

Abstract
Purpose
Diabetes leads to the downregulation of the retinal Kir4.1 channels and Müller cell dysfunction. The insulin receptor substrate-1 (IRS-1) is a critical regulator of insulin signaling in Müller cells. Circadian rhythms play an integral role in normal physiology; however, diabetes leads to a circadian dysrhythmia. We hypothesize that diabetes will result in a circadian dysrhythmia of IRS-1 and Kir4.1 and disturbed clock gene function will have a critical role in regulating Kir4.1 channels.
Methods
We assessed a diurnal rhythm of retinal IRS-1 and Kir4.1 in db/db mice. The Kir4.1 function was evaluated using a whole-cell recording of Müller cells. The rat Müller cells (rMC-1) were used to undertake in vitro studies using a siRNA.
Results
The IRS-1 exhibited a diurnal rhythm in control mice; however, with diabetes, this natural rhythm was lost. The Kir4.1 levels peaked and troughed at times similar to the IRS-1 rhythm. The IRS-1 silencing in the rMC-1 led to a decrease in Kir4.1 and BMAL1. The insulin treatment of retinal explants upregulated Kir4.1 possibly via upregulation of BMAL1 and phosphorylation of IRS-1 and Akt-1.
Conclusions
Our studies highlight that IRS-1, by regulating BMAL1, is an important regulator of Kir4.1 in Müller cells and the dysfunctional signaling mediated by IRS-1 may be detrimental to Kir4.1.
Keywords: diabetic retinopathy, Müller cell, circadian rhythm, insulin receptor substrate 1, Kir4.1 channels
Diabetic Retinopathy (DR) is the most common complication of both type 1 and type 2 diabetes (T1D and T2D), leading to distorted vision and even blindness. Due to its increasing prevalence around the world, it is important to understand DR and its risk factors that may affect its prognosis.1 Several studies suggest that diabetes-induced changes to neurons and glia can lead to vascular injury.2,3 Notably, diabetes-induced changes in retinal neurons and glia precede the onset of clinically evident vascular injury.4–9 Müller cells are at the core of this neurovascular unit, acting as a principal glia of the retina.2 Due to their role as glial cells, Müller cells produce numerous cytokines, neurotransmitters, support factors, and display a variety of ion channels and transporters.10 Previous studies have shown that diabetes leads to a decrease in Kir4.1 expression and potassium currents, resulting in a Müller cell swelling, as well as an altered distribution of the Kir4.1 protein.10 The K+ concentration in Müller cells is largely regulated via the Kir4.1 channels, and the transcellular water transport is facilitated by the aquaporin-4 (AQP4) water channel.11 Furthermore, diabetes is associated with Müller cell gliosis, due to an increase of glial fibrillary acidic protein (GFAP) and an upregulation of proinflammatory molecules.10,12
Insulin receptor substrate 1 (IRS-1) is an important regulator of insulin signaling and low levels of IRS-1 are regarded as a marker of insulin resistance in clinical studies.13,14 Upon insulin binding to its receptor, IRS-1 is phosphorylated at tyrosine residues. This phosphorylated tyrosine residue then becomes a major docking point for proteins like PI 3-kinase that mediate the action of insulin,15 such as cell cycle progression and maturation.16,17 The phosphorylation of Tyr608 and Tyr 628 in rats generates the major docking sites for the PI 3-kinase.18 In particular, in the retina, the IRS-1 deletion promotes Müller cell apoptosis.19 Furthermore, it has been shown in retinal Müller cells phosphorylation of Ser307 in IRS-1 can lead to inhibition of insulin signaling.20 Insulin-mediated signaling has been shown to be critical for optimum Kir4.1 expression21; however, the precise mechanism(s) remains unknown.
Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL) or BMAL1 is a protein in humans encoded by the ARNTL gene. BMAL1 is an important component of autoregulatory transcription-translation negative feedback loop, which is responsible for generating molecular circadian rhythms. Previous studies demonstrated that clock disrupted Bmal1-knockout mice lack a natural rhythm of insulin release, develop resistance to insulin action, and obesity.22 Importantly, restoration of Bmal by a constitutively expressed promoter corrects insulin resistance and obesity in Bmal knockout mice.23 We previously reported that Müller cells exhibit a diurnal rhythm of Kir4.1 and that the BMAL1 directly modulates Kir4.1 rhythm,24 possibly by interacting with E-box elements.
The IRS-1 displays a diurnal rhythm in adipocytes25; however, it is not yet known whether retinal IRS-1 exhibits a similar rhythm. In this, study using an animal model of T2D (i.e., db/db mice, we demonstrate that the IRS-1 displays a diurnal rhythm in the retina). Furthermore, we found that IRS-1 regulates Kir4.1 channels via clock gene Bmal, thereby suggesting an important role of circadian rhythms and Müller cell dysfunction observed in diabetes.
Methods
Animal Studies
The B6.BKS (D)-Leprdb/J db/db and control Leprdb/+ db/m mice were purchased from The Jackson Laboratory. All animal care and experimental procedures were in accordance with the guiding principles in the care and use of animals (NIH) and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The animal protocol was approved by the institutional animals care and use committee (IACUC) at Indiana University (Indianapolis, IN, USA). The animals were maintained under a 12-hour light/12-hour dark condition. The zeitgeber time (ZT) corresponds to the turning of lights on (ZT-0 = 7 AM) and off (ZT-12 = 7 PM) in animal facilities. The animals were euthanized at 6 months of diabetes and the retinas were processed for, Müller cell isolation, Western blot, and agarose embedding.
