

Abstract
Pancreatic islets of type 2 diabetes patients have altered DNA methylation, contributing to islet dysfunction and the onset of type 2 diabetes. The cause of these epigenetic alterations is largely unknown. We set out to test whether (i) islet DNA methylation would change with aging and (ii) early postnatal overnutrition would persistently alter DNA methylation. We performed genome-scale DNA methylation profiling in islets from postnatally over-nourished (suckled in a small litter) and control male mice at both postnatal day 21 and postnatal day 180. DNA methylation differences were validated using quantitative bisulfite pyrosequencing, and associations with expression were assessed by RT-PCR. We discovered that genomic regions that are hypermethylated in exocrine relative to endocrine pancreas tend to gain methylation in islets during aging (R2 = 0.33, P < 0.0001). These methylation differences were inversely correlated with mRNA expression of genes relevant to β cell function [including Rab3b (Ras-related protein Rab-3B), Cacnb3 (voltage-dependent L-type calcium channel subunit 3), Atp2a3 (sarcoplasmic/endoplasmic reticulum calcium ATPase 3) and Ins2 (insulin 2)]. Relative to control, small litter islets showed DNA methylation differences directly after weaning and in adulthood, but few of these were present at both ages. Surprisingly, we found substantial overlap of methylated loci caused by aging and small litter feeding, suggesting that the age-associated gain of DNA methylation happened much earlier in small litter islets than control islets. Our results provide the novel insights that aging-associated DNA methylation increases reflect an epigenetic drift toward the exocrine pancreas epigenome, and that early postnatal overnutrition may accelerate this process.
Keywords: epigenetics, DNA methylation, islet aging, aging, type II diabetes, overnutrition, programing, programming, endocrine pancreas; development; islet
Introduction
As of 2017, 9.3% of the U.S. population are afflicted with diabetes, >90% of which is type 2 diabetes (T2D) [1]. Pancreatic islet failure is a critical component of T2D pathogenesis [2]. The development of T2D is influenced by both genetic and environmental factors, suggesting the likely involvement of epigenetic mechanisms which have the potential to both influence gene expression and be affected by environment [3]. Indeed, genome-wide studies have shown significantly altered DNA methylation in islets from T2D patients [4, 5]. A critical outstanding question is what causes these DNA methylation alterations.
During critical ontogenic periods, transient nutritional and other environmental influences can affect developmental outcomes, with lifelong consequences. Extensive human [6] and animal model data [7, 8] indicate that such “developmental programing” may help explain the increasing prevalence of T2D in recent decades [9]. In rodents, early postnatal overnutrition due to suckling in small litters (SLs) induces persistent alterations in circulating insulin concentrations [7, 10]. In vitro studies demonstrated impaired glucose-stimulated insulin secretion and stable gene expression changes in SL islets, suggesting that the endocrine pancreas is the primary repository of the persistent effect [10]. Induced alterations in epigenetic regulation could mediate such developmental programing in the endocrine pancreas. In particular, methylation of cytosine-phosphate-guanine (CpG)-dinucleotides in DNA is recognized as the most stable epigenetic mark, and therefore a prime candidate to mediate the lifelong persistence that is the hallmark of developmental programing [9, 11].
To our knowledge only one previous study has employed DNA methylation profiling in islets to examine epigenetic responses to an early nutritional exposure [12]. In pancreatic islets of 7-week old rats that had been subjected to intrauterine growth retardation, over 1400 loci showed DNA methylation changes compared to controls. That study, however, did not assess whether these changes were actually present at the end of the “exposure” period (i.e. birth) and persisted to adulthood [12]. No previous study has used a genome-scale approach to test whether early postnatal nutrition induces persistent epigenetic changes in the endocrine pancreas.
In addition to genetics and nutrition, aging is a major etiologic factor in T2D. Both glucose tolerance and in vitro glucose-stimulated insulin secretion decline with age in both humans and rodents [13, 14], and age-associated epigenetic changes have been linked to disrupted β cell function [15–17]. Therefore, both developmental programing and aging may contribute to islet epigenetic alterations that lead to endocrine dysfunction.
Accordingly, the main goal of this study was to test the hypothesis that both aging and early postnatal overnutrition cause DNA methylation alterations in endocrine pancreas. Our data show age-associated epigenetic changes in the endocrine pancreas, which appear to be functional and are strongly associated with DNA methylation differences that distinguish endocrine from exocrine pancreas. By examining islets from SL mice, we found that although suckling-period overnutrition results in subtle increases in islet DNA methylation at weaning, these group differences generally do not persist to adulthood. More notably, our cross-sectional study design also indicated that the DNA methylation increments caused by SL feeding resemble those associated with aging, suggesting that early postnatal overnutrition causes accelerated epigenetic aging in islets.
Results
Substantial DNA Methylation Differences between Exocrine and Endocrine Pancreas
Genome-scale DNA methylation analysis was performed by methylation specific amplification and next-generation sequencing (MSA-seq) [18] which interrogates a subset of SmaI/XmaI intervals in the genome. Prior to sequencing experimental samples, MSA specificity was verified using sex-specific PCR amplification (Supplementary Fig. S1), which showed methylated X chromosomal loci only in females. To document the sensitivity of MSA-seq, we began by profiling DNA methylation differences between endocrine (islets) and exocrine pancreas. As expected, MSA-seq detected extensive methylation differences between exocrine vs. endocrine pancreas at postnatal day 21 (P21) (Fig. 1a), so we applied a fairly stringent cutoff (P < 0.005) to identify hits. There were 2311 SmaI/XmaI intervals (regions) with higher methylation in exocrine than endocrine pancreas (i.e. exocrine hypermethylated) and just 456 exocrine hypomethylated regions (Fig. 1a and Data Set). Interestingly, whereas regions of exocrine hypermethylation were not enriched at genes (Fig. 1b), regions of exocrine hypomethylation were, particularly at transcription start sites (TSSs) (Fig. 1b, Supplementary Table S1). Gene ontology (GO) analysis (Fig. 1c, Supplementary Table S2) showed that exocrine vs. endocrine methylation differences were associated with biological processes such as “protein binding,” “cytoskeleton organization” and “amino acid metabolism,” of potential relevance to regulated hormone secretion.
