

Abstract
Serotonin syndrome (SS) (also referred to as serotonin toxicity) is a potentially life-threatening drug-induced toxidrome associated with increased serotonergic activity in both the peripheral (PNS) and central nervous systems (CNS). It is characterised by a dose-relevant spectrum of clinical findings related to the level of free serotonin (5-hydroxytryptamine [5-HT]), or 5-HT receptor activation (predominantly the 5-HT1A and 5-HT2A subtypes), which include neuromuscular abnormalities, autonomic hyperactivity, and mental state changes. Severe SS is only usually precipitated by the simultaneous initiation of 2 or more serotonergic drugs, but the syndrome can also occur after the initiation of a single serotonergic drug in a susceptible individual, the addition of a second or third agent to long-standing doses of a maintenance serotonergic drug, or after an overdose. The combination of a monoamine oxidase inhibitor (MAOI), in particular MAO-A inhibitors that preferentially inhibit the metabolism of 5-HT, with serotonergic drugs is especially dangerous, and may lead to the most severe form of the syndrome, and occasionally death. This review describes our current understanding of the pathophysiology, clinical presentation and management of SS, and summarises some of the drugs and interactions that may precipitate the condition. We also discuss the newer novel psychoactive substances (NPSs), a growing public health concern due to their increased availability and use, and their potential risk to evoke the syndrome. Finally, we discuss whether the inhibition of tryptophan hydroxylase (TPH), in particular the neuronal isoform (TPH2), may provide an opportunity to pharmacologically target central 5-HT synthesis, and so develop new treatments for severe, life-threatening SS.
Keywords: Serotonin syndrome, serotonin toxicity, novel psychoactive substances, neuroleptic malignant syndrome, toxidromes, tryptophan hydroxylase inhibitors
Introduction
Epidemics of ‘convulsive ergotism’ were widespread and well described east of the Rhine river from 1085 to 1927.1 Given that ergot alkaloids, a known contaminant of human cultivated grains, are now recognised to induce serotonin syndrome (SS), this suggests that the syndrome may have been a public health problem long before its more recent ‘discovery’ as a complication of modern pharmacology.1
5-Hydroxytryptamine (5-HT), a monoamine neurotransmitter, first recognised in 1948,2 has a wide range of functions in the central nervous system (CNS), including modulation of attention, cognition, behaviour, memory, and thermoregulation, as well as in the peripheral nervous system (PNS), where it regulates, for example, gastrointestinal (GI) motility, uterine contraction, vasoconstriction, and bronchoconstriction.3 The first report of a clinical picture consistent with what nowadays is termed SS was in 1960,4 which described the coadministration of L-tryptophan (the substrate of the rate-limiting enzyme, tryptophan hydroxylase [TPH], in the biochemical synthesis of serotonin) with a monoamine oxidase inhibitor (MAOI) inducing delirium. The first use of the term SS came 20 years later, describing the characteristic features in rats of tremor, rigidity, hypertonicity, hind-limb abduction, Straub tail, lateral head shaking, hyperactivity to auditory stimuli, myoclonus, generalised seizures, and various autonomic responses such as salivation, penile erection, and ejaculation.5 Two years later, the first contemporaneous case in humans was reported by Insel et al,6 followed by numerous case reports and reviews.7-11
The aim of this review is to summarise the pathophysiology of SS in relation to the 5-HT signalling pathway in the CNS, clinical features, diagnosis, management, and prognosis.
Epidemiology
Serotonin syndrome is observed across the full range of age groups, from neonates all the way through to the elderly, with an increasing incidence likely to represent the increasing use of serotonergic drugs in clinical practice.7,12,13 The percentage of adults taking antidepressants in the United States nearly doubled between 1999 and 2010, increasing from 6% to 10.4%.14 In 2016, the Toxic Exposure Surveillance System, which receives case descriptions from emergency departments, inpatient settings, and office-based practices, reported 54 410 incidences of exposures to selective serotonin reuptake inhibitors (SSRIs; 43% of which were single exposures), with 102 deaths (the ninth most common cause of fatality in drug overdoses in the United States in this period). This represented an 18% increase in cases between 2002 and 2016 and an 8% increase in the number of deaths.15-17 Large case series suggest that moderate SS occurs in approximately 15% of poisonings with SSRIs.18 A recent study found that close to half of U.S. Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) SS reports involved a single drug (depending on the diagnostic criteria used), which is, perhaps, a little surprising, given the common perception of a multidrug aetiology.19
The true incidence of SS, however, is unknown, as is the number of cases that are mild, moderate, or severe. There are a number of reasons for this: it is a relatively uncommon condition that cannot be easily picked up in randomised clinical control trials,20 and the condition is under-recognised and under-reported by physicians (85% of general physicians [GPs] were not familiar with the condition in one survey,21 and mild cases are often dismissed or self-limiting).20
Pathophysiology and Molecular Mechanisms
The serotonin signalling pathway
The cells of the raphe nuclei located in the midline of brainstem (from the midbrain to the medulla) are the source of neuronal 5-HT in the CNS.22 The 9 raphe nuclei (B1-B9) are centred in the reticular formation, and axons from the serotonergic neurons in this region form a neurotransmitter system that functionally affects most parts of the CNS. The rostral group of nuclei project into multiple cortical and subcortical structures contributing to the regulation of wakefulness, attention, affective behaviour (depression and anxiety), sexual behaviour, appetite, thermoregulation, and migraine.22 The caudal group project into the spinal regions and are involved in motor tone as well as nociception.22 In the periphery 5-HT, produced in the enterochromaffin (EC) cells of the GI tract, is involved in the regulation of GI motility as well a number of roles in vascular biology.22 These include control of blood pressure through vasoconstriction or vasodilation depending on the expressed receptor type in the vessel wall, as well as control of haemostasis and platelet function. Serotonin is secreted during platelet activation, playing an important role in their aggregation and concomitant vasoconstriction of the surrounding blood vessels during haemostasis. Platelets lack enzymes to synthesise serotonin,23 and are therefore reliant on taking up serotonin from the plasma via the serotonin transporter. Selective serotonin reuptake inhibitors can inhibit the uptake of serotonin by platelets leading to decreased aggregation responses and thereby increasing bleeding time.24 For this reason, SSRIs should be used in caution in those patients on anticoagulants or at higher risk of bleeding. Intriguingly, there is emerging evidence that SSRI treatment may lower myocardial infarction risk though their inhibition of platelet aggregation.25,26 The occurrence of disseminated intravascular coagulation in severe SS27 may be due to an exaggerated platelet aggregation response driven by excess peripheral serotonin.
The initial and rate-limiting step in the biosynthesis of 5-HT, the decarboxylation and hydroxylation of the essential amino acid tryptophan, is catalysed by TPH.28 There are 2 isoforms of this enzyme, TPH1 and TPH2, which are highly homologous but differ in their kinetic properties and their tissue distribution.29,30 Tryptophan hydroxylase 1, the source of most peripheral 5-HT, is predominantly expressed in the EC cells of the gut and other peripheral tissues, such as the pancreas, lung, and adipose tissue, though is also expressed in the pineal gland where 5-HT serves as the precursor molecule for melatonin biosynthesis.29,31,32 Tryptophan hydroxylase 2, the source of the central neurotransmitter pool of 5-HT, is predominantly expressed in the raphe projection neurons of the brainstem,29,33 though is also found in the myenteric neurons of the gut.32,34 Work with knockout animals suggests that TPH1 expression in the pineal gland does not significantly contribute to the central 5-HT pool,35 and similarly, TPH2 expression in the myenteric plexus does not significantly contribute to the peripheral 5-HT pool.36 Once synthesised from tryptophan, 5-HT is then stored in presynaptic vesicles until it is required for neurotransmission and released into the synaptic cleft. Serotonergic signalling is a tightly regulated process characterised by a combination of feedback loops, multiple reuptake mechanisms, and metabolising enzymes (Figure 1). Depolarisation of the presynaptic axon leads to the release of serotonin into the synaptic cleft, which leads to multiple downstream effects; activation of presynaptic serotonin autoreceptors inhibits exocytosis of further vesicles, while binding to postsynaptic 5-HT receptors effects neurotransmission. Serotonergic signalling is terminated primarily by the uptake of 5-HT from the synaptic cleft back into the presynaptic neuron. This is accomplished predominantly by the serotonin reuptake transporter protein (SERT) on the presynaptic membrane, though there is evidence that the plasma membrane monoamine transporter (PMAT) may also contribute.37 5-Hydroxytryptamine is then metabolised into 5-hydroxyindoleacetic acid (5-HIAA) primarily by monoamine oxidase A (MAO-A).
Figure 1.
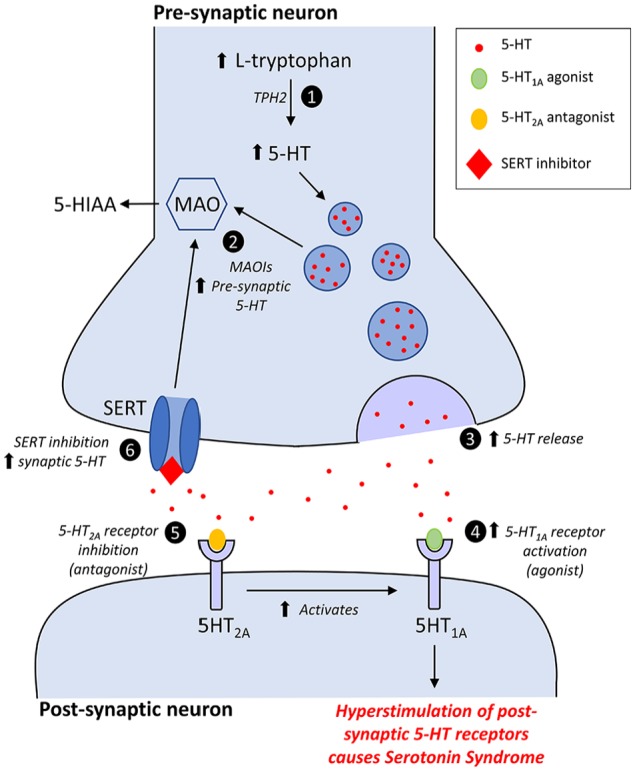
Mechanisms of serotonin syndrome: (1) Increased levels of L-tryptophan will lead to increased levels of endogenous 5-HT; a step catalysed by the enzyme tryptophan hydroxylase 2 (TPH2). (2) Increased presynaptic concentrations of 5-HT due to inhibition of serotonin metabolism by MAOIs. (3) Increased 5-HT release by drugs including amphetamines and their derivatives, cocaine, MDMA, and levodopa. (4) Direct or indirect activation of postsynaptic 5-HT1A receptors. (5) Direct or indirect antagonism of postsynaptic 5-HT2A receptors is thought to enhance the effect of 5-HT1A agonists. (6) Increased synaptic levels of 5-HT due to inhibition of the SERT by reuptake inhibitors such as SSRIs, and TCAs. MAOIs indicate monoamine oxidase inhibitors; MDMA, 3,4-methylenedioxy-methamphetamine; SERT, serotonin reuptake transporter protein; SSRIs, selective serotonin reuptake inhibitors; TCAs, tricyclic antidepressant.