Electrophysiologic Recordings
The retinas were dissected and incubated in Ringer's solution, containing 0.3 mg/mL of papain and 2.5 mM l-cysteine for 30 minutes at 37°C. After short incubation in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 0.2 mg/mL DNase 1 at room temperature, the tissue was gently triturated. The triturated cells were layered over a discontinuous Percoll gradient, consisting of layers of 15%, 30%, and 45% Percoll and centrifuged at 800g for 5 minutes. The Müller cells enriched fraction on top of the 30% Percoll was collected, washed with DMEM containing 10% FBS, and transferred to a Poly-L-Lysine laminin coated coverslip allowing cells to adhere. The whole-cell voltage-clamp recordings for Kri4.1 currents were measured as described previously.26
Immunofluorescence Staining of Retinal Sections
The retinas from db/m and db/db mice were fixed in 4% paraformaldehyde for 15 minutes at room temperature followed by washing in PBS. The fixed retinas were embedded in 3% agarose and sectioned using a vibratome. The agarose slices were washed in a PBS buffer and blocked with 5% goat serum in a washing buffer (PBS containing 3% DMSO and 0.3% Triton X-100) at room temperature for 2 hours, and then incubated overnight at 4°C in primary antibodies diluted in blocking solution. The slices were washed in a washing buffer and incubated 1 hour in appropriate secondary antibodies diluted in a blocking buffer. The following primary antibodies were used: Kir4.1 (Cat. # APC-035, Alomone Labs, 1:200), glutamine synthetase GS-1 (Cat. # MAB302, 1:200; Millipore), or IRS-1 (Cat. # 2382, 1:200; Cell Signaling Technology). The immunofluorescence images were taken using a confocal microscope (Zeiss LSM 510 META, Carl Zeiss MicroImaging GmbH, Jena, Germany).
The Culture of rMC-1 Cells
The rat Müller cells (rMC-1) were obtained as a gift from Vijay Sarthy, Northwestern University, Chicago, Illinois, United States. Cells were cultured in DMEM media that was supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and 1% of antibiotic-antimycotic (Thermo Fisher Scientific). The rMC-1 cells were authenticated using short tandem repeat analysis, contamination for interspecies and mycoplasma (IDEXX Bioresearch, Columbia, MO, USA). For insulin study, cells were serum starved in DMEM for 18 hours and then stimulated with 10 nM insulin (Sigma-Aldrich Corp., St. Louis, MO, USA) in the presence or absence of the inhibitor, SecinH3 (Tocris Bioscience, Bristol, UK). DMSO-treated cells (0.2% final concentration of DMSO) were used as a control. Cells were collected, washed with PBS and stored at −80°C.
The Culture of Human Retinal Müller Cells
Fresh donor eyes were obtained from Saving Sight Eye Bank (Kansas City, MO, USA), provided by Weiming Mao, Department of Ophthalmology, Indiana University. Müller cells were isolated as described previously.27 Retinas were minced and incubated in 0.25% trypsin (Thermo Fisher Scientific) at 37°C for 1 hour. The tissue pieces were gently triturated by pipetting and washed in high glucose DMEM/Ham's F-12 medium (1:1 ratio, 2.25 g/L glucose) containing 20% fetal bovine serum and 1% antibiotic-antimycotic. After attachment in a high glucose medium, cells were fed with low glucose DMEM/Ham's F-12 medium (1:1 ration, 0.5 g/L glucose) supplemented with 20% FBS and 1% antibiotic-antimycotic. The Müller cells were characterized using antibodies to vimentin (Cat. #C9080, 1:200; Sigma-Aldrich Corp.), glial fibrillary acidic protein (Cat. #12389S, 1:200; Cell Signaling Technology), and glutamine synthetase (Cat. #MAB302, 1:200; Millipore Corp.). Cells used in this study were of passages 3 to 6.
siRNA Transfection for IRS-1
rMC-1 were grown and transfected with 50 nM IRS-1 siRNA (Thermo Fisher Scientific) using reagent (Lipofectamine RNAiMax; Thermo Fisher Scientific). The cells were harvested 24 hours post-transfection for RNA analysis and protein analysis by Western blot. Cells transfected with negative control siRNA (Thermo Fisher Scientific) were used as a control.
qRT-PCR for mRNA Analysis
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's protocol and 1 μg of RNA was reverse transcribed using cDNA Synthesis Kit (SuperScript VILO; Thermo Fisher Scientific). Gene-specific primers were used along with a master mix (TaqMan Fast Universal; Thermo Fisher Scientific) and respective mRNA levels were determined using quantitative PCR (Viia7; Thermo Fisher Scientific). All genes were normalized to β-actin. Primers used were Kcnj10 (gene for Kir4.1; Rn00581058_m1), IRS-1 (Rn02132493_s1), β-actin (Rn00667869_m1), and Arntl (Rn00577590_m1).
