

Abstract
Background
Ameloblastin (AMBN) is a secreted matrix protein that is critical for the formation of dental enamel and is enamel‐specific with respect to its essential functions. Biallelic AMBN defects cause non‐syndromic autosomal recessive amelogenesis imperfecta. Homozygous Ambn mutant mice expressing an internally truncated AMBN protein deposit only a soft mineral crust on the surface of dentin.
Methods
We characterized a family with hypoplastic amelogenesis imperfecta caused by AMBN compound heterozygous mutations (c.1061T>C; p.Leu354Pro/ c.1340C>T; p.Pro447Leu). We generated and characterized Ambn knockout/NLS‐lacZ (Ambn lacZ/lacZ) knockin mice.
Results
No AMBN protein was detected using immunohistochemistry in null mice. ß‐galactosidase activity was specific for ameloblasts in incisors and molars, and islands of cells along developing molar roots. Ambn lacZ/lacZ 7‐week incisors and unerupted (D14) first molars showed extreme enamel surface roughness. No abnormalities were observed in dentin mineralization or in nondental tissues. Ameloblasts in the Ambn lacZ/lacZ mice were unable to initiate appositional growth and started to degenerate and deposit ectopic mineral. No layer of initial enamel ribbons formed in the Ambn lacZ/lacZ mice, but pockets of amelogenin accumulated on the dentin surface along the ameloblast distal membrane and within the enamel organ epithelia (EOE). NLS‐lacZ signal was positive in the epididymis and nasal epithelium, but negative in ovary, oviduct, uterus, prostate, seminal vesicles, testis, submandibular salivary gland, kidney, liver, bladder, and bone, even after 15 hr of incubation with X‐gal.
Conclusions
Ameloblastin is critical for the initiation of enamel ribbon formation, and its absence results in pathological mineralization within the enamel organ epithelia.
Keywords: Ambn‐/‐Amelx‐/-, amelin, ameloblastin, amelogenin, dental enamel formation, matrix proteins, mineralization, missense mutation, sheath protein, sheathlin
1. INTRODUCTION
Ameloblastin (AMBN) (OMIM *601259) is a phosphorylated glycoprotein secreted by ameloblasts into the enamel matrix of developing teeth (Fincham, Moradian‐Oldak, & Simmer, 1999). AMBN is required for enamel formation, as defects in AMBN cause non‐syndromic inherited enamel malformations in humans (Poulter et al., 2014; Prasad et al., 2016) and in Ambn defective mice (Fukumoto et al., 2004). An intact Ambn is consistently found in vertebrates that make enamel, while Ambn is pseudogenized in mammals that have lost the ability to make teeth, such as baleen whales (Mysticeti; Demere, McGowen, Berta, & Gatesy, 2008; Meredith, Gatesy, Cheng, & Springer, 2011), turtles (Meredith, Gatesy, & Springer, 2013), birds (Meredith et al., 2013), narwhal (Meredith et al., 2013), aardvark (Meredith et al., 2013), and two‐toed sloth (Meredith et al., 2013). Other tooth‐specific genes implicated in the etiology of non‐syndromic amelogenesis imperfecta in humans, including ENAM, AMEL, MMP20, ODAPH, KLK4, and AMTN (Springer et al., 2016), have been shown to be inactivated in one or more toothless vertebrates and/or in vertebrates with enamel‐less teeth.
The spotted gar (Lepisosteus oculatus) is among the most distant extant relatives of tetrapods that make enamel (Sire, 1994, 1995). In the gar, enamel (also called ganoin) forms on both its teeth and scales, and Ambn expression is specifically associated with the formation of these mineralized organs, and is not expressed in gar bone, embryos, or any of the nine organs included in the PhyloFish database (Braasch et al., 2016; Kawasaki et al., 2017). AMBN is a tooth‐specific gene that is required for amelogenesis, although there are some reports that AMBN functions in other tissues, such as bone (Atsawasuwan et al., 2013; Jacques et al., 2014). These claims are inconsistent with (1) the absence of AMBN expressed sequence tags (EST) from bone in the human (Hs.272396; 0/71618) and mouse (Mm.8437; 0/34066) EST databases, (2) Ambn pseudogenization in mammals that continue to make bone but not enamel, and (3) the absence of reported bone phenotypes in persons with amelogenesis imperfecta (AI) caused by biallelic AMBN defects.
1.1. Discovery of Ameloblastin and proteolysis by MMP20
AMBN was first discovered by the characterization of its cleavage products in developing pig enamel. AMBN cleavage products were purified from enamel scraped from developing pig teeth, characterized using N‐terminal sequencing and amino acid composition analyses, and used to raise specific polyclonal antibodies (Fukae & Tanabe, 1987a, 1987b; Fukae, Tanabe, Uchida, Yamakoshi, & Shimizu, 1993; Murakami et al., 1997; Shimizu, 1984). Immunohistochemistry of developing pig enamel using these antibodies outlined the enamel rods, producing a honeycomb pattern throughout the inner and middle enamel layers (Uchida et al., 1991), leading to its designation as “sheath protein” (Uchida et al., 1995), although the concept of an organic sheath surrounding the rods and cementing them together had been previously dismissed (Orams, 1964, 1966). Rat and pig cDNA clones encoding the “sheath protein” were cloned independently from three groups and labeled “ameloblastin” (Krebsbach et al., 1996), “amelin” (Cerny, Slaby, Hammarstrom, & Wurtz, 1996), and “sheathlin” (Hu et al., 1997). With ameloblastin being the first cDNA cloning manuscript to be submitted and published, it became the formal and accepted designation. The N‐terminal sequences of six different non‐amelogenin peptides isolated from fresh secretory stage pig enamel were identified within the AMBN deduced amino acid sequences. MMP20 catalyzed the cleavage of recombinant AMBN in vitro at the exact same positions as were shown to occur in vivo (Chun et al., 2010; Iwata et al., 2007). Similar work had previously demonstrated that MMP20 also cleaves amelogenin (AMEL) in vitro at the same sites known to be cleaved in vivo through the isolation and characterization of matrix proteins (Ryu et al., 1999). AMBN is expressed and secreted by ameloblasts (Lee et al., 1996), and in mouse enamel concentrates near the secretory surface of the ameloblast (Nanci et al., 1998).
1.2. Ambn gene structure
Alternative splicing results in the secretion of two ameloblastin isoforms. Transcript 1 includes (while transcript 2 omits) a 45 nucleotide segment encoding 15 amino acids at the 5' end of Exon 6 (Cerny et al., 1996; Hu et al., 1997). Alignment of 53 functional AMBN sequences, covering over 200 million years (MY) of evolution, identified 80 unchanged and 81 highly conserved amino acid residues (out of 447 residues in the human AMBN protein, inclusive of the signal peptide; Delsuc, Gasse, & Sire, 2015). All 15 amino acids encoded by the 5'‐end of Exon 6 that can be deleted during RNA splicing are highly or absolutely conserved, and this peptide also includes an O‐glycosylation (human secreted protein Ser112), suggesting that there may be an important functional difference between the two ameloblastin isoforms generated by alternative splicing (Kobayashi et al., 2007). Evolutionary analysis suggested that ancestral mammalian AMBN likely possessed 11 exons (corresponding to exons 1 to 7 and 10 to 13 in humans, as Exons 8 and 9 were added in the human line by tandem duplications of Exon 7; Delsuc et al., 2015). All AMBN introns are phase 0 (located between complete codons), so duplication and deletion of exons during evolution, and alternative mRNA splicing, does not shift the downstream reading frame. Virtually all introns in secretory calcium‐binding phosphoprotein genes (SCPPs), including AMBN, are phase 0 (Kawasaki et al., 2017).
A potentially important AMBN structural feature is the presence of two in‐frame, putative translational start sites (TSS; Hu et al., 1997). The 3' TSS (in Exon 2) is in a much better context for translation initiation, having purines in both the −3 and +4 positions, whereas the 5' TSS (in Exon 1) has pyrimidines in both of these positions (Hinnebusch, 2017; Kozak, 1984). Phylogenetic analyses showed that the TSS in Exon 2 is conserved in all AMBN analyzed, while the TSS in Exon 1 appeared recently, in the eutherian (placental mammal) ancestor and was independently lost in various eutherian species (Delsuc et al., 2015). Ribosomes scanning the mRNA from the 5’ end usually skip over the first weak TSS to initiate more frequently at the strong TSS (leaky scanning; Hinnebusch, Ivanov, & Sonenberg, 2016). Initiation from the more 5' TSS (in Exon 1) would add 10 amino acids to the beginning of the signal peptide compared with translation initiation from the more 3' TSS (in Exon 2), with use of either translation start site generating the same secreted protein.
1.3. Etiology of human amelogenesis imperfecta
Although AMBN has long been a candidate gene for the etiology of amelogenesis imperfecta (AI), only two AI‐causing AMBN defects have been reported to date (Figure S2). Both are homozygous recessive mutations, and both are predicted to delete a single exon. The first was a 2,347 bp deletion within AMBN (c.294+139_531+478del; p.Tyr99_Glu177del) inclusive of Exon 6 (Poulter et al., 2014). The second was a splice‐junction mutation at the end of Intron 6 (c.532‐1G>C; p.Leu178_Ser190del) that was predicted to delete Exon 7 (Prasad et al., 2016). Both of these AMBN defects were associated with a non‐syndromic hypoplastic AI (thin enamel) phenotype. Recently an AMBN single allele mutation (NM_016519.5: c.1069C>T; p.Pro357Ser) was reported to cause a dominant condition with both enamel and dentin defects (Lu et al., 2018), but the clinical phenotype exactly matched that of dentinogenesis imperfecta, an autosomal dominant condition caused by mutations in DSPP, a gene linked to AMBN on chromosome 4 that contains a large repetitive region that cannot be characterized using whole exome sequence analyses (Yang et al., 2016).
1.4. Ambn mutant mice
Generating a complete knockout of genes belonging to the secretory calcium‐binding phosphoprotein (SCPP) family (which includes enamel‐specific AMBN, AMELX, and ENAM) is complicated because of the ubiquitous phase‐0 introns. Because of them, alternative splicing always maintains the reading frame and potentially generates internally truncated proteins (Kawasaki & Weiss, 2003). Previously the Amelx−/− mice were generated by deleting only Exon 2, inclusive of the translation initiation codon (Gibson et al., 2001). However, reverse transcription polymerase chain reaction (RT‐PCR) helped detect Amelx mRNA transcripts lacking only the 69 nucleotides encoded by Exon 2 (including the translation initiation codon) in the Amelx− / − mice. Dental enamel in the Amelx− / − mice was dramatically malformed, and no AMEL protein was detected on Western blots using multiple antibodies, so clearly no AMEL was being translated from knockout transcripts lacking Exon 2 (Gibson et al., 2001). An Ambn “knockout” mouse was generated by deleting Exons 5 and 6 and resulted in significant enamel malformations (Fukumoto et al., 2004). Ambn transcripts from this mouse were not detected using RT‐PCR, and ameloblastin protein was not detected using immunohistochemistry (IHC; Fukumoto et al., 2004). Ameloblasts in this knockout mouse differentiated but soon detached from the matrix surface, lost cell polarity, resumed proliferation to form multiple cell layers, and deposited a thin, hypocalcified, unstructured matrix above the dentin surface (Fukumoto et al., 2004). Amelx mRNA was reported to be downregulated, and about 25% of the homozygous mice developed soft tissue tumors in the buccal vestibules of the oral cavity starting at 26 weeks (Fukumoto et al., 2004; Fukumoto, Yamada, Nonaka, & Yamada, 2005). However, a subsequent study of this mouse discovered that a shorter Ambn mRNA that only lacked exons 5 and 6 was expressed (Wazen, Moffatt, Zalzal, Yamada, & Nanci, 2009). The mutant transcript maintained the Ambn reading frame and replaced the two wild‐type 381 and 396 amino acid secreted AMBN isoforms with a single secreted 279 amino acid internally truncated AMBN protein lacking the Tyr41‐Glu157 segment encoded by Exons 5 and 6. The aberrant AMBN protein was detected on Western blot and using immunohistochemistry using antibodies raised against amino acids encoded by Exons 8 to 11 (Smith et al., 2011; Wazen et al., 2009). They redesignated this knockin mouse Ambn Δ5,6/Δ5,6. As the mutant protein might be partially functional or toxic, important additional information about AMBN function could be gained by characterization of a true Ambn null mouse.