There are at least 7 families of 5-HT receptors (5-HT1 to 5-HT7), some of which have multiple subtypes, resulting in a total of 14 structurally and pharmacologically distinct receptors.38
The 5-HT1 receptor family consist of 5 subtypes: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1e, and 5-HT1F receptors are G-protein-coupled receptors (GPCRs) that signal via the inhibition of adenylate cyclase or G-protein-sensitive K+ channels (note that the 5-HT1e receptor is still just recognised as a gene product, since no function has been attributed to this receptor in native tissue). While a number of clinically used drugs affect these receptors, the 5-HT1A receptor provides a pharmacological target for the anxiolytic agent, buspirone, which lacks potential for dependency. However, 5-HT1B/1D (and possibly 5-HT1F) receptors are targeted by the antimigraine ‘triptans’, examples of which include sumatriptan and zolmitriptan (which are also used for cluster headaches).
The 5-HT2 receptor family consists of 3 members: 5-HT2A, 5-HT2B, and 5-HT2C receptors that are excitatory GPCRs signalling via Gq to activate, for example, phospholipase C to generate Inositol 1,4,5-triphosphate (IP3) and diacyl glycerol. Many antipsychotic drugs at relevant clinical concentrations interact with 5-HT2 receptors, with antagonism of the 5-HT2A receptor likely to contribute to therapeutic effects (eg, risperidone). More recently (2012), the 5-HT2C receptor has been targeted selectively with the agonist, lorcaserin, to treat obesity by reducing food intake (although not yet approved in Europe).
The 5-HT3 receptor is a ligand-gated ion channel of the cys-cys loop class that conducts Na+/K+/Ca2+; this excitatory receptor is expressed predominantly by neurons and is responsible for initiating emesis following aggressive anticancer therapy (that releases copious quantities of 5-HT from the EC GI tract) such that antagonists such as ondansetron offer effective antiemetic treatment. The 5-HT3 receptor also mediates prokinetic activity of the GI tract – as occurs in irritable bowel syndrome (IBS) or carcinoid patients; for both patient cohorts, 5-HT3 receptor antagonists are beneficial to reduce diarrhoea, although currently, they are only indicated for IBS (and only in some territories like the United States and Japan).
The 5-HT4 receptor is a GPCR that promotes adenylate cyclase activity via Gs. The receptor increases gastric motility by increasing the activity of, for example, cholinergic neurons in the GI tract and is activated by some drugs used to reverse constipation (eg, prucalopride).
The 5-HT5 receptors are GPCRs and the family has 2 members: 5-HT5A and 5-HT5b receptors. The latter receptor is not functional in humans due to the insertion of a stop codon into the gene that results in a truncated protein. Little is known about the 5-HT5A receptor, and it is not actively targeted for therapeutic benefit.
The 5-HT6 and 5-HT7 receptors are both GPCRs that couple via Gs to promote cAMP synthesis via adenylate cyclase. There has been much interest in these as potential clinical targets, largely due to the known interaction of a wide range of antipsychotic and antidepressant drugs that antagonise these receptors. However, to date, no clinically used drug selectively targets either receptor.
Further structural and functional diversity occurs in 5-HT receptors through allelic polymorphisms, splice variants, and the formation of receptor heterodimers.34,39
Pharmacology of serotonin and cellular toxicity
Serotonin syndrome can result from agonism (either from increased concentrations of 5-HT or medications that act directly as receptor agonists), and/or antagonism, of varying combinations of the 5-HT receptor subtypes. Stimulation of the postsynaptic 5-HT1A and 5-HT2A receptors has been implicated in SS, but no single receptor is likely to be solely responsible. The classical view, mainly supported by evidence from animal studies (with few studies in humans), is that the life-threatening effects of SS, in particular, severe hypertonicity and hyperthermia, are primarily mediated by the activation of 5-HT2A receptors at higher serotonin concentrations.18 The 5-HT1A receptors, which have a higher affinity for 5-HT and are therefore likely to be nearly fully occupied at much lower extracellular 5-HT concentrations, possibly contribute to some of the milder symptoms including anxiety and hyperactivity.40 This may provide a pharmacological explanation for why severe SS is usually only seen with particular combinations of drug classes, in particular MAOIs (that increase 5-HT concentration in the synapse) and SSRIs (that modulate 5-HT receptor signalling at the postsynaptic membrane in addition to inhibiting serotonin reuptake via the SERT).18,41,42
Recently, a bioinformatics approach has demonstrated an association between second-generation antipsychotics (SGAs) and SS.43 Counter to the classical view that 5-HT2A agonism is a significant contributor to severe SS, the authors have suggested a potential role for 5-HT2A antagonism in concert with 5-HT1A agonism as a common mechanism that may explain the SGA-SS association. It is this effect on the serotonergic system that distinguishes the SGAs (‘atypical’ antipsychotics) from the older ‘typical’ antipsychotics (butyrophenones, diphenylbutylpiperidines, and phenothiazines) that predominantly antagonise D2 dopaminergic receptors and are not associated with SS. They postulate that in the presence of SERT inhibition, 5-HT levels are increased throughout serotonergic synapses, and 5-HT2A antagonism then shunts elevated levels of 5-HT to the co-localised 5-HT1A receptor.44,45 This selective activation of the 5-HT2A receptor not only appears to increase its sensitivity to 5-HT46-48 but may also enhance the effects of 5-HT1A agonists.49,50 In addition, there is evidence that 5-HT2A receptor antagonists may elevate 5-HT neurotransmission via a disinhibitory feedback loop, through inhibition of local gamma-aminobutyric acid (GABA) interneurons in the dorsal raphe nucleus.51 This association (and potential mechanistic explanation) is reinforced by a recent meta-analysis of SS cases, in which 3% (10/299) of cases were associated with SGAs as the decisive triggering factor. Eight of these cases occurred in combination with antidepressants and 2 in the context of an SGA swap. Interestingly, in this study, 45.4% of the SS cases had rigidity/hypertonicity, and 14.0% of cases had rhabdomyolysis and symptoms also commonly associated with neuroleptic malignant syndrome (NMS).20 Whether 5-HT2A receptor antagonism in concert with 5-HT1A agonism plays a role in all cases of SS or is the exception in the case of SGAs remains to be determined.
An additional question is whether other neurotransmitters play a role, particularly in the development of severe SS. A full review of the large body of evidence in this area is beyond the scope of this review, though it is important to emphasise that active research is ongoing, and many questions remain to be definitively answered. In animal models, it has been shown that 5-HT can cause the release of noradrenaline from the anterior hypothalamus, a neurotransmitter known to cause CNS hyperexcitability, which may correlate with the clinical outcome.52-54 Gamma-aminobutyric acid, N-methyl-D-aspartate (NMDA), and dopamine may also play a role through their recruitment, in addition to direct serotonin stimulation, in severe SS.52,55 A recent study using a systematic bioinformatics approach to analyse off-target effects of the drugs most commonly involved in SS supports the hypothesis that multiple neurotransmitter pathways may be recruited in addition to direct serotonin stimulation in severe SS.19 As well as highlighting the importance of SERT inhibition (46/71 serotonergic drugs interacted with this protein), there was a higher than expected association of SS cases in the triple receptor drug cohort (drugs that interact at SERT, norepinephrine transporter [NET], and muscarinic receptors).19 Off target receptor activity may help to explain the differences in the severity of the syndrome as well as the spectrum of clinical features. These findings raise the possibility that when the offending drug (or combination of drug classes) has (have) both antidopaminergic and serotonergic activity,20 the resulting toxidrome may share features of both SS and NMS.
Drugs commonly associated with SS
A wide range of drug types and combinations have been implicated in SS, with the final common pathway thought to involve a net increase in serotonergic neurotransmission. The main drug classes classically implicated in SS can be divided into serotonin precursors, inhibitors of serotonin reuptake from the synaptic cleft, inhibitors of serotonin metabolism, direct serotonin receptor agonists, and drugs that sensitise serotonin receptors (Table 1). As discussed in the previous paragraph, there is evidence that serotonin antagonism (in particular 5-HT2A antagonism) may also play a role, based on the observed association between SGAs and SS.43
Table 1.