Figure 1:

DNA methylation differences associated with age and tissue type. (a) Volcano plot of MSA-seq data on exocrine vs. endocrine pancreas. Each dot represents the mean exocrine:endocrine MSA-seq read count ratio at one SmaI/XmaI interval; red and green dots represent intervals with higher and lower methylation in exocrine relative to endocrine pancreas, respectively (P < 0.005; n = 5). (b) Genomic distribution of intervals with higher (red) and lower (green) DNA methylation in exocrine vs. endocrine pancreas [****significant (P < 0.0001) enrichment relative to genome-wide]. (c) GO analysis of genes with exocrine vs. endocrine differences in DNA methylation in the pancreas (see Supplementary Table S2). (d) Volcano plot of MSA-seq data for P180 vs. P21 islets. Each dot represents the mean P180:P21 MSA-seq read count ratio at one SmaI/XmaI interval; red and green dots represent age-related increases and decreases, respectively (P < 0.05; n = 5). (e) Genomic distribution of intervals with increased (red) and decreased (green) DNA methylation with age [**significant (P < 0.01) enrichment or depletion relative to genome-wide]. (f) GO analysis of genes showing methylation increases with age. TES, transcription end site
DNA Methylation in the Endocrine Pancreas Increases with Age, Approaching That of the Exocrine Pancreas
We next profiled P180 vs. P21 endocrine pancreas to identify age-associated DNA methylation changes. Unlike in the exocrine vs. endocrine comparison, age-related changes were relatively subtle, so relatively loose criteria (P < 0.05) were applied to identify hits. Overall, islet methylation tended to increase with age; 528 genomic regions gained and only 44 lost DNA methylation (Fig. 1d and Data Set). Though slightly under-represented at genes (Fig. 1e), age-associated increases in DNA methylation were associated with biological processes of direct relevance to islet function including “regulation of membrane repolarization” (18.4-fold enrichment) and “regulation of insulin secretion” (5.5-fold enrichment) (Fig. 1f and Supplementary Table S3). Regions that lost methylation with age were significantly enriched near TSSs (Fig. 1e) but associated genes were not enriched for any specific ontologies.
We noted that many of the same genes came up in both the age-associated and endocrine vs. exocrine comparisons. Therefore, to test whether the exocrine vs. endocrine methylation differences might be related to those that occur during aging in endocrine pancreas, we focused on all SmaI/XmaI intervals with significant exocrine:endocrine methylation ratios (P < 0.005), and plotted these vs. their P180:P21 ratios in the endocrine pancreas. Among regions with an exocrine:endocrine methylation ratio >1, we found a striking positive correlation (Fig. 2a). There was no such correlation for regions with an exocrine:endocrine methylation ratio <1 (Fig. 2a). A similar result was obtained if we instead focused the analysis on all intervals undergoing methylation changes from P21 to P180 (Supplementary Fig. S2). Overall, 244 intervals showed both a gain of methylation from P21 to P180 and higher methylation in exocrine relative to endocrine pancreas (Data Set). GO analysis of associated genes showed the strongest process enrichments for “regulation of insulin secretion” and “positive regulation of ion transport” (Fig. 2b and Supplementary Table S4). This novel observation suggests that as islets age, locus-specific methylation gradually increases toward that of the exocrine pancreas, with accumulated DNA methylation and potential suppression of genes involved in insulin secretion pathways.
Figure 2:

hypermethylation in exocrine relative to endocrine pancreas predicts increased methylation during islet aging. (a) In regions showing significant exocrine vs. endocrine differences, age-associated methylation changes in islets are plotted vs. the exocrine:endocrine ratio. (b) GO analysis of genes with higher DNA methylation in both P180 vs. P21 islets and exocrine vs. endocrine pancreas
Bisulfite Pyrosequencing Validates the Observed DNA Methylation Differences
To confirm these observations, we selected 11 SmaI/XmaI intervals based on their endocrine/exocrine methylation differences and/or islet associated function (Supplementary Table S5). Bisulfite pyrosequencing validated the MSA-seq results for 9 of these, confirming positive P180:P21 and exocrine:endocrine methylation ratios. For example, evidence suggests that Rab3b (Ras-related protein Rab-3B) [19], Cacnb3 (voltage-dependent L-type calcium channel subunit 3) [20] and Atp2a3 (sarcoplasmic/endoplasmic reticulum calcium ATPase 3) [21] play important roles in regulating insulin secretion. At all three of these, MSA-seq showed age-associated methylation increases, such that the DNA methylation profile of P180 islets approached that of exocrine tissue (Fig. 3a, c and e). Pyrosequencing confirmed these results (Fig. 3b, d and f). At 5 more MSA-seq hits in genes associated with islet function, Rxra (retinoic acid receptor RXR-alpha) [22], Kcnq1 (potassium voltage-gated channel subfamily KQT) [23], Rasa3 (Ras GTPase-activating protein 3) [24], Prkce (protein kinase C epsilon type) [25] and Igfbp2 (insulin-like growth factor-binding protein 2) [26] bisulfite-pyrosequencing confirmed increased methylation during islet aging and in exocrine relative to endocrine pancreas (Supplementary Fig. S3).
Figure 3:

validation of DNA methylation differences detected by MSA-seq in the exocrine and aging endocrine pancreas. (a, c and e) Average MSA-seq data (n = 5) for Rab3b, Cacnb3 and Atp2a3 show increased DNA methylation as islets age (P180 vs. P21) and in exocrine relative to endocrine pancreas. Each tick represents 5 MSA-seq reads; asterisks indicate SmaI/XmaI sites selected for validation (Supplementary Table S5). Bisulfite pyrosequencing confirmed these differences at Rab3b (b) (age P = 0.005, tissue P < 0.0001), (d) Cacnb3 (age P < 0.0001, tissue P < 0.0001), (f) Atp2a3 (age P = 0009, tissue P < 0.0001) and Ins2 (h) (age P < 0.05, tissue P < 0.0001). (The SmaI/XmaI site that was informative at Rab3b is indicated by a gray bar. Shown are mean ± SEM, n = 7. Interrupted lines represent multiple pyrosequencing assays.) (g) Top: the Ins2 proximal promoter region; vertical lines indicate CpG sites analysed by clonal bisulfite sequencing (−182 and −171 bp from the TSS; the only two in this region). Bottom: clonal bisulfite sequencing data in P21 islets, P180 islets and P21 exocrine pancreas. Each line represents a clone; filled and empty circles represent methylated and unmethylated CpG sites, respectively. Average site-specific % methylation values are shown. TES, transcription end site
Although MSA-seq is not informative for the Ins2 (insulin 2) promoter, we asked whether DNA methylation in this region might follow a similar pattern. Indeed, clonal bisulfite sequencing in P180 vs. P21 islets (Fig. 3g) showed a 20–30% increase of DNA methylation at two CpG sites implicated in transcriptional regulation of Ins2 [27]. In exocrine tissue these two sites were almost completely methylated (Fig. 3g). Bisulfite pyrosequencing at these same two sites (Fig. 3h) confirmed increasing DNA methylation as islets age, and even higher methylation in the exocrine pancreas.