To gain a better understanding of Ambn tissue‐specific expression and to improve our understanding of amelogenesis and the role played by AMBN in it, we have generated and extensively characterized a new Ambn knockout/NLS‐lacZ knockin mouse, which we alternately refer to as Ambn lacZ/lacZ or Ambn −/−. We also report a novel AMBN mutation that causes AI in humans.
2. MATERIALS AND METHODS
2.1. Enrollment of human subjects
This study was reviewed and approved by the Institutional Review Boards at the University of Texas Health Science Center at San Antonio and the University of Michigan. Subject enrollment, clinical examinations, and collection of saliva samples were performed with the understanding and written consent of each participant in compliance with the Declaration of Helsinki, described previously (Chan et al., 2011; Kim et al., 2006).
2.2. Whole exome sequencing and bioinformatics analyses
A Hispanic family with hypoplastic enamel trait was recruited. Mother, proband, proband's half‐sister, her husband, and their daughter participated in the study and provided samples. Subject sample preparation, whole exome sequencing, variant annotation, and mutational analyses were conducted based upon established protocols (Zhang et al., 2019). The ABMN mutations were identified by screening of 42 known AI candidate genes (Table S1).
2.3. Generation of Ambn lacZ/lacZ mice
The Ambn knockin construct was designed, fabricated, and used to establish germline Ambn knockout/NLS‐lacZ knockin mice (Ambn lacZ) by Ozgene (Bentley DC, Western Australia, Australia). The Ambn lacZ mice were crossed with C57BL/6 mice for at least eight generations. All mice were maintained on a soft diet (DietGel, ClearH2O), as described previously (Simmer, Hu, Lertlam, Yamakoshi, & Hu, 2009). These mice are available at the Mutant Mouse Resource and Research Center (MMRRC) repository (MMRRC:037503‐UCD).
2.4. RT‐PCR analyses Ambn + and Ambn lacZ expression products
RNA was extracted from enamel organ epithelia (EOE) dissected from maxillary and mandibular first molars of D5 Ambn +/+, Ambn +/lacZ, and Ambn lacZ/lacZ mice. Reverse transcription was performed using the Invitrogen SuperScript III First‐Strand Synthesis System, and polymerase chain reaction (PCR) was conducted using Invitrogen Platinum Hot Start PCR Master Mix (2X), according to the manufacturer's instructions (Invitrogen). The primer sequences are shown in Table S2. The band around 700 bp in Ambn lacZ/lacZ mice amplified by primers in Exons 1 and 11 was isolated from the agarose gel and characterized using DNA sequencing.
2.5. Whole‐mount X‐gal staining
D2 and D6 Ambn +/+, Ambn +/lacZ, and Ambn lacZ/lacZ mice were anesthetized with isoflurane and fixed by cardiac perfusion. Blood was cleared from the vasculature using lactated Ringer's solution (30–45 s) followed by fixation with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS; 135 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM Na2H2PO4; pH 7.3) for 20 min. Following perfusion, the liver, kidneys, submandibular salivary glands, bladder, prostate, ovaries, oviducts, uterus, testis, seminal vesicle, and epididymis were dissected, immersed in the same fixative for 2–3 hr at 4°C, followed by several washes in PBS. Samples were incubated at 45°C for 5 hr in freshly prepared X‐gal solution (0.1 M HEPES, 1 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2% Triton X‐100, 1 mg/ml X‐gal substrate; pH 8.0). After staining, the samples were washed in PBS and photographed using a dissection microscope.
2.6. X‐gal staining of histology sections
Ambn +/+, Ambn +/lacZ, and Ambn lacZ/lacZ mouse heads at D2, D6, D14, and 7 weeks were quickly dissected of skin, cut in half, and fixed by immersion in 4% PFA in PBS overnight at 4°C. The following day, the tissues were washed several times with PBS and decalcified at 4°C in 4.13% disodium ethylenediaminetetraacetic acid (EDTA), pH 7.3. The heads of D2 mice were decalcified for 5 days, D6 mice for 3 weeks, D14 mice for 5 weeks, and 7‐week mice for 7 weeks. Decalcified tissues were immersed in 15% sucrose (1–2 hr) followed by 30% sucrose (3–4 hr) at 4°C for cryoprotection and then embedded in optimal cutting temperature (OCT) compound and stored at −80°C. The blocks were cryosectioned at 8 µm thickness at −20 to −22°C on a Leica cryostat microtome. Slides were stored at −80°C until staining. For X‐gal staining of soft tissues, 7‐week‐old Ambn +/+ and Ambn lacZ/lacZ mice were anesthetized with isoflurane and fixed by cardiac perfusion. Blood was cleared from the vasculature using lactated ringer's solution (30–45 s) followed by 4% PFA in PBS for 20 min. Following perfusion, the liver, kidneys, submandibular salivary glands, bladder, prostate, ovaries, oviducts, uterus, testis, seminal vesicle, and epididymis were dissected, immersed in the same fixative for 2–3 hr at 4°C, followed by several washes in PBS. The tissues were then immersed in 15% sucrose (1–2 hr) followed by 30% sucrose (3–4 hr) at 4°C for cryoprotection, embedded in OCT media and stored at −80°C. The blocks were cryosectioned at 8 µm at −20 to −22°C on a Leica cryostat. Slides were stored at −80°C until staining.
Slides were removed from −80°C and immediately treated with glutaraldehyde fixative [0.1 M 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), 1.25 mM ethylene glycol tetraacetic acid (EGTA), 2 mM MgCl2, 0.5% glutaraldehyde, pH 7.3] and then washed with 0.1 M HEPES, 2 mM MgCl2 three times for 5 min. The slides were stained with X‐gal solution (0.1 M HEPES, 1 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2% Triton X‐100, 1 mg/ml X‐gal substrate; pH 8.0) for 1 hr at 45°C and then washed several times in PBS, and counterstained with 0.1% (w/v) Nuclear Fast Red, coverslipped with Aqua‐Mount (Thermo Scientific) and imaged using a Nikon Eclipse TE300 inverted microscope. A 1 hr staining time was used because a 5 hr staining period caused significant overstaining in developing teeth and bleeding of the stain beyond the nuclei and into adjacent tissue.
2.7. Immunohistochemistry
Slides were baked at 60°C for 1 hr and rehydrated in a gradient ethanol series. Antigen retrieval was performed using the Heat Mediated Antigen Retrieval Solution pH 6.0 (ab973; abcam) following the manufacturer's instructions. Sections were blocked and incubated in primary antibody. The primary antibodies were both polyclonal, raised in rabbits, and used at 1:1000 dilution. The anti‐AMBN antibody was raised against peptide CMRPREHETQQYEYS, which is a remake of the AMBN‐63 antibody (Iwata et al., 2007); the anti‐AMEL antibody was raised against recombinant mouse amelogenin rM179 (Simmer et al., 1994). Sections were incubated in Goat anti‐rabbit IgG(H+L) Secondary Antibody, Alexa Fluor Plus 555 (Invitrogen, A32732) at a concentration of 1:1000, then mounted in Invitrogen™ ProLong™ Gold Antifade Mountant with DAPI (P36935; Invitrogen). Pictures were taken using a Nikon SP8 confocal microscope system (Biomedical Imaging Core, University of Michigan).
2.8. Dissecting microscopy
Ambn +/+, Ambn +/lacZ, and Ambn lacZ/lacZ mice at 7 weeks were anesthetized with isoflurane for facial photos, sacrificed, and perfused with PBS for 10 min. Their mandibles were denuded of soft tissues, postfixed by immersion in 4% PFA overnight, and rinsed with PBS three times, for 5 min each. The teeth were cleaned with 1% bleach (sodium hypochlorite), rinsed with PBS, air dried, displayed on the Nikon SMZ1000 dissection microscope, and photographed using a Nikon DXM1200 digital camera as described previously (Kim et al., 2019).
2.9. Backscattered scanning electron microscopy
The backscattered scanning electron microscopy procedures were described previously (Wang et al., 2015). Briefly, for molar enamel surface evaluation, D14 mandibles were fixed in 4% PFA overnight. The hemi‐mandibles were carefully dissected and submerged in 1% NaClO for 5 min, rinsed, and acetone dehydrated. The hemi‐mandibles were coated with conductive carbon. For incisor enamel surface evaluation, the 7‐week‐old mandibular incisors were carefully freed from the surrounding bony caps and soft tissue. For cross‐section evaluation, 7‐week hemi‐mandibles were embedded in Epon, cross‐sectioned at 1 mm increments, and imaged using a Hitachi S‐3000N microscope.
2.10. Focused ion beam scanning electron microscopy
The sample preparation and Focused ion beam scanning electron microscopy (FIB‐SEM) procedures were described previously (Smith, Hu, Hu, & Simmer, 2016).
3. RESULTS
3.1. Novel AMBN mutations causing AI
We recruited a three‐generation Hispanic family with recessive AI characterized by extremely thin or nonexistent enamel affecting both the primary and secondary dentitions, with no radiopaque layer covering dentin dental radiographs (Figure 1). The enamel defects segregated with the presence of compound heterozygous missense mutations in AMBN (NG_042078.1: g.19190T>C; NM_016519.5: c.1061T>C; p.Leu354Pro and NG_042078.1: g.19469C>T; NM_016519.5: c.1340C>T; p.Pro447Leu). Leu354 is a highly conserved amino acid (Delsuc et al., 2015). The p.Leu354Pro substitution gave a Sorting Intolerant from Tolerant (SIFT; Kumar, Henikoff, & Ng, 2009) score of 0.001 and a Polymorphism Phenotyping v2 (Polyphen2; HDIV; Adzhubei et al., 2010) score of 1. This mutation is rare. Its frequency is 0.042 in the ExAC (Lek et al., 2016) and 0.0176 in the 1000 genomes databases. The mother (II:2) and half‐sister (III:2) of the proband (III:1) were heterozygous for the p.Leu354Pro substitution. Very minor enamel pitting was observed in the two subjects heterozygous for the c.1061T>C/ p.Leu354Pro defect (Figure S1). The second AMBN mutation (p.Pro447Leu) changes the codon of the C‐terminal amino acid and is invariant in all 53 mammalian species expressing a functional ameloblastin protein (Delsuc et al., 2015). The AMBN p.Pro447Leu substitution gave a SIFT score of 0.000 and a Polyphen2 score of 1, supporting a deleterious effect on the protein. This mutation is rare and not listed in the ExAC (0.3.1) or 1000 genomes databases. The p.Pro447Leu variant was apparently inherited from the unrecruited father and was not observed in the other four recruit family members besides the proband. As no simple heterozygote carrying this c.1340C>T/ p.Pro447Leu mutation was recruited, the potential for it causing a heterozygous phenotype could not be assessed. Whole exome sequencing did not help identify any other potentially disease‐causing mutation among our current list of 42 AI candidate genes (Table S1), nor was any other potential disease‐causing candidate gene identified using whole exome analyses. The AMBN genomic reference sequence with the locations of known AI‐causing AMBN mutations is provided in Figure S2.