Drugs associated with serotonin syndrome.
| Mechanism | Drugs associated with SS |
|---|---|
| Increased 5-HT synthesis | Dietary supplements: Tryptophan |
| Inhibition of 5-HT metabolism | MAOIs: include safinamide, selegiline, rasagiline, phenelzine, tranylcypromine, isocarboxazid, moclobemide, linezolid, tedizolid, methylene blue, procarbazine, and Syrian rue [Peganum harmala and harmine] |
| Herbal supplements: St. John’s wort [Hypericum perforatum] | |
| DNRIs: include buspirone | |
| Increased release of 5-HT | Drugs of abuse: cocaine, MDMA (‘Ecstasy’) |
| Amphetamine and derivatives: include phentermine, fenfluramine, and dexfenfluramine | |
| Cold remedies: dextromorphan | |
| Activation of 5-HT1 receptors | DNRIs: buspirone |
| Triptans: include almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, and zolmitriptan | |
| Ergot derivatives: include ergotamine and methylergonovine | |
| Opiates: fentanyl and meperidine | |
| Drugs of abuse: LSD | |
| Antidepressants/mood stabilisers: mirtazapine, trazodone, and lithium | |
| Antagonism of 5-HT2A receptors | Second-generation antipsychotics: include quetiapine, risperidone, olanzapine, clozapine, and aripiprazole20,43 |
| Inhibition of 5-HT uptake from synaptic cleft | Amphetamine and derivatives: include phentermine, fenfluramine, and dexfenfluramine |
| Drugs of abuse: cocaine and MDMA (‘Ecstasy’) | |
| SSRIs: include citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline | |
| SNRIs: include venlafaxine, duloxetine, milnacipran, and desvenlafaxine | |
| TCAs: amitriptyline, amoxapine, clomipramine, desipramine, doxepin, imipramine, maprotiline, nortriptyline, protriptyline, and trimipramine | |
| DNRIs: include buspirone | |
| Opioids: include levomethorphan, levorphanol, meperidine, methadone, pentazocine, pethidine, tapentadol, and tramadol | |
| 5-HT3 receptor antagonists: ondansetron, and granisetron | |
| Antihistamines: chlorphenamine | |
| Herbal supplements: St. John’s wort [Hypericum perforatum] | |
| Cold remedies: dextromorphan |
Abbreviations: 5-HT, 5-hydroxytryptamine; SS, Serotonin syndrome; MAOI, monoamine oxidase inhibitor; DNRI, dopamine-norepinephrine uptake inhibitor; MDMA, 3,4-methylenedioxy-methamphetamine; LSD, lysergic acid diethylamide; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin-norepinephrine reuptake inhibitor; TCA, tricyclic antidepressant.
Pharmacokinetic drug interactions are also implicated through the inhibition of the cytochrome P450 pathway, a pathway that SSRIs themselves inhibit (in particular CYP2D6 and CYP3A4.56,57 At least 25 serotonergic drugs are metabolised by the cytochrome P450 pathway,19 and a recent study has shown that 50% of the top 20 drugs associated with SS have known pharmacokinetic interactions in which coadministration of cytochrome P-450 inhibitors may elevate drug concentrations to toxic levels. These drugs collectively were shown to participate in > 70% of all the reported SS reports in their study.19 Examples include the concomitant use of SSRIs and tramadol,12,58 SSRIs and ciprofloxacin,59 as well as of citalopram and fluconazole.60
Although SS has been described after overdose of a single drug,61 and occasionally from increasing therapeutic doses in susceptible individuals, this only usually results in mild to moderate SS.4,13,62 Severe SS usually only occurs with the concomitant administration of 2 or more serotonergic drugs (even at therapeutic doses) (Table 2), with the combination of serotonergic drugs with MAOIs being especially dangerous, causing serious adverse outcome including death.8,41,42
Table 2.
| Drug class | Drug combinations |
|---|---|
| MAOIs | MAOIs + SSRIs or SNRIs or TCAs or opiates Imipramine + tranylcypromine Phenelzine + meperidine Methylene blue + clomipramine or paroxetine |
| SSRIs | SSRIs + MAOIs or TCAs or SNRIs or opiates or triptans Fluoxetine + carbamazepine or phentermine or fentanyl |
| SNRIs | SNRIs + TCAs or MAOIs or opiates or triptans Venlafaxine + lithium or calcineurin inhibitors or mirtazapine or tranylcypromine |
| Other antidepressants | Mirtazapine + SSRIs Trazadone + amitriptyline + lithium |
| Opiates | Opiates + MAOIs or SSRIs or SNRIs or triptans |
| Cold remedies | Dextromorphan + SSRIs or TCAs or atypical antipsychotics |
| Atypical antipsychotics | Olanzapine + citalopram and lithium Risperidone + paroxetine or fluoxetine |
| Antibiotics/antifungals | Linezolid + SSRIs or tapentadol Fluconazole + citalopram Ciprofloxacin + methadone + venlafaxine |
Abbreviations: MAOI, monoamine oxidase inhibitor; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin-norepinephrine reuptake inhibitor; TCA, tricyclic antidepressant.
Taken together, these observations are consistent with the hypothesis that SS is a dose-dependent spectrum of adverse effects mediated by a combination of elevation of endogenous intrasynaptic or extrasynaptic serotonins and direct or indirect activation of 5-HT receptors.63 However, the fact that different patients present with SS at differing drug dosages and/or combinations suggests that there may also be underlying genetic and pharmacodynamic factors that modulate susceptibility.11 Genetic factors could include polymorphisms in the CYP450 pathway,64 the SERT gene,65 as well as the 5-HT2 receptor.18,13,66
The role of novel psychoactive substances in SS
Novel psychoactive substances (NPSs), commonly described as novel, designer, or synthetic drugs,67 are a growing public health concern due their increasing availability and use. They are often synthetic derivatives and analogues of known compounds currently prohibited under current drug laws, such as amphetamines and cannabis.68 The ease of synthesising and speed of technological innovation has led to an explosion in the number of newly identified NPS, creating significant challenges to the regulatory authorities as well as to the research community investigating the pharmacology of these agents.69-72 In 2014, 101 newly identified NPSs were reported to the European Union early warning system,71 and by 2017, the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) was monitoring more than 620 NPSs,73 with 1 new NPS being identified each week. There is currently limited population-based data detailing NPS use, and a wide range of estimates based on the data that does exist. A 2014/15 Crime Survey for England and Wales estimated that 0.9% of 16 to 59 year olds are users, with this figure rising to 2.8% in the 16- to 24-year-old age group.74 This compared to 6.7% for cannabis, 2.4% for cocaine, 1.7% for 3,4-methylenedioxy-methamphetamine (MDMA), and 0.6% amphetamines.74 Another annual survey reported that 10% of United Kingdom and 22% of Irish 15 to 24 year olds had experimented with NPS.75 A recent review of the presence of NPS in HM Coroners or Police Forces cases in the United Kingdom, showed that over a 2-year period, NPSs were detected in 203 cases; the presence of multiple NPSs and/or other stimulants was a particular feature in some cases, and of the drug deaths (58% of total cases), 7% were solely due to an NPS.76
Classification of these compounds can be based either on their psychotropic effects (stimulant, empathogen/enactogen, hallucinogen, and dissociative)68,77 or based on their chemical family (phenethylamines, amphetamines, cathinones, piperazines, pipradrols/piperidines, aminoindanes, benzofurans, and tryptamines).78 Most of these drugs interact with the monoamine neurotransmitter transporters (dopamine transporter [DAT], SERT, NET), in the CNS to varying degrees altering levels of dopamine, serotonin, and noradrenaline, and therefore, in overdose can produce a range of overlapping toxidromes (serotonergic, sympathomimetic, and so on) depending on their particular receptor specificities. The relative dopaminergic to serotonergic properties in vitro (dopamine/serotonin transporter inhibition ratio) of an NPS have been postulated as a useful marker for its potential clinical psychotropic and acute toxic effects;79 MDMA-like agents are associated with a high serotonin:dopamine transporter-inhibition ratio, whereas more pure amphetamine-like agents have a higher dopamine:serotonin transporter-inhibition ratio.68 This ratio, however, does not capture the full complexity of action of these NPSs, not least because it does not account for the noradrenergic effects that can be a predominant feature in particular with the amphetamine, cathinone, and aminoindane drug classes.
