

Summary
Long non-coding RNA (lncRNA) play critical roles in the occurrence and development of various diseases. The determination of the lncRNA-disease associations thus would contribute to provide new insights into the pathogenesis of the disease, the diagnosis, and the gene treatments. Considering that traditional experimental approaches are difficult to detect potential human lncRNA-disease associations from the vast amount of biological data, developing computational method could be of significant value. In this paper, we proposed a novel computational method named LDASR to identify associations between lncRNA and disease by analyzing known lncRNA-disease associations. First, the feature vectors of the lncRNA-disease pairs were obtained by integrating lncRNA Gaussian interaction profile kernel similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity. Second, autoencoder neural network was employed to reduce the feature dimension and get the optimal feature subspace from the original feature set. Finally, Rotating Forest was used to carry out prediction of lncRNA-disease association. The proposed method achieves an excellent preference with 0.9502 AUC in leave-one-out cross-validations (LOOCV) and 0.9428 AUC in 5-fold cross-validation, which significantly outperformed previous methods. Moreover, two kinds of case studies on identifying lncRNAs associated with colorectal cancer and glioma further proves the capability of LDASR in identifying novel lncRNA-disease associations. The promising experimental results show that the LDASR can be an excellent addition to the biomedical research in the future.
Subject Areas: Bioinformatics, Biocomputational Method, Computational Bioinformatics
Graphical Abstract
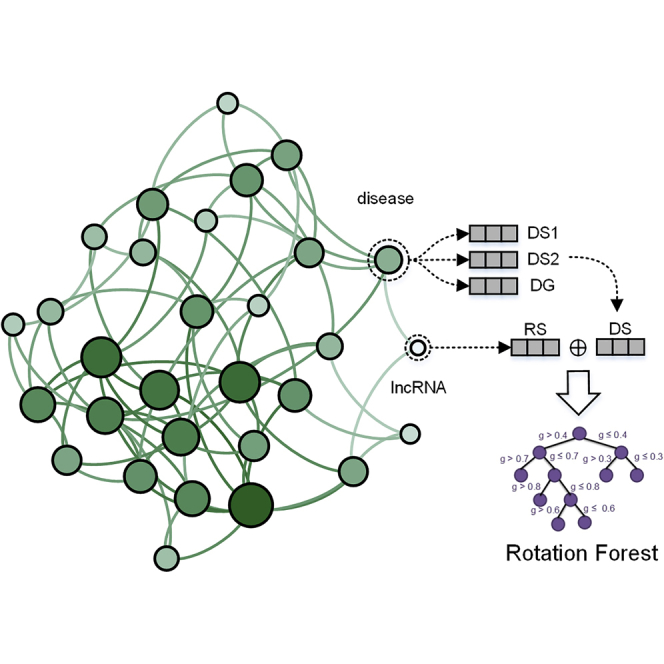
Highlights
-
•
We propose a similarity-based characterization method for RNA-disease associations
-
•
The model automatically captures important association features
-
•
This method determines the prospects of machine learning techniques on such problems
Bioinformatics; Biocomputational Method; Computational Bioinformatics
Introduction
Long non-coding RNAs (lncRNAs) are an important class of transcripts, with the length longer than 200 nt, which participates in various physiological processes, such as immune surveillance, post-translational regulation, cell differentiation, proliferation, apoptosis, and epigenetic regulation. Especially, accumulating studies have indicated that a large number of lncRNAs are involved in numerous complex human diseases, such as various cancers (Chung et al., 2011, Zhang et al., 2012), blood diseases (Congrains et al., 2012, Alvarez-Dominguez and Lodish, 2017, Sallam et al., 2018), and neurodegeneration diseases (Johnson, 2012). Therefore, inferring the potential association between lncRNA and disease is helpful to understand the pathogenesis of complex diseases at the molecular level and provide new insights into the diagnosis, treatment, and prognosis of diseases.
Profit from the development of high-throughput experimental techniques, such as Microarray, Northern blots and qPCR, Fluorescence in situ hybridization, RNA interference, and RNA immunoprecipitation (Yan et al., 2012), a large amount of data about lncRNAs-disease associations have been determined and distributed in different public databases, such as lncRNAdb (Amaral et al., 2010), NRED (Dinger et al., 2008), and NONCODE (Xie et al., 2013). However, although experimentally validated lncRNA-disease associations drive research and development of medical molecular biology, they often have high false positives and false negatives. Moreover, many experimental methods are expensive and time-consuming. Consequently, it is essential to develop a computational prediction approach based on the accumulated biological data to accurately and rapidly find potential lncRNAs-disease associations. Computational method can quantitatively describe the associations between lncRNAs and diseases and efficiently screen out the most promising lncRNA-disease association pairs for further biological experimental validation.
