

Purpose of review
Congenital myasthenic syndromes (CMS) are a group of heterogeneous inherited disorders caused by mutations in genes encoding proteins whose function is essential for the integrity of neuromuscular transmission. This review updates the reader on the expanding phenotypic spectrum and suggested improved treatment strategies.
Recent findings
As next-generation sequencing is taken into the clinic, its use is both continuing to unearth new causative genes in which mutations underlie CMS and also broadening the phenotypic spectrum for known CMS genes. The number of genes in which mutations may cause neuromuscular transmission defects has now passed 30. The defective transmission may be part of an overall more complex phenotype in which there may be muscle, central nervous system or other involvement. Notably, mutations in series of genes encoding proteins located in the presynatic motor bouton have been identified. Rare cases of mutations in basal laminar proteins of the synaptic cleft are coming to light and additional mutations/phenotypic features have been located in some of the larger neuromuscular junction proteins such as AGRN and MUSK, where previously mutation screening by sanger sequencing was time consuming and costly. Finally, there are more reports of the beneficial effects of treatment with β2-adrenergic receptor agonists in patients, and the study of their action in disease models.
Summary
Recent studies of the CMS illustrate the increasing complexity of the genetics and pathophysiological mechanisms involved. With therapy tailored for the underlying disease mechanism treatment, although incomplete, is usually life-transforming. However, treatment for newly identified conditions in which myasthenia is only one component within complex multisystem disorder will prove challenging.
Keywords: congenital myasthenic syndromes, ephedrine, next-generation sequencing, postsynaptic, presynaptic, salbutamol, synaptic
INTRODUCTION
Congenital myasthenic syndromes (CMS) are a group of rare inherited disorders of neuromuscular transmission [1,2]. The syndromes share the clinical feature of fatigable muscle weakness, but the age of onset, presenting symptoms, distribution of weakness and response to treatment vary according to the gene harbouring the mutation(s) and the underlying molecular mechanism that impairs signal transmission at the neuromuscular junction. Typical characteristics of many myasthenic disorders are ptosis, ophthalmoparesis, facial and bulbar weakness, and generalized muscle weakness presenting in the neonatal period or early childhood. However, symptoms can present in later childhood, adolescence or even adulthood and the pattern of muscle weakness vary. In a number of subtypes, there is marked limb-girdle weakness but the eyes and facial muscles are spared, and this can make differential diagnosis more challenging. Due to the more widespread adoption of next-generation sequencing the number of different genes implicated in CMS continues to grow and is now greater than 30 (Table 1). It is likely that mutations in some of these newly recognized genes will be exceptionally rare causes of CMS and will essentially remain isolated case studies, whereas others will warrant inclusion in standard genetic screening panels. Many of the more recently identified mutations causing CMS are in proteins whose expression is not restricted to the neuromuscular junction and, not surprisingly, mutations in these genes may often give rise to disorders with complex phenotypes with varying levels of involvement of impaired neuromuscular transmission.
Table 1.
