

Abstract
Kinetic parameters kex (s–1) and kex/Kd (M–1 s–1) are reported for exchange for deuterium in D2O of the C-6 hydrogen of 5-fluororotidine 5′-monophosphate (FUMP) catalyzed by the Q215A, Y217F, and Q215A/Y217F variants of yeast orotidine 5′-monophosphate decarboxylase (ScOMPDC) at pD 8.1, and by the Q215A variant at pD 7.1–9.3. The pD rate profiles for wildtype ScOMPDC and the Q215A variant are identical, except for a 2.5 log unit downward displacement in the profile for the Q215A variant. The Q215A, Y217F and Q215A/Y217F substitutions cause 1.3–2.0 kcal/mol larger increases in the activation barrier for wildtype ScOMPDC-catalyzed deuterium exchange compared with decarboxylation, because of the stronger apparent side chain interaction with the transition state for the deuterium exchange reaction. The stabilization of the transition state for the OMPDC-catalyzed deuterium exchange reaction of FUMP is ca. 19 kcal/mol smaller than the transition state for decarboxylation of OMP, and ca. 8 kcal/mol smaller than for OMPDC-catalyzed deprotonation of FUMP to form the vinyl carbanion intermediate common to OMPDC-catalyzed reactions OMP/FOMP and UMP/FUMP. We propose that ScOMPDC shows similar stabilizing interactions with the common portions of decarboxylation and deprotonation transition states that lead to formation of this vinyl carbanion intermediate, and that there is a large ca. (19–8) = 11 kcal/mol stabilization of the former transition state from interactions with the nascent CO2 of product. The effects of Q215A and Y217F substitutions on kcat/Km for decarboxylation of OMP are expressed mainly as an increase in Km for the reactions catalyzed by the variant enzymes, while the effects on kex/Kd for deuterium exchange are expressed mainly as an increase in kex. This shows that the Q215 and Y217 side chains stabilize the Michaelis complex to OMP for the decarboxylation reaction, compared with the complex to FUMP for the deuterium exchange reaction. These results provide strong support for the conclusion that interactions which stabilize the transition state for ScOMPDC-catalyzed decarboxylation at a nonpolar enzyme active site dominate over interactions that destabilize the ground-state Michaelis complex.
Introduction
Orotidine 5′-monophosphate decarboxylase (OMPDC) catalyzes the decarboxylation of OMP and 5-fluororotidine 5′-monophosphate (FOMP) to form uridine 5′-monophosphate (UMP) and FUMP, respectively (Scheme 1).1−4 The enzyme mechanism of action has been a subject of intense interest because the protein catalyst provides an enormous 31 kcal/mol stabilization of the decarboxylation transition state, and a large selectivity in binding the reaction transition state with a higher affinity than the 8 kcal/mol binding energy of substrate OMP.5
Scheme 1. Decarboxylation Reactions Catalyzed by OMPDC.

OMPDC also catalyzes the exchange of the C-6 hydrogen of UMP,6,7 5-fluororotidine 5′-monophosphate (FUMP),6,7 or the truncated substrate 1-(β-d-erythrofuranosyl)-5-fluorororotic acid (FEO)8 with deuterium from solvent D2O. A comparison of the kinetic parameters for enzyme-catalyzed decarboxylation and deuterium exchange shows that OMPDC provides strong stabilization of the common UMP vinyl carbanion reaction intermediate. This is consistent with the conclusion that the first step in the deuterium exchange reaction, deprotonation of enzyme-bound UMP or FUMP (Figure 1), is the reverse of protonation of the vinyl carbanion intermediate of the decarboxylation reaction.
Figure 1.

OMPDC-catalyzed decarboxylation of OMP and FOMP, and deuterium exchange reactions of UMP and FUMP, through common UMP or FUMP vinyl carbanion intermediates.
There has been progress toward defining the roles of amino acid side chains at yeast OMPDC (ScOMPDC) in the stabilization of the rate-determining decarboxylation transition state.1,9−14 We have focused on the contribution to catalysis of binding interactions between ScOMPDC and the substrate phosphodianion, which drive the large protein conformational change shown in Figure 2, from an inactive flexible form of ScOMPDC to the stiff active form that provides an optimal stabilization of the decarboxylation transition state.13,15−17 This conformational change is driven by interactions of the OMP phosphodianion with the side chains of Q215 and Y217 from a gripper loop, with the guanidine cation side chain of R235 (Figure 2), and by an intraloop clamping interaction between the S154 and Q215 side chains.18 The contribution of these side chains to the enzymatic rate acceleration was determined for ScOMPDC-catalyzed decarboxylation of OMP,9,11FOMP,9,10 and for decarboxylation of the phosphodianion truncated substrates 1-(β-d-erythrofuranosyl)orotic acid (EO)13 and 1-(β-d-erythrofuranosyl)-5-fluorororotic acid (FEO).19 The results show that the S154, Q215, and R235 side chains provide a total 10 kcal/mol stabilization of the transition state for decarboxylation of OMP, but a < 1 kcal/mol stabilization of the transition state for decarboxylation of EO.13 This shows that the observed transition state stabilization is from interactions of the Q215, Y217, and R235 side chains with bound dianions, and that there is no transition state stabilization from interactions with the distant pyrimidine ring.13 The results provide strong evidence that the side chains function to stabilize the closed form of ScOMPDC that shows a high reactivity toward decarboxylation of OMP.15,20
Figure 2.

Representations of the X-ray crystal structures of ScOMPDC from Saccharomyces cerevisiae (ScOMPDC). The left-hand and middle surface structures show, respectively, the open unliganded form of ScOMPDC (PDB entry 1DQW) and the closed form with 6-hydroxyuridine 5′-monophosphate bound (PDB entry 1DQX). The phosphodianion gripper (residues 202–220) and pyrimidine umbrella loops (residues 151–165) are shaded blue, and the side chain from R235 is shaded green in both structures. The right-hand structure (PDB entry 1DQX) shows the interactions of Q215, Y217, and R235 with the phosphodianion of 6-hydroxyuridine 5′-monophosphate, and the clamping interaction between the Q215 side chain and the S154 side chain from the phosphodianion gripper and the pyrimidine umbrella loops.
The enzyme conformational change has also been proposed to promote decarboxylation through the induction of electrostatic stress into the substrate carboxylate that is relieved at the decarboxylation transition state,21−23 and by distorting the bond to the substrate carboxylate ∼36° out of the plane of the pyrimidine ring.4,24−27 The results of our earlier studies have provided no evidence for the utilization of phosphodianion binding energy to introduce interactions that destabilize enzyme-bound OMP at yeast OMPDC, but they have not focused on the evaluation of ground-state effects.
The relative binding affinity to ScOMPDC reported for OMP, the product UMP,28 and for inhibitors29−31 are difficult to reconcile with the proposal that the Michaelis complex to OMP is destabilized by interactions with ScOMPDC. For example, the ca. 200-fold weaker binding affinity of product UMP compared with substrate OMP shows that the Michaelis complex to OMP is stabilized by interactions between the enzyme and substrate carboxylate group.28 The significance of these binding studies has been challenged,32 and the 50 year old proposal that enzyme-catalyzed decarboxylation is promoted by interactions that destabilize the reaction ground state33−38 remains entrenched, despite being strongly disputed.39−41 The disputes are largely theoretical in nature, and there have been few experimental studies on this problem.