Western Blotting
The retina samples or rMC-1 cells were lysed in a RIPA buffer (R0278, Sigma-Aldrich Corp.) containing protease inhibitor mixture. Protein concentration was estimated using the BCA assay (Pierce, Thermo Fisher Scientific) and equal amounts of proteins were loaded and separated on 4% to 12% Bis-Tris Gel (Novex; Thermo Fisher Scientific). Proteins were transferred to a PVDF membrane (Thermo Fisher Scientific) and blocked with 4% BSA in TBS-T buffer. The following antibodies were used for probing: α-Tubulin (Cat. # T9026, 1:5000; Sigma-Aldrich Corp.), Kir4.1 (Cat. # APC-035, 1:2000, Alomone Labs), Akt (Cat. # 4691, Cell Signaling Technology), p-AktS473 (Cat. #4060, 1:500; Cell Signaling Technology), IRS-1 (Cat. # 2382, 1:200, Cell Signaling Technology), phospho-IRS-1Ser307 (Cat. #2381, 1:200; Cell Signaling Technology), phospho-IRS-1Tyr612 (Cat. # −44-816G, 1:200; Thermo Fisher Scientific), BMAL1 (Cat. #NB100-2288, 1:2000; Novus Biologicals). The bands were visualized using ECL2 Western blotting substrate (Thermo Fisher Scientific) on laser scanner (Typhoon FLA 9500; GE Healthcare Life Sciences, Pittsburgh, PA, USA). The protein bands were quantified using ImageJ.
Statistics
The data were expressed as mean ± SEM and the statistical analysis was performed using graphing software (GraphPad Prism 7; GraphPad Software, La Jolla, CA, USA). The data were analyzed using the 1-way ANOVA followed by the Tukey-Kramer or Newman Keul's test for post hoc analysis unless otherwise specified. The data were considered to be statistically significant when the value of P < 0.05.
Results
Loss of Diurnal Rhythm for IRS-1 in db/db Mice
While the IRS-1 has been reported as a critical regulator of insulin signaling in Müller cells28, it remains unknown whether IRS-1 exhibits any diurnal rhythm. To test this, we examined IRS-1 expression in retinal slices at different time intervals. We observed that IRS-1 expression was at a peak at ZT-9, while at the lowest at ZT-3 (Fig. 1A). In the retinas of diabetic mice, we observed an overall decrease in the levels of IRS-1 and a lack of peak response for IRS-1 (Fig. 1B). In order to see the nature of IRS-1 staining in Müller cells, the agarose-embedded retinal slices were costained with glutamine synthetase (GS-1) and IRS-1 antibodies. The IRS-1 mainly stained around the blood vessels and near the outer plexiform layer. The IRS-1 expression was highest at ZT-9 similar to Western blot data and with diabetes, and we observed an overall decrease in IRS-1 levels (Figs. 1C–E).
Figure 1.

IRS-1 follows a diurnal rhythm in the retina. (A) Western blot showing IRS-1 levels in db/m and db/db mice. (B) Line chart showing a peak increase in IRS-1, retinas of db/db mice did not exhibit any diurnal rhythm. Retinal slices are stained with IRS-1 (green) and glutamine synthetase (GS-1; red), representative photomicrographs showing staining pattern for IRS-1 and GS-1 in (C) db/m, (D) db/db mice, and (E) a line chart showing the quantification of immunofluorescence (n = 3).
BMAL1 Rhythm Reduced in db/db Mice
BMAL1 is known to oscillate in different tissues and we previously reported that BMAL1 regulates Kir4.1 in retinal Müller cells24; however, its oscillatory pattern in db/db mice is not known. To test this, we determined levels of clock regulatory protein BMAL1 in the retinas of db/db and db/m mice using a Western blot. The BMAL1 peaked at ZT-9 in db/m mice, for the db/db mice the peak response was also at ZT-9. There was an overall decrease in BMAL-1 in db/db mice (Figs. 2A, 2B).
Figure 2.
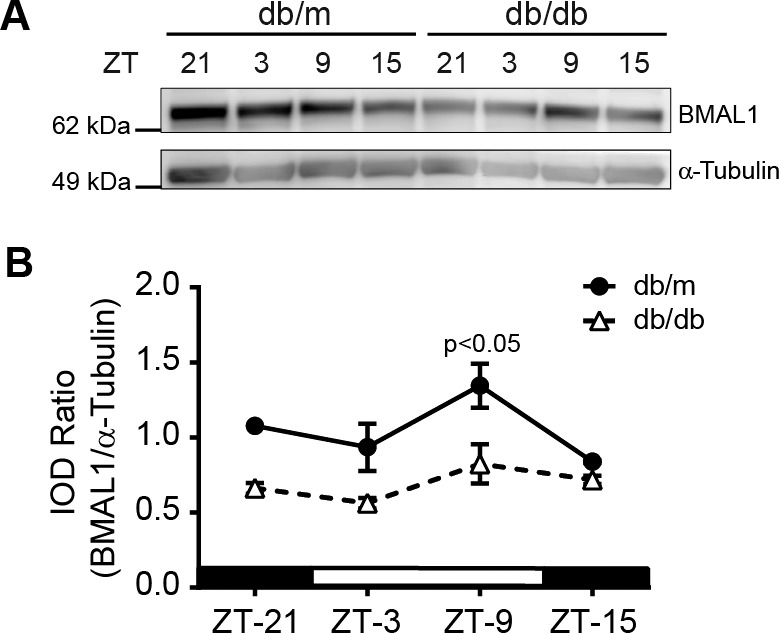
Diurnal rhythm of Bmal1 in db/db mice. (A) Western blot showing BMAL1 levels in the retina at different time points, (B) A line chart showing quantification of BMAL1 at the respective time points (n = 3).