Congenital myasthenic syndromes
| Site of defect | Mechanism | Gene | Protein |
| Presynaptic | Defects in ACh recycling | SLC5A7 | ChT |
| Defects in ACh synthesis | CHAT | ChAT | |
| Defects in loading of ACh in synaptic vesicles | SLC18A3 | VAChT | |
| Defects in synaptic vesicle docking, priming, fusing and exocytosis | SNAP25B | Soluble N-ethylmaleimide-sensitive factor attachment protein receptor 25 | |
| UNC13A | Munc 13 | ||
| SYB1/VAMP1 | Synaptobrevin-1/Vesicle associated membrane protein 1 | ||
| SYT2 | Synaptotagmin-2 | ||
| PREPL | Propyl-endopeptidase-like | ||
| Defects in axonal transport of proteins | MYO9A | Myosin IXA | |
| Synaptic | Acetylcholinesterase deficiency | COLQ | Collagen-tail subunit of acetylcholinesterase |
| Synaptic basement membrane defects | COL13A1 | Collagen type 13 α1 | |
| LAMA5 | Laminin α5 | ||
| LAMB2 | Laminin β2 | ||
| Defects in AChR clustering pathway | AGRN | Agrin | |
| Postsynaptic | Reduced numbers of AChR (AChR deficiency) | CHRNE(CHRNA1CHRNB1CHRND) | AChR subunits |
| Kinetic changes in AChR function (slow channel syndromes) | CHRNA1CHRNB1CHRNDCHRNE | AChR subunits | |
| Kinetic changes in AChR function (fast channel syndromes) | CHRNA1CHRNDCHRNB1CHRNE | AChR subunits | |
| Defects in AChR clustering pathway | LRP4 | LDL-related protein 4 | |
| MUSK | Muscle-specific tyrosine kinase | ||
| DOK7 | Downstream of kinase 7 | ||
| RAPSN | Rapsyn | ||
| Defect in skeletal muscle voltage-gated sodium channel | SCN4A | Sodium voltage-gated channel α4 | |
| Plectin deficiency | PLEC | Plectin | |
| Pre + post | Defective glycosylation | ALG2 | α-1,3-Mannosyltransferase |
| ALG14 | UDP-N-acetylglucosaminyltransferase | ||
| DPAGT1 | Dolichyl-phosphate N-acetyl-glucosaminephosphotransferase 1 | ||
| GFPT1 | Glutamine-fructose-6-phosphate transaminase 1 | ||
| GMPPB | GDP-mannose pyrophosphorylase |
ACh, acetylcholine; AChR, acetylcholine receptor; ChAT, choline acetyltransferase; ChT, choline transporter; COL13A1, collagen type XIII alpha 1; LRP4, LDL receptor-related protein 4; VAChT, vesicular acetylcholine transporter.
In this review, we report on the expanding number of ‘causative-genes’ in which mutations that underlie impaired neuromuscular transmission have been identified, we update on the growing phenotypic spectrum for both new and better established CMS subtypes, and review recent thinking on treatment strategies.
Box 1.

no caption available
PRESYNAPTIC CONGENITAL MYASTHENIC SYNDROMES
It has become apparent that the presynaptic forms of CMS can be subdivided into two main categories; first, those involved in the synthesis and recycling of acetylcholine (ACh) and second, those involved in vesicle docking and transmitter release from the nerve terminal. Genes harbouring CMS-causing mutations in the ACh-recycling pathway include SLC5A7, CHAT and SLC18A3, which encode the high-affinity choline uptake transporter, cholineacetyltransferase and the vesicular ACh transporter, respectively [3–5]. Mutations in CHAT have long been established as a cause of a CMS characterized by episodic apneas and respiratory crises early in life that are often induced by infections or stress [4]. Between crises symptoms may be relatively mild and the number of life-threatening crises reduces with age so that by adolescence and adulthood occurrence is very rare. SLC5A7 and SLC18A3 CMS subtypes in general are more severe than choline acetyltransferase (CHAT)-CMS, and can lead to fatal foetal akinesia during pregnancy, and they share the same life-threatening apnoeic crises in early life. In addition, patients are reported to have marked ptosis, ophthalmoplegia, muscle fatigability, and in some cases [3,5] of both SLC5A7 and SLC18A3 CMS learning difficulties have been noted, which, in the absence of hypoxic damage, rarely if ever occurs in CHAT CMS. The reported phenotype for SLC5A7 CMS varies with many showing arthrogryposis/joint contractures at birth and limited survival, but other are relatively mild. Patients with mutations in the ACh recycling pathway respond to cholinesterase medication although in the severe cases the effect is minimal. It is of note that distal hereditary motor neuropathy type VII is caused by dominant mutations in SLC5A7[6,7], but surprisingly there appears to be little phenotypic overlap between the two disorders.