The role of ground-state destabilization in decarboxylation catalyzed by ScOMPDC is examined here by comparing the effect of amino acid substitutions at dianion gripper side chains Q215 and Y217 (Figure 2) on the kinetic parameters for the enzyme-catalyzed decarboxylation of OMP, where ground state effects are proposed to be important, with kinetic parameters for the deuterium exchange reaction of FUMP (Figure 1) that lacks the −CO2–. Substitutions of dianion gripper side chains that introduce destabilizing interactions into bound OMP should reduce ground-state destabilization, and result in decreases in kcat and in Km for ScOMPDC-catalyzed decarboxylation, but there can be no ground-state effects on the corresponding kinetic parameters for the ScOMPDC-catalyzed deuterium exchange reactions of FUMP.
We report the effect of Q215A, Y217F and Q215A/Y217F substitutions on the kinetic parameters for ScOMPDC-catalyzed deuterium exchange reactions of FUMP. These results build upon an earlier study of the effect of the R235A substitution.12 A comparison of the effect of these substitutions on catalysis of the decarboxylation and deuterium exchange reactions shows that each protein substitution results in a (1.3–2.0)-kcal/mol larger increase in the activation barrier to the deuterium exchange compared to decarboxylation reaction. This is surprising, because the ca 19 kcal/mol smaller total stabilization of the deuterium exchange compared with the decarboxylation transition state is consistent with weaker transition state stabilization from interactions with the protein catalyst.6,7 Our analysis strongly supports the conclusion that the transition states for ScOMPDC-catalyzed decarboxylation of OMP/FOMP and for deprotonation of UMP/FUMP show similar stabilizing interactions with the protein catalyst over the shared portions of these substrates, and differ because OMPDC shows robust binding interactions with the nascent CO2 product at the decarboxylation transition state. This conclusion is generalized to other enzyme-catalyzed decarboxylation reactions, where the first step is transfer of the substrate −CO2– from water to a hydrophobic protein binding pocket.38,42−46
Experimental Section
Materials
Glycylglycine (GlyGly, >99%) was obtained from USB. 3-(N-Morpholino)propanesulfonic acid (MOPS, ≥99.5%) was purchased from Fluka. The following deuterium labeled compounds were purchased from Cambridge Isotope Laboratories: D2O (99.9%), DCl (35 wt %, 99.9% D), and NaOD (30 wt %, 98%D). The water was distilled and purified on a Milli-Q water purification system. The triethylammonium salt of FUMP was synthesized as described in earlier work, and was converted to the free acid by passage over Amberlite IR120 resin (H+-form) in methanol.6 All other chemicals were reagent grade and were used without further purification.
Preparation of Solutions
Solution pH and pD was determined at 25 °C using an Orion model 720A pH meter equipped with a Radiometer pHC4006-9 combination electrode that was standardized at pH 7.00 and 10.00 at 25 °C. The pD of buffers in D2O was obtained by adding 0.4 to the reading on the pH meter.47 The acidic protons of GlyGly were exchanged for deuterium by dissolving the buffer in D2O, followed by evaporation and drying under vacuum at 55 °C. Buffered solutions of imidazole were prepared by dissolving the buffer base in D2O and adjusting to the required pD using DCl. Buffered solutions of MOPS and GlyGly were prepared by dissolving the commercial buffer in D2O and adjusting to the required pD using NaOD.
Protein Variants of ScOMPDC
The plasmid pScODC-15b containing the gene encoding ScOMPDC from Saccharomyces cerevisiae with a N-terminal His6- or His10-tag was available from previous studies.14,48 The procedures for the preparation of the Q215A,14 Y217F,13 and Q215A/Y217F13 variants were described in earlier work. In all cases the N-terminal His6- or His10-tag was removed by the action of thrombin (1 unit/mg ScOMPDC) at room temperature for ca. 16 h, as described in the Supporting Information to ref (14). The protein variants of ScOMPDC were stored at −80 °C. These enzymes were thawed and then dialyzed at 7 °C against 10 mM MOPS at pH 7.1 and I = 0.10 (NaCl). This was followed by exhaustive dialysis [at least 3 changes in dialysis buffer] at 7 °C in D2O against 5–10 mL of the following buffers (I = 0.1, NaCl): pD 7.1, 50 mM imidazole; pD 7.4, 50 mM MOPS; pD 7.7, 50 mM MOPS; pD 8.1, 50 mM GlyGly; pD 9.3; 50 mM GlyGly. The dialysis was with a D-tube dialyzer (10 kDa MWCO, Novagen) placed inside a narrow vessel that was isolated from atmospheric moisture using parafilm. The concentration of stock solutions of protein variants of ScOMPDC was determined from the absorbance at 280 nm using an extinction coefficient of 29 900 M–1 cm–1, calculated using the ProtParam tool available on the ExPASy server.49,50 The activity of these protein variants was determined by monitoring the decrease in absorbance at 279 nm during the enzyme-catalyzed decarboxylation of OMP.11
Deuterium Exchange at C-6 of FUMP Monitored by 19F NMR
The exchange of the C-6 proton of FUMP for deuterium from solvent D2O catalyzed by Q215A, Y217F, Q215A/Y217F variants of ScOMPDC at 25 °C and I = 0.1 (NaCl) was monitored by following formation of deuterium labeled product (d-FUMP). The reaction mixtures (1–2 mL in D2O) were prepared by mixing the stock enzyme solution with the appropriate buffer and NaCl to give the desired enzyme, buffer and salt concentrations. The reactions were initiated by the addition of FUMP in D2O. The following are the final reaction solutions for the Q215A variant at (I = 0.1, NaCl): pD 7.1, 50 mM imidazole, 80–120 μM ScOMPDC at 1.0–10 mM FUMP; pD 7.4, 50 mM MOPS, 40–100 μM ScOMPDC at 0.75–7.5 mM FUMP; pD 7.7, 50 mM MOPS, 20–80 μM ScOMPDC at 0.50–7.1 mM FUMP; pD 8.1, 50 mM GlyGly, 20–60 μM ScOMPDC at 0.48–7.5 mM FUMP; pD 9.3, 50 mM GlyGly, 15–100 μM ScOMPDC at 0.25–7.5 mM FUMP. The final solutions for the other protein variants were at pD 8.1 (50 mM GlyGly, I = 0.10 NaCl): Y217A variant, 10–20 μM ScOMPDC at 0.75–7.5 mM FUMP; Q215A/Y217A variant, 50–100 μM ScOMPDC at 2.5–5.0 mM FUMP.
At timed intervals aliquots of 100–500 μL were withdrawn from the reaction solutions and quenched with 20 μL of neat HCO2D. The ScOMPDC was removed by ultracentrifugation using an Amicon Ultrafiltration device (10K MWCO). The volume of the filtrate was adjusted to 700 μL in D2O and transferred to an NMR tube for analyses. The reactions were followed for up to several days, during which time no decrease (<10%) in enzyme activity was observed. For selected reactions at high [FUMP] the pD of the filtrate was determined, and in all cases was within 0.1 unit of the starting pD.