The Retinas of Diabetic Mice Lack a Diurnal Rhythm of Kir4.1
Next, we determined the levels of Kir4.1 at time points similar to IRS-1. The Kir4.1 protein levels were peaked at ZT-9, while these levels were at the lowest at ZT-15 (Fig. 3A). The retinas of diabetic mice exhibited an overall decrease in Kir4.1 levels with an apparent lack of any rhythm (Fig. 3B). Next, we evaluated whether Kir4.1 in Müller cells also display a similar diurnal rhythm. To test this, we stained retinal sections with GS-1 and Kir4.1 antibodies. The Kir4.1 mainly stained internal limiting membrane and near the blood vessels, similar to previous reports.2,10 In db/m mice, the Kir4.1 was peaked at ZT-9 (mostly near the blood vessels) similar to Western blot studies (Fig. 3C). The db/db mice displayed an overall decrease in Kir4.1 levels without any diurnal rhythm (Figs. 3D, 3E).
Figure 3.

The lack of diurnal rhythm of Kir4.1. (A) Western blot showing the Kir4.1 protein levels in db/m and db/db mice. (B) Quantification of Kir4.1 protein showing a peak increase at ZT-9. The retinal slices were stained with glutamine synthetase (GS-1; red) and Kir4.1 antibodies (green), representative images showing Kir4.1 and GS-1 staining pattern in (C) db/m and (D) db/db mice. (E) Line chart showing quantification of immunofluorescence (n = 3).
The Decrease in Kir4.1 Currents in db/db Mice
To determine the functional defect, we freshly isolated Müller cells at two-time points, ZT-3 and ZT-15. The Kir4.1 currents were determined using whole cell patch clamp. The currents were elicited by applying 50 ms depolarizing potentials ranging from −140 mV to +30 mV from a holding potential of −60 mV in increments of 10 mV. We observed a biphasic response to Kir4.1 currents, at ZT-3 the response for Kir4.1 currents was higher when compared to ZT-15 time point. The db/db mice exhibited a similar response to Kir4.1 currents; however, there was an overall decrease in a current amplitude (Figs. 4A, 4B).
Figure 4.
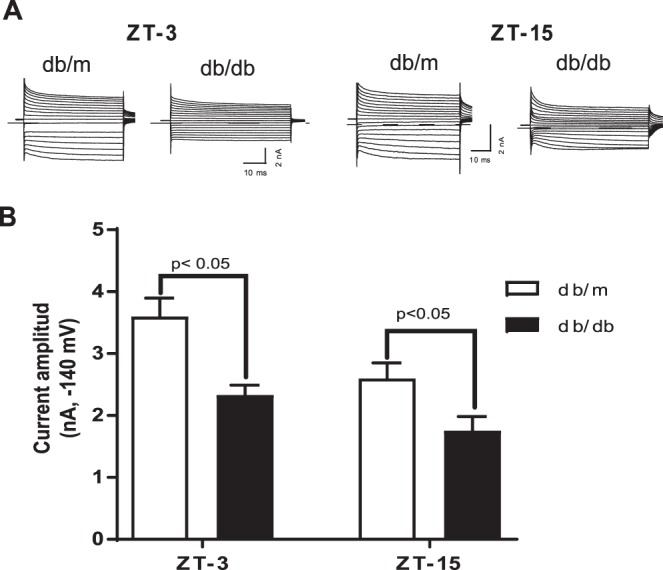
Biphasic response of Kir4.1 currents and decrease in diabetes. (A) Representative current traces of freshly isolated Müller cells from db/m and db/db mice. (B) Mean amplitude of the inward potassium currents elicited at −140 mv (db/m, n = 15; db/db, n = 13).
IRS-1 Regulates Kir4.1 in Retinal Müller Cells
Based on previous studies, which suggest that insulin is necessary for optimum levels of Kir4.121 and our observation of a parallel rhythm for Kir4.1 and IRS-1, we hypothesized that IRS-1 regulates Kir4.1 in retinal Müller cells. To test this, we first transfected rMC-1 cells with an IRS-1 siRNA or a scrambled control siRNA. The siRNA transfection resulted in a 1.5-fold decrease in IRS-1 levels (Fig. 5A). IRS-1 silencing resulted in a decrease in IRS-1 and phospho-AktSer473 (Figs. 5B–D). The IRS-1 silencing led to a 1.5-fold decrease in Bmal1 mRNA (Fig. 5E), a similar change was observed in BMAL1 protein levels (Figs. 5F, 5G). The IRS-1 silencing led to a 1.25-fold decrease in Kcnj10 levels (Fig. 5H; P < 0.05). The Kir4.1 protein was reduced by a 2-fold following siRNA silencing of IRS-1 (Figs. 5I, 5J). To see whether a similar correlation between IRS-1, KIr4.1, and Bmal exists in human Müller cells, we cultured Müller cells from human retinas. The human Müller cells stained brightly for vimentin and were negative for GS-1 and GFAP (Supplementary Fig. S1). The IRS-1 silencing of human Müller cells resulted in a downregulation BMAL1 and Kir4.1 similar to rMC-1 cells (Supplementary Fig. S2). Taken together this suggests that IRS-1, BMAL1, and Kir4.1 are in synchrony in the Müller cells.