A second group of presynaptic syndromes features proteins involved in neurotransmitter release from the presynaptic nerve terminal. The soluble N-ethylmaleimide-sensitive factor attachment protein (SNARE) complex governs vesicle membrane docking, fusion and transmitter release from the presynaptic terminal. Synaptobrevin (VAMP1), syntaxin and synaptosomal-associated protein 25B (SNAP25B) form the core of the SNARE complex. Synaptotagmin 2 (SYT2) acts as a sensor detecting the influx of calcium following activation of voltage-gated calcium channels and interacts with the SNARE complex to trigger transmitter release. MUNC13 (UNC13A) is involved in assembly of the SNARE complex. Mutations in VAMP1[8,9], SNAP25B[10], UNC13A[11] and SYT2[12,13] have been found underlie syndromes in which synaptic transmission at the neuromuscular junction is impaired. However, SNARE-mediated membrane fusion is an essential feature of eukaryotic organisms and occurs at multiple sites and thus impaired neuromuscular transmission is often only likely to be one component within a wider multisystem disorder. For example, a de-novo SNAP25B mutation gave rise to a case of reduced quantal release from the motor nerve terminal, but also severe developmental delay, and ataxic gait and dysarthria. By contrast with the majority of CMS, SYT2 mutations give rise to a dominant disorder with features of a motor neuropathy and a neuromuscular transmission disorder resembling the autoimmune Lambert–Eaton myasthenic syndrome in which autoantibodies are directed against the presynaptic voltage-gated calcium channels. Like the Lambert–Eaton myasthenic syndrome, the syndromes arising from mutations in vesicle docking and neurotransmitter release mechanisms most commonly show increment of compound muscle action potential amplitude with high-frequency repetitive nerve stimulation. A more detailed review of the molecular mechanisms and electrophysiology of the presynaptic CMS is given in [14▪▪].
BASAL LAMINA-ASSOCIATED SYNDROMES
Mutations in the collagen-like tail subunit (COLQ) that anchors the asymmetric form of acetylcholinesterase (AChE) to the basal lamina in the synaptic cleft of the neuromuscular junction are an established major cause of CMS. Some laminins subtypes are selectively expressed at the neuromuscular synapse and mouse models indicate a role in both synaptic structure and signalling but CMS resulting from mutations in laminin subunits are exceedingly rare. Single case reports have identified a homozygous mutation in laminin α5 (LAMA5) [15] and laminin β2 (LAMB2) [16]. In each case, the neuromuscular junction defect was reported in association with additional nonmyasthenic phenotypic features, for LAMA5 it was myopia and facial tics, for LAMB2 it is Pierson syndrome [17] characterized by chronic renal failure and neurodevelopmental problems. Collagen type XIII alpha 1 (COL13A1) is a single-pass type II transmembrane protein with a triple-helical collagenous ectodomain. It has main basic forms, one anchored in the plasma membrane via the transmembrane domain, and the second results from proteolytic cleavage of the ectodomain to produce a soluble product that interacts with components of the basal lamina. Mouse models have shown that loss of COL13A1 affects early maturation of neuromuscular junction structures on both pre and postsynaptic sides, and endplates remained small immature and fragmented [18]. In keeping with these studies of COL13A1 function a report on 16 cases from 11 independent kinships found clinical presentation mostly at birth with hypotonia, and breathing and feeding difficulties [19▪▪]. Respiratory crises related to recurrent apnoea usually triggered by infections were common. Bilateral nonfatigable ptosis in adulthood and marked weakness of neck flexion were characteristic features. Patients respond to treatment with salbutamol and 3,4-diaminopyridine in their early years, but, whether treated or not, disease severity improves over time so that in some cases by adulthood muscle strength may be normal [19▪▪,20]. Studies of COL13A1 mutations emphasis the role of extracellular matrix proteins that are not part of the AGRN–LDL receptor-related protein 4 (LRP4)–MUSK–DOK7 signalling pathway in maturation of the mature synapse.