DNA Methylation Differences Are Correlated with Expression
Focusing on Rab3b, Cacnb3, Atp2a3 and Ins2 we next asked whether these age-related increases in DNA methylation are associated with changes in gene expression. Analysing across young and old islets and exocrine tissue we found a strong inverse correlation between DNA methylation and mRNA expression for Rab3b (Fig. 4a), Cacnb3 (Fig. 4b), Atp2a3 (Fig. 4c) and Ins2 (Fig. 4d). In all four cases, expression in P180 islets was intermediate between that of P21 islets and exocrine tissue (Fig. 4a–d and Supplementary Fig. S4). Overall, our data indicate that as islets age, increases in DNA methylation occur at regions normally hypermethylated in exocrine relative to endocrine pancreas, likely contributing to the age-associated deterioration of islet function.
Figure 4:

age- and tissue type-specific differences in DNA methylation correlate with expression at (a) Rab3b (R2 = 0.56, P < 0.0001), (b) Canb3 (R2 = 0.76, P < 0.0001), (c) Atp2a3 (R2 = 0.58, P < 0.0001) and (d) Ins2 (R2 = 0.67, P < 0.0001). Each point represents average % methylation at each locus and mRNA expression (averaged across triplicate measurements), n = 7 per age/tissue type
Early Postnatal Overnutrition Accelerates Aging-Associated DNA Methylation in Pancreatic Islets
Lastly, we asked if early postnatal overnutrition affects islet DNA methylation. As described previously [28], suckling-period overnutrition was achieved by cross fostering mice on P1 to SLs (4 pups) or control (C – 9 pups). At weaning, SL mice are ∼20% heavier and fatter than C mice [28], showing that they are substantially over-nourished during the suckling period. Extensive research with the SL rodent model over the last several decades shows that early postnatal overnutrition leads to permanent impairment of glucose-stimulated insulin secretion. Importantly, impaired glucose-stimulated insulin secretion is found both in vivo [7, 10] and in vitro [10], indicating that early postnatal overnutrition induces stable functional changes intrinsic to the islets, and suggesting the potential involvement of epigenetic mechanisms. To examine if the SL feeding altered islet DNA methylation, we first determined that SL feeding did not alter the β cell percentage, and only slightly reduced the α cell percentage (by 5%) (Fig. 5). This indicates that any DNA methylation differences detected between SL and C islets likely reflect epigenetic alterations within specific cell types (most likely β cells – the predominant islet cell type) rather than changes in the islet cellular composition.
Figure 5:

islet cellular composition is not substantially altered in SL mice. (a, b) Representative immunohistochemistry of P21 C and SL islets of Langerhans [green: insulin (β cells), red: glucagon (α cells); blue: 4′,6-diamidino-2-phenylindole (nuclei)]. (c) Quantitation of cellular composition (α and β cells) in C and SL islets. There is no change in the proportion of β cells, but α cell proportion is slightly lower in SL islets (*P < 0.05). (Shown are mean ± SEM, n = 6 mice per group, 10 islets per mouse)
The theoretical construct of “metabolic imprinting” [29] was proposed to guide mechanistic studies of developmental programing. A definitive characteristic of a primary imprint (a molecular change that mediates effect persistence) is that it is detectable directly after the imprinting period and persists to adulthood. To test whether DNA methylation serves as a primary imprint in this model, we used MSA-seq to perform DNA methylation profiling of islets from male SL and C mice at both P21 and P180 (n = 5 per group at each age, all from different foster litters). Although no SL vs. C group differences reached genome-wide significance, we focused our analysis and attempted validation on those with an unadjusted P < 0.05. Volcano plots (Fig. 6a) indicated SL vs. C hypermethylation at both P21 and P180 (330 and 444 SmaI/XmaI intervals, respectively, Data Set). However, only 7 genes showed evidence of persistently increased DNA methylation (SL > C at both ages) and only three of these [Ttyh3 (protein tweety homolog 3), Igf2bp1 (insulin-like growth factor 2 mRNA-binding protein) and Tmem145 (transmembrane protein 145)] involved the same SmaI/XmaI interval at both ages.
Figure 6:
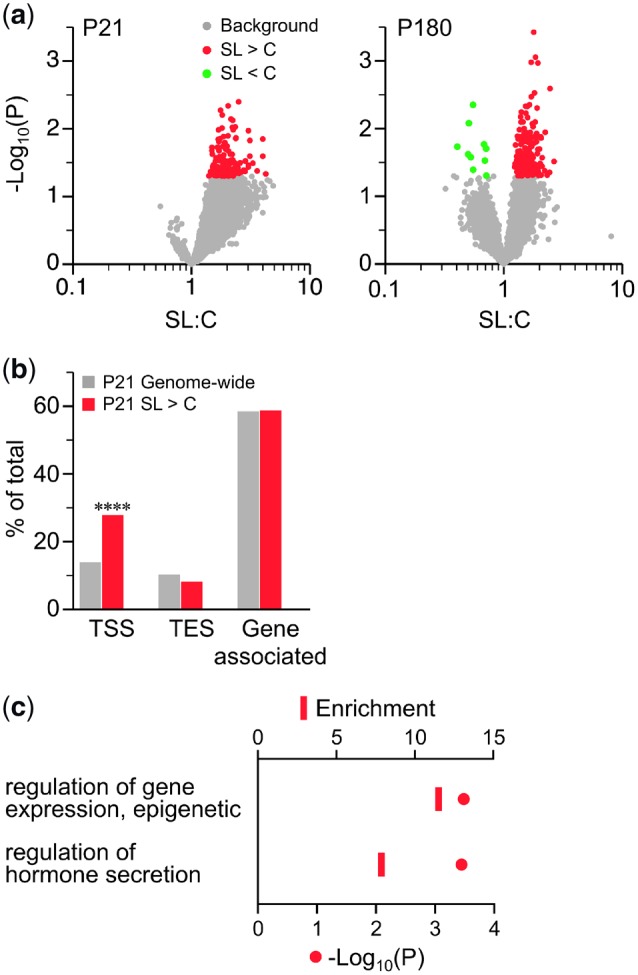
SL islets are modestly hypermethylated at both P21 and P180. (a) Volcano plots of SL vs. C MSA-seq data of islets at P21 (left) and P180 (right) (n = 5 per group at each age). Each dot represents the mean SL:C MSA-seq read count ratio at one SmaI/XmaI interval; red and green dots represent intervals with SL > C and SL < C, respectively (P < 0.05). At both ages, SL > C predominates. (b) Genomic distribution of intervals showing SL > C at P21 (red) vs. genome-wide (gray) (****P < 0.0001). (c) GO analysis of TSS-associated intervals that showed SL > C at P21 (see Supplementary Table S6). TES, transcription end site
Despite the loose cutoff of our screen, genomic regions showing SL > C methylation differences at P21 were strongly and highly-significantly enriched at TSSs (Fig. 6b). Interestingly, GO process analysis of these indicated associations with genes involved in “regulation of gene expression, epigenetic” and “regulation of hormone secretion” (Fig. 6c, Supplementary Tables S6 and S7). Of 13 selected SmaI/XmaI intervals with evidence of SL > C at both P21 and P180 (Supplementary Table S8), only 3 were validated by bisulfite-pyrosequencing. These were located within intronic regions of Akt1 (RAC-alpha serine/threonine-protein kinase) (Fig. 7a and b), Cacna1i (calcium channel, voltage-dependent, alpha 1I) (Fig. 7c and d) and Scn10a (sodium channel protein type 10 subunit alpha) (Fig. 7e and f).