Figure 5.

IRS-1 inhibition downregulates Kir4.1 and Bmal1 in Müller cells. The rMC-1 cells were transfected with an IRS-1 siRNA or a scrambled control siRNA (NC), bar chart showing mRNA levels of (A) IRS-1. (B) Representative Western blot showing a decrease in IRS-1 and phospho-AktSer473. Bar chart showing a quantification of (C) phospho-AktSer473 and (D) IRS-1. (E) IRS-1 siRNA-transfected rMC-1 cells showing a decrease in Bmal1 mRNA, (F, G) BMAL1 protein, (H) Kcnj10 mRNA, and (I, J) Kir4.1 protein (n = 4).
Restoration of IRS-1 Signaling Corrects Kir4.1 Levels
Considering the critical role of IRS-1 in regulating Kir4.1, we reasoned whether restoration of IRS-1 signaling would modulate Kir4.1 channels. We first tested our hypothesis using rMC-1 cells treated with insulin, a well-established modulator of IRS-1. We observed that Kir4.1 levels were at the peak in between 20 and 30 minutes after treatment with insulin, the increase in Kir4.1 corresponded with a gradual increase in BMAL1 and phosphorylation of IRS-1Tyr612 and AktSer473 (Fig. 6A). The Akt phosphorylation continued to climb up over 60 minutes; however, the BMAL1 and IRS-1Tyr612 were peaked at 20 and 30 minutes, respectively, and then declined over the course of time points studied (Fig. 6B).
Figure 6.

Restoration of IRS-1 signaling corrects Kir4.1 levels. (A) rMC-1 cells were treated with 10 nM insulin, Western blot showing an increase in phosphorylation of IRS-1Tyr612, AktSer473, BMAL-1, and Kir4.1. (B) Line chart showing quantification of the respective protein. The retinal explants from db/m and db/db mice were treated with insulin. (C) Western blot. (D) Bar chart showing an increase in phosphorylation of IRS-1Ser307. (E) Western blot showing IRS-1Tyr612 phosphorylation after insulin treatment and (F) quantification of phospho-IRS-1Tyr612. (G) Western blot showing a concurrent increase in BMAL1 and phosphorylation of AktSer473. (H) Bar chart showing a quantification of phospho-AktSer473 and (I) BMAL1. (J) Western blot showing Kir4.1 protein levels in insulin-treated explants. (K) Bar chart showing the quantification of Kir4.1 (n = 5).
To further study whether IRS-1 regulates Kir4.1, we treated the rMC-1 cells with a pharmacologic inhibitor of IRS-1, Secin H3.29 The rMC-1 cells were treated with Secin H3 (10 and 50 μM) and protein expression of phosphorylated IRS-1, BMAL1, AktSer473, and Kir4.1 were determined. The Secin H3 treatment inhibited IRS-1Tyr612 phosphorylation and downstream activation of Bmal1-Kir4.1 as well as AktS473 phosphorylation (Supplementary Fig. S3).
In the next phase of the study, we treated retinal explants of db/m and db/db mice with insulin ex vivo. The insulin treatment increased phosphorylation of IRS-1Ser307 in both db/m and db/db mice; however, this difference did not reach statistical significance (Figs. 6C, 6D). Interestingly, the insulin treatment of retinas exhibited a profound increase in IRS-1Tyr612 in both db/m and db/db mice (Figs. 6E, 6F). There was an increase in AktSer473 phosphorylation following an insulin treatment (Figs. 6G, 6H). The BMAL1 was upregulated in both groups; however, for db/m mice, this change was greatest (Figs. 6G, 6I). Finally, the insulin treatment resulted in an upregulation of Kir4.1 in both db/m and db/db mice (Figs. 6J, 6K).
Discussion
In the present study, we demonstrate that insulin signaling mediated by IRS-1 is critical to Kir4.1 channel function in Müller cells and that the diurnal rhythm of Kir4.1 is in sync with the IRS-1 rhythm. In retinas of db/db mice, the IRS-1 levels were decreased irrespective of the time and BMAL1 was dysrhythmic. One of the salient features of our study is a decrease in Kir4.1 channel function, as demonstrated by a decrease in Kir4.1 currents in db/db mice. Circadian regulatory protein BMAL1 serves as an important linkage between IRS-1 and Kir4.1. Overall, our studies unravel a novel mechanism of Kir4.1 decrease in diabetes and shed new light on understanding how circadian dysrhythmia is involved in Müller cell dysfunction observed in diabetes.