AGRN is a large extracellular matrix proteoglycan. It has several isoforms, and it is its neural isoform that plays a key role in maintaining synaptic structure through binding the LRP4 which in turn binds and activates MUSK [21]. Each of these three proteins is relatively large, with many exons, various splicing isoforms and multiple functional domains. The size and the number of exons within these genes made them challenging to screen using standard PCR and Sanger sequencing techniques. Next-generation sequencing has greatly facilitated mutation detection within these genes and led to the a broadening of the phenotypic spectrum for CMS due to either AGRN [22,23] or MUSK mutations including variants in MUSK giving rise to a late onset limb girdle CMS [24▪] isolated vocal cord paralysis [25] or pregnancy-associated respiratory failure [26]. It is likely that there will be increasing pick up of variants within these genes but defining pathogenicity will often require time-consuming and quite complex functional studies.
GLYCOSYLATION PATHWAY CONGENITAL MYASTHENIC SYNDROMES
The asparagine-linked (N-linked) glycosylation pathway is a ubiquitous process present in all eukaryotic cells where there is sequential attachment of sugar moieties to a membrane lipid (dolichol) that are then transferred to a protein. The addition and processing of these glycans are crucial for the folding, assembly, stability and intracellular transport of proteins. Mutations in the genes which encode components of this pathway are often devastating causing severe multisystem disorders that may be fatal. However, a group of CMS, characterized by a limb girdle pattern on muscle weakness with little ocular or facial involvement, are found to have mutations in proteins that contribute to the early steps of this pathway [27–30]. Mutations within the same in genes, for example DPAGT1, can lead to either a severe multisystem disorder or a myasthenic disorder in which fatigable muscle weakness is the only presenting symptom. An article by Dong et al.[31▪▪] had shed light on what may underlie these very different manifestations. DPAGT1 encodes dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetylglucosaminephosphotransferase 1 and is the enzyme responsible for the first step in the assembly of the core glycan (Glc3Man9GlcNAc2) on the lipid dolichol. Crystallization of DPAGT1 in its apo state, when bound to UDP-GlcNac or tunicamycin and functional studies of DPAGT1 enzymatic activity showed that mutations the cause the multisystem disorder (CDG1j) are either null (frameshift or nonsense mutations) or affects catalytic activity, whereas those causing CMS tend to be located further from the catalytic site and may reduce protein expression/dimerization or modestly decrease catalytic activity. The neuromuscular junction is known to be very heavily glycosylated and it seems likely that it is sensitive to perturbations in function caused by modest impairment of the early steps of the N-linked glycosylation pathway. Uncovering the molecular structure of DPAGT1 should help predict the pathogenicity and severity of genetic variants detected in the DPAGT1[31▪▪].
UPDATE ON TREATMENT STRATEGIES
The current repertoire of drugs for use in CMS includes as follows:
Drugs that increase ACh release, such as potassium blockers (3,4-diaminopyridine); drugs that maintain high ACh concentrations within the synaptic cleft, such as AChE inhibitors (mainly pyridostigmine); β2-adrenergic receptor agonists (ephedrine, salbutamol); and ACh receptor (AChR) open-channel blockers (fluoxetine, quinidine) [1,32]. A diagrammatic representation of the neuromuscular junction and the site of action for these drugs are shown in Fig. 1. The response to treatment depends upon the subtype of CMS and the underlying pathogenic molecular mechanism. A number of recent reports have reviewed literature on treatment and provided potential algorithms for treatments strategies [32–34].
FIGURE 1.