Figure 7:

limited evidence of persistent DNA methylation increases in SL islets. (a, c and e) MSA-seq data (n = 5) for Akt1, Cacna1i and Scn10a, indicating increased DNA methylation at P21 and P180 in SL vs. C islets. Each tick represents 5 MSA-seq reads; asterisks indicate SmaI/XmaI sites selected for validation (Supplementary Table S8). Bisulfite pyrosequencing results at P21 and P180 for (b) Akt1 (P = 0.02 and P = 0.50, respectively), (d) Cacna1i (P = 0.02 and P = 0.0006, respectively) and (f) Scn10a (P = 0.01 and P = 0.02, respectively). Grey bars indicate informative SmaI/XmaI sites (mean ± SEM, n = 7)
It therefore appears that although numerous biologically relevant DNA methylation changes are observed in SL islets at P21, most of these do not persist into adulthood. As we already showed, however, even without early postnatal overnutrition, islet DNA methylation dynamically changes (mostly increasing) from P21 to P180. This suggests two possible reasons to explain why the P21 SL > C differences do not persist to P180: either (i) the P21 hypermethylation at these loci reverts by P180 or (ii) the P21 hypermethylation in SL islets persists, but by P180 is matched by age-associated increases in C islets. To distinguish between them, we compared 790 regions showing SL:C > 1.3 (regardless of P value) with the P180 > P21 hits. Strikingly, of the 790 P21 SL > C hits, 401 are also P180 > P21 hits (P < 0.0001 by χ2 test) (Fig. 8a). GO analysis (Supplementary Fig. S5) shows that the top biological process associated with these 401 regions is “regulation of receptor-mediated endocytosis,” which is important to β cell function and increases with age [30]. Thus, our interpretation is that much of the hypermethylation induced by early postnatal overnutrition does persist to adulthood. The apparent reason for the lack of SL vs. C differences at P180 is that, at many of these same loci, C islets eventually gain DNA methylation to similar levels as those of SL islets by P180. Indeed, by plotting all 401 overlapped hits from P21 to P180, most intervals gain DNA methylation in C islets (Fig. 8c) and remain unchanged or slightly decrease in SL islets (Fig. 8c). Furthermore, among these hits, the DNA methylation levels in SL islets at P21 are more comparable to those of C islets at P180 rather than P21. Taken together, our data indicate that early postnatal overnutrition accelerates age-related increases in DNA methylation in pancreatic islets.
Figure 8:
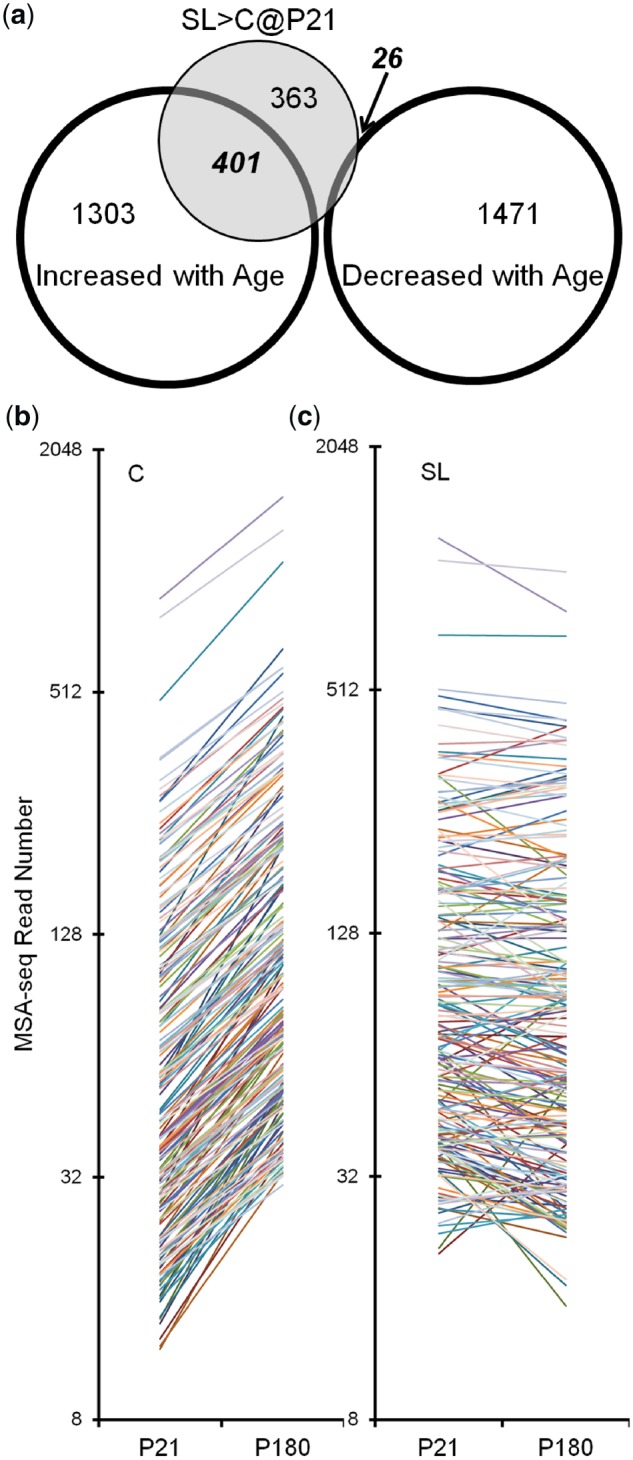
significant overlap of methylated loci caused by aging and SL feeding. (a) Numbers of loci that show increased/decreased DNA methylation from P21 to P180, and those showing higher methylation in SL islets at P21. Out of 790 P21 SL > C loci, 401 also gain DNA methylation with aging (P < 0.0001 with χ2 test). (b) All 401 overlapping loci were plotted according to their MSA-seq read numbers from P21 to P180 in C (left) and SL islets (right). An obvious trend of increasing DNA methylation can be seen only in C islets.