The proposed computational method for predicting lncRNA-disease association can be roughly divided into three categories. Methods in the first category uncover ncRNA-disease associations based on the idea of network or link prediction. The underlying assumption is that lncRNAs associated with the same or similar diseases are more likely to have similar functions. Liao et al. constructed a coding-non-coding gene co-expression network based on public microarray expression profiles to discover the potential functions of lncRNA (Liao et al., 2011). Yang et al. applied a propagation algorithm to predict lncRNA-disease associations by constructing a coding-non-coding gene-disease bipartite network based on known associations between diseases and disease-causing genes (Yang et al., 2014). Chen et al. came up with the model called IRWRLDA to identify potential associations by integrating known lncRNA-disease associations, disease semantic similarity, and various lncRNA similarity measures (Chen et al., 2016). Huang et al. proposed a model called PBMDA to predict microRNA (miRNA)-disease associations by integrating known human miRNA-disease associations, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity (You et al., 2017). Methods in the second category utilize matrix factorization to identify potential lncRNA-disease associations. The basic assumption is that unknown association information can be derived from other known association information. Fu et al. predicted lncRNA-disease associations by decomposing data matrices of heterogeneous data sources into low-rank matrices (Fu et al., 2017). Lu et al. developed a method called SIMCLDA for potential lncRNA-disease association prediction based on inductive matrix completion (Lu et al., 2018). These two types of methods are based on specific assumptions, but these assumptions are not unanimously accepted. Relevant studies have shown that in many cases bio macromolecules with similar structures or ligands do not have the same functions. Matrix factorization approaches will experience dramatic performance degradation when the known associated information is insufficient. In addition, these methods both cannot mine the similarity feature of lncRNA and disease, and consider the inherent logic of the association between lncRNA and disease from the perspective of data-driven. Machine learning models are used in the third category to discover the unknown lncRNA-disease associations. Lan et al. proposed a method called LDAP to identify latent associations between lncRNAs and diseases by using a bagging support vector machine (SVM) classifier based on lncRNA similarity and disease similarity (Lan et al., 2016). Since these methods are the beginning of machine learning application for lncRNA-disease association prediction, there is still much room for improvement in the prediction performance, prediction accuracy of such methods can be still greatly improved by increasing training samples and using more appropriate and advanced learning algorithms. Recently, the accumulation of association data between lncRNA and disease and the development of machine learning technology provide a better opportunity for predicting the association between lncRNA and disease using supervised learning model.
Instead of using network-based and matrix factorization-based methods to compute association scores directly, we explored to extract association features from lncRNA-disease pairs by multiple similarity matrices and trained machine learning models in a supervised manner to predict their association. In this study, we proposed a novel supervised computational method named (LDASR) for large-scale lncRNA-disease association prediction based on collaborative filtering and machine learning technologies. First, the feature vectors of the lncRNA-disease pairs were obtained by integrating lncRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity. Second, autoencoder neural network was employed to low the feature dimension and get the optimal feature subspace from the original feature set. Finally, considering the size of training samples and the possible non-linear relationship in input, we trained rotating forest to carry out prediction of LncRNA-Disease Association. The flow of LDASR is represented in Figure 1. In leave-one-out cross-validation (LOOCV) and five cross-validation to evaluate test data, the proposed LDASR model achieved better results than some previous methods, with AUC of 0.9502 and 0.9428, respectively. The test results show that supervised learning model can achieve better performance.
Figure 1.

Flowchart of LDASR
Step 1: Building three similarity matrices for disease by combining semantic information and Gaussian kernel information. Step 2: Building 1 similarity matrix for lncRNA. Step 3: Extraction of similarity feature vectors for disease and lncRNA from disease similarity matrix and lncRNA similarity matrix. Step 4: Extracting the same number of positive and negative samples from the adjacency matrix to construct the dataset used in this paper. Step 5: Selecting the most valuable features and reducing feature noise by using autoencoder. Step 6: more discriminant feature vectors were put into Rotation Forest ensemble classifier for training, verification, and prediction. The construction of disease semantic matrix can see also Figure S1.