The empathogenic NPSs are of particular interest in relation to SS, given that the effects of MDMA, the prototypic empathogen, are generally considered to depend on its serotonergic effects (driving increased 5-HT efflux into the synaptic cleft).68,80 3,4-Methylenedioxy-methamphetamine has been consistently associated with serotonergic toxicity and SS.81-83 In addition, MDMA and other amphetamine-like drugs such as fenfluramine, are direct agonists of the 5-HT2B receptor and may cause fibrosis in the periphery (eg, cardiac valvulopathy). Therefore, NPS drug classes (cathinones, aminoindanes, piperazines, and phenylethylamines) that have similar empathogenic effects to MDMA are likely to similarly lead to increased levels of serotonin, and a similar risk of SS at higher doses. In support of this, there are numerous case reports, as well as toxicity studies in animal models, documenting the development of SS with abuse of those drug classes with a high serotonin:dopamine transporter-inhibition ratio (cathinones,84,85 phenylethylamines,67,86-90 aminoindanes,91-93 as well as mixed serotonergic/sympathomimetic toxidromes in those where the dopamine:serotonin transporter-inhibition ratio is higher).77,86,94
The difficulty in identifying the true levels of usage of these NPSs, as well the associated clinical presentation in acute toxicity, is highlighted in a recent study that reviewed what data are currently being collected in Europe on emergency department presentations with acute toxicity related to NPSs and classical drugs of abuse.95 This revealed that of the 27 countries included, more than half (52%) did not perform any registration of such data, while only 37% collected systematic clinical data on NPS at a national level. The lack of knowledge about these NPSs is reflected in the lack of confidence among the health care team in managing acute NPS toxicity in the emergency setting.96 Current research into the effects of these NPSs, however, suggests that toxicity associated with these agents broadly falls into 3 broad groups similar to that seen with classical drugs (stimulant, hallucinogenic, and depressant). Future training should therefore focus on recognising and managing the particular toxidrome(s) that an individual presents with, rather than the particular agent (which is often never identified) that the patient has taken.94,97 The reader is directed to the recent review on the practical management of toxicity associated with NPS.94
Clinical Features
Given that the diagnosis of SS is made solely on clinical grounds, a detailed history and comprehensive physical and neurological examinations are essential. The presentation is highly variable ranging from mild symptoms to a severe life-threatening syndrome, a spectrum that likely reflects a combination of the degree of excess serotonergic activity in the CNS, or the particular 5-HT receptor subtype(s) that is (are) activated (directly or indirectly). Given the difficulty in determining the exact point at which serotonergic signs associated with therapeutic drug administration meet the criteria for SS, has led some experts in the field to prefer the use of the term serotonin toxicity, as they feel this more accurately represents the wide range of clinical features of varying severity.18,41 Traditionally, it is said that, in contrast to the onset of NMS, symptoms in SS usually begin within 24 hours of ingestion of the causative agent(s) and that most patients seek medical advice within 6 hours.11 However, these claims appear to be based on one review of 41 cases of SS from 1995 to 1999,12 and in a more recent meta-analysis, just more than 60% presented within 6 hours of ingestion (usually where pro-serotonergic drugs were administered quickly or in large doses including overdose), while around one quarter of patients presented later than 24 hours (usually when medication were more gradually titrated and/or cross-tapered).20 This temporal disparity may depend on the context in which the syndrome occurs, as well as pharmacokinetic and dynamic factors; an example would be the delayed onset of linezolid-associated SS in the elderly.98
The classic triad of clinical features (Figure 2) are altered mental status (including anxiety, agitation, and confusion), autonomic nervous system overactivity (including diaphoresis, tachycardia, hyperthermia, hypertension, vomiting, and diarrhoea), and neuromuscular hyperactivity (muscle rigidity, hyperkinesis including myoclonus and tremor, hyperreflexia, and bilateral Babinski sign).12 The acute onset of these features should alert the physician to the potential diagnosis, and trigger a search for a toxic cause, while also excluding other mimics such as encephalitis (infectious or autoimmune), alcohol and/or drug withdrawal, epileptic, and nonepileptic seizures.8 In milder cases, the patients are usually normothermic, with mild autonomic symptoms and neuromuscular signs. In moderate SS, in addition to hyperthermia (>40°C), patients usually develop eye movement abnormalities, agitation, pressured speech, and hypervigilance. Patients may also startle easily or develop an unusual movement disorder characterised by repetitive head rotation with the neck held in moderate extension.7 In severe cases, as well as the above features, patients usually have a temperature greater than 41.1°C, haemodynamic/autonomic instability, hyperactive bowel sounds, delirium, and muscle rigidity. Complications in severe cases include seizures, renal failure, metabolic acidosis, rhabdomyolysis, disseminated intravascular coagulation, acute respiratory distress syndrome, respiratory failure, and even death.7 Interestingly, the hyperreflexia and clonus are usually more prominent in the lower limbs,7 and this gives a clue as to the underlying diagnosis.
Figure 2.

The spectrum of clinical features in serotonin syndrome. GCS indicates Glasgow Coma Scale.
Source: Buckley et al.8
Diagnosis
Serotonin syndrome is a diagnosis of exclusion with no single diagnostic test that confirms the syndrome.7,11,99 It is an abstract construct combining various concepts, clinical signs, and symptoms that aims to relate signs of CNS hyperexcitability with purported drug-induced serotonin excess.20 Given the protean ways that CNS hyperexcitability can present, combined with the ability of multiple nonserotonergic pathways to manifest similar symptoms, makes it difficult to establish a gold standard to confirm SS. A plausible diagnosis of SS can therefore only be made when there is a high Bayesian prior probability (high index of clinical suspicion) in the setting of starting, increasing (or overdose) of a relevant serotonergic drug, or shortly after a second serotonergic drug is added.8 Linked to this, it is critically important that there is a strict definition for what is classed as a serotonergic drug; namely, it causes a generalised increase in endogenous 5-HT and/or activates 5-HT receptors directly or indirectly, as well as producing clinical signs that are specific for SS.18
Therefore, a diagnosis of SS is entirely clinical, based on a careful history and examination, with a detailed enquiry about the use of prescription, over-the-counter, and illicit drugs (stimulants such as cathinones and other synthetic stimulants, ecstasy, amphetamines, or cocaine), as well as any dietary supplements (such as St John’s wort, ginseng, tryptophan, and pharmaceutical adulterants in appetite suppressants).7,8 The rate of change of symptoms from onset, as well as development of new symptoms should also be reviewed. The presence of certain comorbidities, including long-term pain and depression, should alert to the clinician to the possibility of the use of serotonergic drugs,11 and the presence of renal disease may predispose those on certain SSRIs (such as sertraline) to developing SS.100 The clinical examination should focus on eliciting any signs of muscle rigidity, increased deep-tendon reflexes, as well as autonomic signs such as the presence of diaphoresis, increased bowel sounds, and mydriasis.7 There must be a high index of suspicion for the diagnosis in patients with any combination of the aforementioned classical clinical features and a supporting drug history, so treatment can be rapidly initiated to prevent the morbidity and mortality associated with this condition. Misdiagnosis can be an issue however; in severe cases, SS can be confused with NMS, while the nonspecific symptoms in mild cases can be confused with other causes such as infection. Symptoms such as anxiety and akathisia can be misattributed to the patient’s mental state, while hypertension, diarrhoea, or tremor can easily be dismissed as part of a systemic or metabolic disorder unrelated to drug therapy.101
There are 3 diagnostic classification systems available, the Sternbach (SC), Radomski (RC), and Hunter Serotonin Toxicity Criteria (HSTC), all of which try to reflect symptoms and symptom constellations that are felt to be characteristic of serotonin toxicity and therefore SS. The most recent and commonly used criteria are the HSTC, which are set of decision rules that focus on the classic features of generalised clonus (inducible, spontaneous, and ocular), agitation, diaphoresis, tremor, and hyperreflexia41 (Figure 3). Spontaneous clonus is characterised by rhythmic, large muscle contractions (which helps to differentiate it from myoclonic jerks), and as is often triggered by minor movement. Ocular clonus refers to abnormal involuntary, fine or coarse, oscillatory eye movements that can either be continuous or triggered by eye movement.8 Rarer eye movements seen include periodic alternating gaze deviation (so-called ‘ping-pong’ gaze).8
Figure 3.

The Hunter Serotonin Toxicity Criteria: for diagnosing serotonin syndrome. Note that the requirement for the presence of some form of neuromuscular excitation, the sine qua non for a diagnosis of serotonin syndrome. *The presence of temperature ⩾38.5°C and/or marked hypertonia or rigidity (especially truncal) indicates severe SS with a risk of progression with respiratory compromise.
Compared to the original SC the HSTC are simpler to use, more sensitive (84% vs 75%), and marginally more specific (97% vs 96%).41 A recent study analysing SS using a systematic bioinformatics approach applied to the FAERS database, found that the number of identified SS cases varied considerably depending on which diagnostic criteria were used.19 The SC identified an additional 1140 cases suggesting that they may indeed be less specific compared to the HSTC and likely picks up milder cases where off-target neurotransmitter effects contribute to a broader nonspecific toxicity presentation.19 It should be noted, however, that the purported superiority of the HSTC is based on one study only.20 Other criticisms of the HSTC include their validity/generalisability to nonoverdose cases, given the criteria were derived solely from single SSRI overdoses; that a percentage of the cases in the derivation data set was also present in the test data set in the original study, which could potentially lead to an overestimation of the validity of the criteria20; and finally that they may not perform well in patients with concomitant neurological disease. The cardinal signs of hyperreflexia and clonus may be masked, both in patients with peripheral neuropathy,102 as well as those with severe SS who have substantial muscle rigidity.7
In addition to truncal rigidity, in severe, life-threatening cases of SS rapidly rising temperatures above 38°C and peripheral hypertonicity are usually key features. Investigations are of little diagnostic value, though are useful to help identify complications of hyperthermia and muscle rigidity (rhabdomyolysis, disseminated intravascular coagulation, and multiorgan failure); abnormalities that are often seen include elevated creatine kinase, liver enzymes, deranged renal function, and low bicarbonate levels.11 It is important to also exclude cardiological effects of drugs in overdose with an electrocardiogram (ECG), as well as alternative diagnoses such as encephalitis and cerebral vasculitis, where relevant, with magnetic resonance imaging (MRI), electroencephalogram (EEG), and lumbar puncture.18 In intentional overdoses, a toxicology screen should be sent including for paracetamol and salicylates. There is no role for testing of blood concentrations of serotonin, as there is no correlation with clinical severity.12
Differential diagnosis
The differential diagnosis for SS includes other toxidromes such as NMS, malignant hyperthermia, and anticholinergic toxicity (Table 3), as well conditions such as meningitis, encephalitis (in particular autoimmune encephalitis), central hyperthermia, and heatstroke.
Table 3.