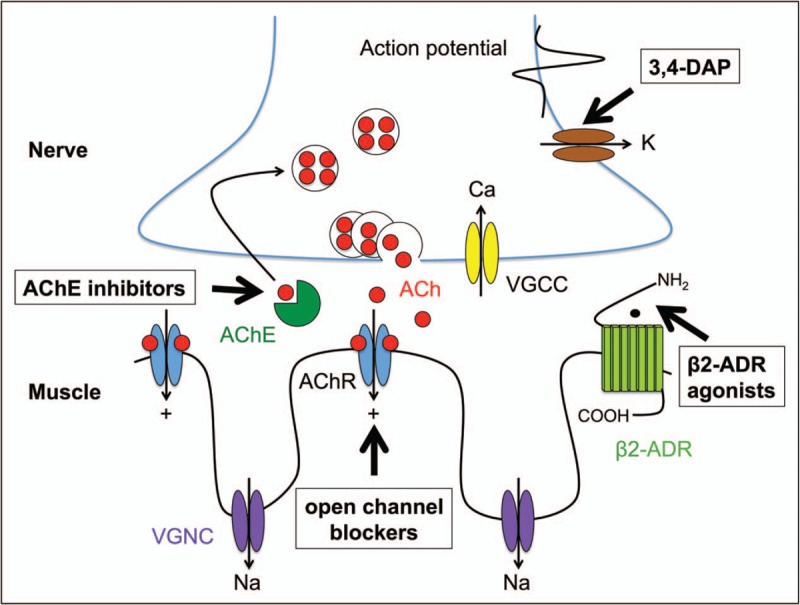
Diagrammatic representation for the sites of action for commonly used drugs for congenital myasthenic syndromes AChE, acetylcholinesterase inhibitors (neostigmine, pyridostigmine); 3,4-DAP, 3,4-diaminopyridine; open channel blockers (quinidine, fluoxetine); β2-ADR agonists, β2-adrenergic receptor agonists (salbutamol, ephedrine). VGCC, voltage-gated calcium channel; VGNC, voltage-gated sodium channel.
For many years reversible, competitive AChE inhibitors, such as pyridostigmine [35], were the mainstay of treatment for myasthenia. By blocking the action of AChE, the presence of ACh within the synapse is prolonged thus giving a greater probability of reaching the depolarization threshold for generation of the muscle action potential. Although effective for many CMS in others there was no clear response and in some it was positively harmful. Now, with greater understanding of the mutations and molecular mechanisms underlying CMS treatments can be tailored for the specific syndrome and depending on diseases severity and patient response this can include utilizing different combinations of the drugs. Pyridostigmine is quite clearly contraindicated for endplate AChE deficiency due to mutations in COLQ[36] as there is already a deficit of AChE function, and similarly in the dominant slow channel syndrome increasing the level and duration of ACh within the synaptic cleft is only likely to exacerbate the disorder. The use of AChR open channel blockers, fluoxetine or quinidine, can be remarkably effective for some slow channel mutations but the response is less marked for others. Adrenergic agonists, in particular ephedrine, have been used for the treatment of myasthenia, since the 1930s [37,38], but were largely replaced once AChE inhibitors were found to be effective in symptomatically treating myasthenia gravis [39]. Over the last 10–15 years, β2-agonists have re-entered as a mainstream option in treatment. Clearly an alternative to cholinesterase inhibitors was required for endplate AChE deficiencies, and in these patients a beneficial response to ephedrine was reported [40]. However, it was following the identification of DOK7 mutations as a major cause of CMS and their slow but remarkable improvement with β2-agonist medication that provided the impetus for its more widespread adoption. It is now apparent that not only DOK7-CMS but all the forms of CMS resulting from mutations in the AGRN–LRP4–MUSK–DOK7 signalling pathway for clustering AChR respond well to β2-agonists although the response for AGRN-CMS tends to be less pronounced. AChR deficiency patients on long-term anticholinesterase medication have also been found to benefit from β2-agonists. Long-term anticholinesterase has been shown to affect neuromuscular transmission and motor endplate fine structure [41], and so over time this treatment may become less effective. It appears that β2 agonists can alleviate these detrimental effects. Studies of ‘knock out’ mouse models of the neuromuscular junction identified that neurotransmission itself act to disperse AChR and disrupt synaptic structures and that this is countered by the input from the AGRN–LRP4–MUSK–DOK7 signalling pathway [42,43] (Fig. 2). A plausible way that β2 agonists have their beneficial effect is through enhancing the signal downstream that comes from the AGRN pathway and thus enhances and stabilize neuromuscular junction structure.
FIGURE 2.