Discussion
Both early postnatal overnutrition [7, 10] and aging [31, 32] have long been implicated in islet dysfunction. The underlying molecular mechanisms, however, remain largely unknown. Our study—the first genome-scale screen for persistent methylation changes in SL islets—yielded two major findings. First, our results revealed extensive DNA methylation alterations caused by aging and/or SL feeding, indicating both aging and early overnutrition as potential causes of epigenetic dysregulation in islets. Second, and more intriguing, we discovered significant overlap between regions affected by aging and SL feeding, suggesting accelerated epigenetic aging in the islets of SL mice.
One obvious weakness of our study is that the profiling technique used (MSA-seq) does not provide an unbiased assessment of genomic methylation. To achieve a more complete picture of DNA methylation changes and associated pathways caused by aging or early overnutrition, a truly genome-wide approach such as whole-genome bisulfite sequencing [33] would be advantageous. Nonetheless, although MSA-seq assays only a subset of CpG sites in the genome, our extensive independent validation by bisulfite pyrosequencing demonstrates that the technique is reliable. We reported overlapping DNA methylation changes associated with aging and early postnatal overnutrition, but determining whether these epigenetic alterations actually cause impaired insulin secretion will require more sophisticated approaches such as epigenome editing [34]. Also, our current data do not elucidate how postnatal overnutrition leads to islet hypermethylation. De novo DNA methylation in the endocrine pancreas is at least partially mediated by DNA methyltransferase 3A (Dnmt3a) [35]. At this point, however, we do not know precisely when during the suckling period the hypermethylation occurs in SL mice. Hence, testing whether P21 islet hypermethylation in SL mice is caused by alterations in Dnmt expression and/or activity will require measuring Dnmt expression at earlier stages such as P7 and P14 (which will be a goal of future studies). Lastly, we assessed DNA methylation in whole islets. Although predominantly β cells, islets additionally contain α, and δ cells and also endothelial and mesenchymal cells. Importantly, we showed that postnatal overnutrition does not markedly change the relative proportions of α and β cells (Fig. 5), so the methylation differences we detected in SL islets are not attributable to changes in cell proportion. Nonetheless, from the current data we cannot definitively identify the islet cell type(s) most affected by either postnatal overnutrition or aging. To perform DNA methylation profiling of insulin-secreting β cells will require isolating pure β cells using either a combination of surface markers [36] or autofluorescence [37]. Alternatively, emerging single-cell sequencing approaches such as single-cell bisulfite sequencing [38] may enable deconvolution of DNA methylation signals from major cell types within intact islets.
Our study was designed to test for metabolic imprinting [29] of DNA methylation as a potential explanation for the persistent endocrine pancreas dysfunction in the rodent SL model. At the end of the suckling period (P21), we detected widespread increases in DNA methylation in SL islets. Comparing SL vs. C DNA methylation differences at both P21 and P180 indicated that only a few persisted to P180, including a subset of genes important for insulin secretion: Akt1 [39], Cacna1i and Scn10a [40]. This comparison approach, however, is valid only under the assumption that there is no DNA methylation change in C islets. We did, however, find age-associated methylation increases in C islets. Moreover, by comparing the hypermethylation caused by early postnatal overnutrition and that associated with aging, we found a substantial overlap. This indicated that the lack of DNA methylation difference at P180 was caused not by loss of DNA methylation in SL islets, but rather by gain of DNA methylation in C islet. It further suggested that early postnatal overnutrition accelerated epigenetic aging in the endocrine pancreas. Obesity has been reported to constrict T-cell diversity by accelerating age-related thymic involution [41], and increase brain age by 10 years measured by cerebral white matter atrophy [42]. Furthermore, epigenetic aging has been shown to be accelerated by obesity in the liver [43] and blood cells [44]. To the best of our knowledge, our current data provide the first evidence supporting accelerated epigenetic aging by early postnatal overnutrition, suggesting a new mechanistic basis for metabolic imprinting.
In support of our finding of early postnatal overnutrition accelerating epigenetic aging, aged mice and adult SL mice share similar special phenotypes. Despite solid evidence showing impaired glucose-stimulated insulin secretion in aged [45, 46] and SL mice [7], glucose tolerance is not negatively affected by either aging [45, 46] or postnatal overnutrition [7, 10]. Such paradoxical findings suggest that other mechanisms, such as glucose clearance in liver, muscle, or adipocytes, may compensate for the islet dysfunction.
Additionally, our data provide the novel insight that, as islets age, their methylation profile undergoes an “epigenetic drift” toward that of the exocrine pancreas. Stochastic, gradual decreases and increases in DNA methylation as a result of age are referred to as epigenetic drift [47]. This phenomenon may be a fundamental process of aging [48] and appears to be involved in the etiology of cancer [49] and age-associated diseases. An overall framework for why certain genomic regions are targeted for epigenetic drift has yet to be proposed. However, epigenetic drift is often tissue-specific and is more prevalent in mitotically active tissues [47].
Aging is one of the major risk factors for development of T2D [31], and age-related epigenetic changes in pancreatic islets have been associated with impairment of islet function and proliferation [15–17]. Our study, the first genome-scale analysis of age-associated DNA methylation changes in the endocrine pancreas, yielded the novel insight that methylation increases during islet aging tend to occur in genomic regions that are normally methylated in exocrine relative to endocrine pancreas (Fig. 2a). We considered whether this correlation might simply be an artifact due to islets isolated from older mice being more contaminated with exocrine tissue. In this case, however, the correlation would also be observed at genomic regions that are hypomethylated in exocrine pancreas relative to islets. This was not the case (Fig. 2a). We also asked whether this gain of DNA methylation might be explained by a shift in cellular composition as islets age. In rodents, however, age-associated increases in the number of β cells per islet [50] are accompanied by commensurate increases in non-β endocrine cell number [51], so overall cellular composition of islets is fairly stable from P21 to P180. Despite the general increase in DNA methylation from P21 to P180 in C islets, little aging-associated gain of DNA methylation was observed in SL islets. At this point we do not know why, but one potential explanation is that the hypermethylated loci in SL islets already reached nominally maximal levels by P21, leaving little room for further increase from P21 to P180. Indeed, the P21 SL vs. C differentially methylated loci we validated (Fig. 7b, d and f) were heavily methylated, consistent with this explanation.