The 19F NMR spectra were obtained as described in previous work, and the chemical shifts referenced to the value of −78.5 ppm for a neat solution of the trifuoroacetic acid external standard.12 The integrated peak areas for the doublet with area (AH) at −165.36 ppm for h-FUMP and the broad upfield-shifted apparent singlet with area (AD) at −165.66 ppm for d-FUMP were recorded, and the initial reaction velocity (vi, eq 1) for enzyme-catalyzed conversion of up to 10% of h-FUMP to d-FUMP was determined as the slopes of linear plots of 3 or 4 values of fP[FUMP]o against time during disappearance of 3–10% of total h-FUMP, where fP = AD/(AH + AD) is the fraction of total FUMP labeled with deuterium at the C-6 position, and [FUMP]o is the initial concentration of h-FUMP.
| 1 |
Results
The ScOMPDC-catalyzed deuterium exchange reactions of FUMP were monitored by 19F NMR spectroscopy at 470 MHz, as described in an earlier study.12Figure 3 shows the dependence of vi/[E] on [FUMP], where vi is the initial velocity of the deuterium exchange reactions catalyzed by the Q215A variant at several different pD and a constant ionic strength of 0.1 (NaCl). Figure 4A,B show the dependence of vi/[E] on [FUMP] for the deuterium exchange reactions catalyzed by the Y217A and Q215A/Y217A variants of ScOMPDC, respectively, at pD 8.1 (20 mM GlyGly) and I = 0.1 (NaCl). The data from Figures 3 and 4A were fit to eq 2, derived for Scheme 2, to give values for the kinetic parameters kex and Kd. The data from Figure 4B were fit to a linear form of eq 2 (Kd ≫ [FUMP]) to give the value of kex/Kd. Table 1 reports these kinetic parameters at pD 8.1, and previously determined values of kcat and Km for wild type and variant forms ScOMPDC-catalyzed decarboxylation of OMP at pH 7.1.11 The parameter Kd (Scheme 2) is treated as a thermodynamic dissociation constant, because formation of the Michaelis complex to h-UMP is effectively reversible with respect to its slow (kex ≤ 10–2 s–1, Table 1) conversion to d-UMP.6 The binding of OMP to OMPDC is partly irreversible for decarboxylation catalyzed by the wild type enzyme (kcat = 16 s–1).28 The parameter Km is used here to describe the stability of the Michaelis complex. The values of the kinetic parameters for Q215A variant ScOMPDC-catalyzed deuterium exchange reactions of FUMP at several different pD are reported in Table 2, along with previously determined values of the kinetic parameters for the wild type ScOMPDC-catalyzed deuterium exchange reactions.6
| 2 |
Figure 3.

Dependence of vi/[E] on [FUMP] for Q215A variant ScOMPDC-catalyzed deuterium exchange reactions at I = 0.1 (NaCl). Key: (●), pD 7.1 (50 mM imidazole buffer); (▼), pD 7.4 (50 mM MOPS buffer); (⧫), pD 7.7 (50 mM MOPS buffer); (▲), pD 8.1 (50 mM GlyGly buffer); (■) pD 9.3 (50 mM GlyGly buffer).
Figure 4.

Dependence of vi/[E] on [FUMP] for ScOMPDC-catalyzed deuterium exchange reactions at pD 8.1 (50 mM GlyGly) and I = 0.1 (NaCl). (A) Y217F variant; (B) Q215A/Y217A variant.
Scheme 2. Kinetic Mechanism for OMPDC-Catalyzed Deuterium Exchange.

Table 1. Kinetic Parameters at 25 °C for Wild Type and Variant Forms of ScOMPDC-Catalyzed Deuterium Exchange Reactions of FUMP at pD 8.1 and Decarboxylation Reactions of OMP at pH 7.1a.
|
FUMPb |
OMPc |
|||||
|---|---|---|---|---|---|---|
| enzyme | kex (s–1) | Kd (mM) | kex/Kd (M–1 s–1) | kcat (s–1) | Km (M) | kcat/Km (M–1 s–1) |
| WT | 4.4 × 10–2 | 0.11 | 400 | 15 | 1.4 × 10–6 | 1.1 × 107 |
| Q215A | (4.3 ± 0.08) × 10–4 | 0.42 ± 0.03 | 1.0 | 24 | 0.94 x10–4 | 2.6 × 105 |
| Y217F | (2.3 ± 0.14) × 10–3 | 5.8 ± 0.15 | 0.40 | 20 | 1.1 × 10–4 | 1.8 × 105 |
| Q215A/Y217F | (3.9 ± 0.1) × 10–3 | 4.8 | 1.4 × 10–3 | 3.4 × 103 | ||
| R235Ad | 9.3 × 10–6 | 5.0 × 10–3 | 1.9 × 10–3 | 1.0 | 1.1 × 10–3 | 910 |
For ScOMPDC-catalyzed deuterium exchange reactions at pD 8.1 (50 mM GlyGly) and I = 0.1 (NaCl) or ScOMPDC-catalyzed decarboxylation reactions at pH 7.1 (30 mM MOPS) and I = 0.10 (NaCl).
Kinetic parameters defined by Scheme 2. The uncertainty in these values is the standard error from the least-squares fit of the kinetic data to the appropriate kinetic equation.
Data from ref (11).
The data for the R235A variant ScOMPDC-catalyzed deuterium exchange reaction is from ref (12).
Table 2. Kinetic Parameters at 25 °C for Deuterium Exchange Reactions of FUMP Catalyzed by Q215A Variant ScOMPDC at Several pDa.
| Q215A ScOMPDCb,c |
wild type ScOMPDCb |
||||||
|---|---|---|---|---|---|---|---|
| pDd | kex (s–1) | Kd (mM) | kex/Kd (M–1 s–1) | kex (s–1) | Kd (mM) | kex/Kd (M–1 s–1) | ΔΔG‡e kcal/mol |
| 7.1 (7.03) | (2.7 ± 0.1) × 10–4 | 2.0 ± 0.2 | 0.13 | 1.7 × 10–2 | 0.51 | 33 | 3.3 |
| 7.4 (7.37) | (3.6 ± 0.1) × 10–4 | 0.70 ± 0.08 | 0.51 | 2.4 × 10–2 | 0.17 | 140 | 3.3 |
| 7.7 (7.64) | (4.1 ± 0.08) × 10–4 | 0.38 ± 0.04 | 1.08 | 3.2 × 10–2 | 0.11 | 300 | 3.3 |
| 8.1 (8.15) | (4.3 ± 0.08) × 10–4 | 0.42 ± 0.03 | 1.02 | 4.4 × 10–2 | 0.11 | 400 | 3.5 |
| 9.3 (9.33) | (3.0 ± 0.1) × 10–4 | 0.54 ± 0.03 | 0.56 | 3.8 × 10–2 | 0.19 | 200 | 3.6 |
For ScOMPDC-catalyzed deuterium exchange reactions at I = 0.1 (NaCl) at: pD 7.1, 50 mM imidazole buffer; pD 7.4. 50 mM MOPS buffer; pD 7.7, 50 mM MOPS buffer; pD 8.1, 50 mM GlyGly buffer; and, pD 9.3, 50 mM GlyGly buffer.
The uncertainty in these values is the standard error from the least-squares fit of the kinetic data to the appropriate kinetic equation.
The pDs in parentheses for the previous study on the wild type ScOMPDC-catalyzed deuterium exchange reaction were reported to three significant figures.6
The effect of the point substitution on the activation barrier ΔG‡ for kex/Kd.
Discussion
Figure 5A,B show pD rate profiles for kex/Kd (M–1 s–1) and kex (s–1), respectively, for wild type and Q215A variant ScOMPDC-catalyzed deuterium exchange reactions at C-6 of FUMP in D2O, constructed using data from Table 2. There is a constant difference of ΔΔG‡ = 3.45 ± 0.15 kcal/mol (Table 2) between the activation barriers for the deuterium exchange reactions catalyzed by wild type and Q215A variant ScOMPDC.6 This shows that the Q215A substitution has no detectable effect on the apparent pKas of the active site side chains that govern the shape of these pD profiles. However, the downward break at low pD is not well-defined for the pD profile of kcat values for the Q215A variant, because we were unable to obtain kinetic parameters for this slow deuterium exchange reaction at pD = 6.45. The kinetic data for the pD rate profiles for wild type and Q215A variant ScOMPDC-catalyzed deuterium exchange reactions at C-6 of FUMP in D2O reported in Figure 5A,B were fit to the kinetic Scheme previously described in a study on the exchange reaction catalyzed by wildtype OMPDC, to give the same apparent values for the ionization constants of enzyme catalytic side chains.6
Figure 5.