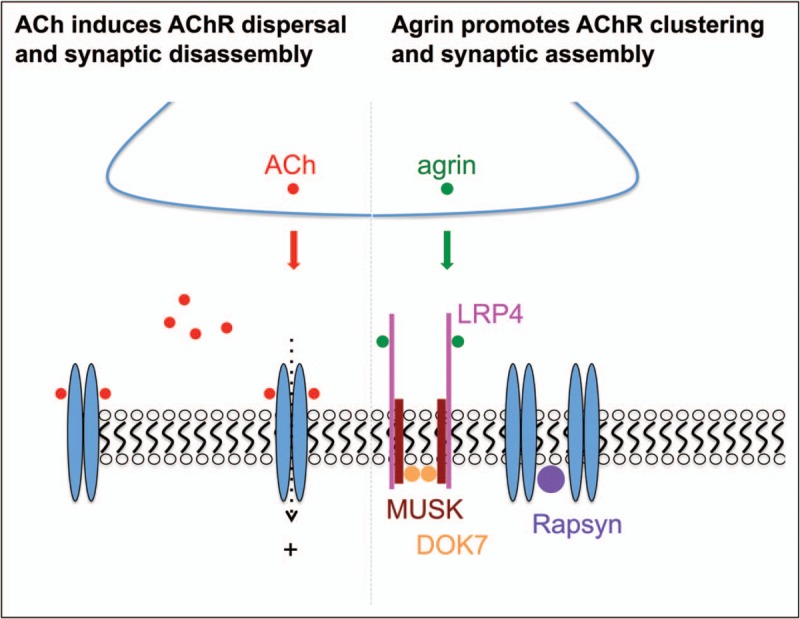
Pathways of synaptic disassembly and assembly at the neuromuscular junctions. Acetylcholine, released from the nerve terminal following a nerve stimulus, activates acetylcholine receptors. In addition to instructing muscle contraction, acetylcholine receptor activation is thought to disperse the tightly packed acetylcholine receptor away from the nerve terminal, which initiates synaptic disassembly. The agrin-induced acetylcholine receptor clustering pathway promotes synaptic assembly and thus counteracts the negative effect of acetylcholine on synaptic structure. Agrin is released from the nerve terminal, then binds LDL receptor-related protein 4, which activates MUSK and DOK7 and ultimately results in aggregation of postsynaptic acetylcholine receptors and maturation of the synaptic apparatus.
Little is known at the molecular level about how β2 agonists are exerting their effect at the neuromuscular junction, but a number of reports have begun to address this. Recently β2-adrenergic receptors were found to be present in higher densities at the postsynaptic membrane [44,45▪]. In addition, it is now thought that neuromuscular junctions might receive direct sympathetic innervation [44,46]. β2-adrenergic agonists do not have an immediate functional effects at the neuromuscular junction at doses clinically attainable [47] but fresh studies, using cell models, zebrafish or mouse models of myasthenia, all show that the β2-adrenergic receptor has a role in maintenance or enhancement of the postsynaptic structure of the neuromuscular junction [45▪,48,49▪].
CONCLUSION
Many cases of CMS can be given effective symptomatic treatment with the drugs that are currently available once an understanding of the disease mechanism resulting from the mutation(s) is known. This often requires a balance between medication that directly enhances neuromuscular transmission and medication that helps maintain synaptic structure. However, clinical application of next-generation sequencing is revealing mutations in which myasthenia is only one component in a much wider disease phenotype. Symptomatic treatment of the neuromuscular junction is not always effective in severe cases of CMS and may not be appropriate for disorders in which myasthenia is the minor component in a multisystem disorder. We may now be reaching the time when it is apt to explore how novel gene therapies might be applied to these rare genetic disorders.