Results
Leave-One-Out Cross-Validation
For LOOCV, each sample in the dataset is selected for testing in turn, and the remaining samples are used as the training set to construct the prediction model. As we have mentioned, 1,765 lncRNA-disease associations, which have been experimental verified, were regarded as positive samples. Then we randomly picked 1,765 lncRNA-disease associations in the remaining associations as negative samples. The total number of datasets was 3,530, so we trained and tested 3,530 times according to the LOOCV method to get the final experimental result. At the same time, we drew ROC (receiver operating characteristic curve) and calculated AUC (area under curve) under LOOCV as shown in Figure 2 to quantify the prediction results and facilitate comparison with other methods. For LOOCV, LDASR obtained AUCs of 0.9502, indicating that the model combining various similarities and rotation forest had a strong ability to distinguish the difference between positive and negative samples.
Figure 2.

The ROC and AUC of LDASR in LOOCV Based on the v2017 Dataset (3,530 lncRNA-Disease Associations)
Five-fold Cross-Validation
For 5-fold cross-validation, the entire dataset is randomly divided into five mutually exclusive subsets of roughly equal size, each of which is used in turn as a test set for evaluation, and the remaining four subsets served as training sets to build the model. To better verify the performance of our method and save computing resources, LDASR was further evaluated by 5-fold cross-validation. For 5-fold cross-validation, LDASR obtained mean AUC of 0.9428 in the end as shown in Figure 3.
Figure 3.

The ROCs and AUCs of LDASR in 5-Fold Cross-validation Based on the v2017 Dataset (3,530 lncRNA-Disease Associations)
To more comprehensively evaluate our model, we used a broader range of evaluation criteria, including accuracy (Acc.), sensitivity (Sen.), specificity (Spec.), precision (Prec.), and MCC. The prediction performance is listed in Table 1. The results of average Acc., Sen., Spec., Prec., MCC, and AUC were 85.72%, 90.14%, 81.31%, 82.86%, 71.74%, and 94.28% when using the proposed method to predict lncRNA-disease associations. The standard deviations of these values were 1.59%, 0.77%, 2.69%, 2.11%, 3.05%, and 0.94%, respectively. For 5-fold cross-validation shown in Figure 5, LDASR obtained high mean AUC of 0.9428. The high AUCs showed that LDASR combining multiple similarities and rotation forest was feasible and effective to predict lncRNA disease associations. At the same time, the lower standard deviation of these standards implied that the proposed model was robust and stable.
Table 1.
Five-fold Cross-validation Results Performed by LDASR on the v2017 Dataset (3,530 lncRNA-Disease Associations)
| Fold | Acc. (%) | Sen. (%) | Spec. (%) | Prec. (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| 0 | 83.85 | 90.08 | 77.62 | 80.10 | 68.24 | 93.11 |
| 1 | 85.27 | 88.95 | 81.59 | 82.85 | 70.73 | 93.19 |
| 2 | 84.42 | 89.80 | 79.04 | 81.07 | 69.24 | 94.78 |
| 3 | 88.10 | 91.22 | 84.99 | 85.87 | 76.35 | 95.27 |
| 4 | 86.97 | 90.65 | 83.29 | 84.43 | 74.14 | 95.08 |
| Average | 85.72 ± 1.59 | 90.14 ± 0.77 | 81.31 ± 2.69 | 82.86 ± 2.11 | 71.74 ± 3.05 | 94.28 ± 0.94 |
Figure 5.

Under the v2012 Dataset (586 lncRNA-Disease Associations), LDASR and LRLSLDA, LRLSLDA-LNCSIM1, LRLSLDA-LNCSIM2 Were Compared between the AUCs Obtained under LOOCV
Compared with Other Classifiers
To assess the performance of Rotation Forest: In this section, we compared Rotation Forest with several common classifiers in 5-fold cross-validation, including Random Forest, Logistic Regression, Naive Bayes, and SVM. To be fair, all settings except classifiers are default and the same dataset is used. The ROC curves implemented by five classifiers are summarized in Figure 4. As seen in Figure 4, it is obvious that the Rotation Forest achieves the best results. The effectiveness mainly has the following factors: (1) Based on the idea of collaborative filtering, there might be cases of non-independence between feature attributes generated by the similarity between lncRNA and disease. This affects the predictive capability of Naive Bayes. (2) The performance of the SVM classifier is more sensitive to data. Proper selection of the parameters and the correct choice of the kernel function will result in a large training cost. (3) The prediction performance of the logistic regression is limited by the assumption that the feature and the target must be linearly separable, so the lower AUC was obtained. (4) Tree-based assemble algorithms such as Random Forest and Rotating Forest are not affected by the nonlinear relationships in the data, so they have achieved excellent results in the five-fold cross-validation. Compared with the Random Forest, Rotating forest randomly combines the sample attribute sets before each subsample is extracted, and Principal Component Analysis (PCA) is utilized to transform the data between the divided sets of sub-attributes. This operation not only makes each sub-sample different, but also plays a certain role in data pre-processing, thereby improving the accuracy and difference of each base classifier to obtain excellent assemble effect.