We observed that Kir4.1 and IRS-1 levels peaked at ZT-9 in our study for control mice, with diabetes there was a loss of diurnal rhythm for both proteins. The diurnal rhythm of IRS-1 has been shown previously in adipocytes, liver and heart, our study extends this process to the retina by showing a diurnal rhythm for IRS-1. Interestingly, the peak time of IRS-1 gene is different in these tissues; for example, in the liver the IRS-1 peaks at ZT-24,30 in the heart at ZT-22,31 and in the lung at ZT-6. We previously reported a peak increase in the retinal Kir4.1 at ZT-1824 in the rat retina while in this study the Kir4.1 levels were peaked at ZT-9. A couple of possible explanations of this anomaly could be a species difference (mouse versus rat) and age of the animals, the mice used in this study were 7 to 8 months old while the rat study was performed on younger animals (8 weeks). Previous studies suggest that aging dramatically affects the rhythmic pattern of clock genes.32
While DR is primarily a microangiopathy, the reactive changes in Müller cells attributed to dysfunctional ion channels and transporters might play a critical role in the pathogenesis of DR. The Kir4.1 channels are important for Müller cell health; in situ hybridization, immunofluorescence, and immunogold studies demonstrate that the large enrichment of Kir4.1 channels to Müller cell's end feet and processes enveloping the retinal blood vessels.21,33 Our studies demonstrate a decrease in Kir4.1 channels in perivascular regions and altered K+ conductance, this could lead to the development of DR. Reduced function of Kir4.1 channels in diabetes could potentially cause an imbalance in K+ concentration leading to neuronal excitation, glutamate toxicity, and neuronal death.34,35 This mechanism could be particularly important since neurodegeneration is reported to precede clinical evident DR.36 As the water transport in Müller cells is coupled to K+ currents, the downregulation of K+ channels by retinal Müller cells may impair transglial water transport, a common feature observed in DR. A decrease in Kir4.1 channels may cause the accumulation of K+ ions within the Müller cells resulting in an increase in osmotic pressure and Müller cell swelling.10 The Kir4.1 decrease in our studies was coupled with a downregulation of IRS-1 and BMAL1; previously the IRS-137 and Bmal38,39 decrease are individually linked in the pathogenesis of the DR. Taken together our studies highlight the interplay of IRS-1, Bmal1 and Kir4.1 in Müller cell dysfunction and which subsequently may lead to the development of DR.
Previous studies demonstrate that a change in circadian timing may lead to short-duration and poor quality of sleep, triggering obesity and the development of T2D.40 Our study further highlights timely expression of Kir4.1 is lost in db/db retinas, the dampened rhythm of Kir4.1 expression was coupled with a decrease in Kir4.1 channel function as demonstrated by a decrease in Kir4.1 currents. This may have potential relevance in the development of DR. Kir.4.1 channels being the major bidirectional K+ channels involved in potassium siphoning by Müller cells.34 Several studies suggest that the retinal Müller cells buffer extracellular retinal potassium concentration by siphoning excessive potassium ions in the vitreous humor.41,42 Thus, vitreous humor serves as a sink for the excessive potassium ions and alteration of the diurnal rhythm of Kir4.1 channels might affect this potassium clearance. Moreover, the potassium activity is vastly susceptible to the light stimulus43,44; the increase in K+ levels in inner and outer plexiform layers occurs shortly after the light stimuli, while the subretinal increase of K+ occurs when the lights are off. Therefore, timely expression of Kir 4.1 channels in Müller cells is necessary in the uptake of K+ released by neurons in response to light and in siphoning these K+ out to the vitreous and vasculature.45 We speculate that the effects of altered diurnal rhythm of Kir4.1 channels are multitude the Müller cells may remain10 consistently swollen throughout the course of diabetes resulting in an increase in oxidative stress and arachidonic acid metabolism; decreased light exposure (e.g., cataract) may lead to a decrease in potassium clearance in the vitreous; finally, change in vitreal composition in diabetes46,47 may independently affect the K+ homeostasis and normal retinal function, contributing together to the pathogenesis of DR. Our study certainly paves a way for pursuing these possible pathogenic mechanisms of DR in future.
The mice used in this study were leptin receptor resistant, the leptin plays an important role in signaling properties in the brain for control of energy homeostasis. Due to the mutation in leptin receptor, the db/db mice exhibit a variety of circadian rhythm defects such as an alteration of sleep regulation, arousal and sleep time, which may, in turn, affect energy metabolism.48 We speculate that dysfunctional leptin signaling may also have a potential role in altered diurnal rhythm of Kir4.1 and IRS-1, in additional to the diabetic milieu.
One of the striking findings of our study is that Kir4.1 rhythm is synchronized with signaling mediated by insulin. The IRS-1 is a critical regulator of insulin signaling in Müller cells.28 The low amount of IRS-1 and decreased phosphorylation of IRS-1 is reported in diabetic individuals. The previous study in insulin-resistant models and humans demonstrate the importance of insulin signaling via the IRS-1/Pi3-K pathway.49 Indeed, disruption of IRS-1 leads to diabetic ketoacidosis.50,51 The tyrosine phosphorylation of the IRS provides a binding site for numerous signaling molecules, including phosphoinositide 3 kinase (PI3K), which play a major role in insulin function, mainly via the activation of the Akt/PKB and the PKCζ cascades.17 The Akt activates a variety of downstream targets involved in cell survival, metabolism, and apoptosis.52 Our studies suggest that IRS-1 blockade inhibits Kir4.1 indirectly via downregulation of IRS1-Akt-BMAL1 axis. However, there is insufficient infrormation about the role of BMAL-1 in Kir4.1 regulation, it is likely that BMAL1 directly binds Kcnj10 based on the presence of an E-box in a promoter region of Kcnj10 and our previous studies demonstrating that Bmal1 silencing indeed downregulates Kir4.1.24 Further studies are necessary to substantiate this assertion.