Figure 1.
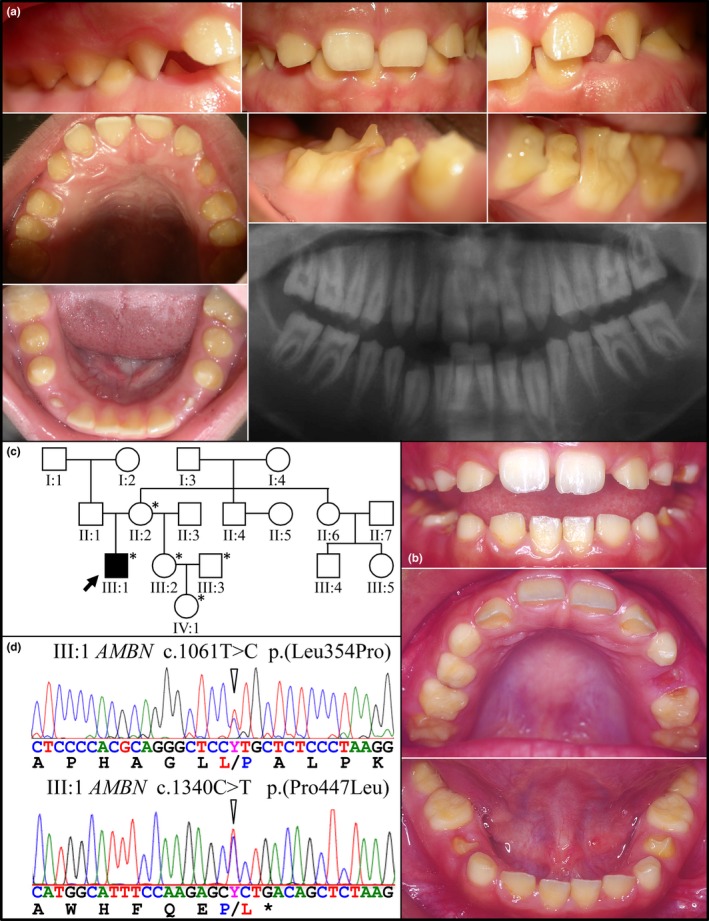
The AI family with compound heterozygous AMBN mutations. (a) Intraoral photos of the proband (III:1) at age 12 revealing dental attrition and extreme enamel hypoplasia in the secondary (permanent) dentition. No enamel is detected on the panoramic radiograph. (b) Intraoral images of proband at age 10 showing enamel hypoplasia and attrition in the primary dentition during the mixed dentition stage. (c) Pedigree of the AMBN AI family with five individuals recruited (asterisks). Only the proband was affected, which is consistent with a recessive pattern of inheritance. (d) Chromatography showing the AMBN compound heterozygous mutations: NG_042078.1:g.19190T>C; NM_016519.5:c.1061T>C; p.(Leu354Pro) and NG_042078.1:g.19469C>T; NM_016519.5:c.1340C>T; p.(Pro447Leu)
3.2. Generating Ambn lacZ/lacZ knockin mice
An Ambn knockout/NLS‐lacZ knockin mouse was generated using a knockin construct (Figure 2a) to delete 4,204 bp segment from the 5' end of Ambn in the C57BL/6J mouse background. The knockin insert started at the evolutionarily invariant strong translation initiation codon in Exon 2 and ended in Intron 4 (Figure S3). The deleted Ambn segment was replaced with a 3,419 bp knockin sequence so that the ATG translation start site of NLS‐lacZ precisely replaced the ATG of the strong Ambn translation start site in Exon 2 (Figure S4), which, like Ambn in the wild‐type mouse, was also positioned in‐frame with the weak translation start site in Exon 1. To minimize transcription of the remaining downstream Ambn exons, two transcription termination signals were included in the 3' untranslated region following the NLS‐lacZ translation termination codon (TAA). Whole‐mount X‐gal staining of D2 (Figure S5) and D6 (Figure S6) mice showed strong X‐gal staining (for ß‐galactosidase activity) restricted to the incisor/molar regions with only trace staining in these areas in the wild‐type mice. X‐gal staining of D2, D6, and D14 mouse head sagittal sections showed strong ß‐galactosidase activity specific for ameloblasts in molars (Figures S7–S9) and incisors (Figures S10, S11), with spotty ß‐galactosidase activity along the roots (Figures S8, S9) in Ambn heterozygous (Ambn +/lacZ) and Ambn lacZ/lacZ mice. X‐gal staining demonstrated that NLS‐lacZ knockin was strongly expressed from the Ambn promoter in Ambn +/lacZ and Ambn lacZ/lacZ mice, but was not expressed in wild‐type mice. These studies provide strong evidence that NLS‐lacZ knockin is expressed from the Ambn promoter, and nuclear X‐gal staining provides an accurate and sensitive signal reporting its tissue‐specific expression.
Figure 2.
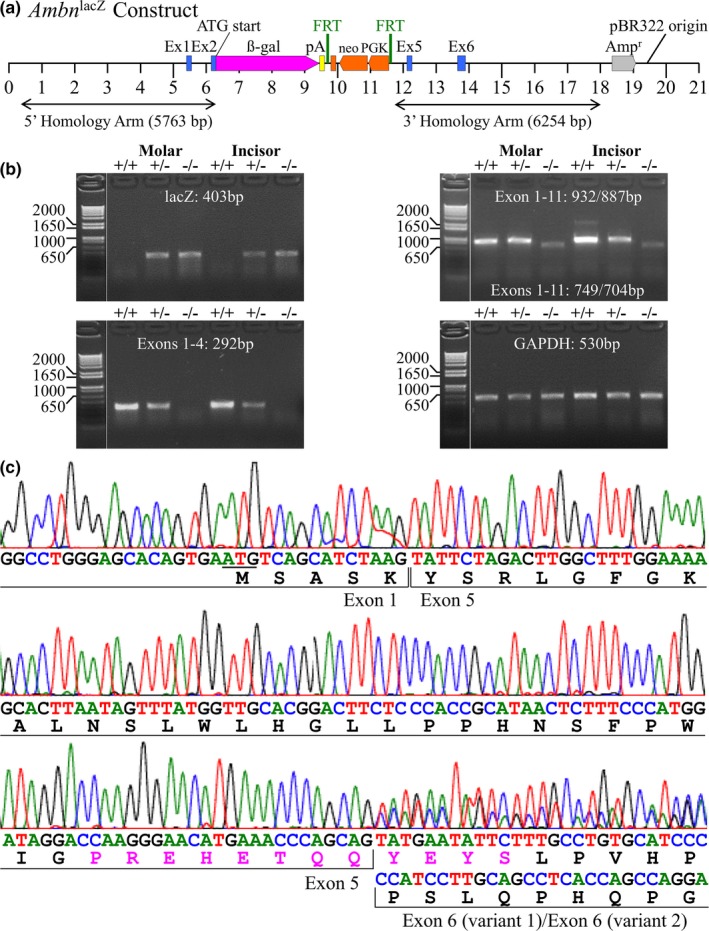
Ameloblastin knockout/NLS‐lacZ knockin mouse (Ambn lacZ/lacZ) generation and RT‐PCR verification. (a) Diagram of the Ambn lacZ construct used to delete 4,204 bp from the 5' end of the mouse C57BL/6J Ambn starting with the translation initiation codon in Exon 2 and ending in Intron 4. The deleted segment encoded 56 amino acids including the signal peptide, signal peptide cleavage site (after Ala26) and key phosphorylated residues, Ser20 and Ser22. This 4,204 bp segment was replaced with NLS‐lacZ encoding ß‐galactosidase with a nuclear localization signal (NLS) followed by two downstream polyadenylation signals (pA) and an FRT‐PGK‐neo‐FRT selection cassette [that expressed a neomycin (neo) resistance gene driven by the mouse phosphoglucokinase (PGK) gene promoter]. After the selection cassette was excised by Flp recombinase, the size of the insert was 3,419 bp. The sequences of Ambn+/+ and Ambn lacZ are provided in Figures S3, S4 respectively. (b) Ethidium bromide stained agarose gels showing RT‐PCR amplified mRNA extracted from D5 mouse first molar enamel organ epithelium. (c) Chromatography showing partial sequence of Ambn lacZ/lacZ mouse cDNA amplified by primers in Exon 1 and 11. Amino acids colored in magenta indicate epitope used to generate AMBN antibody
3.3. Verifying the Ambn knockout
To assess the possibility of AMBN protein expression from our Ambn lacZ knockin allele, we performed RT‐PCR analyses using mRNA isolated from the enamel organ epithelia (EOE) of developing molars extracted from Ambn +/+, Ambn +/lacZ, and Ambn lacZ/lacZ mice. Amplification using primers that annealed to Ambn Exon 1 and lacZ in the modified Exon 2 generated the expected 403 bp RT‐PCR product in both the Ambn lacZ/lacZ and Ambn+ /lacZ mice, demonstrating that NLS‐lacZ knockin mRNA was correctly expressed (Figure 2b). RT‐PCR amplification of mRNA isolated from D5 first molar enamel organ epithelia (EOE) using primers that annealed to Ambn Exons 1 and 4 produced no product in the Ambn lacZ/lacZ mouse (which lacks Ambn Exons 4 annealing site). These primers produced the expected 292 bp RT‐PCR product in wild‐type controls, demonstrating that no wild‐type Ambn transcripts were expressed in the Ambn lacZ/lacZ mouse (Figure 2b). RT‐PCR amplification using primers that annealed to Ambn Exons 1 and 11 produced a single band in Ambn +/+ mice that contained the expected 932 and 887 bp RT‐PCR products corresponding to the two alternatively spliced wild‐type Ambn transcripts (Ambn transcript variant 1, NM_001303431.1 and transcript variant 2, NM_009664.2; Figure 2b). A faint amplification band lower on the gel was also detected in the Ambn lacZ/lacZ mouse. This band was characterized using DNA sequencing and contained a mixture of two (749 bp and 704 bp) amplification products corresponding to alternatively spliced Ambn transcripts missing Ambn Exons 2 through 4, and thus lacking the Ambn coding region for the strong translation initiation site, signal peptide, signal peptide cleavage site, and the N‐terminal 40 amino acids from the secreted AMBN protein (Figure 2c). It was apparent that this amplicon was generated by amplification of rare Ambn lacZ transcripts that failed to terminate transcription at the two transcription termination signals at the 3' end of NLS‐lacZ knockin, so transcription sometimes continued all the way to the 3' end of Ambn and generated transcripts that were spliced from Ambn Exon 1 to Exon 5, and then alternatively spliced to include or delete the 5' end of Exon 6 (as occurs in wild‐type transcripts). Inclusion of the 5' end of Exon 6 was favored over its deletion, based upon the relative sizes of the superimposed peaks on the DNA sequencing chromatogram; Figure 2c). SignalP 4.1 predicted that these transcripts, if translated from the weak translation start site in Exon 1, would not have generated a functional signal peptide (Petersen, Brunak, von Heijne, & Nielsen, 2011). Immunohistochemistry of the continuously growing mandibular incisors was performed to determine if any AMBN protein could be detected in Ambn lacZ/lacZ mice.