Figure 4.

Comparison with Random Forest, Logistic Regression, Naive Bayes, and SVM in 5-Fold Cross-validation Based on the v2017 Dataset (3,530 lncRNA-Disease Associations)
Compared with Other Methods
The LDASR was further tested by comparing it with other three state-of-the-art methods involving LRLSLDA (Chen and Yan, 2013), LRLSLDA-LNCSIM1 (Chen et al., 2015), and LRLSLDA-LNCSIM2 (Chen et al., 2015). The comparison of the obtained AUC between LDASR and previous methods in LOOCV is shown in Figure 5. The results on 2012v dataset showed obviously that our proposed method made some progress. As a result, the proposed method achieved 0.1013, 0.0643, and 0.0575 improvements in terms of AUC compared with other three approaches in terms of AUC under LOOCV. Because the Leave-one-out cross-validation uses one observation as the validation set and the remaining observations as the training set, the model under LOOCV will always be a very stable solution. Therefore, it was difficult to add error bars to Figure 5. Compared with traditional computational methods for the prediction of lncRNAs-diseases associations, machine learning can consider similarity information from the perspectives of probability, statistics, approximation, and convex analysis and iteratively and optimally grasp the essential associations rule between RNA and disease. And this data-driven approach will show a stronger advantage as data are accumulated. The improved prediction performance produced by this method further validates the potential of machine learning algorithms on such problems. Although 5% does not seem to be a considerable improvement, we hope to draw the attention of relevant researchers and open a novel perspective on solving problems by machine learning strategies.
Case Study
To evaluate the capability of the model in practical application, we applied LDASR to predict Colorectal Cancer, Glioma, and Prostate cancer as two kinds of case studies. For the purpose of simulating the real environment and ensuring the fairness of case studies, the associations of LncRNADisease database (v2017) were used to train the model and the remaining four additional databases, including Lnc2Cancer (Ning et al., 2015), MNDR (Cui et al., 2017), CRlncRNA (Wang et al., 2018), and LncRNAWiki (Ma et al., 2014), were used to verify the results.
For the first kind of case study, Colorectal Cancer was chosen as the research subject. In this case study, all 1,765 associations in LncRNADisease database (v2017) were used as positive samples. The negative samples of the same size as positive samples are generated by random selection in the rest. Therefore, the test set was constructed by connecting colorectal cancer diseases to all lncRNAs in the other three databases, respectively. As a result, a total of 881 lncRNA-colorectal cancer pairs were verified as test samples. Finally, samples with a predicted probability greater than 0.5 are screened out and sorted according to the probability values from large to small. Recent results in biological experiments confirmed that Colorectal Cancer was related to gene XIST (Lassmann et al., 2007), AB073614 (Xue et al., 2018, Wang et al., 2017), and SNHG3 (Huang et al., 2017). They were all in the top of the list, but they were not included in the LncRNADisease database. Then, we ranked all the 881 lncRNAs based on their predicted association scores and validated the top 10 lncRNAs in LncRNADisease, CRlncRNA, MNDR 2.0, and Lnc2Cancer. The results are shown in the Table 2.
Table 2.