A proinflammatory cytokine tumor necrosis factor alpha (TNF-α) is known to be elevated in diabetic retina. We previously reported that the treatment of Müller cells with TNF-α results in a downregulation of the Kir4.1 channels. Interestingly, TNF-α is known to activate the suppression of cytokine signaling pathway (SOCs3) pathway and is involved in repressing the IRS-1 signaling in Müller cells.20,28 While not studied in this manuscript, we predict that the inflammatory milieu of TNF-α may further influence IRS-1-Kir4.1 signaling via the involvement of the SOCs3 pathway.
Previous reports21 and studies performed in our laboratory suggest that insulin is necessary for optimum levels of Kir4.1. This study further reiterates the importance of insulin signaling in restoring Kir4.1 channels in db/db mice. In conclusion, our study identified a novel role of IRS-1 in regulating Kir4.1 levels in retinal Müller cells. By demonstrating BMAL1 as an important trigger, we further highlighted the critical role of circadian rhythms in Müller cell dysfunction observed in diabetes.
Supplementary Material
Acknowledgments
The authors thank Weiming Mao, PhD, (Department of Ophthalmology, Indiana University) for providing human retinal tissue for the isolation of human Müller cells and Deepa Mathew, PhD, (Department of Ophthalmology, Indiana University) for discussions on circadian rhythm studies.
Supported by a funding from International Retinal Research Foundation, NIH-National Eye Institute (R01EY027779), and Pilot & Feasibility award from Center for Diabetes and Metabolic Research at the Indiana University to ADB.
Disclosure: Q. Luo, None; Y. Xiao, None; A. Alex, None; T.R. Cummins, None; A.D. Bhatwadekar, None
References
- 1.Liu E, Craig JE, Burdon K. Diabetic macular oedema: clinical risk factors and emerging genetic influences. Clin Exp Optom. 2017;100:569–576. doi: 10.1111/cxo.12552. [DOI] [PubMed] [Google Scholar]
- 2.Bringmann A, Pannicke T, Grosche J, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Puro DG. Diabetes-induced dysfunction of retinal Muller cells. Trans Am Ophthalmol Soc. 2002;100:339–352. [PMC free article] [PubMed] [Google Scholar]
- 4.Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012;366:1227–1239. doi: 10.1056/NEJMra1005073. [DOI] [PubMed] [Google Scholar]
- 5.Gowda K, Zinnanti WJ, LaNoue KF. The influence of diabetes on glutamate metabolism in retinas. J Neurochem. 2011;117:309–320. doi: 10.1111/j.1471-4159.2011.07206.x. [DOI] [PubMed] [Google Scholar]
- 6.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chronopoulos A, Tang A, Beglova E, Trackman PC, Roy S. High glucose increases lysyl oxidase expression and activity in retinal endothelial cells: mechanism for compromised extracellular matrix barrier function. Diabetes. 2010;59:3159–3166. doi: 10.2337/db10-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chronopoulos A, Trudeau K, Roy S, Huang H, Vinores SA, Roy S. High glucose-induced altered basement membrane composition and structure increases trans-endothelial permeability: implications for diabetic retinopathy. Curr Eye Res. 2011;36:747–753. doi: 10.3109/02713683.2011.585735. [DOI] [PubMed] [Google Scholar]
- 9.Tien T, Barrette KF, Chronopoulos A, Roy S. Effects of high glucose-induced Cx43 downregulation on occludin and ZO-1 expression and tight junction barrier function in retinal endothelial cells. Invest Ophthalmol Vis Sci. 2013;54:6518–6525. doi: 10.1167/iovs.13-11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pannicke T, Iandiev I, Wurm A, et al. Diabetes alters osmotic swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes. 2006;55:633–639. doi: 10.2337/diabetes.55.03.06.db05-1349. [DOI] [PubMed] [Google Scholar]
- 11.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Xu G, Ling Q, Da C. Expression of aquaporin 4 and Kir4.1 in diabetic rat retina: treatment with minocycline. J Int Med Res. 2011;39:464–479. doi: 10.1177/147323001103900214. [DOI] [PubMed] [Google Scholar]
- 13.Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krook A, Bjornholm M, Galuska D, et al. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49:284–292. doi: 10.2337/diabetes.49.2.284. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa W, Matozaki T, Kasuga M. Role of binding proteins to IRS-1 in insulin signalling. Mol Cell Biochem. 1998;182:13–22. [PubMed] [Google Scholar]
- 16.Tamemoto H, Kadowaki T, Tobe K, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- 17.Brady MJ. IRS2 takes center stage in the development of type 2 diabetes. J Clin Invest. 2004;114:886–888. doi: 10.1172/JCI23108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 19.Walker RJ, Anderson NM, Bahouth S, Steinle JJ. Silencing of insulin receptor substrate-1 increases cell death in retinal Muller cells. Mol Vis. 2012;18:271–279. [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Biswas SK, Steinle JJ. Serine 307 on insulin receptor substrate 1 is required for SOCS3 and TNF-alpha signaling in the rMC-1 cell line. Mol Vis. 2014;20:1463–1470. [PMC free article] [PubMed] [Google Scholar]