Our interpretation is that epigenetic drift in islets essentially causes their epigenome to drift toward that of the exocrine pancreas. This may indicate a gradual reversal of innate processes of pancreatic development and cellular fate maintenance. Morphological analyses of pancreatic development indicate that endocrine and exocrine cells originate from a common pool of progenitor cells in the gut endoderm [52]. Due to their common origin, mature exocrine cells induced to express a set of β cell specific transcription factors can be reprogramed into insulin-expressing endocrine like cells [53]. Analogously, epigenetic drift during islet aging may represent a form of stochastic epigenetic reprograming toward the exocrine lineage. Future studies will be required to determine if epigenetic drift in other tissues likewise mirrors a reversal of cellular differentiation.
Importantly, we show that these methylation differences associated with age and tissue type correlate with alterations in expression of genes important to β cell function (Fig. 4). Rab3b encodes the Ras-related GTPase, which functions in rapid insulin granule refilling [19]. Since regulation of membrane Ca2+ potential plays a central role in insulin exocytosis, we were intrigued to find coordinated changes in methylation and expression at the voltage-gated Ca+ channel β3 subunit (Cacnb3) and Ca2+-ATPase (Atp2a3). Ion channels serve an important role in the regulation of insulin secretion [14, 54]. Hence, DNA methylation alterations affecting concentration of intracellular Ca2+ provide a plausible link between aging and T2D.
Additional genes with importance for normal islet function showed increased methylation during islet aging: Rxra [22], Kcnq1 [23], Prkce [25], Rasa3 [24], Igfbp2 [26] and Ins2. In particular, the age-associated changes in DNA methylation we observed at the Ins2 promoter merit special consideration. Data from both mice and humans indicate that insulin gene expression is regulated by DNA methylation at its promoter [27]. In patients with T2D, increased DNA methylation at the INS promoter is associated with decreased expression [55]. In rodents, Ins2 expression decreases with age [10] and the mouse and human insulin promoter is hypomethylated specifically in islets [27]. Consistent with these findings, we found that the Ins2 promoter is hypermethylated in exocrine relative to endocrine pancreas. We discovered that during islet aging, increased methylation at the Ins2 promoter (Fig. 3g and h) is associated with decreased expression (Fig. 4d). In vitro studies indicate that the Ins2 promoter is hypermethylated in mouse β cell progenitors and becomes demethylated only during the late stages of β cell differentiation [27], consistent with our conjecture that epigenetic drift during islet aging represents a reversal of differentiation.
Collectively, our data indicate that locus-specific increases in DNA methylation may be a fundamental molecular mechanism contributing to the progressive decline in islet function with age, and that early postnatal overnutrition accelerates this process. If similar mechanisms can be confirmed in humans, this may enable new strategies to prevent and treat T2D. Moreover, given that obesity is associated with inflammation [56] and inflammation with hypermethylation [57], epigenetic drift may provide a unifying mechanism linking obesity, inflammation, and endocrine pancreas dysfunction.
Methods
Animals
Newborn FVB/NJ mice (The Jackson Laboratory, Maine) were cross-fostered at P1 in litters of 9 (C) or 4 (SL), as previously described [28]. Only males were used for islet studies and the endocrine vs. exocrine pancreas MSA-seq analysis. At P21, 1 male offspring from each litter of both groups was used for islet isolation, and the rest were weaned onto a fixed-formula, soy protein free diet (2020X; Harlan Teklad, IN) and aged to P180. At P180, another male offspring from each litter was used for islet isolation. All animals were handled in accordance with the Animal Care and Use Committee of Baylor College of Medicine.
Immunocytochemistry and Cell Counting
Paraffin sections (4-µm thick) were prepared from P21 pancreata. Sections were double stained for insulin and glucagon (Abcam #7842 and #18461, respectively), and nuclei stained with 4′,6-diamidino-2-phenylindole. The numbers of α (red), β (green) and other (non-stained) cells were manually counted in a blinded fashion. Percentage of α or β cells was calculated as the number of red or green cells divided by the total 4′,6-diamidino-2-phenylindole counting within the islet area. These studies included at least 10 different islets from each mouse and included 6 mice per group.
Islet Isolation
Islet isolation was performed as previously described [58]. Briefly, the pancreas was perfused with collagenase P and digested at 37°C for 10 min. Islets of high purity were obtained with gradient centrifugation in Histopaque 1100 (Sigma-Aldrich, MO) followed by three rounds of manual picking under a dark field stereomicroscope (Supplementary Fig. S6). Islets were immediately stored at −80°C.
Genome-Scale DNA Methylation Profiling
Genome-scale DNA methylation analysis was performed by MSA-seq, as previously described [18]. GO analysis was performed using the GO enrichment analysis and visualization tool (Gorilla) [59]; the reference set was composed of genes associated with potentially informative SmaI/XmaI intervals (those yielding a total of at least 100 MSA-seq reads across all experimental groups). GO terms associated with at least 4 genes were considered important. Raw MSA-seq reads are available in GEO (http://www.ncbi.nlm.nih.gov/geo/) (accession #GSE63811).
Bisulfite-Pyrosequencing
Validation of site-specific DNA methylation differences was performed by bisulfite pyrosequencing [60]. Genomic DNA (500 ng) was bisulfite treated and amplified using biotinylated primers (Supplementary Tables S5 and S8) before sequencing on a PyroMark MD (Qiagen, CA). Each assay’s sensitivity and quantitative accuracy were confirmed by running genomic DNA methylation standards (Supplementary Tables S5 and S8). A SmaI/XmaI interval was considered validated if either of the SmaI/XmaI sites showed a methylation difference in the direction indicated by the MSA-seq data.
Clonal Bisulfite Sequencing
Bisulfite converted DNA was amplified with Ins2 specific primers designed using MethPrimer (http://www.urogene.org/methprimer/index1.html) [61] (sense primer: TTTAAGTGGGATA TGGAAAGAGAGATA and antisense primer: ACTACAATTTCCAA ACACTTCCCTAATA) and subcloned into pCR2.1 TOPO-TA (Invitrogen, CA). Sequence data were analysed using the Quantification tool for Methylation Analysis (http://quma.cdb.riken.jp/, QUMA, Riken Institute, Japan) [62]. Clones with <95% of C to T conversion at non-CpG cytosines were excluded.