Top 10 Colorectal Cancer-Associated lncRNAs Predicted by LDASR
| Num | lncRNA | Confirmed Database |
|---|---|---|
| 1 | snhg3 | LncRNAWiki |
| 2 | linc00237 | Unconfirmed |
| 3 | kcna2 | Unconfirmed |
| 4 | xist | LncRNADisease/MNDR 2.0 |
| 5 | cahm | LncRNADisease/CRlncRNA |
| 6 | bx649059 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 7 | ab073614 | Lnc2Cancer |
| 8 | bx648207 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 9 | ak123657 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 10 | fas-as1 | Unconfirmed |
In the second kind of case study, Glioma and Prostate Cancer were the subjects of the study. A glioma is a tumor that begins with glial cells in the brain or spine (Mamelak and Jacoby, 2007). The experimental design for case study 2 is as follows: For positive sample set, we removed all Glioma-related associations in the positive sample set. There were 42 positive samples related to Glioma here, so the number of positive samples was 1,723. Like case study 1, we used the same method to select 1,723 negative samples and 881 test samples. The test sample set was put into the classifier after training with positive and negative samples. In the end, we found that XIST and CYTOR were both at the top of the list, but they were not included in the LncRNADisease database. Recent results in biological experiments confirmed that Glioma was related to XIST (Yao et al., 2015) and CYTOR (Yu et al., 2017). Then, we ranked all the 881 lncRNAs based on their predicted association scores and validated the top 10 lncRNAs in LncRNADisease, CRlncRNA, MNDR 2.0, and Lnc2Cancer. The results can be seen in Table 3.
Table 3.
Top 10 Glioma-Associated lncRNAs Predicted by LDASR
| Num | lncRNA | Confirmed Database |
|---|---|---|
| 1 | zfat-as1 | Unconfirmed |
| 2 | xist | CRlncRNA/MNDR 2.0 |
| 3 | spry4-it1 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 4 | cytor | MNDR 2.0 |
| 5 | neat1 | LncRNADisease/CRlncRNA |
| 6 | meg3 | LncRNADisease/CRlncRNA |
| 7 | malat1 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 8 | cdkn2b-as1 | LncRNADisease |
| 9 | h19 | LncRNADisease/CRlncRNA |
| 10 | hotair | LncRNADisease/CRlncRNA/MNDR 2.0 |
For Prostate Cancer, all the experimental steps are the same as Glioma. For the positive sample set, we removed all Prostate Cancer-related associations in the positive sample set. There were 55 positive samples related to Glioma here, so the number of positive samples was 1,710. Like case study Glioma, we used the same method to select 1,710 negative samples and 881 test samples. The list of the validated top 10 lncRNAs are listed in Table 4.
Table 4.
Top 10 Prostate Cancer-Associated lncRNAs Predicted by LDASR
| Num | lncRNA | Confirmed Database |
|---|---|---|
| 1 | pcat29 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 2 | tug1 | Unconfirmed |
| 3 | malat1 | LncRNADisease/CRlncRNA/MNDR 2.0 |
| 4 | hif1a-as2 | Unconfirmed |
| 5 | h19 | LncRNADisease/CRlncRNA |
| 6 | dleu1 | LncRNADisease/MNDR 2.0 |
| 7 | dgcr5 | Unconfirmed |
| 8 | cytor | Unconfirmed |
| 9 | cdkn2b-as3 | Unconfirmed |
| 10 | cdkn2b-as11 | LncRNADisease/CRlncRNA/MNDR 2.0/Lnc2Cancer |
Discussion
Accumulating evidences have highlighted the positive role of developing a powerful machine-learning-based method to predict potential associations between lncRNAs and diseases, which could significantly help people to understand the pathogenesis of complex diseases at the molecular level and provide new insights into the diagnosis, treatment, and prognosis of diseases. In this paper, we proposed a novel computational method, LDASR, to predict the unknown lncRNA-disease associations by integrating multiple similarity information. Compared with previous methods, we embed this task into a machine learning framework to better understand the essential law of the association between lncRNA and diseases. First, we extracted feature vectors for lncRNA and disease from multiple similarity matrices and constructed the feature vector of RNA-disease pairs by connecting features of lncRNA to that of disease. Then, autoencoder neural network was employed to reduce the feature dimension and improve the efficiency and accuracy of classifier. Finally, we applied rotation forest to carry out prediction. LDASR first shows its good performance by LOOCV and 5-fold cross-validation experiments. Furthermore, the comparison test shows that LDASR has a powerful prediction ability to distinguish positive and negative samples, which is obviously better than the other state-of-the-art methods. Finally, the analyses of case studies further prove that LDASR holds significant value in inferring potential lncRNA-disease associations in practice. As a novel computational method, it is anticipated that LDASR has potential value in biomedical research for comprehending the pathogenesis of diseases, which can further advance the quality of disease diagnostics, therapy, prognosis, and prevention. As a novel computational approach, LDASR could not only play a positive role in rapidly understanding the pathogenesis of disease and improving the quality of disease diagnosis, treatment, prognosis, and prevention but also confirms the great potential of machine learning in predicting the relationship between RNA and disease.