- 21.Ishii M, Horio Y, Tada Y, et al. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Muller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;17:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23:372–381. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassan I, Luo Q, Majumdar S, Dominguez JM, II, Busik JV, Bhatwadekar AD. Tumor necrosis factor alpha (TNF-alpha) Disrupts Kir4.1 channel expression resulting in muller cell dysfunction in the retina. Invest Ophthalmol Vis Sci. 2017;58:2473–2482. doi: 10.1167/iovs.16-20712. [DOI] [PubMed] [Google Scholar]
- 25.Aoyagi T, Shigeki S, Tezuka M. Characteristics of circadian gene expressions in mice white adipose tissue and 3T3-L1 adipocytes. J Health Sci. 2005;51:21–32. [Google Scholar]
- 26.Thompson K, Chen J, Luo Q, Xiao Y, Cummins TR, Bhatwadekar AD. Advanced glycation end (AGE) product modification of laminin downregulates Kir4.1 in retinal Muller cells. PLoS One. 2018;13:e0193280. doi: 10.1371/journal.pone.0193280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusner LL, Sarthy VP, Mohr S. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: a role in high glucose-induced apoptosis in retinal Muller cells. Invest Ophthalmol Vis Sci. 2004;45:1553–1561. [PubMed] [Google Scholar]
- 28.Jiang Y, Pagadala J, Miller D, Steinle JJ. Reduced insulin receptor signaling in retinal Muller cells cultured in high glucose. Mol Vis. 2013;19:804–811. [PMC free article] [PubMed] [Google Scholar]
- 29.Hafner M, Schmitz A, Grune I, et al. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature. 2006;444:941–944. doi: 10.1038/nature05415. [DOI] [PubMed] [Google Scholar]
- 30.Hughes ME, DiTacchio L, Hayes KR, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsimakouridze EV, Straume M, Podobed PS, et al. Chronomics of pressure overload-induced cardiac hypertrophy in mice reveals altered day/night gene expression and biomarkers of heart disease. Chronobiol Int. 2012;29:810–821. doi: 10.3109/07420528.2012.691145. [DOI] [PubMed] [Google Scholar]
- 32.Wyse CA, Coogan AN. Impact of aging on diurnal expression patterns of CLOCK and BMAL1 in the mouse brain. Brain Res. 2010;1337:21–31. doi: 10.1016/j.brainres.2010.03.113. [DOI] [PubMed] [Google Scholar]
- 33.Nagelhus EA, Horio Y, Inanobe A, et al. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 34.Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem. 2008;107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coughlin BA, Feenstra DJ, Mohr S. Muller cells and diabetic retinopathy. Vision Res. 2017;139:93–100. doi: 10.1016/j.visres.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sohn EH, van Dijk HW, Jiao C, et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci U S A. 2016;113:E2655–E2664. doi: 10.1073/pnas.1522014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiter CE, Wu X, Sandirasegarane L, et al. Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes. 2006;55:1148–1156. doi: 10.2337/diabetes.55.04.06.db05-0744. [DOI] [PubMed] [Google Scholar]
- 38.Busik JV, Tikhonenko M, Bhatwadekar A, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lahouaoui H, Coutanson C, Cooper HM, Bennis M, Dkhissi-Benyahya O. Diabetic retinopathy alters light-induced clock gene expression and dopamine levels in the mouse retina. Mol Vis. 2016;22:959–969. [PMC free article] [PubMed] [Google Scholar]
- 40.Taheri S. The link between short sleep duration and obesity: we should recommend more sleep to prevent obesity. Arch Dis Child. 2006;91:881–884. doi: 10.1136/adc.2005.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newman EA. Distribution of potassium conductance in mammalian Muller (glial) cells: a comparative study. J Neurosci. 1987;7:2423–2432. [PMC free article] [PubMed] [Google Scholar]
- 43.Steinberg RH, Oakley B, II, Niemeyer G. Light-evoked changes in [K+]0 in retina of intact cat eye. J Neurophysiol. 1980;44:897–921. doi: 10.1152/jn.1980.44.5.897. [DOI] [PubMed] [Google Scholar]
- 44.Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newman E, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–312. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Lu Q, Lu P. Quantitative proteomics analysis of vitreous body from type 2 diabetic patients with proliferative diabetic retinopathy. BMC Ophthalmol. 2018;18:151. doi: 10.1186/s12886-018-0821-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barba I, Garcia-Ramirez M, Hernandez C, et al. Metabolic fingerprints of proliferative diabetic retinopathy: an 1H-NMR-based metabonomic approach using vitreous humor. Invest Ophthalmol Vis Sci. 2010;51:4416–4421. doi: 10.1167/iovs.10-5348. [DOI] [PubMed] [Google Scholar]
- 48.Laposky AD, Bradley MA, Williams DL, Bass J, Turek FW. Sleep-wake regulation is altered in leptin-resistant (db/db) genetically obese and diabetic mice. Am J Physiol Regul Integr Comp Physiol. 2008;295:R2059–R2066. doi: 10.1152/ajpregu.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes. 2006;55:2392–2397. doi: 10.2337/db06-0391. [DOI] [PubMed] [Google Scholar]
- 50.Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- 51.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 52.Lebrun P, Mothe-Satney I, Delahaye L, Van Obberghen E, Baron V. Insulin receptor substrate-1 as a signaling molecule for focal adhesion kinase pp125(FAK) and pp60(src) J Biol Chem. 1998;273:32244–32253. doi: 10.1074/jbc.273.48.32244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.