3.4. AMBN and AMEL immunohistochemistry
AMBN immunohistochemistry was performed on 7‐week‐old Ambn+ / + and Ambn lacZ/lacZ littermates using an affinity‐purified antipeptide antibody recognizing an epitope encoded by Ambn Exons 5 and 6 (Iwata et al., 2007). In Ambn+/+ incisors, AMBN signal was specific for ameloblasts and their associated extracellular matrix and was significantly stronger in the secretory, relative to the maturation stage (Figures 3a‐e). No AMBN immunofluorescence was observed in odontoblasts, pulp, or bone. In the Ambn lacZ/lacZ incisors, there was no AMBN immunofluorescence in ameloblasts or in the enamel matrix (Figure 3f‐k). The only positive signal was cross‐reaction with material in vascular spaces in the thick pathological epithelium covering the enamel space, which was characteristic of the pathology evident in the enamel organ epithelia Ambn lacZ/lacZ mice. As no AMBN could be detected, we conclude that the enamel phenotype in Ambn lacZ/lacZ mice is due to the absence of AMBN protein.
Figure 3.
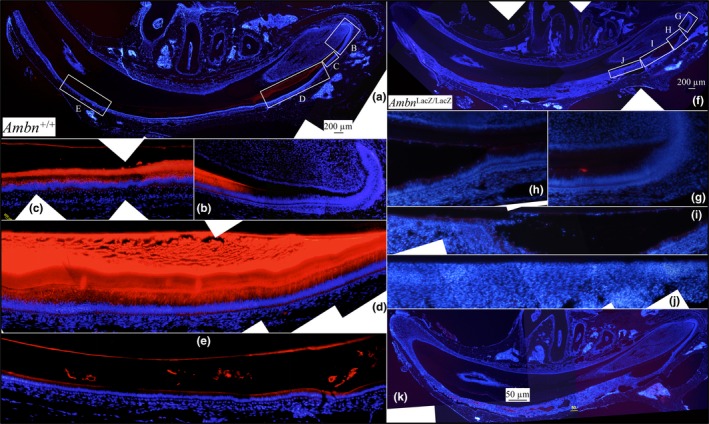
Immunohistochemistry of hemi‐mandible longitudinal sections of 7‐week Ambn +/+ (a‐e) and Ambn lacZ/lacZ (f‐k) incisors using an antibody raised against the AMBN peptide CMRPREHETQQYEYS. Red stain is AMBN; blue is nuclei stained with 4',6‐diamidino‐2‐phenylindole (DAPI). (a) Low magnification montage of WT section. Boxes labeled B‐E show positions of high magnification montages below. (b‐d) Strong AMBN signal is observed in secretory stage ameloblasts and the enamel extracellular matrix. (e) Much weaker AMBN signal is observed in maturation stage ameloblasts in the maturing enamel matrix. (f, k) Low magnification montages of Ambn lacZ/lacZ mouse sections. Boxes in f labeled g‐j indicate positions of the high magnification montages below. (h‐j) AMBN signal is absent in the Ambn lacZ/lacZ mouse mandibular incisor sections. (k) A second longitudinal section from Ambn lacZ/lacZ mouse showed minor autofluorescence within blood vessels in pathological areas. Similar autofluorescence was observed in this region using the amelogenin antibody (Figure 4)
Histologically, the Ambn lacZ/lacZ enamel organ epithelia showed extensive pathology (Figure 3f‐k), which was particularly evident following AMEL immunostaining (Figure 4f‐m). Differentiating ameloblasts and preameloblasts appear to be normal and secrete AMEL onto the dentin surface, some of which penetrated into the dentin layer (Figure 4g). The ameloblasts degenerated and detached from the dentin surface (Figure 4h‐j). Separation appeared to occur within the unmineralized enamel matrix, leaving distinct layers of accumulated AMEL on the dentin surface and also along the detached distal surface of now pathological EOE (Figure 4h‐i). Amelogenin seemed to accumulate in pockets between the degenerating ameloblasts and the stratum intermedium (Figure 4h). A cyst‐like structure formed between the dentin surface and the EOE (Figure 3h‐j), which compressed the epithelial layer as the enamel organ epithelia progressively lost its characteristic tissue organization evident in wild‐type mice (Figures 4b‐e). Amelogenin continued to be secreted, but within the disorganized epithelium, generating numerous AMEL‐rich spherical nodules. The pathology in the epithelial layer covering dentin in the Ambn lacZ/lacZ incisors was similar to that previously described in the Ambn Δ5,6/Δ5,6 mice, with two apparent differences: the Ambn Δ5,6/Δ5,6 mice expressed an internally truncated AMBN protein that accumulated in the soft tissue nodules (Wazen et al., 2009), and amelogenin expression appeared to be suppressed (Fukumoto et al., 2004). In contrast, our Ambn lacZ/lacZ mice were negative for AMBN expression, but continued secreting large amounts of amelogenin (Figure 4f‐m).
Figure 4.
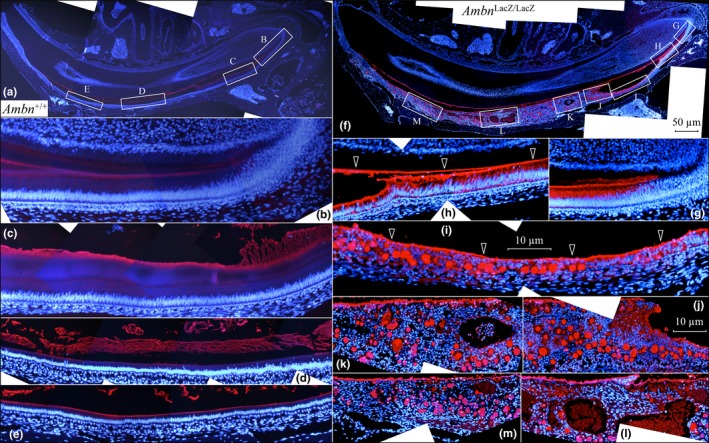
Immunohistochemistry of hemi‐mandible longitudinal sections of 7‐week Ambn +/+ (a‐e) and Ambn lacZ/lacZ (f‐m) incisors using an antibody raised against the recombinant mouse amelogenin rM179 (Simmer et al., 1994). Red stain is AMEL; blue is nuclei stained with 4',6‐diamidino‐2‐phenylindole (DAPI). (a) Low magnification montage of Ambn +/+ section. Boxes labeled B‐E show positions of high magnification montages shown below. (b, c) Amelogenin signal is detected in the enamel matrix during the secretory stage and in the early dentin matrix. (d, e) Amelogenin signal diminishes in the enamel matrix during the maturation stage. (f) Low magnification montages of Ambn lacZ/lacZ mouse section. Boxes in f labeled g‐m indicate positions of the high magnification montages below. (g) AMEL stain is strong in ameloblasts and in the matrix along their distal membrane. Some amelogenin has penetrated into dentin. (h‐j) Amelogenin accumulates along the dentin surface and stratum intermedium (arrowheads). A cyst has formed beneath the ameloblasts and is associated with the onset of severe ameloblast pathology and circular pockets of amelogenin within the enamel organ epithelia. (k‐l) Brown autofluorescence is observed in sinuses containing blood elements
3.5. Enamel surface in Ambn lacZ/lacZ mice
Malformation of the enamel layer was evident by inspection under a dissection microscope of the mandibular incisors and molars of Ambn lacZ homozygous and heterozygous mice (Figure 5) relative to those of the wild‐type (Figure S12). The Ambn lacZ/lacZ maxillary and mandibular incisors at 7 weeks were chalky‐white with rough surfaces and no detectable enamel layer. The Ambn lacZ/lacZ molar crowns also lacked enamel and underwent rapid attrition. Ambn +/lacZ mice displayed a well‐defined enamel layer. Their maxillary incisors appeared normal in color, whereas the mandibular incisors were chalky‐white and sometimes chipped. The molars look essentially normal, but their surface texture was slightly rougher than that of wild‐type molars. Radiographs revealed no abnormalities in root number, size, or shape in the Ambn lacZ/lacZ mice.
Figure 5.
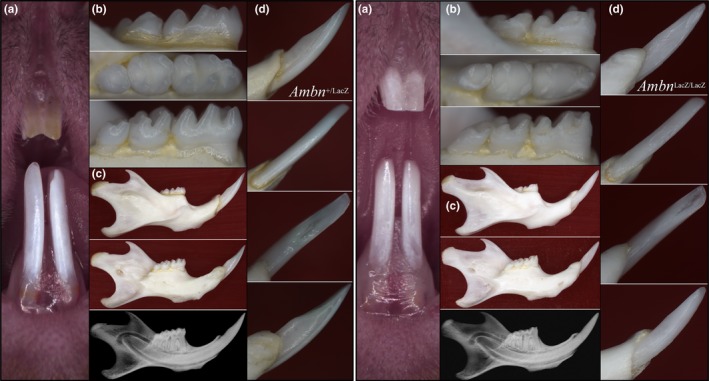
Dissecting microscope photographs of 7‐week‐old Ambn +/lacZ, and Ambn lacZ/lacZ mice. (a) Frontal view of maxillary and mandibular incisors. (b) Buccal (upper), occlusal (middle), and lingual (lower) views of mandibular molars. As shown in the radiographs, no appreciable abnormality in root number, size, and shape was observed in the Ambn +/lacZ and Ambn lacZ/lacZ mice. (c) Lateral (top), medial (middle) views, and radiograph (bottom) of hemi‐mandibles. (d) Lingual (top), labial, lingual, and buccal (bottom) views of mandibular incisors. Comparable images from a 7‐week‐old Ambn +/+ mouse are shown in Figure S12
To get a closer look at fully formed enamel surfaces prior to their alteration in the oral cavity, nearly erupted D14 mandibular first molars were characterized using backscattered scanning electron microscopy (bSEM) following removal of the thin overlying soft tissue (Figure 6 and Figure S13). The Ambn +/lacZ mandibular first molar crowns were indistinguishable from wild‐type crowns except for a slightly rougher surface texture. The Ambn lacZ/lacZ mandibular first molars, in contrast, had thin cusps, and their crowns were covered by irregular masses of crusty mineral. The enamel surfaces of carefully extracted 7‐week mandibular incisors were inspected using bSEM (Figure 7). The wild‐type (Figure 7a) and the Ambn +/lacZ mandibular incisors (Figure 7b) looked similar with respect to surface roughness and degree of mineralization (grayscale). The Ambn lacZ/lacZ incisors were hypomineralized (dark), rough‐surfaced (Figure 7c), and still retained the fragile crusty mineral nodules on their unerupted, unabraded surfaces.
Figure 6.