Acknowledgements
We would like to acknowledge the support of the Wellcome Trust and the MRC for the funding that underlies this review. D.B. holds MRC programme grant MR/M006824. A.E.V. was a Wellcome Trust Clinical Training Fellow.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Engel AG. Congenital myasthenic syndromes in 2018. Curr Neurol Neurosci Rep 2018; 18:46. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez Cruz PM, Palace J, Beeson D. The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int J Mol Sci 2018; 19:1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauche S, O’Regan S, Azuma Y, et al. Impaired presynaptic high-affinity choline transporter causes a congenital myasthenic syndrome with episodic apnea. Am J Hum Genet 2016; 99:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohno K, Tsujino A, Brengman JM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci U S A 2001; 98:2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Grady GL, Verschuuren C, Yuen M, et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016; 87:1442–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barwick KE, Wright I, Al-Turki S, et al. Defective presynaptic choline transporter underlies hereditary motor neuropathy. Am J Hum Genet 2012; 91:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salter CG, Beijer D, Hardy H, et al. Truncating SLC5A7 mutations underlie a spectrum of dominant hereditary motor neuropathies. Neurol Genet 2018; 4:e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salpietro V, Lin W, Delle Vedove A, et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol 2017; 81:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen XM, Scola RH, Lorenzoni PJ, et al. Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann Clin Trans Neurol 2017; 4:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia and intellectual disability. Neurology 2014; 83:2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel AG, Selcen D, Shen XM, et al. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet 2016; 2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrmann DN, Horvath R, Sowden JE, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert–Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet 2014; 95:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whittaker RG, Hermann DN, Bansagi B, et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 2015; 85:1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14▪▪.Nicolau S, Milone M. The electrophysiology of presynaptic congenital myasthenic syndromes with and without facilitation: from electrodiagnostic findings to molecular mechanisms. Front Neurol 2019; 10:257. [DOI] [PMC free article] [PubMed] [Google Scholar]; A clear and very helpful summary of the diagnostic features that may point in the direction of a presynaptic congenital myasthenic syndrome.
- 15.Maselli RA, Arredondo J, Vázquez J, et al. A presynaptic congenital myasthenic syndrome attributed to a homozygous sequence variant in LAMA5. Ann N Y Acad Sci 2018; 1413:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maselli RA, Ng JJ, Anderson JA, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet 2009; 46:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matejas V, Hinkes B, Alkandari F, et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 2010; 31:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haronen H, Zainul Z, Tu H, et al. Collagen XIII secures pre and postsynaptic integrity of the neuromuscular synapse. Hum Mol Genet 2017; 26:2076–2090. [DOI] [PubMed] [Google Scholar]
- 19▪▪.Rodríguez Cruz PM, Cossins J, Estephan EP, et al. The clinical spectrum of the congenital myasthenic syndrome resulting from COL13A1 mutations. Brain 2019; 142:1547–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive collection of patients that provide an insight into the spectrum of the disease phenotype associated with collagen type XIII alpha 1 mutations that should facilitate early recognition of this treatable disorder.
- 20.Dusl M, Moreno T, Munell F, et al. Congenital myasthenic syndrome caused by novel COL13A1 mutations. J Neurol 2019; 266:1107–1112. [DOI] [PubMed] [Google Scholar]
- 21.Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol 2013; 5:a009167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xi J, Yan C, Liu WW, et al. Novel SEA and LG2 Agrin mutations causing congenital myasthenic syndrome. Orphanet J Rare Dis 2017; 12:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudell JB, Maselli RA, Yarov-Yarovoy V, Ferns MJ. Pathogenic effects of agrin V1727F mutation are isoform-specific and decrease its expression and affinity for HSPGs and LRP4. Hum Mol Genet 2019; doi: 10.1093/hmg/ddz081. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24▪.Owen D, Töpf A, Preethish-Kumar V, et al. Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am J Med Genet A 2018; 176:1594–1601. [DOI] [PubMed] [Google Scholar]; Expands the phenotype associated with MUSK mutations and makes the point that MUSK mutations should be considered for some later onset cases of congenital myasthenic syndromes.