RNA Isolation and RT-PCR Analysis
Pancreata of P21 mice were injected in situ with RNAlater (Ambion, NY), excised and immediately placed in a petri dish containing RNAlater. Adipose tissue was quickly removed and RNA purified using RNA Stat-60 (Tel-Test, Inc., TX). Following reverse transcription, quantitative PCR was performed in triplicate using TaqMan assays (Life Technologies, NY) for Rab3b (Mm00772238_m1), Cacnb3 (Mm00432233_m1), Ins2 (Mm00731595_gH) and Atp2a3 (Mm00443898_m1); β-actin (Mm00607939_s1) was used as an endogenous control. Relative gene expression was quantitated using the 2−ΔΔCt method.
Statistical Methods
Enrichment of SmaI/XmaI cut sites relative to gene regions was analysed by χ2 test. Linear regression analysis was performed using GraphPadInStat software (GraphPad Software Inc., CA). Differences in DNA methylation by pyrosequencing were analysed using repeated-measures analysis of variance (SAS Proc Mixed), with multiple CpG sites within each gene region treated as repeated measures. MSA-seq (SmaI/XmaI interval-specific read counts) and mRNA expression data were analysed by Student’s t-test (two-tailed, equal variance).
Supplementary Material
Acknowledgements
We thank Adam Gillum (USDA/ARS CNRC) for assistance with figure design.
Funding
This work was supported by grants from NIH/NIDDK (1R01DK081557 and 1R01DK111831) and USDA (CRIS 3092-5-001-059) to R.A.W., and from the Thrasher Research Fund (NR-0136) to G.L.
Contribution Statement
G.L. and T.D.P. performed research and wrote the manuscript. E.L., M.S.B., and S.Z. performed research. N.K. conducted bioinformatic analyses. R.A.W. designed the study and wrote the manuscript. All authors have approved the manuscript for publication. R.A.W. is responsible for the integrity of the work as a whole.
Data Availability
Raw MSA-seq reads are available in the GEO database (http://www.ncbi.nlm.nih.gov/geo/; accession #GSE63811).
Conflict of interest statement. None declared.
References
- 1. Centers for Disease Control and Prevention. National diabetes statistics report, 2017. U.S. Dept of Health and Human Services, 2017.
- 2. Prentki M, Nolan CJ.. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006;116:1802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Jesus DF, Kulkarni RN.. Epigenetic modifiers of islet function and mass. Trends Endocrinol Metab 2014;25:628–36. [DOI] [PubMed] [Google Scholar]
- 4. Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, Calonne E, Volkmar U, Igoillo-Esteve M, Naamane N et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 2012;31:1405–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Volkov P, Bacos K, Ofori JK, Esguerra JLS, Eliasson L, Rönn T, Ling C.. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 2017;66:1074–85. [DOI] [PubMed] [Google Scholar]
- 6. Yajnik CS, Deshpande SS, Jackson AA, Refsum H, Rao S, Fisher DJ, Bhat DS, Naik SS, Coyaji KJ, Joglekar CV et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: the Pune Maternal Nutrition Study. Diabetologia 2007;51:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aubert R, Suquet JP, Lemonnier D.. Long-term morphological and metabolic effects of early under- and over-nutrition in mice. J Nutr 1980;110:649–61. [DOI] [PubMed] [Google Scholar]
- 8. Hahn P. Effect of litter size on plasma cholesterol and insulin and some liver and adipose tissue enzymes in adult rodents. J Nutr 1984;114:1231–4. [DOI] [PubMed] [Google Scholar]
- 9. Gluckman PD. Epigenetics and metabolism in 2011: epigenetics, the life-course and metabolic disease. Nat Rev Endocrinol 2011;8:74–6. [DOI] [PubMed] [Google Scholar]
- 10. Waterland RA, Garza C.. Early postnatal nutrition determines adult pancreatic glucose-responsive insulin secretion and islet gene expression in rats. J Nutr 2002;132:357–64. [DOI] [PubMed] [Google Scholar]
- 11. Waterland RA, Michels KB.. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007;27:363–88. [DOI] [PubMed] [Google Scholar]
- 12. Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM.. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem 2010;285:15111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stumvoll M, Goldstein BJ, van Haeften TW.. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333–46. [DOI] [PubMed] [Google Scholar]
- 14. Gunasekaran U, Gannon M.. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY) 2011;3:565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park JH, Stoffers DA, Nicholls RD, Simmons RA.. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 2008;118: 2316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, Jones RH, Marquez VE, Cairns W, Tadayyon M et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl Acad Sci USA 2011;108:5449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen H, Gu X, Su I-H, Bottino R, Contreras JL, Tarakhovsky A, Kim SK.. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 2009;23:975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li G, Zhang W, Baker MS, Laritsky E, Mattan-Hung N, Yu D, Kunde-Ramamoorthy G, Simerly RB, Chen R, Shen L et al. Major epigenetic development distinguishing neuronal and non-neuronal cells occurs postnatally in the murine hypothalamus. Hum Mol Genet 2014;23:1579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cazares VA, Subramani A, Saldate JJ, Hoerauf W, Stuenkel EL.. Distinct actions of Rab3 and Rab27 GTPases on late stages of exocytosis of insulin. Traffic 2014;15:997–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berggren P-O, Yang S-N, Murakami M, Efanov AM, Uhles S, Köhler M, Moede T, Fernström A, Appelskog IB, Aspinwall CA et al. Removal of Ca2+ channel beta3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell 2004;119:273–84. [DOI] [PubMed] [Google Scholar]
- 21. Beauvois MC, Merezak C, Jonas J-C, Ravier MA, Henquin J-C, Gilon P.. Glucose-induced mixed [Ca2+]c oscillations in mouse beta-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am J Physiol Cell Physiol 2006;290:C1503–11. [DOI] [PubMed] [Google Scholar]
- 22. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 2011;60:1528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamagata K, Senokuchi T, Lu M, Takemoto M, Fazlul Karim M, Go C, Sato Y, Hatta M, Yoshizawa T, Araki E et al. Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 beta-cell line. Biochem Biophys Res Commun 2011;407:620–5. [DOI] [PubMed] [Google Scholar]
- 24. Schurmans S, Pouillon V, Marechal Y.. Regulation of B cell survival, development and function by inositol 1,4,5-trisphosphate 3-kinase B (Itpkb). Adv Enzyme Regul 2011;51:66–73. [DOI] [PubMed] [Google Scholar]
- 25. Mendez CF, Leibiger IB, Leibiger B, Høy M, Gromada J, Berggren P-O, Bertorello AM.. Rapid association of protein kinase C-epsilon with insulin granules is essential for insulin exocytosis. J Biol Chem 2003;278:44753–7. [DOI] [PubMed] [Google Scholar]
- 26. Hoeflich A, Wu M, Mohan S, Föll J, Wanke R, Froehlich T, Arnold GJ, Lahm H, Kolb HJ, Wolf E et al. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology 1999;140:5488–96. [DOI] [PubMed] [Google Scholar]