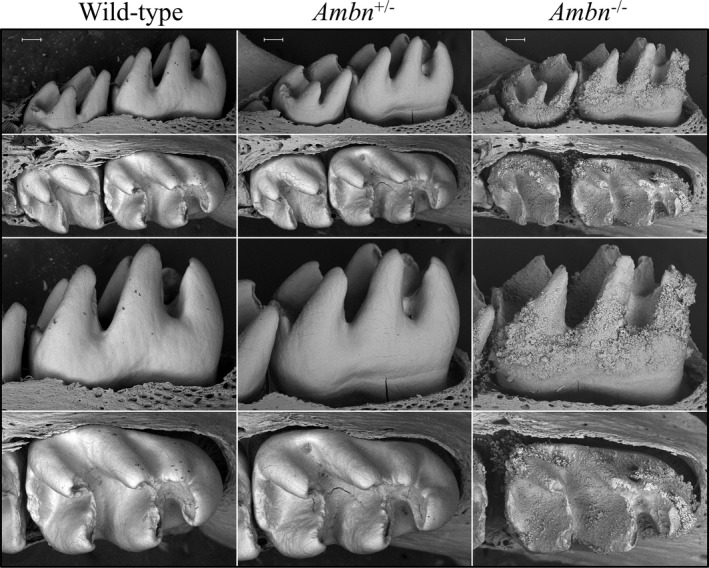
Backscattered SEMs of D14 mandibular molars of Ambn +/+ (Wild‐type), Ambn +/lacZ (Ambn ±) heterozygous, and Ambn lacZ/lacZ (Ambn −/−) null mice following soft tissue removal. These images show the molars immediately prior to their eruption into occlusion. The top and third rows are lingual views; the second and bottom rows are occlusal views. The null molars show thin cusps and surface accumulation of an irregular crusty material. Mild surface roughness of the heterozygous molar relative to the wild‐type is most evident from the occlusal views. Another set of molars is shown in Figure S13. Scale bars = 500 µm
Figure 7.
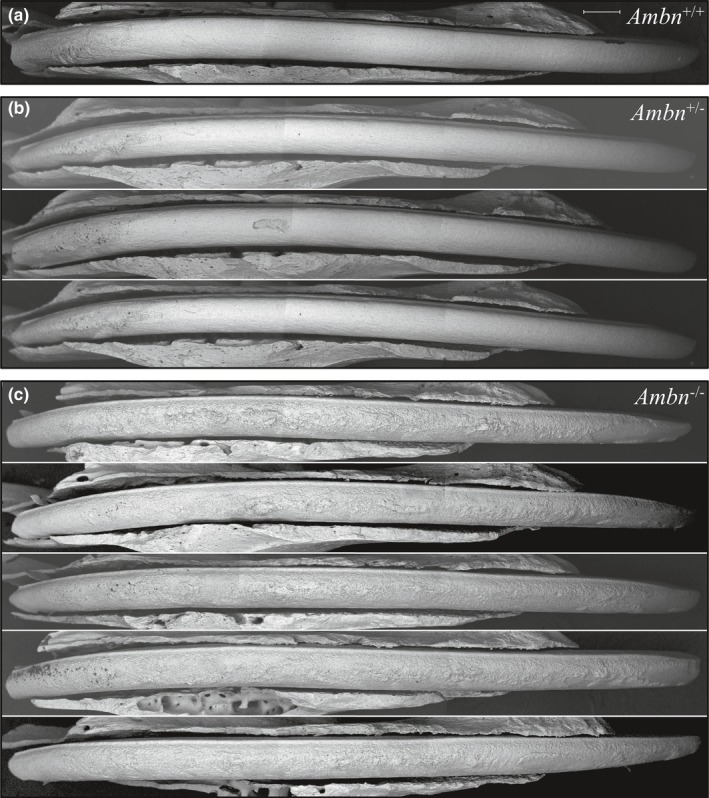
bSEM montages of 7‐week mandibular incisors denuded of overlying tissues. The labial surfaces are shown, from the basal (left) to erupted (right) ends of the incisors. Dark areas contain less surface mineral than white areas. The surface of secretory stage enamel (left ~1/4 of the incisor length) appears rough in all genotypes. The maturation stage enamel surface of the Ambn +/+ incisor (a) was similar to those from the Ambn +/lacZ mice (b) whereas the Ambn lacZ/lacZ homozygous incisors (c) exhibited very rough, poorly mineralized (dark), crusty surfaces with protruding nodules. Scale bar = 500 µm
3.6. bSEM of mandibular incisor cross‐sections
To gain information about the formation of the crusty mineral surface, Ambn lacZ/lacZ incisors were cross‐sectioned at 1 mm increments and imaged using bSEM (Figure 8). Dentin formation was comparable in the wild‐type and Ambn lacZ/lacZ mice, while the entire enamel layer was pathological in the Ambn lacZ/lacZ incisors. During what corresponded to the secretory stage in wild‐type incisors (levels 1–3 located 1 to 3 mm from the apical loop), an irregular mineral crust formed on the surface of dentin that was already evident at level 2. The level 2 crust was less mineralized than dentin and contained unmineralized voids within it, evidently caused by the trapping of soft tissue (epithelia). The crust was continuous along the dentin surface but varied significantly in thickness throughout, averaging about 15 µm. By level 3 (nearing the end of what would be the secretory stage in wild‐type mice), the crust on the dentin surface had expanded to about 20 µm in thickness, while a large number of spherical mineral foci, often fused together, appeared within the epithelial layer at various distances from the irregular crusty surface. Some of these mineral foci showed concentric rings in cross‐section and looked like concretions. The concretions were more abundant laterally and appeared to expand in size incisally, being larger at levels 6 through 8 when compared to the nodules in levels 3 through 5. These detached nodules correlated with the presence of amelogenin‐filled pockets within the EOE that characterized the histological sections of Ambn lacZ/lacZ mice immunostained for amelogenin (Figure 4). In wild‐type incisors, the enamel was constant in thickness from levels 4 through 8 (being about 114 µm thick at the height of contour) as ions excreted by maturation stage ameloblasts do not continue to add mineral ions to the tips of the enamel crystals (which would progressively thicken the enamel layer as a whole), but instead the ions penetrate into the enamel matrix and adsorb exclusively onto the sides of the enamel crystals (Smith, 1998).
Figure 8.
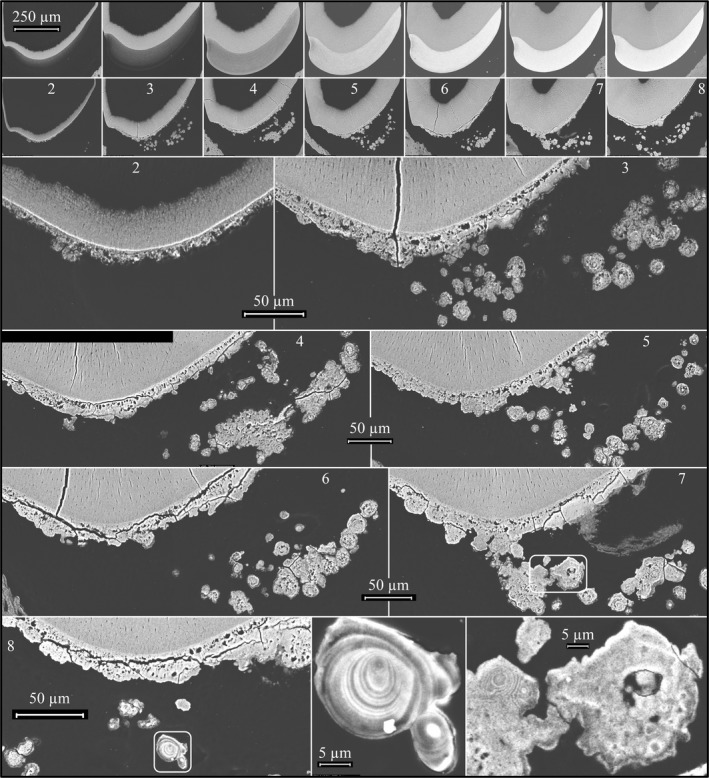
bSEM images of a wild‐type and an Ambn lacZ/lacZ homozygous 7‐week mandibular incisor cross‐sections at 1 mm increments. Numbers in panels indicate distance in mm from apical loop. The higher the number the more advanced is the dentin and enamel development. Top: Lower magnifications views of wild‐type above the corresponding Ambn lacZ/lacZ homozygous mouse incremental sections. Note that dentin formation is comparable in the wild‐type and Ambn null mice, but that enamel formation is not. Below: Higher magnification bSEM images of sections 2 through 8 and details of boxed concretions
3.7. FIB‐SEM
To view events on the surface of dentin at the point on the mandibular incisor where enamel normally initiates as oriented mineral ribbons (Smith et al., 2016), an Ambn lacZ/lacZ incisor was cross‐sectioned 1 mm from the basal loop and characterized using FIB‐SEM, with overlapping images taken at 5000x, 10000x, 20000x, and 35000x (Figure S14–S23). The series spanned 155 µm of developing tooth along the ameloblast distal border. The earliest developmental stage on the section was the most lateral and shows the distal part of the ameloblasts intimately associated with pre‐dentin matrix following the breakdown of the basement membrane and onset of amelogenin secretion but prior to the onset of dentin mineralization (Figure S14). The series progressed to show the onset of dentin mineralization within collagen fibers oriented parallel to the long axis of the ameloblast and terminating against its distal membrane (Figure S15). Mineralization of the collagen‐rich pre‐dentin matrix expanded toward the ameloblast membrane and also in the opposite direction toward the odontoblasts, while coalescing into a continuous mineral mass of steadily increasing density. At this early stage pathology was evident in the form of droplets of enamel matrix, presumed to be comprised of amelogenin, that accumulated along the ameloblast membrane where the enamel ribbons normally initiate (Figures S16, S17). The dentin mineral increased in density and thickness, but no enamel ribbons formed (Figure S18). As the process of dentin mineralization continued normally, no enamel ribbons formed in the Ambn lacZ/lacZ incisor. Instead, enamel proteins (particularly amelogenin) accumulated between the mineralized dentin surface and the nearby ameloblast distal membrane (Figures S18–S23).
To test our interpretation that the pathological accumulations in the Ambn lacZ/lacZ were amelogenin, we crossed the Ambn lacZ/lacZ and Amelx ‐/‐ mice to generate Ambn −/− Amelx −/− double null mice. We extensively characterized the mandibular incisors in the double null in the region where enamel ribbons should have formed (Figures [Link], [Link]), starting prior to dentin coalescing into a continuous mineral layer (Figure S24), and continuing long past the normal onset of enamel formation and on to a point where overt cell pathology was evident and crusty mineral started to form on dentin (Figures S38–S40, S42). The accumulations of enamel protein observed between ameloblasts and the underlying dentin surface in Ambn lacZ/lacZ mice were absent in the Ambn −/− Amelx −/− mice, demonstrating that the material accumulating on the dentin surface was largely amelogenin, which was supported by the finding of strong amelogenin immunostaining on the dentin surface using light microscopy (Figure 4).
The series of high magnification FIB‐SEM images detailing the early dentin and enamel formation in the Ambn lacZ/lacZ and Ambn −/− Amelx −/− double null mice strongly support the interpretation that dentin forms normally in the absence of ameloblastin, but the mineralization front along the ameloblast distal membrane was defective and no enamel ribbons formed. Instead, a very thin (~1 µm), sparse layer of curled mineral was deposited on dentin (Figure 9) well after the normal onset of enamel mineralization, when dentin mineralization was well underway and ameloblasts had started to deteriorate. The curled ribbons were not oriented from the mineralized dentin to the ameloblast distal membrane and did not elongate (Figure 9). For comparison, a full series of wild‐type FIB‐SEM images covering the onset of dentin mineralization through formation of the initial enamel are provided (Figures S45–S66). Enam ‐/‐ mice also fail to form enamel ribbons (lack enamel), but do not accumulate deposits of amelogenin between the ameloblasts and dentin (Smith et al., 2016). Amelx ‐/‐ null mice initiate enamel ribbon formation almost normally, but the process degenerates rapidly and the ribbons convert into octacalcium phosphate and branch into fan‐like structures, forming plates (Hu et al., 2016).