Differentiating serotonin syndrome from other toxidromes.
| Toxidrome | Causative agent | Onset, resolution | Vital signs | Pupils | Mental state | Other clinical features |
|---|---|---|---|---|---|---|
| Serotonin syndrome | Serotonergic drugs | Sudden <24 h Most resolve with 24 h with treatment (though 25% develop symptoms after >24 h) |
Hyperthermia (>41.1°C), tachycardia, hypertension, and tachypnoea | Mydriasis | Delirum, agitation, and coma | Neuromuscular hyperactivity (tremor, myoclonus, hyperreflexia, and clonus), diaphoresis, and hyperactive bowel sounds |
| Neuroleptic malignant syndrome | Dopamine antagonists and dopamine withdrawal | Slower onset (days to weeks) Up to 10 days to resolve with treatment |
Hyperthermia (>41.1°C), tachycardia, hypertension, and tachypnoea | Normal or mydriasis | Delirium, agitation | Neuromuscular hypoactivity (‘lead-pipe’ rigidity and bradykinesia) Hypoactive bowel sounds |
| Anticholinergic toxicity | Anticholinergic agents | Sudden <24 h Resolves hours to days with treatment |
Hyperthermia (usually <38.8°C), tachycardia, hypertension (mild), and tachypnoea | Mydriasis | Hyper-vigilance, agitation, hallucination, delirium with mumbling speech, and coma | Normal muscle tone and reflexes Dry flushed skin and mucous membranes Hypoactive bowel sounds Urinary retention Hyperkinesis (myoclonus, choreoathetosis, and picking behaviour) Seizures (rare) |
| Malignant hyperthermia | Inhalational anaesthetics and depolarising muscle relaxants (succinylcholine) | Very sudden (mins to hours) Resolves 24-48 h with treatment |
Hyperthermia (can be as high as 46°C), tachycardia, hypertension, and tachypnoea | Normal | Agitation | Rigour-mortis like rigidity Hyporeflexia Hypoactive bowel sounds Rising end-tidal CO2 Mottled skin with flushing and cyanosis |
Neuroleptic malignant syndrome is a fulminant and life-threatening idiosyncratic drug reaction to therapeutic doses of dopamine antagonists. Although the exact pathophysiology is not completely understood, central dopaminergic blockade is thought to play a pivotal role, with sympathoadrenal dysfunction also potentially involved.103 Although NMS is often confused with SS, a detailed history focusing on time course of onset and medication usage, examination findings and clinical course allow differentiation of the 2 conditions. While SS is characterised by neuromuscular hyperactivity (hyperreflexia with clonus, myoclonus, and tremor), in NMS, there is usually decreased neuromuscular activity with extrapyramidal features (hypokinesia and lead-pipe rigidity). Importantly in NMS, hyperreflexia and clonus are rarely feature104 something reflected in the primacy of clonus in establishing a diagnosis of SS in the HTSC.41 Established orthodoxy is that NMS develops over days to weeks while SS develops more rapidly (usually within 24 h), though this has been challenged recently by a group that have shown that 25% of patients in their study developed SS later than 24 hours after ingestion of the causative agent.20 In those patients where serotonergic medications were gradually titrated and cross-tapered, the percentage presenting after 24 hours increased to more than 60.4%. Hyperthermia, muscle rigidity, altered mental status, rhabdomyolysis, and deranged blood tests (leucocytosis, elevated hepatic transaminases, elevated creatine kinase, and metabolic acidosis) can be seen in severe cases of both syndromes. Care must therefore be taken in weighting the diagnosis too heavily based these findings as well as speed of onset.
In anticholinergic toxicity, muscular tone and reflexes are normal, with hypoactive bowel sounds and dry, erythematous skin; all features that help distinguish it from SS. They classically present in addition with hyperthermia, agitation, altered mental status, dry mucous membranes, and mydriasis.18
Malignant hyperthermia occurs in genetically susceptible individuals within minutes of exposure to inhalational halogenated anaesthetic agents and depolarising muscle relaxants (such as succinylcholine). The syndrome is characterised by hyperthermia, tachycardia, rigour-mortis-like muscle rigidity, metabolic acidosis, and increasing concentrations of end-tidal carbon dioxide.105
With regards to the other differential diagnoses, SS can usually be distinguished from them based on increased neuromuscular activity; patients with CNS infections or sympathomimetic toxidromes are less likely to exhibit this increased activity.106
Management
The 2 main components for optimal management of SS are first good supportive care (especially in severe cases) and second risk assessment and close monitoring of those with mild to moderate SS to avoid progression to severe life-threatening SS.18 The first step is to identify and stop the offending serotonergic medication(s), with supportive care to stabilise vital signs (maintenance of oxygen saturations, intravenous fluids, and continuous temperature cardiac monitoring), sedation with benzodiazepines (such as diazepam or midazolam), and in more severe cases, consideration of serotonin antagonists alongside muscle paralysis. When paralysis is indicated, strong consideration should be given to the initiation of continuous EEG, if available, to assess for electrographic seizure activity that cannot be clinically manifest while chemically paralysed. Alongside early recognition, it is essential the clinician recognises the potentially rapid rate of progression of SS and instigates immediate aggressive treatment in those who were previously being treated conservatively that deteriorate.7,13,107
The intensity of treatment depends on the severity of the toxicity (Figure 2). In mild cases, discontinuation of the offending medication, sedation with benzodiazepines and supportive care is often sufficient, whereas in moderate SS, there needs to be more aggressive treatment of haemodynamic/autonomic instability with consideration of adding in a serotonin antagonist (such as cryproheptadine). Severe SS is a medical emergency often complicated by severe hyperthermia, rhabdomyolysis, disseminated intravascular coagulation, and acute respiratory distress syndrome (ARDS), requiring sedation, muscle paralysis, and intubation in the intensive care setting.106 Most cases of SS usually resolve with 24 to 72 hours, though this may take longer in those on drugs with longer elimination half-lives, active metabolites, or a longer duration of action.7 Irreversible MAOIs carry the greatest risk with symptoms lasting for several days, while patients can still be at risk of SS up to several weeks after the discontinuation of SSRIs. Fluoxetine is a particular risk, with a half-life as long as 7 days after long-term admission and its metabolite norfluoxetine up to 2.5 weeks.108
Agitation
Chemical restraint is preferred to sedating agitated patients, due to the risk of severe lactic acidosis and hyperthermia from physical restraints causing isometric muscle contractions. Of the sedating agents, benzodiazepines are commonly used first line; not only do they control agitation, but they also blunt the hyperadrenergic component, helping to correct mild increases in blood pressure and heart rate.107 There is evidence in animal models of SS that pretreatment with diazepam and chlormethiazole prolongs survival,55 though there is no evidence currently in humans. Clinicians should titrate the benzodiazepine to clinical effect, with the goal of patient sedation and maintenance of normal vital signs.106
Autonomic instability
In severe SS, patients can often have large and rapid changes in heart rate and blood pressure, making management difficult.7 If benzodiazapines are not sufficient to lower blood pressure, then patients should be treated with short-acting agents such as esmolol or nitroprusside.41 Longer acting agents such as propranolol, which can cause hypotension and mask tachycardia (a sign which is useful in monitoring patient response and improvement), should be avoided.7,41 Hypotension can be a particular problem in those patients who have taken MAOIs, and preferred treatment in this situation is with low doses of direct-acting sympathomimetic amines (such as norepinephrine, epinephrine, or phenylephrine).7 Indirect sympathomimetic agents such as dopamine should be avoided, due to theoretical risk of an exaggerated haemodynamic response in the setting of MAOI inhibition.
Hyperthermia
In severe SS, preventing hyperthermia and consequent multiorgan failure is a key goal. Lowering temperature in animal models has also been shown to indirectly downregulate 5-HT2A receptors in the CNS and reduce overall serotonin levels.109 Given the increase in body temperature in these patients is due to an increase in muscular activity, rather than an increase in the hypothalamic set point (that is seen in pyrexia), antipyretic agents have little effect and are not indicated.7,48 Key components of management include sedation (oral diazepam or a midazolam infusion) to reduce muscle activity, active cooling (including fans, water sprays, ice packs, cooled crystalloids, or cooling blankets), and in severe cases muscle paralysis and ventilation on the intensive care unit (ITU). In the severe cases where the temperature is greater than 41.1°C, patients should be immediately paralysed and intubated, with nondepolarizing agents such as vecuronium to maintain paralysis. It is important for clinicians to be aware that in rare cases succinylcholine can worsen rhabdomyolysis and put the patient at risk of hyperkalaemic-induced arrythmias.7 Fentanyl should also be avoided due its serotonergic effect. Adequate sedation with a benzodiazepine while the patient is paralysed should be maintained. The optimum timing for stopping of neuromuscular paralysis is not currently known, but there is evidence from case reports that premature termination can lead to recrudescence of the hyperthermia7; as such, the clinician should anticipate this rebound and have a low threshold for restarting muscle paralysis.
Antidote(s)
If the use of the aforementioned strategies fails to control the agitation and correct vital signs, then there is some evidence for the use of serotonin antagonists. Animal studies have suggested that 5-HT2A receptor antagonists may reduce hyperthermia and other severe manifestations of SS.18,109,110 Both chlorpromazine and cyproheptadine in high doses have been used, though given chlorpromazine can cause severe hypotension, as well as case reports that antipsychotics can themselves precipitate SS, means cyproheptadine is usually favoured first line.106 There is some evidence from case reports that support its role in reducing symptoms in mild to moderate SS, though there its role in severe SS is less clear.111-115 Cyproheptadine is a histamine-1 receptor antagonist with nonspecific 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT6, and 5-HT7 receptor antagonistic properties as well as some weak anticholinergic effects.111 The purported role of 5-HT2A antagonism in mediating cyproheptadine’s beneficial effect is interesting, given that there appears to be an association between 5-HT2A antagonists themselves in the form of SGAs and the risk of SS.43 Given that it antagonises a number of different serotonin receptors in addition to 5-HT2a, it may be that concomitant antagonism of 5-HT1A receptors (as well as others) at higher doses accounts for this apparent contradiction, though evidence for this is currently lacking.
An initial loading dose of 12 mg orally or crushed via a nasogastric tube is recommended, followed by 2 mg every 2 hours until clinical improvement is seen.11 An alternative dosing regimen involves switching to 8 mg every 6 hours once symptoms are controlled.7 Cyproheptadine is only available in an oral form, but the tablets may be crushed and given through a nasogastric or orogastric tube. It can cause a decrease in vascular tone with a consequent transient hypotension, though this is usually responsive to intravenous fluids.