Limitations of the Study
There are several limitations in the current model. First, in the stage of characterizing lncRNA, we hope to fully combine and utilize a variety of information such as the sequence of lncRNA in the future instead of using only the Gaussian Interaction Profile Kernel Similarity. Second, Gaussian Interaction Profile Kernel Similarity is a traditional network representation learning method widely used in the embedding of bipartite graph nodes. With the rise of deep learning, novel network representation learning methods emerge in an endless stream, which can more effectively characterize the behavior of nodes and the structure of the entire network. We hope to take advantage of deep learning to improve prediction ability in the future. Third, all parameters are default in the process of constructing the model, and we believe that the performance of models can achieve a visible progress through the adjustment of the parameters.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work is supported in part by the National Natural Science Foundation of China (Grant nos. 61722212 and 61572506) and in part by the Pioneer Hundred Talents Program of the Chinese Academy of Sciences. The authors would like to thank the editors and anonymous reviewers for their constructive advice.
Author Contributions
Z.-H.G., Z.-H.Y., and Y.-B.W. considered the algorithm, arranged the datasets, and performed the analyses. H.-C.Y. and Z.-H.C. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.030.
Contributor Information
Zhu-Hong You, Email: zhuhongyou@ms.xjb.ac.cn.
Yan-Bin Wang, Email: wangyanbin15@mails.ucas.ac.cn.
Supplemental Information
References
- Alvarez-Dominguez J.R., Lodish H.F. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood. 2017;130:1965–1975. doi: 10.1182/blood-2017-06-788695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral P.P., Clark M.B., Gascoigne D.K., Dinger M.E., Mattick J.S. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2010;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Yan C.C., Luo C., Ji W., Zhang Y., Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 2015;5:11338. doi: 10.1038/srep11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Yan G.-Y. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics. 2013;29:2617–2624. doi: 10.1093/bioinformatics/btt426. [DOI] [PubMed] [Google Scholar]
- Chen X., You Z.-H., Yan G.-Y., Gong D.-W. IRWRLDA: improved random walk with restart for lncRNA-disease association prediction. Oncotarget. 2016;7:57919. doi: 10.18632/oncotarget.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S., Nakagawa H., Uemura M., Piao L., Ashikawa K., Hosono N., Takata R., Akamatsu S., Kawaguchi T., Morizono T. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011;102:245–252. doi: 10.1111/j.1349-7006.2010.01737.x. [DOI] [PubMed] [Google Scholar]
- Congrains A., Kamide K., Oguro R., Yasuda O., Miyata K., Yamamoto E., Kawai T., Kusunoki H., Yamamoto H., Takeya Y. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis. 2012;220:449–455. doi: 10.1016/j.atherosclerosis.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Cui T., Zhang L., Huang Y., Yi Y., Tan P., Zhao Y., Hu Y., Xu L., Li E., Wang D. MNDR v2. 0: an updated resource of ncRNA–disease associations in mammals. Nucleic Acids Res. 2017;46:D371–D374. doi: 10.1093/nar/gkx1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger M.E., Pang K.C., Mercer T.R., Crowe M.L., Grimmond S.M., Mattick J.S. NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 2008;37:D122–D126. doi: 10.1093/nar/gkn617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu G., Wang J., Domeniconi C., Yu G. Matrix factorization-based data fusion for the prediction of lncRNA–disease associations. Bioinformatics. 2017;34:1529–1537. doi: 10.1093/bioinformatics/btx794. [DOI] [PubMed] [Google Scholar]
- Huang W., Tian Y., Dong S., Cha Y., Li J., Guo X., Yuan X. The long non-coding RNA SNHG3 functions as a competing endogenous RNA to promote malignant development of colorectal cancer. Oncol. Rep. 2017;38:1402–1410. doi: 10.3892/or.2017.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. Long non-coding RNAs in Huntington's disease neurodegeneration. Neurobiol. Dis. 2012;46:245–254. doi: 10.1016/j.nbd.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Lan W., Li M., Zhao K., Liu J., Wu F.-X., Pan Y., Wang J. LDAP: a web server for lncRNA-disease association prediction. Bioinformatics. 2016;33:458–460. doi: 10.1093/bioinformatics/btw639. [DOI] [PubMed] [Google Scholar]