Figure 9.
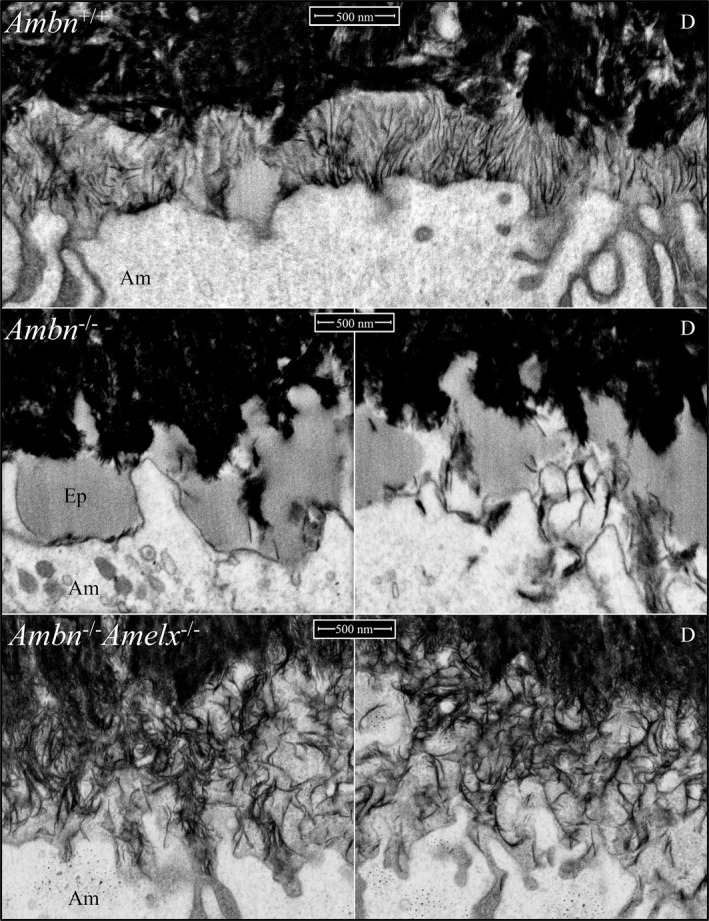
FIB‐SEM images of corresponding dentin surfaces in wild‐type (Ambn +/+), Ambn null (Ambn lacZ/lacZ), and Ambn‐Amelx double null (Ambn lacZ / lacZ Amelx −/−) mice. Top: Formation of a characteristic field of initial enamel ribbons extending from dentin mineral to the ameloblast membrane. Middle: Accumulation of enamel protein, mostly amelogenin, on the dentin surface and absence of a field of enamel ribbons in Ambn lacZ/lacZ mice. Bottom: Ambn‐Amelx double null mice show an absence of amelogenin accumulation and no field of enamel deposition. Key: Am, ameloblast; D, dentin; Ep, enamel protein
3.8. NLS‐lacZ expression in non‐dental tissues
Because NLS‐lacZ was inserted at the Ambn Exon 2 translation initiation codon (TIC) while deleting the Ambn coding region from the TIC through Exon 4, the Ambn lacZ knockin mice allowed us to assess tooth formation in the absence of Ambn expression and also detect ß‐galactosidase reporter expression from the Ambn promoter using the sensitive X‐gal staining assay. X‐gal staining of tissues containing developing teeth helped confirm that the Ambn lacZ/lacZ mouse was established properly. Strong X‐gal staining of ameloblasts was observed with 1 hr incubations (Figures S6–S12). Trace expression was detected in the molar junctional epithelium at D28, but only after incubation overnight (15.5 hr; Figure S67). We sought to detect even trace expression in non‐dental tissues by using 15.5 hr (overnight) X‐gal incubations. To assess cranial tissues and calvaria, we cross‐sectioned D6 Ambn lacZ/lacZ mouse heads at five different levels (Figure 10). X‐gal staining was intense in the ameloblasts of developing teeth and strong in the nasal epithelium, but no staining was detected in the calvarium or the frontal suture. The calvarium and sutures were also negative in D2, D6, and D14 whole‐mounts (Figure 10c). Soft tissue organs were also tested for X‐gal staining. Duct cells in the tail of the epididymis were positive after 1 hr of X‐gal staining (Figure S68). All other organs were negative even after 15.5 hr of X‐gal staining, including the ovary and oviduct, uterus, prostate, seminal vesicle, and testis (Figure S69), as well as the submandibular salivary gland, kidney, liver, and bladder (Figure S70).
Figure 10.
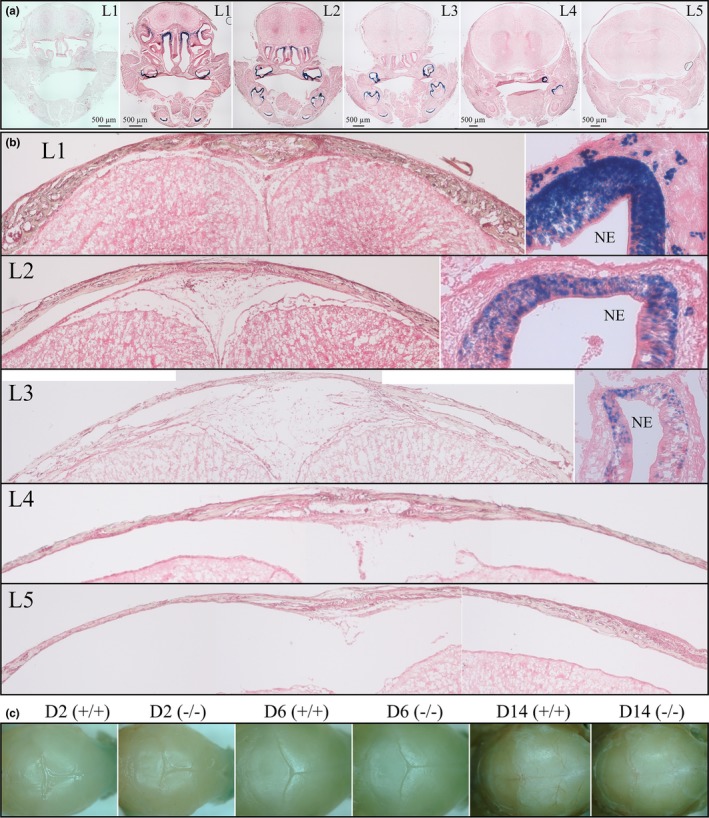
LacZ staining of mouse heads. Slides were stained in X‐gal solution for 1 hr at 45°C and then counterstained with Nuclear Fast Red. (a) Successive coronal plane sections (ventral L1 to L5 dorsal) of mouse heads. (b) Sagittal suture at L1 to L5 and nasal epithelium (NE) at L1 to L3 under higher magnification. (c) Whole‐mount lacZ staining of mouse heads from D2, D6 and D14 wild‐type (Ambn+/+) and null (Ambn lacZ/lacZ) demonstrating no appreciable NLS‐lacZ positive staining. Scale bars = 500 µm
4. DISCUSSION
4.1. AMBN and amelogenesis imperfecta
Despite the early identification of AMBN cleavage products in developing enamel (Fukae & Tanabe, 1987a, 1987b; Shimizu, 1984), the cloning and characterization of Ambn cDNA from multiple organisms, the early recognition of AMBN as a major constituent of developing enamel (Fincham et al., 1999), its association with dramatic enamel defects in Ambn mutant mice (Fukumoto et al., 2004), and linkage of AMBN to a chromosomal region associated with human enamel defects (MacDougall et al., 1997), only two AMBN mutations have been found to cause amelogenesis imperfecta (AI). A homozygous genomic deletion (c.294+139_531+478del; p.Tyr99_Glu177del; Poulter et al., 2014) and a homozygous splice‐junction defect predicted to cause a deletion by exon skipping during RNA splicing (c.532‐1G>C; p.Leu178_Ser190del; Prasad et al., 2016) are the only AMBN mutations shown to cause AI. Both of these mutations caused autosomal recessive AI (ARAI), with no defects reported besides those in enamel. Here we report two AMBN missense mutations (c.1061T>C; p.Leu354Pro/ c.1340C>T; p.Pro447Leu) that caused severely hypoplastic ARAI in compound heterozygotes, with minor pitting in single heterozygotes for the AMBN c.1061T>C; p.Leu354Pro defect. In contrast, over 20 ENAM mutations have been reported to cause AI (Zhang et al., in press).
4.2. The Ambn lacZ/lacZ enamel phenotype and tissue‐specific expression
The most fundamental feature of dental enamel formation is the deposition (on dentin mineral) and elongation of thin mineral ribbons along the secretory surface of the ameloblast distal membrane. This occurs in all vertebrates that make true enamel, and existed 450 MY ago in the most recent common ancestor of humans and gar. Ameloblastin (Ambn) and enamelin (Enam) are expressed during tooth and scale formation in the gar, whereas amelogenin (Amel) is not (Braasch et al., 2016; Kawasaki et al., 2017). Amelogenin apparently evolved shortly thereafter, as it is found in sarcopterygian genomes, such as the coelacanth (Kawasaki & Amemiya, 2014), but not in gar or teleosts (Kawasaki, 2018). No enamel ribbons form in Enam ‐/‐ mice. Enamel ribbons form in Amelx ‐/‐ mice, but the ribbons are short, elongate more slowly than normal (Hu et al., 2008; Smith et al., 2016), and develop into a thin layer of octacalcium phosphate plates (Hu et al., 2016). Here we demonstrate that no field of initial enamel ribbons form in the absence of ameloblastin (Figure 9). Dentin forms normally, but enamel ribbons do not form on its surface. This is true even though amelogenin accumulates on the dentin surface where the enamel ribbons would normally initiate.
A remarkable feature observed in both the Enam ‐/‐ and Ambn lacZ/lacZ mice, in addition to the complete failure to deposit a field of initial enamel ribbons (Figure 9), is the pathology and ectopic mineral evident within the enamel organ epithelia (Hu et al., 2011; Figure 4). Histologically, the Ambn lacZ/lacZ ameloblasts degenerate and detach from the dentin surface following their failure to deposit the initial enamel. Amid this pathology, an irregular mineral crust is deposited on the molar (Figure 6) and incisor (Figures 7 and 8) surfaces, which does not form through the normal process of enamel mineral ribbon deposition (compare WT Figures S58–S65 to Ambn lacZ/lacZ Figures S36–S44). The mineral crust covering dentin in Ambn lacZ/ lacZ mice was readily observed in the bSEM analyses of the D14 first molar (Figure 6) and 7‐week incisor (Figure 7) surfaces, and the 7‐week incisor cross‐sections (Figure 8). The crust underwent rapid attrition after the teeth erupted into occlusion. A unique feature of the failed amelogenesis in the Ambn lacZ/lacZ mice was the accumulation of organic material on the dentin surface where the enamel ribbons failed to form (Figure 9; Figures S16–S23). These accumulations were absent in the Ambn lacZ/lacZ Amelx ‐/‐ double null (Figures S26–S36), indicating that the deposits were comprised principally of amelogenin. While amelogenin deposits are normally associated with the formation and elongation of enamel ribbons along the ameloblast membrane, the accumulations in Ambn lacZ/lacZ mice were larger than those is observed in wild‐type and Enam ‐/‐ mice (Smith et al., 2016). We do not infer that amelogenin expression increases, simply that amelogenin accumulates more than normal on the dentin surface as a feature of the failed amelogenesis in Ambn lacZ/lacZ mice. Without ribbon elongation, the ameloblasts do not retreat and expand the enamel volume, so the amelogenin must accumulate on the mineralized dentin after it is secreted.