Historically, therapeutics targeting the serotonergic system centrally have been focused on 5-HT transporters and receptors, to modulate conditions such as depression, migraine, or psychosis.116-118 There is currently a lack of novel therapeutics targeting serotonin synthesis in the CNS, drugs which if developed, might offer a therapeutic option in SS. An interesting avenue of research comes from work in the carcinoid syndrome. Carcinoid syndrome describes a constellation of clinical symptoms including diarrhoea, wheezing, bronchoconstriction, cutaneous flushing, cardiac valve disease, and mesenteric fibrosis.119 It is usually caused by neuroendocrine tumours, a clinically and genetically heterogeneous group of GI, bronchial, and more rarely pancreatic, kidney, or ovarian tumours. These tumours can release a cocktail of vasogenic amines (predominantly 5-HT), as well as prostaglandins, and other polypeptides which induce the clinical syndrome. Gastrointestinal NETs account for up to 80% of cases of carcinoid syndrome usually in the setting of hepatic metastases.120 The inability of 5-HT to cross the blood brain barrier opens up the opportunity to pharmacologically target the central and peripheral 5-HT systems independently. Since the functional classification of TPH isoforms in 2003,28 TPH1, the peripheral isoform, has been established as a pharmacological target for the treatment of peripheral 5-HT-associated diseases such as carcinoid. Early inhibitors developed to treat carcinoid syndrome, the low molecular weight phenylalanine analogues p-chlorophenylalanine (fenclonine, PCPA) and p-ethynylphenylalanine (PEPA), showed promise in terms of lowering peripheral 5-HT. However irreversible inhibition of TPH2 centrally, limited their therapeutic use, due to the significant reduction in brain 5-HT concentrations.121-123 This off-target effect may however offer an opportunity to develop specific, reversible TPH2 inhibitors that could play a role in treatment of SS, through inhibition of synthesis leading to depletion of endogenous CNS serotonin. More recently, reversible TPH inhibitors such as telotristat (Masib) derived from administration of the relatively inactive prodrug telotristat ethyl have been developed. Telotristat is indicated in combination with somatostatin analogues (eg, octreotide) for the treatment of diarrhoea in carcinoid patients. Due to significant active-site structural homology, telotristat inhibits both TPH1 and TPH2 with similar affinity in vitro, yet the inability to penetrate the blood-brain barrier prevents reduction in central serotonin levels.28
Prognosis
As emphasised previously, the SS is a spectrum of dose-dependent clinical features ranging from very mild to life-threatening severe toxicity. Prognosis is generally favourable if the syndrome is recognised early, the causative agent is stopped, and complications are treated. This requires the clinician to have a high clinical suspicion for SS, and even if the diagnosis is unclear, discontinue any serotonergic agents and start supportive care. In severe cases, aggressive management on ITU is required, whereas those with moderate features need admitting for observation and vital system monitoring until symptoms resolve, with a low threshold for escalating management should there be any early signs of deterioration. These patients can usually be monitored for up to 6 hours, and if vital signs and mental state remain normal, with no evidence of neuromuscular hyperexcitability (in particular clonus or hyperreflexia), then they can be discharged home with close follow-up.106 In most cases of SS, symptoms usually resolve within 24 hours of stopping the drug, though in a minority of cases where the drug has active metabolites or a longer half-life, symptoms can last for longer than this.7 In milder cases, it is necessary for the clinician, in close collaboration with the patient, to weigh up risks and the benefits to decide whether the patient should restart the serotonergic agent (preferably at a lower dose).
Prevention
Prevention of SS requires a multitiered approach including education of both physicians and patients regarding potent serotonergic drug interactions, and the clinical signs and symptoms of SS to look out for, modification of prescribing practices to integrate automated e-prescribing algorithms to flag potential drug interactions, and ongoing pharmacogenetic research.
Although there are numerous systematic reviews focused on identifying which drug interactions can precipitate severe SS,63,124-128 there has been a proliferation in spurious drug associations, in part driven by the nonspecific clinical features in milder forms of the condition, and a (some might argue too) broad definition of what entails a serotonergic agent.8 Currently, clinicians prescribing an SSRI, or patients looking for information online, can be presented with up to 1000 interacting drugs, with a significant number of these warning of the potential for SS. While some of these interactions are potentially lethal and should be avoided (for example, SSRIs and MAOIs),8 many are likely to only result in minor effects, and for some drugs (including carbamazepine, triptans,127 most tricyclic antidepressants [TCAs], and atypical antidepressants),63,129 there is no or little evidence to support precipitating SS with coadministration. For the diagnosis of SS to be clinically meaningful, if the classification of a serotonergic agent is broadened to include any agent that has any modulatory effect on the serotonergic system in the CNS, then the clinical features need to be highly specific for the condition. Conversely, if we accept that the clinical features in the current criteria do not meet this threshold of specificity, then there must be evidence with a particular drug, or combination of drugs, that they directly lead to excessive serotonergic activity18 rather than simply a poorly defined modulatory action on the serotonergic pathway. Ongoing drug surveillance to identify potent serotonergic effects and integration of the results into physician and patient education is also critical, both to maintain awareness and also to keep these groups abreast of developments in the field.21 Patients also need to be aware that over-the-counter medications and herbal remedies, such as St John’s wort, can have serotonergic activity and can lead to serious interactions with prescribed serotonergic drugs such as SSRIs and MAOIs.8
Footnotes
Funding:The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by QEHB Charity, Birmingham, UK.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: WJS, LJH, ACW and NMB drafted, proofed and submitted the final manuscript.
ORCID iDs: William Johnson Scotton  https://orcid.org/0000-0003-0607-3190
https://orcid.org/0000-0003-0607-3190
Lisa J Hill
https://orcid.org/0000-0001-8431-7029
References
- 1. Eadie MJ. Convulsive ergotism: epidemics of the serotonin syndrome. Lancet Neurol. 2003;2:429-434. [DOI] [PubMed] [Google Scholar]
- 2. Rapport MM, Green AA, Page IH. Serum vasoconstrictor, serotonin: isolation and characterization. J Biol Chem. 1948;176:1243-1251. [PubMed] [Google Scholar]
- 3. Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oates JA, Sjoerdsma A. Neurologic effects of tryptophan in patients receiving a monoamine oxidase inhibitor. Neurology. 1960;10:1076-1078. [DOI] [PubMed] [Google Scholar]
- 5. Gerson SC, Baldessarini RJ. Motor effects of serotonin in the central nervous system. Life Sci. 1980;27:1435-1451. [DOI] [PubMed] [Google Scholar]
- 6. Insel TR, Roy BF, Cohen RM, Murphy DL. Possible development of the serotonin syndrome in man. Am J Psychiatry. 1982;139:954-955. [DOI] [PubMed] [Google Scholar]
- 7. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120. [DOI] [PubMed] [Google Scholar]
- 8. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. [DOI] [PubMed] [Google Scholar]
- 9. Bodner RA, Lynch T, Lewis L, Kahn D. Serotonin syndrome. Neurology. 1995;45:219-223. [DOI] [PubMed] [Google Scholar]
- 10. Sporer KA. The serotonin syndrome. Implicated drugs, pathophysiology and management. Drug Saf. 1995;13:94-104. [DOI] [PubMed] [Google Scholar]
- 11. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13:533-540. [PMC free article] [PubMed] [Google Scholar]
- 12. Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore). 2000;79:201-209. [DOI] [PubMed] [Google Scholar]
- 13. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713. [DOI] [PubMed] [Google Scholar]
- 14. Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications. J Clin Psychiatry. 2014;75:169-177. [DOI] [PubMed] [Google Scholar]
- 15. Watson WA, Litovitz TL, Klein-Schwartz W, et al. 2003 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2004;22:335-404. [DOI] [PubMed] [Google Scholar]
- 16. Watson WA, Litovitz TL, Rodgers GC, Jr, et al. 2002 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353-421. [DOI] [PubMed] [Google Scholar]
- 17. Gummin DD, Mowry JB, Spyker DA, Brooks DE, Fraser MO, Banner W. Annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 34th annual report. Clin Toxicol (Phila). 2017;55:1072-1252. [DOI] [PubMed] [Google Scholar]
- 18. Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol. 2005;28:205-214. [DOI] [PubMed] [Google Scholar]
- 19. Culbertson VL, Rahman SE, Bosen GC, Caylor ML, Echevarria MM, Xu D. Implications of off-target serotoninergic drug activity: an analysis of serotonin syndrome reports using a systematic bioinformatics approach. Pharmacotherapy. 2018;38:888-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Werneke U, Jamshidi F, Taylor DM, Ott M. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mackay FJ, Dunn NR, Mann RD. Antidepressants and the serotonin syndrome in general practice. Br J Gen Pract. 1999;49:871-874. [PMC free article] [PubMed] [Google Scholar]
- 22. Saper CB. Brainstem modulation of sensation, movement, and consciousness. In: Kandel ER, Schwartz JH, Jessell TM, eds. Principles in Neural Science. 4th ed. New York: McGraw-Hill; 2000:896. [Google Scholar]
- 23. Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol. 2006;33:575-583. [DOI] [PubMed] [Google Scholar]
- 24. Carneiro AMD, Cook EH, Murphy DL, Blakely RD. Interactions between integrin alphaIIbbeta3 and the serotonin transporter regulate serotonin transport and platelet aggregation in mice and humans. J Clin Invest. 2008;118:1544-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sauer WH, Berlin JA, Kimmel SE. Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation. 2003;108:32-36. [DOI] [PubMed] [Google Scholar]
- 26. Taylor CB, Youngblood ME, Catellier D, et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry. 2005;62:792-798. [DOI] [PubMed] [Google Scholar]
- 27. Miller F, Friedman R, Tanenbaum J, Griffin A. Disseminated intravascular coagulation and acute myoglobinuric renal failure: a consequence of the serotonergic syndrome. J Clin Psychopharmacol. 1991;11:277-279. [PubMed] [Google Scholar]
- 28. Matthes S, Bader M. Peripheral serotonin synthesis as a new drug target. Trends Pharmacol Sci. 2018;39:560-572. [DOI] [PubMed] [Google Scholar]
- 29. Walther DJ, Bader M. A unique central tryptophan hydroxylase isoform. Biochem Pharmacol. 2003;66:1673-1680. [DOI] [PubMed] [Google Scholar]
- 30. McKinney J, Knappskog PM, Haavik J. Different properties of the central and peripheral forms of human tryptophan hydroxylase. J Neurochem. 2005;92:311-320. [DOI] [PubMed] [Google Scholar]
- 31. Amireault P, Sibon D, Cote F. Life without peripheral serotonin: insights from tryptophan hydroxylase 1 knockout mice reveal the existence of paracrine/autocrine serotonergic networks. ACS Chem Neurosci. 2013;4:64-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cote F, Thevenot E, Fligny C, et al. Disruption of the nonneuronal TPH1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U S A. 2003;100:13525-13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gutknecht L, Waider J, Kraft S, et al. Deficiency of brain 5-HT synthesis but serotonergic neuron formation in Tph2 knockout mice. J Neural Transm (Vienna). 2008;115:1127-1132. [DOI] [PubMed] [Google Scholar]
- 34. Neal KB, Parry LJ, Bornstein JC. Strain-specific genetics, anatomy and function of enteric neural serotonergic pathways in inbred mice. J Physiol. 2009;587:567-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alenina N, Kikic D, Todiras M, et al. Growth retardation and altered autonomic control in mice lacking brain serotonin. Proc Natl Acad Sci U S A. 2009;106:10332-10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savelieva KV, Zhao S, Pogorelov VM, et al. Genetic disruption of both tryptophan hydroxylase genes dramatically reduces serotonin and affects behavior in models sensitive to antidepressants. Bartolomucci A, ed. PLoS ONE. 2008;3:e3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou M, Engel K, Wang J. Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem Pharmacol. 2007;73:147-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083-1152. [DOI] [PubMed] [Google Scholar]
- 39. Hoyer D, Clarke DE, Fozard JR, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev. 1994;46:157-203. [PubMed] [Google Scholar]
- 40. Mignon L, Wolf WA. Postsynaptic 5-HT1A receptors mediate an increase in locomotor activity in the monoamine-depleted rat. Psychopharmacology (Berl). 2002;163:85-94. [DOI] [PubMed] [Google Scholar]
- 41. Dunkley EJC, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96:635-642. [DOI] [PubMed] [Google Scholar]
- 42. Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol. 2004;42:277-285. [DOI] [PubMed] [Google Scholar]
- 43. Racz R, Soldatos TG, Jackson D, Burkhart K. Association between serotonin syndrome and second-generation antipsychotics via pharmacological target-adverse event analysis. Clin Transl Sci. 2018;11:322-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Celada P, Puig M, Amargos-Bosch M, Adell A, Artigas F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci. 2004;29:252-265. [PMC free article] [PubMed] [Google Scholar]
- 45. Artigas F. Serotonin receptors involved in antidepressant effects. Pharmacol Ther. 2013;137:119-131. [DOI] [PubMed] [Google Scholar]
- 46. Stahl SM. Basic psychopharmacology of antidepressants, part 1: antidepressants have seven distinct mechanisms of action. J Clin Psychiatry. 1998;59:5-14. [PubMed] [Google Scholar]
- 47. Duggal HS, Fetchko J. Serotonin syndrome and atypical antipsychotics. Am J Psychiatry. 2002;159:672-673. [DOI] [PubMed] [Google Scholar]
- 48. Dvir Y, Smallwood P. Serotonin syndrome: a complex but easily avoidable condition. Gen Hosp Psychiatry. 2008;30:284-287. [DOI] [PubMed] [Google Scholar]
- 49. Backus LI, Sharp T, Grahame-Smith DG. Behavioural evidence for a functional interaction between central 5-HT2 and 5-HT1A receptors. Br J Pharmacol. 1990;100:793-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marek GJ, Carpenter LL, McDougle CJ, Price LH. Synergistic action of 5-HT2A antagonists and selective serotonin reuptake inhibitors in neuropsychiatric disorders. Neuropsychopharmacology. 2003;28:402-412. [DOI] [PubMed] [Google Scholar]
- 51. Boothman LJ, Sharp T. A role for midbrain raphe gamma aminobutyric acid neurons in 5-hydroxytryptamine feedback control. Neuroreport. 2005;16:891-896. [DOI] [PubMed] [Google Scholar]
- 52. Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Memantine, an NMDA antagonist, prevents the development of hyperthermia in an animal model for serotonin syndrome. Pharmacopsychiatry. 2004;37:57-62. [DOI] [PubMed] [Google Scholar]
- 53. Isbister GK, Whyte IM. Serotonin toxicity and malignant hyperthermia: role of 5-HT2 receptors. Br J Anaesth. 2002;88:603; author reply 603-604. [DOI] [PubMed] [Google Scholar]
- 54. Done CJ, Sharp T. Biochemical evidence for the regulation of central noradrenergic activity by 5-HT1A and 5-HT2 receptors: microdialysis studies in the awake and anaesthetized rat. Neuropharmacology;33:411-421. [DOI] [PubMed] [Google Scholar]
- 55. Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int. 2003;43:155-164. [DOI] [PubMed] [Google Scholar]
- 56. Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, eds. Goodman and Gilman’s – The Pharmacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996. [Google Scholar]
- 57. Mitchell PB. Drug interactions of clinical significance with selective serotonin reuptake inhibitors. Drug Saf. 1997;17:390-406. [DOI] [PubMed] [Google Scholar]
- 58. Lange-Asschenfeldt C, Weigmann H, Hiemke C, Mann K. Serotonin syndrome as a result of fluoxetine in a patient with tramadol abuse: plasma level-correlated symptomatology. J Clin Psychopharmacol. 2002;22:440-441. [DOI] [PubMed] [Google Scholar]
- 59. Lee J, Franz L, Goforth HW. Serotonin syndrome in a chronic-pain patient receiving concurrent methadone, ciprofloxacin, and venlafaxine. Psychosomatics. 2009;50:638-639. [DOI] [PubMed] [Google Scholar]
- 60. Levin TT, Cortes-Ladino A, Weiss M, Palomba ML. Life-threatening serotonin toxicity due to a citalopram-fluconazole drug interaction: case reports and discussion. Gen Hosp Psychiatry. 2008;30:372-377. [DOI] [PubMed] [Google Scholar]
- 61. Gill M, LoVecchio F, Selden B. Serotonin syndrome in a child after a single dose of fluvoxamine. Ann Emerg Med. 1999;33:457-459. [DOI] [PubMed] [Google Scholar]
- 62. Asch DA, Parker RM. The Libby Zion case. N Engl J Med. 1988;318:771-775. [DOI] [PubMed] [Google Scholar]
- 63. Gillman PK. A review of serotonin toxicity data: implications for the mechanisms of antidepressant drug action. Biol Psychiatry. 2006;59:1046-1051. [DOI] [PubMed] [Google Scholar]
- 64. Stanford SC, Stanford BJ, Gillman PK. Risk of severe serotonin toxicity following co-administration of methylene blue and serotonin reuptake inhibitors: an update on a case report of post-operative delirium. J Psychopharmacol. 2010;24: 1433-1438. [DOI] [PubMed] [Google Scholar]
- 65. Fox MA, Jensen CL, Gallagher PS, Murphy DL. Receptor mediation of exaggerated responses to serotonin-enhancing drugs in serotonin transporter (SERT)-deficient mice. Neuropharmacology. 2007;53:643-656. [DOI] [PubMed] [Google Scholar]
- 66. Murphy GM, Jr, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry. 2003;160:1830-1835. [DOI] [PubMed] [Google Scholar]
- 67. Logan BK, Mohr ALA, Friscia M, et al. Reports of adverse events associated with use of novel psychoactive substances, 2013-2016: a review. J Anal Toxicol. 2017;41:573-610. [DOI] [PubMed] [Google Scholar]
- 68. Baumeister D, Tojo LM, Tracy DK. Legal highs: staying on top of the flood of novel psychoactive substances. Ther Adv Psychopharmacol. 2015;5:97-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fraser F. New Psychoactive Substances – Evidence Review. Edinburgh: The Scottish Government Social Research; 2014. [Google Scholar]
- 70. United Nations Office on Drugs and Crime. The Challenge of New Psychoactive Substances. Vienna: United Nations Office on Drugs and Crime; 2013. https://www.unodc.org/documents/scientific/NPS_2013_SMART.pdf. Accessed February 9, 2019.
- 71. EMCDDA. New Psychoactive Substances in Europe: An Update from the EU Early Warning System March 2015. Lisbon: EMCDDA; http://www.emcdda.europa.eu/attachements.cfm/att_235958_EN_TD0415135ENN.pdf. Accessed February 9, 2019; 2015. [Google Scholar]
- 72. EMCDDA. Health Responses to New Psychoactive Substances. Luxembourg: EMCDDA; http://www.emcdda.europa.eu/system/files/publications/2812/TD0216555ENN.pdf. Accessed February 9, 2019; 2016. [Google Scholar]
- 73. EMCDDA. European Drug Report 2017: Trends and Developments (2017). Luxembourg: Publications Office of the European Union; http://www.emcdda.europa.eu/system/files/publications/4541/TDAT17001ENN.pdf. Accessed February 9, 2019. [Google Scholar]
- 74. Drug Misuse: Findings from the 014/15 Crime Survey for England and Wales; 2015:1-49. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/462885/drug-misuse-1415.pdf.
- 75. European Commission. Flash Eurobarometer 401. Young People and Drugs; 2014. http://ec.europa.eu/commfrontoffice/publicopinion/flash/fl_401_en.pdf. Accessed 9 February 9, 2019.