- 25.Murali C, Li D, Grand K, et al. Isolated vocal cord paralysis in two siblings with compound heterozygous variants in MUSK: expanding the phenotypic spectrum. Am J Med Genet A 2019; 179:655–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wadwekar V, Pillai RR, Sesh S, et al. Pregnancy-associated respiratory failure in muscle specific kinase congenital myasthenic syndrome. Muscle Nerve 2019; 59:E24–E26. [DOI] [PubMed] [Google Scholar]
- 27.Senderek J, Müller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 2011; 88:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belaya K, Finlayson S, Slater CR, et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet 2012; 91:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 2013; 136:944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belaya K, Rodriguez Cruz PM, Liu WW, et al. Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 2015; 138:2493–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31▪▪.Dong YY, Wang H, Pike ACW, et al. Structures of DPAGT1 explain glycosylation disease mechanisms and advance TB antibiotic design. Cell 2018; 175:1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the crystallization and determination of the protein structure of DPAGT1 which is used to demonstrate how differing missense mutations can cause very different disease severities.
- 32.Lee M, Beeson D, Palace J. Therapeutic strategies for congenital myasthenic syndromes. Ann N Y Acad Sci 2018; 1412:129–136. [DOI] [PubMed] [Google Scholar]
- 33.Farmakidis C, Pasnoor M, Barohn RJ, Dimachkie MM. Congenital myasthenic syndromes: a clinical and treatment approach. Curr Treat Options Neurol 2018; 20:36. [DOI] [PubMed] [Google Scholar]
- 34.Thompson R, Bonne G, Missier P, Lochmüller H. Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerg Top Life Sci 2019; 3:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehndiratta MM, Pandey S, Kuntzer T. Acetylcholinesterase inhibitor treatment for myasthenia gravis. Cochrane Database Syst Rev 2014; CD006986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A 1998; 95:9654–9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgeworth H. A report of progress on the use of ephedrine in a case of myasthenia gravis. J Am Med Assoc 1930; 94:1136. [Google Scholar]
- 38.Edgeworth H. The effect of ephedrine in the treatment of myasthenia gravis: second report. J Am Med Assoc 1933; 100:1401. [Google Scholar]
- 39.Rowland LP. Prostigmine-responsiveness and the diagnosis of myasthenia gravis. Neurology 1955; 5:612–623. [DOI] [PubMed] [Google Scholar]
- 40.Bestue-Cardiel M, Saenz de Cabezon-Alvarez A, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology 2005; 65:144–146. [DOI] [PubMed] [Google Scholar]
- 41.Engel AG, Lambert EH, Santa T. Study of long-term anticholinesterase therapy – effects on neuromuscular-transmission and on motor endplate fine-structure. Neurology 1973; 23:1273–1281. [DOI] [PubMed] [Google Scholar]
- 42.Lin W, Dominguez B, Yang J, et al. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 2005; 46:569–579. [DOI] [PubMed] [Google Scholar]
- 43.Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol 2006; 16:74–82. [DOI] [PubMed] [Google Scholar]
- 44.Khan MM, Lustrino D, Silveira WA, et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci U S A 2016; 113:746–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45▪.McMacken GM, Spendiff S, Whittaker RG, et al. Salbutamol modifies the neuromuscular junction in a mouse model of ColQ myasthenic syndrome. Hum Mol Genet 2019; doi: 10.1093/hmg/ddz081. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]; Provides evidence that salbutamol modifies the structure of the neuromuscular junction when in a diseased/disrupted state.
- 46.Straka T, Vita V, Prokshi K, et al. Postnatal development and distribution of sympathetic innervation in mouse skeletal muscle. Int J Mol Sci 2018; 19:1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milone M, Engel AG. Block of the endplate acetylcholine receptor channel by the sympathomimetic agents ephedrine, pseudoephedrine, and albuterol. Brain Res 1996; 740:346–352. [DOI] [PubMed] [Google Scholar]
- 48.McMacken G, Cox D, Roos A, et al. The beta-adrenergic agonist salbutamol modulates neuromuscular junction formation in zebrafish models of human myasthenic syndromes. Hum Mol Genet 2018; 27:1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49▪.Clausen L, Cossins J, Beeson D. Beta-2 adrenergic receptor agonists enhance AChR clustering in C2C12 myotubes: implications for therapy of myasthenic disorders. J Neuromuscul Dis 2018; 5:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]; Suggests that β2-agonists have a clear postsynaptic affect at the neuromuscular junction.