The phenotypes of the Ambn lacZ/lacZ mice are similar to those of the Ambn Δ5,6/Δ5,6 mice, including ameloblast detachment, lack of enamel formation, ectopic mineral formation in the epithelium, and normal dentin formation (Fukumoto et al., 2004; Wazen et al., 2009). The Ambn Δ5,6/Δ5,6 mice however, showed reduced Amelx expression and developed odontogenic tumors, which were not observed in Ambn lacZ/lacZ mice. The absence of enamel formation in the Ambn lacZ/lacZ and Ambn Δ5,6/Δ5,6 mice indicates that ABMN function is dependent upon inclusion of the peptide segment encoded by Exons 5 and 6. We observed strong ectopic expression of amelogenin in the EOE of Ambn lacZ/lacZ mice (Figure 4) that correlated with the formation of ectopic mineral in the EOE (Figure 8). The importance of amelogenin in forming this ectopic mineral is supported by formation of ectopic mineral in the Ambn Δ5,6/Δ5,6 mice (Fukumoto et al., 2004), but not in Ambn Δ5,6/Δ5,6 Amelx−/− double mutants (Hatakeyama et al., 2009). An increasing body of evidence suggests that amelogenin does not play a critical role in the shaping of enamel mineral ribbons, including the absence of amelogenin in gar (which forms enamel ribbons; Prostak, Seifert, & Skobe, 1989; Sire, 1995), formation of enamel ribbons in Amelx− / − mice (Smith et al., 2016), enamel mineral adopting its ribbon shape before it is crystalline (and therefore cannot be shaped by amelogenin binding to selected crystal faces; Beniash, Metzler, Lam, & Gilbert, 2009), and ectopic mineral deposited in amelogenin‐laded spaces in Ambn lacZ/lacZ mice that is not comprised of enamel‐like ribbon‐shaped crystals (Figure 8). Amelogenin is a critical matrix constituent, but it can only function properly in the context of a functioning mineralization front apparatus that initiates and shapes the enamel mineral ribbons, which requires both enamelin (ENAM) and ameloblastin (AMBN; Smith et al., 2016).
The NLS‐lacZ reporter expressed nuclear localized ß‐galactosidase activity strongly in ameloblasts, with spotty expression along the developing molar roots. Mildly positive tissues included the tail of the epididymis and nasal epithelium, and trace, spotty signal was detected in the junctional epithelium. Except for enamel deposited by the strongly positive ameloblast layer, no phenotypic differences were observed in any other tissues, including the nasal epithelium, epididymis, and junctional epithelium where reporter signal was detected. We conclude that ameloblastin functions at the mineralization front along the secretory surface of the ameloblast membrane to establish and extend enamel mineral ribbons, a specialized function it has performed for over 450 MY in all vertebrates that produce true enamel, and does not serve any critical function in any other tissues.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
We thank the family for their participation in this study. We thank the Facility for Electron Microscopy Research at McGill University, Montreal, Canada for their technical support. This study was supported by NIDCR/NIH grants DE‐015846 (JH), DE‐025758 (YHC), and DE‐026769 (YHC); by National Taiwan University Hospital (NTUH) (Grant 106‐N3424), the Ministry of Science and Technology in Taiwan (MOST) (Grant 107‐2314‐B‐002‐014); and the National Research Foundation of Korea, funded by the Korean government (NRF‐2017R1A2A2A05069281).
Liang T, Hu Y, Smith CE, et al. AMBN mutations causing hypoplastic amelogenesis imperfecta and Ambn knockout‐NLS‐lacZ knockin mice exhibiting failed amelogenesis and Ambn tissue‐specificity. Mol Genet Genomic Med. 2019;7:e929 10.1002/mgg3.929
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsawasuwan, P. , Lu, X. , Ito, Y. , Chen, Y. , Gopinathan, G. , Evans, C. A. , … Diekwisch, T. (2013). Expression and function of enamel‐related gene products in calvarial development. Journal of Dental Research, 92(7), 622–628. 10.1177/0022034513487906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beniash, E. , Metzler, R. A. , Lam, R. S. , & Gilbert, P. U. (2009). Transient amorphous calcium phosphate in forming enamel. Journal of Structural Biology, 166(2), 133–143. 10.1016/j.jsb.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch, I. , Gehrke, A. R. , Smith, J. J. , Kawasaki, K. , Manousaki, T. , Pasquier, J. , … Postlethwait, J. H. (2016). The spotted gar genome illuminates vertebrate evolution and facilitates human‐teleost comparisons. Nature Genetics, 48(4), 427–437. 10.1038/ng.3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerny, R. , Slaby, I. , Hammarstrom, L. , & Wurtz, T. (1996). A novel gene expressed in rat ameloblasts codes for proteins with cell binding domains. Journal of Bone and Mineral Research, 11(7), 883–891. 10.1002/jbmr.5650110703 [DOI] [PubMed] [Google Scholar]
- Chan, H. C. , Estrella, N. M. , Milkovich, R. N. , Kim, J. W. , Simmer, J. P. , & Hu, J. C. (2011). Target gene analyses of 39 amelogenesis imperfecta kindreds. European Journal of Oral Sciences, 119(Suppl 1), 311–323. 10.1111/j.1600-0722.2011.00857.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, Y. H. , Yamakoshi, Y. , Yamakoshi, F. , Fukae, M. , Hu, J. C. , Bartlett, J. D. , & Simmer, J. P. (2010). Cleavage site specificity of MMP‐20 for secretory‐stage ameloblastin. Journal of Dental Research, 89(8), 785–790. 10.1177/0022034510366903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc, F. , Gasse, B. , & Sire, J. Y. (2015). Evolutionary analysis of selective constraints identifies ameloblastin (AMBN) as a potential candidate for amelogenesis imperfecta. BMC Evolutionary Biology, 15, 148 10.1186/s12862-015-0431-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demere, T. A. , McGowen, M. R. , Berta, A. , & Gatesy, J. (2008). Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Systematic Biology, 57(1), 15–37. 10.1080/10635150701884632 [DOI] [PubMed] [Google Scholar]
- Fincham, A. G. , Moradian‐Oldak, J. , & Simmer, J. P. (1999). The structural biology of the developing dental enamel matrix. Journal of Structural Biology, 126(3), 270–299. 10.1006/jsbi.1999.4130 [DOI] [PubMed] [Google Scholar]
- Fukae, M. , & Tanabe, T. (1987a). 45Ca‐labeled proteins found in porcine developing dental enamel at an early stage of development. Advances in Dental Research, 1(2), 261–266. 10.1177/08959374870010021501 [DOI] [PubMed] [Google Scholar]
- Fukae, M. , & Tanabe, T. (1987b). Nonamelogenin components of porcine enamel in the protein fraction free from the enamel crystals. Calcified Tissue International, 40(5), 286–293. 10.1007/BF02555263 [DOI] [PubMed] [Google Scholar]
- Fukae, M. , Tanabe, T. , Uchida, T. , Yamakoshi, Y. , & Shimizu, M. (1993). Enamelins in the newly formed bovine enamel. Calcified Tissue International, 53(4), 257–261. 10.1007/bf01320911 [DOI] [PubMed] [Google Scholar]
- Fukumoto, S. , Kiba, T. , Hall, B. , Iehara, N. , Nakamura, T. , Longenecker, G. , … Yamada, Y. (2004). Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. Journal of Cell Biology, 167(5), 973–983. 10.1083/jcb.200409077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto, S. , Yamada, A. , Nonaka, K. , & Yamada, Y. (2005). Essential roles of ameloblastin in maintaining ameloblast differentiation and enamel formation. Cells Tissues Organs, 181(3–4), 189–195. 10.1159/000091380 [DOI] [PubMed] [Google Scholar]
- Gibson, C. W. , Yuan, Z.‐A. , Hall, B. , Longenecker, G. , Chen, E. , Thyagarajan, T. , … Kulkarni, A. B. (2001). Amelogenin‐deficient mice display an amelogenesis imperfecta phenotype. Journal of Biological Chemistry, 276(34), 31871–31875. 10.1074/jbc.M104624200 [DOI] [PubMed] [Google Scholar]
- Hatakeyama, J. , Fukumoto, S. , Nakamura, T. , Haruyama, N. , Suzuki, S. , Hatakeyama, Y. , … Kulkarni, A. B. (2009). Synergistic roles of amelogenin and ameloblastin. Journal of Dental Research, 88(4), 318–322. 10.1177/0022034509334749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch, A. G. (2017). Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends in Biochemical Sciences, 42(8), 589–611. 10.1016/j.tibs.2017.03.004 [DOI] [PubMed] [Google Scholar]
- Hinnebusch, A. G. , Ivanov, I. P. , & Sonenberg, N. (2016). Translational control by 5'‐untranslated regions of eukaryotic mRNAs. Science, 352(6292), 1413–1416. 10.1126/science.aad9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, C.‐C. , Fukae, M. , Uchida, T. , Qian, Q. , Zhang, C. H. , Ryu, O. H. , … Simmer, J. P. (1997). Sheathlin: Cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel sheath proteins. Journal of Dental Research, 76(2), 648–657. 10.1177/00220345970760020501 [DOI] [PubMed] [Google Scholar]
- Hu, J. C. , Hu, Y. , Smith, C. E. , McKee, M. D. , Wright, J. T. , Yamakoshi, Y. , … Simmer, J. P. (2008). Enamel defects and ameloblast‐specific expression in Enam knock‐out/lacz knock‐in mice. Journal of Biological Chemistry, 283(16), 10858–10871. 10.1074/jbc.M710565200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. C. , Lertlam, R. , Richardson, A. S. , Smith, C. E. , McKee, M. D. , & Simmer, J. P. (2011). Cell proliferation and apoptosis in enamelin null mice. European Journal of Oral Sciences, 119(Suppl 1), 329–337. 10.1111/j.1600-0722.2011.00860.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Smith, C. E. , Cai, Z. , Donnelly, L. A. , Yang, J. , Hu, J. C. , & Simmer, J. P. (2016). Enamel ribbons, surface nodules, and octacalcium phosphate in C57BL/6 Amelx‐/‐ mice and Amelx+/‐ lyonization. Molecular Genetics and Genomic Medicine, 4(6), 641–661. 10.1002/mgg3.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, T. , Yamakoshi, Y. , Hu, J. C. , Ishikawa, I. , Bartlett, J. D. , Krebsbach, P. H. , & Simmer, J. P. (2007). Processing of ameloblastin by MMP‐20. Journal of Dental Research, 86(2), 153–157. 10.1177/154405910708600209 [DOI] [PubMed] [Google Scholar]
- Jacques, J. , Hotton, D. , Asselin, A. , De la Dure‐Molla, M. , Coudert, A. E. , Isaac, J. , & Berdal, A. (2014). Ameloblastin as a putative marker of specific bone compartments. Connective Tissue Research, 55(Suppl 1), 117–120. 10.3109/03008207.2014.923849 [DOI] [PubMed] [Google Scholar]
- Kawasaki, K. (2018). The origin and early evolution of SCPP genes and tissue mineralization in vertebrates In Endo K., Kogure T., & Nagasawa H. (Eds.), Biomineralization from molecular and nanostructural analyses to environental science (pp. 157–164). Singapore: Springer Open. [Google Scholar]