- 76. Elliott S, Evans J. A 3-year review of new psychoactive substances in casework. Forensic Sci Int. 2014;243:55-60. [DOI] [PubMed] [Google Scholar]
- 77. Liechti M. Novel psychoactive substances (designer drugs): overview and pharmacology of modulators of monoamine signaling. Swiss Med Wkly. 2015;145:w14043. [DOI] [PubMed] [Google Scholar]
- 78. Hill SL, Thomas SHL. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49:705-719. [DOI] [PubMed] [Google Scholar]
- 79. Simmler L, Buser T, Donzelli M, et al. Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol. 2013;168:458-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681-698. [DOI] [PubMed] [Google Scholar]
- 81. Liechti ME, Kunz I, Kupferschmidt H. Acute medical problems due to ecstasy use. Case-series of emergency department visits. Swiss Med Wkly. 2005;135:652-657. [DOI] [PubMed] [Google Scholar]
- 82. Liechti ME. ‘Ecstasy’ (MDMA): pharmacology, toxicology, and treatment of acute intoxication. Dtsch Med Wochenschr. 2003;128:1361-1366. [DOI] [PubMed] [Google Scholar]
- 83. Simmler LD, Hysek CM, Liechti ME. Sex differences in the effects of MDMA (ecstasy) on plasma copeptin in healthy subjects. J Clin Endocrinol Metab. 2011;96:2844-2850. [DOI] [PubMed] [Google Scholar]
- 84. Caffrey CR, Lank PM. When good times go bad: managing ‘legal high’ complications in the emergency department. Open Access Emerg Med. 2017;10:9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones. J Med Toxicol. 2012;8:33-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hassan Z, Bosch OG, Singh D, et al. Novel psychoactive substances-recent progress on neuropharmacological mechanisms of action for selected drugs. Front Psychiatry. 2017;8:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Burish MJ, Thoren KL, Madou M, Toossi S, Shah M. Hallucinogens causing seizures? A case report of the synthetic amphetamine 2,5-dimethoxy-4-chloroamphetamine. Neurohospitalist. 2015;5:32-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Halberstadt AL. Pharmacology and toxicology of N-benzylphenethylamine (‘NBOMe’) hallucinogens. Curr Top Behav Neurosci. 2017;32:283-311. [DOI] [PubMed] [Google Scholar]
- 89. Hieger MA, Rose SR, Cumpston KL, Stromberg PE, Miller S, Wills BK. Severe poisoning after self-reported use of 2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine, a novel substituted amphetamine: a case series. Am J Emerg Med. 2015;33:1843.e1-1843.e3. [DOI] [PubMed] [Google Scholar]
- 90. Srisuma S, Bronstein AC, Hoyte CO. NBOMe and 2C substitute phenylethylamine exposures reported to the National Poison Data System. Clin Toxicol (Phila). 2015;53:624-628. [DOI] [PubMed] [Google Scholar]
- 91. Nichols DE, Brewster WK, Johnson MP, Oberlender R, Riggs RM. Nonneurotoxic tetralin and indan analogues of 3,4-(methylenedioxy)amphetamine (MDA). J Med Chem. 1990;33:703-710. [DOI] [PubMed] [Google Scholar]
- 92. Palenicek T, Lhotkova E, Zidkova M, et al. Emerging toxicity of 5,6-methylenedioxy-2-aminoindane (MDAI): pharmacokinetics, behaviour, thermoregulation and LD50 in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2016;69:49-59. [DOI] [PubMed] [Google Scholar]
- 93. Gatch MB, Dolan SB, Forster MJ. Locomotor, discriminative stimulus, and place conditioning effects of MDAI in rodents. Behav Pharmacol. 2016;27:497-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dignam G, Bigham C. Novel psychoactive substances: a practical approach to dealing with toxicity from legal highs. BJA Educ. 2017;17:172-177. [Google Scholar]
- 95. Heyerdahl F, Hovda KE, Giraudon I, et al. Current European data collection on emergency department presentations with acute recreational drug toxicity: gaps and national variations. Clin Toxicol (Phila). 2014;52:1005-1012. [DOI] [PubMed] [Google Scholar]
- 96. Wood DM, Ceronie B, Dargan PI. Healthcare professionals are less confident in managing acute toxicity related to the use of new psychoactive substances (NPS) compared with classical recreational drugs. QJM. 2016;109:527-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wood DM, Hill SL, Thomas SHL, Dargan PI. Using poisons information service data to assess the acute harms associated with novel psychoactive substances. Drug Test Anal. 2014;6:850-860. [DOI] [PubMed] [Google Scholar]
- 98. Morales-Molina JA, Mateu-de Antonio J, Marin-Casino M, Grau S. Linezolid-associated serotonin syndrome: what we can learn from cases reported so far. J Antimicrob Chemother. 2005;56:1176-1178. [DOI] [PubMed] [Google Scholar]
- 99. Houlihan DJ. Serotonin syndrome resulting from coadministration of tramadol, venlafaxine, and mirtazapine. Ann Pharmacother. 2004;38:411-413. [DOI] [PubMed] [Google Scholar]
- 100. Chander WP, Singh N, Mukhiya GK. Serotonin syndrome in maintenance haemodialysis patients following sertraline treatment for depression. J Indian Med Assoc. 2011;109:36-37. [PubMed] [Google Scholar]
- 101. Uddin M, Alweis R, Shah SR, et al. Controversies in serotonin syndrome diagnosis and management: a review. J Clin Diagn Res. 2017;11:OE05-OE07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Prakash S, Gosai F, Brahmbhatt J, Shah C. Serotonin syndrome in patients with peripheral neuropathy: a diagnostic challenge. Gen Hosp Psychiatry. 2014;36:450.e9-450.e11. [DOI] [PubMed] [Google Scholar]
- 103. Dosi R, Ambaliya A, Joshi H, Patell R. Serotonin syndrome versus neuroleptic malignant syndrome: a challenging clinical quandary. BMJ Case Rep. 2014;2014:bcr2014204154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13:763-783. [DOI] [PubMed] [Google Scholar]
- 105. Ali SZ, Taguchi A, Rosenberg H. Malignant hyperthermia. Best Pract Res Clin Anaesthesiol. 2003;17:519-533. [DOI] [PubMed] [Google Scholar]
- 106. Boyer EW, Traub SJ, Grayzel J. Serotonin Syndrome. Waltham, MA: UpToDate; 2010. [Google Scholar]
- 107. Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol. 1999;13:100-109. [DOI] [PubMed] [Google Scholar]
- 108. Martin TG. Serotonin syndrome. Ann Emerg Med. 1996;28:520-526. [DOI] [PubMed] [Google Scholar]
- 109. Krishnamoorthy S, Ma Z, Zhang G, Wei J, Auerbach SB, Tao R. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107:830-841. [DOI] [PubMed] [Google Scholar]
- 110. Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust. 2007;187:361-365. [DOI] [PubMed] [Google Scholar]
- 111. Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med;16:615-619. [DOI] [PubMed] [Google Scholar]
- 112. Baigel GD. Cyproheptadine and the treatment of an unconscious patient with the serotonin syndrome. Eur J Anaesthesiol. 2005;20:586-588. [DOI] [PubMed] [Google Scholar]
- 113. Horowitz BZ, Mullins ME. Cyproheptadine for serotonin syndrome in an accidental pediatric sertraline ingestion. Pediatr Emerg Care. 1999;15:325-327. [DOI] [PubMed] [Google Scholar]
- 114. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331:1021-1022. [DOI] [PubMed] [Google Scholar]
- 115. McDaniel WW. Serotonin syndrome: early management with cyproheptadine. Ann Pharmacother. 2001;35:870-873. [DOI] [PubMed] [Google Scholar]
- 116. Stahl SM, Lee-Zimmerman C, Cartwright S, Morrissette DA. Serotonergic drugs for depression and beyond. Curr Drug Targets. 2013;14:578-585. [DOI] [PubMed] [Google Scholar]
- 117. Werner F-M, Covenas R. Safety of antipsychotic drugs: focus on therapeutic and adverse effects. Expert Opin Drug Saf. 2014;13:1031-1042. [DOI] [PubMed] [Google Scholar]
- 118. Derry CJ, Derry S, Moore RA. Sumatriptan (all routes of administration) for acute migraine attacks in adults – overview of Cochrane reviews. Cochrane Database Syst Rev. 2014;5:CD009108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Molina-Cerrillo J, Alonso-Gordoa T, Martinez-Saez O, Grande E. Inhibition of peripheral synthesis of serotonin as a new target in neuroendocrine tumors. Oncologist. 2016;21:701-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Masab M, Saif MW. Telotristat ethyl: proof of principle and the first oral agent in the management of well-differentiated metastatic neuroendocrine tumor and carcinoid syndrome diarrhea. Cancer Chemother Pharmacol. 2017;80:1055-1062. [DOI] [PubMed] [Google Scholar]
- 121. Engelman K, Lovenberg W, Sjoerdsma A. Inhibition of serotonin synthesis by para-chlorophenylalanine in patients with the carcinoid syndrome. N Engl J Med. 1967;277:1103-1108. [DOI] [PubMed] [Google Scholar]
- 122. Kvols LK. Metastatic carcinoid tumors and the malignant carcinoid syndrome. Ann N Y Acad Sci. 1994;733:464-470. [DOI] [PubMed] [Google Scholar]
- 123. Zimmer L, Luxen A, Giacomelli F, Pujol J-F. Short- and long-term effects of p-ethynylphenylalanine on brain serotonin levels. Neurochem Res. 2002;27:269-275. [DOI] [PubMed] [Google Scholar]
- 124. Gillman PK. Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br J Anaesth. 2005;95:434-441. [DOI] [PubMed] [Google Scholar]
- 125. Gillman PK. Is there sufficient evidence to suggest cyclobenzaprine might be implicated in causing serotonin toxicity? Am J Emerg Med. 2009;27:509-510; author reply 510. [DOI] [PubMed] [Google Scholar]
- 126. Gillman PK. CNS toxicity involving methylene blue: the exemplar for understanding and predicting drug interactions that precipitate serotonin toxicity. J Psychopharmacol. 2011;25:429-436. [DOI] [PubMed] [Google Scholar]
- 127. Evans RW, Tepper SJ, Shapiro RE, Sun-Edelstein C, Tietjen GE. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin-norepinephrine reuptake inhibitors: American Headache Society position paper. Headache. 2010;50:1089-1099. [DOI] [PubMed] [Google Scholar]
- 128. Ramsey TD, Lau TT, Ensom MH. Serotonergic and adrenergic drug interactions associated with linezolid: a critical review and practical management approach. Ann Pharmacother. 2013;47:543-560. [DOI] [PubMed] [Google Scholar]
- 129. Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737-748. [DOI] [PMC free article] [PubMed] [Google Scholar]