Logarithmic rate profiles of kinetic parameters for ScOMPDC-catalyzed exchange for deuterium of the C-6 proton of FUMP in D2O at 25 °C and I = 0.1 (NaCl). (A) Second-order rate constants kex/Kd (M–1 s–1) for reactions catalyzed by wild type ScOMPDC (●) and the Q215A variant (▼). (B) First order rate constants kex (s–1) for reactions catalyzed by wild type ScOMPDC (●) and the Q215A variant (■). The solid lines show that fits of these data to the kinetic Scheme described previously for the reaction catalyzed by wild type OMPDC.6
ScOMPDC-Catalyzed Decarboxylation and Deuterium Exchange Reactions: Relative Rate Acceleration and Transition State Stabilization
The rate acceleration for the decarboxylation of OMP catalyzed by ScOMPDC at a standard state of 1 M OMPDC is 4 × 1022-fold. This is the ratio of kcat/Km = 1.1 × 107 M–1 s–1 for decarboxylation at the pH optimum of 7.1,28,51 and an estimated k0 = 2.8 × 10–16 s–1 for uncatalyzed nonenzymatic decarboxylation of OMP in water.5 This corresponds to (ΔΔG‡)dc = 31 kcal/mol for the stabilization of the transition state for nonenzymatic decarboxylation.6 The second-order rate constant of kHO = 0.71 M–1 s–1 for HO–-catalyzed deprotonation of FUMP gives knon = [0.71 M–1 s–1][1.3 × 10–7 M] = 9 × 10–8 s–1 for nonenzymatic deprotonation of FUMP at pH 7.1.6 Combining this with kex/Kd = 33 M–1 s–1 for the deuterium exchange reaction of FUMP catalyzed by ScOMPDC at pD 7.1, gives an enzymatic rate acceleration of 4 × 109 fold and a transition state stabilization of (ΔΔG‡)ex = 12 kcal/mol. The 5-F of FUMP results in similar 5.0 and 4.8 kcal/mol stabilization of the transition states for DO– and ScOMPDC-catalyzed deprotonation of UMP, so that ScOMPDC provides a similar stabilization of the transition states for the two nonenzymatic reactions.6,10 This 12 kcal/mol transition state stabilization is 19 kcal/mol smaller than the 31 kcal/mol transition state stabilization estimated for ScOMPDC-catalyzed decarboxylation of OMP. [This 19 kcal/mol transition state stabilization is calculated from a comparison of rate constants for the enzymatic and nonenzymatic deprotonation reactions determined a pH or pD of 7.1. No attempt has been made to correct for the uncertain deuterium isotope effects on these reactions. By comparison, an estimate of 17 kcal/mol for this transition state stabilization was reported in an earlier paper, using the second order rate constant of kE = 2300 M–1 s–1 for OMPDC with the catalytic side chains in their most reactive protonation states.6 The 2 kcal/mol difference between these two estimates does not affect the conclusion that OMPDC provides a substantially greater stabilization of the decarboxylation compared with the proton transfer transition state.]
This analysis demonstrates a large difference in the stabilization of the transition states for OMPDC-catalyzed decarboxylation and deuterium exchange reactions. We view the 19 kcal/mol estimate for this difference as qualitative rather than quantitative because of uncertainties in the estimates of knon obtained by the long extrapolation of rate date to 25 °C.5 This difference in transition state stabilization for these two reactions reflects one interaction that provides specific stabilization of the transition state for decarboxylation of OMP, and a second interaction that destabilizes the transition state for the deuterium exchange reaction compared with direct deprotonation of FUMP.
-
(1)
There is little or no activation barrier for addition of CO2 to the vinyl carbanion intermediate of OMPDC-catalyzed decarboxylation,52 so that the reaction transition state is early for CO2 addition and late for the reverse cleavage reaction. It is possible that the barrier for “CO2 reversion” is so small that some type of motion of CO2 away from the vinyl carbanion is rate determining for decarboxylation, in which case CO2 will be fully formed at the rate-determining transition state.52,53 This late CO2-like transition state for decarboxylation is stabilized by binding interactions with the CO2/-CO2– of OMP at the hydrophobic binding pocket described below, but CO2 is not present during OMPDC-catalyzed the deuterium exchange reaction of FUMP (Figure 6). The values of Kd = 6–36 μM for CO2 that have been reported for fixation of carbon dioxide catalyzed by D-ribulose-1,5-bisphosphate carboxylase (RuBisCO)54−56 correspond to a 6–7 kcal/mol CO2 binding energy. This observed CO2 binding energy will underestimate the intrinsic CO2 binding energy,35 by the binding energy utilized to reduce the translational and rotational entropy of solvated CO2 upon transfer to the active site of RuBisCO.57,58 Entropic costs to the reaction of free CO2 of (3–7) kcal/mol may be estimated from comparisons of the kinetic parameters for reactions of whole phosphodianion substrates and [phosphite + truncated substrate] pieces for several enzymatic reactions.58−61 This sets 9–14 kcal/mol as the range of stabilization for the decarboxylation transition state from interactions between OMPDC and the nascent CO2.
-
(2)
The barrier to krot for C–N bond rotation at −CH2–NL3+ of the K93 side chain, which exchanges the positions of ammonium cation hydrons, is added to the barrier for the deuterium exchange but not the decarboxylation reaction (Figure 7).62,63 This is demonstrated by the observation that the deuterium enrichment of the products of OMPDC-catalyzed decarboxylation of OMP or FOMP in 50/50 H2O/D2O is equal to the 50% enrichment of solvent;62,63 this gives a product deuterium isotope effect (PDIE) of 1.0.62,63 The expected selectivity for the reaction of -H compared with -D from a primary DIE is not observed, because bond rotation by krot, which would allow for this selection is so slow relative to protonation of the intermediate (kp ≫ krot, Figure 7) that the H/D product enrichment is equal to the initial H/D enrichment of the -C-NL3+ side chain. Note that kp from Figure 7 is the rate constant for the microscopic reverse of substrate deprotonation (kdp) and that the fractionation factor for hydron exchange between LOH and -C-NL3+ is 1.0.62,64,65 We therefore concluded that the rate constant for deuterium exchange (kex) is smaller than for substrate deprotonation (kdp) because the vinyl carbanion reaction intermediate undergoes fast protonation to generate h-UMP, and only rarely undergoes bond rotation (kp ≫ krot) that leads to deuterium exchange.6,62,63 By, comparison, this rotational barrier does not contribute to the barrier to OMPDC-catalyzed decarboxylation of OMP or FOMP, because the reaction intermediate is directly protonated by the cationic K93 side chain (Figure 7).
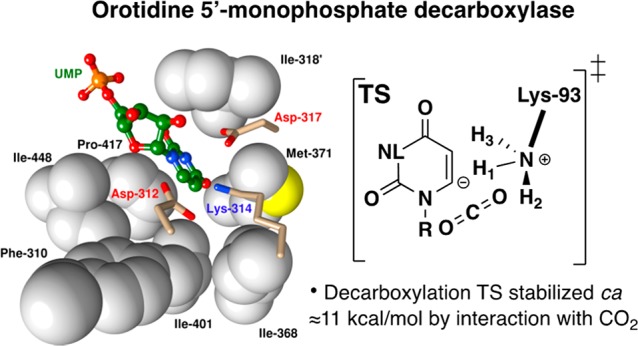
Figure 6.

Diagram which illustrates the hypothetical interactions between ScOMPDC and late vinyl carbanion-like transition states for decarboxylation of OMP and deprotonation of FUMP. These diagrams were constructed using X-ray crystallographic data for the complex to the 6-hydroxyuridine 5′-monophosphate (BMP) transition state analog (PDB entry 1DQX), and assuming similar contacts for the stable ligand and hypothetical transition states. Not shown is stabilization of nascent CO2 by interactions with hydrophobic amino acid side chains (see below).
Figure 7.

Stepwise OMPDC-catalyzed decarboxylation of OMP (X= H, bottom reaction) and the exchange reaction of -H from FUMP for -D from solvent D2O (X = F, top reaction). The inequality kp ≫ krot for partitioning of the vinyl carbanion intermediate between proton transfer and bond rotation at the K93 side chain ensures that the deuterium enrichment of product of OMPDC-catalyzed decarboxylation of OMP is equal to the initial 50% enrichment of the mixed H2O/D2O solvent,62,63 and that the barrier to the deprotonation of FUMP (ΔGdp‡) is smaller than the overall barrier for the deuterium exchange reaction.
Only a small barrier (kp ≈ 1011 s–1) is expected for strongly thermodynamically favorable protonation of the vinyl carbanion intermediate of OMPDC-catalyzed reactions.66−68 The positioning of the side chains for Asp-91, Lys-93, and Asp-96 revealed by the X-ray crystal structure of ScOMPDC complexed with BMP (Figure 6) provides strong evidence that the −CH2–NL3+group of Lys-93 is immobilized by hydrogen bonds to the carboxylate groups of Asp-91 and Asp-96 (Figure 7),18 so that krot ≪ 1011 s–1 for unhindered bond rotation and ΔΔGex‡ = ΔΔGdp – ΔGrot‡, where ΔΔGex and ΔΔGdp‡ are the enzymatic stabilization of the transition states for the OMPDC-catalyzed deprotonation and deuterium exchange reactions of FUMP, relative to a common barrier for nonenzymatic deprotonation of FUMP in water.
We estimate, crudely, that the requirement for cleavage of hydrogen bonds to allow side-chain rotation results in an increase in the barrier to deuterium exchange of ca. 4 kcal/mol/H-bond. If so, then the barrier to deprotonation of FUMP will be ΔGrot‡ = (2)(4) = 8 kcal/mol lower than the barrier to the deuterium exchange reaction, and the overall stabilization of the transition stated for substrate deprotonation will be (ΔΔG‡)dp = [(ΔΔG‡)ex + 2(4)] = 20 kcal/mol, where (ΔΔG‡)ex = 12 kcal/mol is the transition state stabilization calculated above for deuterium exchange into FUMP. This gives (ΔΔG‡)dc – (ΔΔG‡)dp = 31–20 = 11 kcal/mol as the corrected difference in the stabilization of the transition states for OMPDC-catalyzed decarboxylation of OMP and deprotonation of FUMP.
The corrected 11 kcal/mol difference in transition state stabilization is in the range of (9–14 kcal/mol) suggested above for stabilization of a late CO2-like decarboxylation transition state by interactions with CO2. This analysis provides support for the conclusion that the transition states for ScOMPDC-catalyzed decarboxylation of OMP/FOMP and for deprotonation of UMP/FUMP show similar stabilizing interactions with the protein catalyst over the shared portions of these substrates (Figure 6), and differ largely or entirely because OMPDC shows robust binding interactions with nascent CO2 at the decarboxylation transition state.
Effect of Protein Substitutions on Kinetic Parameters for ScOMPDC-Catalyzed Decarboxylation and Deuterium Exchange Reactions
The value of Km for ScOMPDC-catalyzed decarboxylation of OMP is 80-fold smaller than for Kd for the deuterium exchange reaction of FUMP (Table 3), but Km is only (4–50)-fold smaller than Kd for variant ScOMPDC-catalyzed reactions (Table 1). We conclude that these protein substitutions act to destabilize the Michaelis complex to OMP compared to FUMP, instead of the relative stabilization of the Michaelis complex to OMP predicted for amino acid substitutions that relieve destabilizing interactions between the protein and the carboxylate of substrate OMP.35
Table 3. Effect of Q215A, Y217F, and R235A Substitutions on the Activation Barriers for ScOMPDC-Catalyzed Decarboxylation of OMP at pH 7.1 and for Exchange of the C-6 Hydrogen of FUMP for Deuterium in D2O at pD 8.1.
These results are consistent with a model developed in earlier work where12 (i) The value for the observed Michaelis constant Km or Kd is equal to (Km)o[1/(1 + Kc)] where (Km)o is the disassociation constant for breakdown of the Michaelis complex to the inactive open form of ScOMPDC and Kc is the equilibrium constant for conversion of the inactive open complex to the reactive closed complex. (ii) The loop-closed form of ScOMPDC is stabilized by interactions with the carboxylate group of OMP, so that (Kc ≫ 1.0) for decarboxylation of OMP and (Kc ≤ 1.0) for the deuterium exchange reaction of FUMP. (iii) The effect of amino acid substitutions that perturb the value of Kc are expressed mainly as a change in Km for the decarboxylation reaction, where Kc ≫ 1.0, and mainly as a change in kex where Kc ≤ 1.0.11,12
Table 3 shows the effects of substitutions of Q215, Y217, or R235 on the activation barriers for ScOMPDC-catalyzed decarboxylation [ΔΔGOMP‡] and D-exchange [ΔΔGFUMP] reactions, and the difference in these changes in activation barrier [ΔΔGFUMP‡ – ΔΔGOMP]. We conclude that the Q215, Y217, and R235 side chains provide a 1.3–1.7 kcal/mol greater stabilization of the transition state for the D-exchange compared to decarboxylation reaction (Table 3), even though the total stabilization of the former transition state from all interactions is ca. 11 kcal/mol [(19–8) kcal/mol, above] smaller than the decarboxylation transition state. This trend, which is illustrated in Figure 8, is consistent with the conclusion above that the transition states for decarboxylation and D-exchange reactions show similar stabilizing interactions with the dianion gripper side chains of ScOMPDC over the shared portions of these reactants.
Figure 8.

Hypothetical free-energy profiles for wild type and protein variants of ScOMPDC-catalyzed decarboxylation of OMP and deprotonation of UMP to form UMP vinyl carbanion intermediates UMP-, which are drawn to show the difference in the stabilization of the respective transition states by interactions with the protein catalyst. These profiles were constructed using kinetic data from Table 1, and assuming that the changes in protein structure effect similar changes in the barriers for ScOMPDC-catalyzed deprotonation of FUMP and UMP.6 The diagrams show: (1) The ca. 11 kcal/mol difference in the stabilization of the transition states for wildtype ScOMPDC-catalyzed decarboxylation of OMP (ΔΔG‡)dc and deprotonation of UMP (ΔΔG‡)dp [(ΔΔG‡)dc – (ΔΔG‡)dp = 11 kcal/mol, Figure 7], that we propose is due to stabilization of the former transition state by interactions with the nascent CO2 product. (2) The difference between the effects of Q215A or Y217F substitutions on ΔΔGUMP‡ (relatively large) and ΔΔGOMP [smaller, Table 3]. (3) The larger effect of amino acid substitutions on the stability of the Michaelis complexes to OMP (ΔΔGOMPB) compared with UMP/FUMP (ΔΔGFUMP). The ca 8 kcal/mol barrier to krot, which is required for the deuterium exchange reaction of UMP (kex) but not for decarboxylation of OMP (kdc) or deprotonation of UMP (kdp), is not shown. The barriers to the two enzymatic reactions have not been scaled to show the thermodynamic driving force to decarboxylation of OMP to form UMP, because this driving force is not known.
We suggest the following explanation for the larger effects of amino acid substitutions on ΔΔGFUMP‡ compared with ΔΔGOMP (Table 3). The single Q215A, Y217F and R235A substitutions at wildtype OMPDC result mainly in the loss of stabilizing interactions with the excised side chain, and in minimal changes in the interactions of peripheral side chains at the structured decarboxylation transition state,9,11 so that ΔGOMP‡ provides an estimate for the excised interactions. (ii) Elimination of tightly bound CO2–/CO2 from this transition state loosens interactions of the remaining side chains at the transition state for the D-exchange reaction of FUMP. Single amino acid substitutions of ScOMPDC further erode these interactions, and this results in elevated values for ΔΔGFUMP compared with ΔΔGOMP‡. In other words, the value of ΔΔGFUMP for the single variants reflects both the loss of the stabilizing interactions of the excised side chain, plus a weakening of transition state stabilization by the remaining side chains.12
Interactions between OMPDC and −CO2–/CO2
The CO2 binding pocket at OMPDC was identified by analysis of the X-ray crystal structure of human OMPDC (HsOMPDC) liganded by UMP.27 This structure of HsOMPDC shows good superposition (Figure 9), of active site side chains, with the structure of ScOMPDC complexed to 6-aza uridine 5′-monophosphate (6-azaUMP) and small differences in the orientation of the pyrimidine rings.69 The hydrophobic CO2 binding pocket lies on the opposite side of the pyrimidine ring from the essential K314 (K93 at ScOMPDC) and is lined by the hydrophobic side chains of F310, I401, and I448 (F89, I183, and I232 at ScOMPDC).6,27 The carboxylate side chain of D312/D91 is close to the position of a hypothetical OMP carboxylate, so that the interactions of these carboxylates might destabilize the Michaelis complex to substrate. However, there is an overall 3 kcal/mol stabilization of the Michaelis complex to OMP compared to UMP(28) and, as discussed above, an even larger ca. 11 kcal/mol stabilization of the transition state for decarboxylation of OMP (kdc) compared with deprotonation of UMP (kdp) to form enzyme-bound UMP vinyl carbanions (Figure 8).
Figure 9.

Representation, on the left, of the X-ray crystal structure for HsOMPDC liganded by UMP (PDB entry 2QCD) and, on the right, for ScOMPDC liganded by 6-azaUMP (PDB entry 3GDL). There is good superposition between the active site residues D312, K314, and D317 at HsOMPDC with D91, K93, and D96 at ScOMPDC, and of the hydrophobic side chains F310, I401, and I448 at the CO2 binding pocket with F89, I183 and I232.
The transfer of carboxylate substrates from an aqueous to a nonpolar solvent results in large increases in the rate constants for nonenzymatic decarboxylation.33,34,36,37 These rate accelerations have been attributed to ground-state effects, where desolvation of the carboxylate group that accompanies this transfer results in an increase in the substrate reactivity toward decarboxylation.35 The transfer from an aqueous to an organic solvent is mimicked by the binding of decarboxylation substrates, including OMP, to hydrophobic binding pockets at enzymes which catalyze decarboxylation reactions.38,42−46,70,71 In one case the polarity of the active site of a thiamine pyrophosphate dependent yeast pyruvate decarboxylase was estimated from the medium-dependent fluorescence wavelength maximum of bound thiochrome diphosphate to be similar to solvents with dielectric constants of 13–15.72
We propose an alternative model, where the dominant effect of the change in medium polarity on the activation barrier for enzymatic and nonenzymatic decarboxylation reactions is stabilization of the late CO2-like decarboxylation transition state by interactions with the hydrophobic solvent for nonenzymatic decarboxylation, or with the hydrophobic enzyme binding cavity. In the case of OMPDC, any increase in Km for decarboxylation of OMP and FOMP from the requirement for desolvation of −CO2– during substrate binding is smaller that the increase in kcat from transition state stabilization by interactions with the nascent CO2 at the hydrophobic CO2 binding pocket (Figure 9). We estimate that the net effect of these interactions at the hydrophobic binding pocket is a 108-fold increase in the kinetic parameter kcat/Km from the 11 kcal/mol stabilizing interactions between OMPDC and the nascent CO2.
The binding of OMP to inactive protein variants of OMPDC results in distortion of the bond to substituents at C-6 from planarity with respect to the pyrimidine ring.25−27,73,74 For example, the D312N variant of HsOMPDC shows a 36° distortion of the bond to the substrate carboxylate. Similar out of plane distortions have been reported for the −CN group of 6-cyanouridine 5′-monophosphate bound to ScOMPDC and for C-6 substituents at other pyrimidine nucleotides bound to HsOMPDC.25,73,74 However, there is no apparent destabilization of this Michaelis complex by substituents at C-6.28 We suggest that relatively little bond angle strain energy is associated with these distortions. The distortions appear to track the early stages of a decarboxylation reaction coordinate, where the acidic K93 side chain approaches one face of the pyrimidine ring as CO2 is lost from the opposite face. However, the product deuterium isotope effect of 1.0 for OMPDC-catalyzed decarboxylation of OMP and FOMP in 50/50 H2O/D2O shows that the hydron provides no electrophilic push to the loss of CO2 that is characteristic of a fully coupled-concerted electrophilic displacement reaction.62,63
Evolution of Hydrophobic Binding Pockets at Decarboxylases
Hydrophobic binding pockets at decarboxylases mimic hydrophobilic solvents that strongly accelerate nonenzymatic decarboxylation relative to water.33,34,37 The observation of these binding pockets prompted the proposal that enzymatic rate accelerations for decarboxylation are promoted by destabilization of the enzyme-bound carboxylate, which moves the energy of the reaction ground state closer to the decarboxylation transition state.35 This proposal has been criticized40,41 and has not received a large amount of direct experimental support.
It is important to emphasize that there is no pressure early in enzyme evolution to select for catalysts that introduce destabilizing interactions into decarboxylation substrates, which are relieved at the decarboxylation transition state, since such ground state interactions have no effect on the kinetic parameter kcat/Km for the overall efficiency of the catalyst.35 Pressure for enzymatic specificity in transition state binding will only be observed as enzymatic catalysis becomes so efficient that the expression of the large transition state binding energy in substrate or product binding results in rate determining ligand binding steps.17,20,75 We propose that the evolutionary pressure results in the selection of nonpolar binding pockets that provide optimal stabilization of late CO2-like decarboxylation transition states. Once this transition state stabilization from CO2 binding interactions has been optimized, it becomes impossible to select for destabilizing interactions between the protein and the substrate carboxylate that do not result in a decrease in kcat/Km by perturbing the preoptimized hydrophobic interactions. In the case of OMPDC, rate-determining substrate binding or product release is avoided through utilization of a substantial fraction of the 31 kcal/mol intrinsic substrate binding energy to drive a thermodynamically favorable enzyme conformational change from the flexible open form, to the stiff Michaelis complex.17,76,77
Acknowledgments
We acknowledge the National Institutes of Health Grants GM39754 and GM116921 for generous support of this work.
Glossary
Abbreviations
- OMPDC
orotidine 5′-monophosphate decarboxylase
- OMP
orotidine 5′-monophosphate
- UMP
uridine 5′-monophosphate
- FOMP
5-fluoroorotidine 5′-monophosphate
- FUMP
5-fluorouridine 5′-monophosphate
- EO
1-(β-d-erythrofuranosyl)orotic acid
- EU
1-(β-d-erythrofuranosyl)uracil
- FEO
1-(β-d-erythrofuranosyl)-5-fluoroorotic acid
- FEU
1-(β-d-erythrofuranosyl)-5-fluorouracil
- MOPS
3-(N-morpholino)propanesulfonic acid
- GlyGly
glycyl glycine
- AP
alkaline phosphatase
The authors declare no competing financial interest.
References
- Richard J. P.; Amyes T. L.; Reyes A. C. Orotidine 5′-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis. Acc. Chem. Res. 2018, 51, 960–969. 10.1021/acs.accounts.8b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Wolfenden R. Catalytic proficiency: the unusual case of OMP decarboxylase. Annu. Rev. Biochem. 2002, 71, 847–885. 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- Callahan B. P.; Miller B. G. OMP decarboxylase - An enigma persists. Bioorg. Chem. 2007, 35, 465–469. 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Fujihashi M.; Mnpotra J. S.; Mishra R. K.; Pai E. F.; Kotra L. P. Orotidine Monophosphate Decarboxylase - A Fascinating Workhorse Enzyme with Therapeutic Potential. J. Genet. Genomics 2015, 42, 221–234. 10.1016/j.jgg.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. A proficient enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J. Am. Chem. Soc. 2012, 134, 14580–14594. 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J. Am. Chem. Soc. 2008, 130, 1574–1575. 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. OMP Decarboxylase: Phosphodianion Binding Energy Is Used To Stabilize a Vinyl Carbanion Intermediate. J. Am. Chem. Soc. 2011, 133, 6545–6548. 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Plache D. C.; Koudelka A. P.; Amyes T. L.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Breaking Down the Catalytic Cage that Activates Orotidine 5′-Monophosphate Decarboxylase for Catalysis. J. Am. Chem. Soc. 2018, 140, 17580–17590. 10.1021/jacs.8b09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Role of a Guanidinium Cation–Phosphodianion Pair in Stabilizing the Vinyl Carbanion Intermediate of Orotidine 5′-Phosphate Decarboxylase-Catalyzed Reactions. Biochemistry 2013, 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry 2012, 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 2008, 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc. 2019, 141, 3320–3331. 10.1021/jacs.8b10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Catalysis by Orotidine 5′-Monophosphate Decarboxylase: Effect of 5-Fluoro and 4′-Substituents on the Decarboxylation of Two-Part Substrates. Biochemistry 2013, 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 2017, 30, 157–165. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Byun K. L.; Kluger R. Catalysis by enzyme conformational change. Top. Curr. Chem. 2004, 238, 113–136. 10.1007/b94541. [DOI] [Google Scholar]
- Gao J. Catalysis by enzyme conformational change as illustrated by orotidine 5′-monophosphate decarboxylase. Curr. Opin. Struct. Biol. 2003, 13, 184–192. 10.1016/S0959-440X(03)00041-1. [DOI] [PubMed] [Google Scholar]
- Wu N.; Mo Y.; Gao J.; Pai E. F. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2017–2022. 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihashi M.; Ishida T.; Kuroda S.; Kotra L. P.; Pai E. F.; Miki K. Substrate distortion contributes to the catalysis of orotidine 5′-monophosphate decarboxylase. J. Am. Chem. Soc. 2013, 135, 17432–17443. 10.1021/ja408197k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihashi M.; Wei L.; Kotra L. P.; Pai E. F. Structural characterization of the molecular events during a slow substrate-product transition in orotidine 5′-monophosphate decarboxylase. J. Mol. Biol. 2009, 387, 1199–1210. 10.1016/j.jmb.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Gillon W.; Pai E. F. Mapping the Active Site-Ligand Interactions of Orotidine 5′-Monophosphate Decarboxylase by Crystallography. Biochemistry 2002, 41, 4002–4011. 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- Wittmann J. G.; Heinrich D.; Gasow K.; Frey A.; Diederichsen U.; Rudolph M. G. Structures of the human orotidine-5′-monophosphate decarboxylase support a covalent mechanism and provide a framework for drug design. Structure 2008, 16, 82–92. 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 2000, 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Wolfenden R. Indiscriminate Binding by Orotidine 5′-Phosphate Decarboxylase of Uridine 5′-Phosphate Derivatives with Bulky Anionic C6 Substituents. Biochemistry 2007, 46, 13331–13343. 10.1021/bi700796t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B. P.; Wolfenden R. OMP Decarboxylase: An Experimental Test of Electrostatic Destabilization of the Enzyme-Substrate Complex. J. Am. Chem. Soc. 2004, 126, 14698–14699. 10.1021/ja0450049. [DOI] [PubMed] [Google Scholar]
- Thirumalairajan S.; Mahaney B.; Bearne S. L. Interrogation of the active site of OMP decarboxylase from Escherichia coli with a substrate analogue bearing an anionic group at C6. Chem. Commun. 2010, 46, 3158. 10.1039/b926894d. [DOI] [PubMed] [Google Scholar]
- Fujihashi M.; Mito K.; Pai E. F.; Miki K. Atomic Resolution Structure of the Orotidine 5′-Monophosphate Decarboxylase Product Complex Combined with Surface Plasmon Resonance Analysis. J. Biol. Chem. 2013, 288, 9011–9016. 10.1074/jbc.M112.427252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby J.; Stone R.; Lienhard G. E. Mechanisms of thiamine-catalyzed reactions. Decarboxylation of 2-(1-carboxy-l-hydroxyethyl)-3,4-dimethylthiazolium chloride. J. Am. Chem. Soc. 1970, 92, 2891–2900. 10.1021/ja00712a048. [DOI] [PubMed] [Google Scholar]
- Kemp D. S.; Paul K. Decarboxylation of benzisoxazole-3-carboxylic acids. Catalysis by extraction of possible relevance to the problem of enzymic mechanism. J. Am. Chem. Soc. 1970, 92, 2553–2554. 10.1021/ja00711a061. [DOI] [Google Scholar]
- Jencks W. P. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Catalan J.; Diaz C.; Garcia-Blanco F. Effects of Medium on Decarboxylation Kinetics: 3-Carboxybenzisoxazoles and Their Potential Use as Environmental Probes in Biochemistry. J. Org. Chem. 2000, 65, 3409–3415. 10.1021/jo9918798. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Wolfenden R. Orotic Acid Decarboxylation in Water and Nonpolar Solvents: A Potential Role for Desolvation in the Action of OMP Decarboxylase. Biochemistry 2009, 48, 8738–8745. 10.1021/bi901085m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L. K.; Brooks H. B.; Myers D. P.; Phillips M. A. Ornithine decarboxylase promotes catalysis by binding the carboxylate in a buried pocket containing phenylalanine 397. Biochemistry 2003, 42, 2933–2940. 10.1021/bi026795z. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Florian J.; Strajbl M.; Villa J. Circe effect versus enzyme preorganization: what can be learned from the structure of the most proficient enzyme?. ChemBioChem 2001, 2, 109–111. . [DOI] [PubMed] [Google Scholar]
- Warshel A.; Strajbl M.; Villa J.; Florian J. Remarkable Rate Enhancement of Orotidine 5′-Monophosphate Decarboxylase Is Due to Transition-State Stabilization Rather Than to Ground-State Destabilization. Biochemistry 2000, 39, 14728–14738. 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- Schütz A.; Sandalova T.; Ricagno S.; Hübner G.; König S.; Schneider G. Crystal structure of thiamindiphosphate-dependent indolepyruvate decarboxylase from Enterobacter cloacae, an enzyme involved in the biosynthesis of the plant hormone indole-3-acetic acid. Eur. J. Biochem. 2003, 270, 2312–2321. 10.1046/j.1432-1033.2003.03601.x. [DOI] [PubMed] [Google Scholar]
- Jordan F.; Patel H. Catalysis in Enzymatic Decarboxylations: Comparison of Selected Cofactor-Dependent and Cofactor-Independent Examples. ACS Catal. 2013, 3, 1601–1617. 10.1021/cs400272x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins A. A.; Morar M.; Kappock T. J.; Mathews I. I.; Zaugg J. B.; Barder T. E.; Peng P.; Okamoto A.; Ealick S. E.; Stubbe J. N5-CAIR Mutase: Role of a CO2 Binding Site and Substrate Movement in Catalysis. Biochemistry 2007, 46, 2842–2855. 10.1021/bi602436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.-C.; Menetret J.-F.; Tsuruta H.; Allen K. N. The origin of the electrostatic perturbation in acetoacetate decarboxylase. Nature 2009, 459, 393–397. 10.1038/nature07938. [DOI] [PubMed] [Google Scholar]
- Patel H.; Nemeria N. S.; Brammer L. A.; Meyers C. L. F.; Jordan F. Observation of thiamin-bound intermediates and microscopic rate constants for their interconversion on 1-deoxy-D-xylulose 5-phosphate synthase: 600-fold rate acceleration of pyruvate decarboxylation by D-glyceraldehyde-3-phosphate. J. Am. Chem. Soc. 2012, 134, 18374–18379. 10.1021/ja307315u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasoe P. K.; Long F. A. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 1960, 64, 188–190. 10.1021/j100830a521. [DOI] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry 2009, 48, 5510–5517. 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud A.; Wilkins M. R.; Appel R. D.; Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. Proteomics Protocols Handbook 2005, 571–607. 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–8. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K. K.; Gerlt J. A.; Richard J. P. An Examination of the Relationship between Active Site Loop Size and Thermodynamic Activation Parameters for Orotidine 5′-Monophosphate Decarboxylase from Mesophilic and Thermophilic Organisms. Biochemistry 2009, 48, 8006–8013. 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger R. Decarboxylation, CO2 and the reversion problem. Acc. Chem. Res. 2015, 48, 2843–2849. 10.1021/acs.accounts.5b00306. [DOI] [PubMed] [Google Scholar]
- Major D. T.; Gao J. An Integrated Path Integral and Free-Energy Perturbation-Umbrella Sampling Method for Computing Kinetic Isotope Effects of Chemical Reactions in Solution and in Enzymes. J. Chem. Theory Comput. 2007, 3, 949–960. 10.1021/ct600371k. [DOI] [PubMed] [Google Scholar]
- Satagopan S.; Spreitzer R. J. Substitutions at the Asp-473 Latch Residue of Chlamydomonas Ribulosebisphosphate Carboxylase/Oxygenase Cause Decreases in Carboxylation Efficiency and CO2/O2 Specificity. J. Biol. Chem. 2004, 279, 14240–14244. 10.1074/jbc.M313215200. [DOI] [PubMed] [Google Scholar]
- Chen Z. X.; Chastain C. J.; Al-Abed S. R.; Chollet R.; Spreitzer R. J. Reduced CO2/O2 specificity of ribulose-bisphosphate carboxylase/oxygenase in a temperature-sensitive chloroplast mutant of Chlamydomonas. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 4696–4699. 10.1073/pnas.85.13.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing W. A.; Ogren W. L.; Hageman R. H. Bicarbonate stabilization of ribulose 1,5-diphosphate carboxylase. Biochemistry 1975, 14, 2269–2275. 10.1021/bi00681a035. [DOI] [PubMed] [Google Scholar]
- Jencks W. P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4046–50. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. Enzyme Architecture: Self-Assembly of Enzyme and Substrate Pieces of Glycerol-3-Phosphate Dehydrogenase into a Robust Catalyst of Hydride Transfer. J. Am. Chem. Soc. 2016, 138, 15251–15259. 10.1021/jacs.6b09936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Rescue of K12G mutant TIM by NH4+ and alkylammonium cations: The reaction of an enzyme in pieces. J. Am. Chem. Soc. 2010, 132, 13525–13532. 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood M. B.; Gerlt J. A.; Richard J. P. Activation of R235A Mutant Orotidine 5′-Monophosphate Decarboxylase by the Guanidinium Cation: Effective Molarity of the Cationic Side Chain of Arg-235. Biochemistry 2010, 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. Enzyme Architecture: Optimization of Transition State Stabilization from a Cation–Phosphodianion Pair. J. Am. Chem. Soc. 2015, 137, 5312–5315. 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Product Deuterium Isotope Effects for Orotidine 5′-Monophosphate Decarboxylase: Effect of Changing Substrate and Enzyme Structure on the Partitioning of the Vinyl Carbanion Reaction Intermediate. J. Am. Chem. Soc. 2010, 132, 7018–7024. 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Product Deuterium Isotope Effect for Orotidine 5′-Monophosphate Decarboxylase: Evidence for the Existence of a Short-Lived Carbanion Intermediate. J. Am. Chem. Soc. 2007, 129, 12946–12947. 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowen K. B.; Schowen R. L. Solvent isotope effects on enzyme systems. Methods Enzymol. 1982, 87, 551–606. 10.1016/S0076-6879(82)87031-6. [DOI] [PubMed] [Google Scholar]
- Jarret R. M.; Saunders M. Use of various nuclei as probes in a new NMR method for obtaining proton/deuteron fractionation data. J. Am. Chem. Soc. 1986, 108, 7549–53. 10.1021/ja00284a018. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Williams G.; Gao J. Experimental and Computational Determination of the Effect of the Cyano Group on Carbon Acidity in Water. J. Am. Chem. Soc. 1999, 121, 715–726. 10.1021/ja982692l. [DOI] [Google Scholar]
- Amyes T.; Richard J. Substituent Effects on Carbon Acidity in Aqueous Solution and at Enzyme Active Sites. Synlett 2017, 28, 1407–1421. 10.1055/s-0036-1588778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Williams G.; O’Donoghue A. C.; Amyes T. L. Formation and Stability of Enolates of Acetamide and Acetate Anion: An Eigen Plot for Proton Transfer at α-carbonyl Carbon. J. Am. Chem. Soc. 2002, 124, 2957–2968. 10.1021/ja0125321. [DOI] [PubMed] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry 2009, 48, 5518–5531. 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.; Snell E. E.; Hackert M. L. Pyruvoyl-dependent histidine decarboxylase. Active site structure and mechanistic analysis. J. Biol. Chem. 1989, 264, 12737–12743. [PubMed] [Google Scholar]
- Zhang S.; Liu M.; Yan Y.; Zhang Z.; Jordan F. C2-alpha-lactylthiamin diphosphate is an intermediate on the pathway of thiamin diphosphate-dependent pyruvate decarboxylation. Evidence on enzymes and models. J. Biol. Chem. 2004, 279, 54312–54318. 10.1074/jbc.M409278200. [DOI] [PubMed] [Google Scholar]
- Jordan F.; Li H.; Brown A. Remarkable Stabilization of Zwitterionic Intermediates May Account for a Billion-fold Rate Acceleration by Thiamin Diphosphate-Dependent Decarboxylases. Biochemistry 1999, 38, 6369–6373. 10.1021/bi990373g. [DOI] [PubMed] [Google Scholar]
- Heinrich D.; Diederichsen U.; Rudolph M. Lys314 is a Nucleophile in Non-Classical Reactions of Orotidine-5′-Monophosphate Decarboxylase. Chem. - Eur. J. 2009, 15, 6619–6625. 10.1002/chem.200900397. [DOI] [PubMed] [Google Scholar]
- Callahan B. P.; Bell A. F.; Tonge P. J.; Wolfenden R. A Raman-active competitive inhibitor of OMP decarboxylase. Bioorg. Chem. 2006, 34, 59–65. 10.1016/j.bioorg.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Specificity in transition state binding: The Pauling model revisited. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. Enzyme Architecture: Erection of Active Orotidine 5′-Monophosphate Decarboxylase by Substrate-Induced Conformational Changes. J. Am. Chem. Soc. 2017, 139, 16048–16051. 10.1021/jacs.7b08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]