- Kawasaki, K. , & Amemiya, C. T. (2014). SCPP genes in the coelacanth: Tissue mineralization genes shared by sarcopterygians. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution, 322(6), 390–402. 10.1002/jez.b.22546 [DOI] [PubMed] [Google Scholar]
- Kawasaki, K. , Mikami, M. , Nakatomi, M. , Braasch, I. , Batzel, P. , Postlethwait, J. H. , … Ishiyama, M. (2017). SCPP Genes and their relatives in gar: Rapid expansion of mineralization genes in osteichthyans. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution, 328(7), 645–665. 10.1002/jez.b.22755 [DOI] [PubMed] [Google Scholar]
- Kawasaki, K. , & Weiss, K. M. (2003). Mineralized tissue and vertebrate evolution: The secretory calcium‐binding phosphoprotein gene cluster. Proceedings of the National Academy of Sciences USA, 100(7), 4060–4065. 10.1073/pnas.0638023100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. W. , Simmer, J. P. , Lin, B. P. , Seymen, F. , Bartlett, J. D. , & Hu, J. C. (2006). Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. European Journal of Oral Sciences, 114(Suppl 1), 3–12. 10.1111/j.1600-0722.2006.00278.x [DOI] [PubMed] [Google Scholar]
- Kim, J. W. , Zhang, H. , Seymen, F. , Koruyucu, M. , Hu, Y. , Kang, J. , … Hu, J. C. (2019). Mutations in RELT cause autosomal recessive amelogenesis imperfecta. Clinical Genetics, 95(3), 375–383. 10.1111/cge.13487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, K. , Yamakoshi, Y. , Hu, J.‐C. , Gomi, K. , Arai, T. , Fukae, M. , … Simmer, J. P. (2007). Splicing determines the glycosylation state of ameloblastin. Journal of Dental Research, 86(10), 962–967. 10.1177/154405910708601009 [DOI] [PubMed] [Google Scholar]
- Kozak, M. (1984). Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucl. Acids Res., 12(2), 857–872. 10.1093/nar/12.2.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebsbach, P. H. , Lee, S. K. , Matsuki, Y. , Kozak, C. A. , Yamada, K. , & Yamada, Y. (1996). Full‐length sequence, localization, and chromosomal mapping of ameloblastin: A novel tooth‐specific gene. Journal of Biological Chemistry, 271(8), 4431–4435. 10.1074/jbc.271.8.4431 [DOI] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lee, S. K. , Krebsbach, P. H. , Matsuki, Y. , Nanci, A. , Yamada, K. M. , & Yamada, Y. (1996). Ameloblastin expression in rat incisors and human tooth germs. International Journal of Developmental Biology, 40(6), 1141–1150. [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, T. , Li, M. , Xu, X. , Xiong, J. , Huang, C. , Zhang, X. , … Xiong, F. U. (2018). Whole exome sequencing identifies an AMBN missense mutation causing severe autosomal‐dominant amelogenesis imperfecta and dentin disorders. International Journal of Oral Science, 10(3), 26 10.1038/s41368-018-0027-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall, M. , DuPont, B. R. , Simmons, D. , Reus, B. , Krebsbach, P. , Kärrman, C. , … Forsman, K. (1997). Ameloblastin gene (AMBN) maps within the critical region for autosomal dominant amelogenesis imperfecta at chromosome 4q21. Genomics, 41(1), 115–118. 10.1006/geno.1997.4643 [DOI] [PubMed] [Google Scholar]
- Meredith, R. W. , Gatesy, J. , Cheng, J. , & Springer, M. S. (2011). Pseudogenization of the tooth gene enamelysin (MMP20) in the common ancestor of extant baleen whales. Proceedings of the Royal Society B: Biological Sciences, 278(1708), 993–1002. 10.1098/rspb.2010.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith, R. W. , Gatesy, J. , & Springer, M. S. (2013). Molecular decay of enamel matrix protein genes in turtles and other edentulous amniotes. BMC Evolutionary Biology, 13, 20 10.1186/1471-2148-13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, C. , Dohi, N. , Fukae, M. , Tanabe, T. , Yamakoshi, Y. , Wakida, K. , … Uchida, T. (1997). Immunochemical and immunohistochemical study of the 27‐ and 29‐kDa calcium‐binding proteins and related proteins in the porcine tooth germ. Histochemistry and Cell Biology, 107(6), 485–494. 10.1007/s004180050136 [DOI] [PubMed] [Google Scholar]
- Nanci, A. , Zalzal, S. , Lavoie, P. , Kunikata, M. , Chen, W.‐Y. , Krebsbach, P. H. , … Smith, C. E. (1998). Comparative immunochemical analyses of the developmental expression and distribution of ameloblastin and amelogenin in rat incisors. Journal of Histochemistry and Cytochemistry, 46(8), 911–934. 10.1177/002215549804600806 [DOI] [PubMed] [Google Scholar]
- Orams, H. J. (1964). The fine structure of the enamel examined with the electron microscope. Australian Dental Journal, 9(5), 369–373. 10.1111/j.1834-7819.1964.tb02093.x [DOI] [Google Scholar]
- Orams, H. J. (1966). An examination of the prism core, prism sheath and interprismatic substance using the electon microscope. Australian Dental Journal, 11(2), 93–104. [DOI] [PubMed] [Google Scholar]
- Petersen, T. N. , Brunak, S. , von Heijne, G. , & Nielsen, H. (2011). SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nature Methods, 8(10), 785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- Poulter, J. A. , Murillo, G. , Brookes, S. J. , Smith, C. E. L. , Parry, D. A. , Silva, S. , … Mighell, A. J. (2014). Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Human Molecular Genetics, 23(20), 5317–5324. 10.1093/hmg/ddu247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, M. K. , Geoffroy, V. , Vicaire, S. , Jost, B. , Dumas, M. , Le Gras, S. , … Bloch‐Zupan, A. (2016). A targeted next‐generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. Journal of Medical Genetics, 53(2), 98–110. 10.1136/jmedgenet-2015-103302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prostak, K. , Seifert, P. , & Skobe, Z. (1989). Ultrastructure of developing teeth in the gar pike, (Lepisosteus) In Fearnhead R. (Ed.), Tooth enamel V (pp. 188–192). Yokohama, Japan: Florence. [Google Scholar]
- Ryu, O. H. , Fincham, A. G. , Hu, C. C. , Zhang, C. , Qian, Q. , Bartlett, J. D. , & Simmer, J. P. (1999). Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. Journal of Dental Research, 78(3), 743–750. 10.1177/00220345990780030601 [DOI] [PubMed] [Google Scholar]
- Shimizu, M. (1984). Investigation of highly aggregative components in porcine enamel proteins In Fearnhead R. W., & Suga S. (Eds.), Tooth Enamel IV (pp. 156–160). New York: Elsevier Science Publishers. [Google Scholar]
- Simmer, J. P. , Hu, Y. , Lertlam, R. , Yamakoshi, Y. , & Hu, J. C. (2009). Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. Journal of Biological Chemistry, 284(28), 19110–19121. 10.1074/jbc.M109.013623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer, J. P. , Lau, E. C. , Hu, C. C. , Aoba, T. , Lacey, M. , Nelson, D. , … Fincham, A. G. (1994). Isolation and characterization of a mouse amelogenin expressed in Escherichia coli . Calcified Tissue International, 54(4), 312–319. https://link.springer.com/article/10.1007/BF00295956 [DOI] [PubMed] [Google Scholar]
- Sire, J. Y. (1994). Light and TEM study of nonregenerated and experimentally regenerated scales of Lepisosteus oculatus (Holostei) with particular attention to ganoine formation. Anatomical Record, 240(2), 189–207. 10.1002/ar.1092400206 [DOI] [PubMed] [Google Scholar]
- Sire, J. Y. (1995). Ganoine formation in the scales of primitive actinopterygian fishes, lepisosteids and polypterids. Connective Tissue Research, 33(1–3), 213–222. 10.3109/03008209509017006 [DOI] [PubMed] [Google Scholar]
- Smith, C. E. (1998). Cellular and chemical events during enamel maturation. Critical Reviews in Oral Biology and Medicine, 9(2), 128–161. 10.1177/10454411980090020101 [DOI] [PubMed] [Google Scholar]
- Smith, C. E. , Hu, Y. , Hu, J. C. , & Simmer, J. P. (2016). Ultrastructure of early amelogenesis in wild‐type, Amelx‐/‐, and Enam‐/‐ mice: Enamel ribbon initiation on dentin mineral and ribbon orientation by ameloblasts. Molecular Genetics and Genomic Medicine, 4(6), 662–683. 10.1002/mgg3.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. E. , Hu, Y. , Richardson, A. S. , Bartlett, J. D. , Hu, J. C. , & Simmer, J. P. (2011). Relationships between protein and mineral during enamel development in normal and genetically altered mice. European Journal of Oral Sciences, 119(Suppl 1), 125–135. 10.1111/j.1600-0722.2011.00871.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, M. S. , Starrett, J. , Morin, P. A. , Lanzetti, A. , Hayashi, C. , & Gatesy, J. (2016). Inactivation of C4orf26 in toothless placental mammals. Molecular Phylogenetics and Evolution, 95, 34–45. 10.1016/j.ympev.2015.11.002 [DOI] [PubMed] [Google Scholar]
- Uchida, T. , Fukae, M. , Tanabe, T. , Yamakoshi, Y. , Satoda, T. , Murakami, C. , … Shimizu, M. (1995). Immunochemical and immunocytochemical study of a 15 kDa non‐amelogenin and related proteins in the porcine immature enamel: Proposal of a new group of enamel proteins sheath proteins. Biomedical Research, 16(3), 131–140. 10.2220/biomedres.16.131 [DOI] [Google Scholar]
- Uchida, T. , Tanabe, T. , Fukae, M. , Shimizu, M. , Yamada, M. , Miake, K. , & Kobayashi, S. (1991). Immunochemical and immunohistochemical studies, using antisera against porcine 25 kDa amelogenin, 89 kDa enamelin and the 13–17 kDa nonamelogenins, on immature enamel of the pig and rat. Histochemistry, 96(2), 129–138. 10.1007/BF00315983 [DOI] [PubMed] [Google Scholar]
- Wang, S.‐K. , Hu, Y. , Yang, J. , Smith, C. E. , Nunez, S. M. , Richardson, A. S. , … Simmer, J. P. (2015). Critical roles for WDR72 in calcium transport and matrix protein removal during enamel maturation. Molecular Genetics & Genomic Medicine, 3(4), 302–319. 10.1002/mgg3.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wazen, R. M. , Moffatt, P. , Zalzal, S. F. , Yamada, Y. , & Nanci, A. (2009). A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biology, 28(5), 292–303. 10.1016/j.matbio.2009.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Kawasaki, K. , Lee, M. , Reid, B. M. , Nunez, S. M. , Choi, M. , … Hu, J.‐C. (2016). The dentin phosphoprotein repeat region and inherited defects of dentin. Molecular Genetics & Genomic Medicine, 4(1), 28–38. 10.1002/mgg3.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Hu, Y. , Seymen, F. , Koruyucu, M. , Kasimoglu, Y. , Wang, S.‐K. , … Hu, J. C.‐C. (in press). ENAM Mutations and Digenic Inheritance. Molecular Genetics and Genomic Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Koruyucu, M. , Seymen, F. , Kasimoglu, Y. , Kim, J. W. , Tinawi, S. , … Hu, J. C. C. (2019). WDR72 mutations associated with amelogenesis imperfecta and acidosis. Journal of Dental Research, 19, 22034518824571 10.1177/0022034518824571 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials