quality and health Fact Sheet, 2016. Available from: http://www.who.int/media-centre/factsheets/fs313/en/ [last accessed May 2017].
- Willis CL, Camire RB, Brule SA, Ray DE. (2013). Partial recovery of the damaged rat blood-brain barrier is mediated by adherens junction complexes, extracellular matrix remodeling and macrophage infiltration following focal astrocyte loss. Neuroscience. 250:773–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Ogura S, Chen J, et al. (2013). LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. 70:2859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Ha SU, Basnet R. (2016). A review of epidemiological research on adverse neurological effects of exposure to ambient air pollution. Front Public Health. 4:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota C, Sawamura T, Watanabe M, et al. (2016). High levels of soluble lectin-like oxidized low-density lipoprotein receptor-1 in acute stroke: an age- and sex-matched cross-sectional study. J Atheroscler Thromb. 23:1222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehendner CM, Librizzi L, Hedrich J, et al. (2013). Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8:e82823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N. (1992). Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipo- protein E. Science. 258:468–47. [DOI] [PubMed] [Google Scholar]
- Zhang X, Staimer N, Gillen DL, et al. (2016). Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ Res. 150:306–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.