- Lassmann S., Weis R., Makowiec F., Roth J., Danciu M., Hopt U., Werner M. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal-and microsatellite-unstable sporadic colorectal carcinomas. J. Mol. Med. 2007;85:293–304. doi: 10.1007/s00109-006-0126-5. [DOI] [PubMed] [Google Scholar]
- Liao Q., Liu C., Yuan X., Kang S., Miao R., Xiao H., Zhao G., Luo H., Bu D., Zhao H. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res. 2011;39:3864–3878. doi: 10.1093/nar/gkq1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Yang M., Luo F., Wu F.-X., Li M., Pan Y., Li Y., Wang J. Prediction of lncRNA–disease associations based on inductive matrix completion. Bioinformatics. 2018;34:3357–3364. doi: 10.1093/bioinformatics/bty327. [DOI] [PubMed] [Google Scholar]
- Ma L., Li A., Zou D., Xu X., Xia L., Yu J., Bajic V.B., Zhang Z. LncRNAWiki: harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2014;43:D187–D192. doi: 10.1093/nar/gku1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamelak A.N., Jacoby D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601) Expert Opin. Drug Deliv. 2007;4:175–186. doi: 10.1517/17425247.4.2.175. [DOI] [PubMed] [Google Scholar]
- Ning S., Zhang J., Wang P., Zhi H., Wang J., Liu Y., Gao Y., Guo M., Yue M., Wang L. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2015;44:D980–D985. doi: 10.1093/nar/gkv1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam T., Sandhu J., Tontonoz P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function. Circ. Res. 2018;122:155–166. doi: 10.1161/CIRCRESAHA.117.311802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang X., Chen W., Li J., Liu C. CRlncRNA: a manually curated database of cancer-related long non-coding RNAs with experimental proof of functions on clinicopathological and molecular features. BMC Med. Genomics. 2018;11:114. doi: 10.1186/s12920-018-0430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kuang H., Xue J., Liao L., Yin F., Zhou X. LncRNA AB073614 regulates proliferation and metastasis of colorectal cancer cells via the PI3K/AKT signaling pathway. Biomed. Pharmacother. 2017;93:1230–1237. doi: 10.1016/j.biopha.2017.07.024. [DOI] [PubMed] [Google Scholar]
- Xie C., Yuan J., Li H., Li M., Zhao G., Bu D., Zhu W., Wu W., Chen R., Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2013;42:D98–D103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J., Liao L., Yin F., Kuang H., Zhou X., Wang Y. LncRNA AB073614 induces epithelial-mesenchymal transition of colorectal cancer cells via regulating the JAK/STAT3 pathway. Cancer Biomarkers. 2018;21:1–10. doi: 10.3233/CBM-170780. [DOI] [PubMed] [Google Scholar]
- Yan B., Wang Z.-H., Guo J.-T. The research strategies for probing the function of long noncoding RNAs. Genomics. 2012;99:76–80. doi: 10.1016/j.ygeno.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Yang X., Gao L., Guo X., Shi X., Wu H., Song F., Wang B. A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PLoS One. 2014;9:e87797. doi: 10.1371/journal.pone.0087797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Ma J., Xue Y., Wang P., Li Z., Liu J., Chen L., Xi Z., Teng H., Wang Z. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015;359:75–86. doi: 10.1016/j.canlet.2014.12.051. [DOI] [PubMed] [Google Scholar]
- You Z.-H., Huang Z.-A., Zhu Z., Yan G.-Y., Li Z.-W., Wen Z., Chen X. PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017;13:e1005455. doi: 10.1371/journal.pcbi.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Xue Y., Zheng J., Liu X., Yu H., Liu L., Li Z., Liu Y. Linc00152 promotes malignant progression of glioma stem cells by regulating miR-103a-3p/FEZF1/CDC25A pathway. Mol. Cancer. 2017;16:110. doi: 10.1186/s12943-017-0677-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang Z., Hao H., Zhang C., Yang X., He Q., Lin J. Evaluation of novel gene UCA1 as a tumor biomarker for the detection of bladder cancer. Zhonghua Yi Xue Za Zhi. 2012;92:384–387. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.