- 27. Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F, Mullen Y, Pfeifer GP, Ferreri K.. Insulin gene expression is regulated by DNA methylation. PLoS One 2009;4:e6953.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li G, Kohorst JJ, Zhang W, Laritsky E, Kunde-Ramamoorthy G, Baker MS, Fiorotto ML, Waterland RA.. Early postnatal nutrition determines adult physical activity and energy expenditure in female mice. Diabetes 2013;62:2773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waterland RA, Garza C.. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr 1999;69:179–97. [DOI] [PubMed] [Google Scholar]
- 30. Cnop M, Grupping A, Hoorens A, Bouwens L, Pipeleers-Marichal M, Pipeleers D.. Endocytosis of low-density lipoprotein by human pancreatic beta cells and uptake in lipid-storing vesicles, which increase with age. Am J Pathol 2000;156:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang AM, Halter JB.. Aging and insulin secretion. Am J Physiol Endocrinol Metab 2003;284:E7–12. [DOI] [PubMed] [Google Scholar]
- 32. Tschen S-I, Dhawan S, Gurlo T, Bhushan A.. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009;58:1312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunde-Ramamoorthy G, Coarfa C, Laritsky E, Kessler NJ, Harris RA, Xu M, Chen R, Shen L, Milosavljevic A, Waterland RA et al. Comparison and quantitative verification of mapping algorithms for whole-genome bisulfite sequencing. Nucleic Acids Res 2014;42:e43.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R et al. Editing DNA methylation in the mammalian genome. Cell 2016;167:233–47 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dhawan S, Tschen S-I, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A.. DNA methylation directs functional maturation of pancreatic beta cells. J Clin Invest 2015;125:2851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, Vethe H, Paulo JA, Joosten AM, Berney T et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019;567:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van De Winkel M, Pipeleers D.. Autofluorescence-activated cell sorting of pancreatic islet cells: purification of insulin-containing B-cells according to glucose-induced changes in cellular redox state. Biochem Biophys Res Commun 1983;114:835–42. [DOI] [PubMed] [Google Scholar]
- 38. Karemaker ID, Vermeulen M.. Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol 2018;36:952–65. [DOI] [PubMed] [Google Scholar]
- 39. Cui X, Yang G, Pan M, Zhang X-N, Yang S-N.. Akt signals upstream of L-type calcium channels to optimize insulin secretion. Pancreas 2012;41:15–21. [DOI] [PubMed] [Google Scholar]
- 40. Rorsman P, Braun M.. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 2013;75:155–79. [DOI] [PubMed] [Google Scholar]
- 41. Yang H, Youm Y-H, Vandanmagsar B, Rood J, Kumar KG, Butler AA, Dixit VD.. Obesity accelerates thymic aging. Blood 2009;114:3803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ronan L, Alexander-Bloch AF, Wagstyl K, Farooqi S, Brayne C, Tyler LK, Fletcher PC.. Obesity associated with increased brain age from midlife. Neurobiol Aging 2016;47:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, Heits N, Bell JT, Tsai P-C, Spector TD et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA 2014;111:15538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nevalainen T, Kananen L, Marttila S, Jylhävä J, Mononen N, Kähönen M, Raitakari OT, Hervonen A, Jylhä M, Lehtimäki T et al. Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin Epigenet 2017;9:20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gregg T, Poudel C, Schmidt BA, Dhillon RS, Sdao SM, Truchan NA, Baar EL, Fernandez LA, Denu JM, Eliceiri KW et al. Pancreatic beta-cells from mice offset age-associated mitochondrial deficiency with reduced KATP channel activity. Diabetes 2016;65:2700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leiter EH, Premdas F, Harrison DE, Lipson LG.. Aging and glucose homeostasis in C57BL/6J male mice. FASEB J 1988;2:2807–11. [DOI] [PubMed] [Google Scholar]
- 47. Teschendorff AE, West J, Beck S.. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet 2013;22:R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fraga MF, Esteller M.. Epigenetics and aging: the targets and the marks. Trends Genet 2007;23:413–8. [DOI] [PubMed] [Google Scholar]
- 49. Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest 2014;124:24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herbach N, Bergmayr M, Göke B, Wolf E, Wanke R.. Postnatal development of numbers and mean sizes of pancreatic islets and beta-cells in healthy mice and GIPR(dn) transgenic diabetic mice. PLoS One 2011;6:e22814.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Montanya E, Nacher V, Biarnes M, Soler J.. Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes 2000;49:1341–6. [DOI] [PubMed] [Google Scholar]
- 52. Shih HP, Wang A, Sander M.. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol 2013;29:81–105. [DOI] [PubMed] [Google Scholar]
- 53. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA.. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008;455:627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ammon HP, Fahmy A, Mark M, Wahl MA, Youssif N.. The effect of glucose on insulin release and ion movements in isolated pancreatic islets of rats in old age. J Physiol (Lond) 1987;384:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang BT, Dayeh TA, Kirkpatrick CL, Taneera J, Kumar R, Groop L, Wollheim CB, Nitert MD, Ling C.. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia 2011;54:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robbins GR, Wen H, Ting JP.. Inflammasomes and metabolic disorders: old genes in modern diseases. Mol Cell 2014;54:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hartnett L, Egan LJ.. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis 2012;33:723–31. [DOI] [PubMed] [Google Scholar]
- 58. Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS.. A practical guide to rodent islet isolation and assessment. Biol Proced Online 2009;11:3–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z.. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009;10:48.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Waterland RA, Kellermayer R, Rached M-T, Tatevian N, Gomes MV, Zhang J, Zhang L, Chakravarty A, Zhu W, Laritsky E et al. Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum Mol Genet 2009;18:3026–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li LC, Dahiya R.. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002;18:1427–31. [DOI] [PubMed] [Google Scholar]
- 62. Kumaki Y, Oda M, Okano M.. QUMA: quantification tool for methylation analysis. Nucleic Acids Res 2008;36:W170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw MSA-seq reads are available in the GEO database (http://www.ncbi.nlm.nih.gov/geo/; accession #GSE63811).
Conflict of interest statement. None declared.