

Abstract
Mutation and recombination are key evolutionary processes governing phenotypic variation and reproductive isolation. We here demonstrate that biodiversity within all globally known strains of Schizosaccharomyces pombe arose through admixture between two divergent ancestral lineages. Initial hybridization was inferred to have occurred ∼20–60 sexual outcrossing generations ago consistent with recent, human-induced migration at the onset of intensified transcontinental trade. Species-wide heritable phenotypic variation was explained near-exclusively by strain-specific arrangements of alternating ancestry components with evidence for transgressive segregation. Reproductive compatibility between strains was likewise predicted by the degree of shared ancestry. To assess the genetic determinants of ancestry block distribution across the genome, we characterized the type, frequency, and position of structural genomic variation using nanopore and single-molecule real-time sequencing. Despite being associated with double-strand break initiation points, over 800 segregating structural variants exerted overall little influence on the introgression landscape or on reproductive compatibility between strains. In contrast, we found strong ancestry disequilibrium consistent with negative epistatic selection shaping genomic ancestry combinations during the course of hybridization. This study provides a detailed, experimentally tractable example that genomes of natural populations are mosaics reflecting different evolutionary histories. Exploiting genome-wide heterogeneity in the history of ancestral recombination and lineage-specific mutations sheds new light on the population history of S. pombe and highlights the importance of hybridization as a creative force in generating biodiversity.
Keywords: hybridization, structural variation, epistasis, reproductive isolation, transgression, Schizosaccharomyces pombe
Introduction
Mutation is the ultimate source of biodiversity. In sexually reproducing organisms it is assisted by recombination shuffling mutations of independent genomic backgrounds into millions of novel combinations. This widens the phenotypic space upon which selection can act and thereby accelerates evolutionary change (Muller 1932; Fisher 1999; McDonald et al. 2016). This effect is enhanced for heterospecific recombination between genomes of divergent populations (Abbott et al. 2013). Novel combinations of independently accumulated mutations can significantly increase the overall genetic and phenotypic variation, even beyond the phenotypic space of parental lineages (transgressive segregation [Nolte and Sheets 2005; Lamichhaney et al. 2017]). Yet, if mutations of the parental genomes are not compatible to produce viable and fertile offspring, hybridization is a dead end. Phenotypic variation then remains within the confines of genetic variation of each reproductively isolated, parental lineage.
It is increasingly recognized that hybridization is commonplace in nature, and constitutes an important driver of diversification (Mallet 2005; Abbott et al. 2013). Ancestry components of hybrid genomes can range from clear dominance of alleles from the more abundant species (Dowling et al. 1989; Taylor and Hebert 1993), over a range of admixture proportions (Lamichhaney et al. 2017; Runemark et al. 2018) to the transfer of single adaptive loci (Heliconius Genome Consortium 2012). The final genomic composition is determined by a complex interplay of demographic processes, heterogeneity in recombination (e.g., induced by genomic rearrangements) (Wellenreuther and Bernatchez 2018) and selection (Sankararaman et al. 2014; Schumer et al. 2016). Progress in sequencing technology, now allows charactering patterns of admixture and illuminatiing of underlying processes (Payseur and Rieseberg 2016). Yet, research has largely focused on animals (Turner and Harr 2014; Vijay et al. 2016; Meier et al. 2017; Jay et al. 2018) and plants (Twyford et al. 2015), and relatively little attention has been paid to natural populations of sexually reproducing micro-organisms (Leducq et al. 2016; Stukenbrock 2016; Peter et al. 2018; Steenkamp et al. 2018).
The fission yeast Schizosaccharomyces pombe is an archiascomycete haploid unicellular fungus with a facultative sexual mode of reproduction. Despite of its outstanding importance as a model system in cellular biology (Hoffman et al. 2015) and the existence of global sample collections, essentially all research has been limited to a single isogenic strain isolated by Leupold in 1949 (Leupold 972; JB22 in this study). Very little is known about the ecology, origin, and evolutionary history of the species (Jeffares 2018). Global population structure has been described as shallow with no apparent geographic stratification (Jeffares et al. 2015). Genetic diversity (π = 3·10−3 substitutions/site) appears to be strongly influenced by genome-wide purifying selection with the possible exception of region-specific balancing selection (Fawcett et al. 2014; Jeffares et al. 2015). Despite the overall low genetic diversity, S. pombe shows abundant additive genetic variation in a variety of phenotypic traits including growth, stress responses, cell morphology, and cellular biochemistry (Jeffares et al. 2015). The apparent world-wide lack of genetic structure in this species appears inconsistent with the large phenotypic variation between strains and with evidence for postzygotic reproductive isolation between interstrain crosses, ranging from 1% to 90% of spore viability (Kondrat’eva and Naumov 2001; Teresa Avelar et al. 2013; Zanders et al. 2014; Jeffares et al. 2015; Naumov et al. 2015; Marsellach X, 2019).
In this study, we integrate whole-genome sequencing data from three different technologies—sequencing-by-synthesis (Illumina technology data accessed from Jeffares et al. [2015]), single-molecule real-time sequencing (Pacific BioSciences technology, this study) and nanopore sequencing (Oxford Nanopore technology, this study)—sourced from a mostly human-associated, global sample collection to elucidate the evolutionary history of the S. pombe complex. Using population genetic analyses based on single-nucleotide polymorphism (SNP) we show that global genetic variation and heritable phenotype variation of S. pombe results from recent hybridization of two ancient lineages. Twenty-five de novo assemblies from 17 divergent strains further allowed us to quantify segregating structural variation including insertions, deletions, inversions, and translocations. In light of these findings, we retrace the global population history of the species, and discuss the relative importance of genome-wide ancestry and structural mutations in explaining phenotypic variation and reproductive isolation.
Results
Global Genetic Variation in S. pombe Is Characterized by Ancient Admixture
Genetic variation of the global S. pombe collection comprises 172,935 SNPs segregating in 161 strains. Considering SNPs independently, individuals can be substructured into 57 clades that differ by more than 1,900 variants, but are near-clonal within clades (Jeffares et al. 2015). To examine population ancestry further, we divided the genome into 1,925 overlapping windows containing 200 SNPs each and selected one representative from each clade (57 samples in total). Principal component analysis (PCA) conducted on each orthologous window showed a highly consistent pattern along the genome (fig. 1a, supplementary fig. 1, Supplementary Material online): (1) the major axis of variation (PC1) split all samples into two discrete groups explaining 60% ± 13% of genetic variance (fig. 1b). (2) All samples fell into either extreme of the normalized distribution of PC1 scores ( (supplementary figs. 2 and 3, Supplementary Material online) with the exception of strains with inferred changes in ploidy level (see Materials and Methods; supplementary fig. 4, Supplementary Material online). (3) PC2 explained 13% ± 6% of variation and consistently attributed higher variation to one of the two groups. This strong signal of genomic windows separating into two discrete groups suggested that the genomic diversity in this collection was derived from two distinct ancestral populations. However, (4) group membership of strains changed among windows along the genome, reflecting recombination between these two well defined groups. Forward population simulations followed by PCA showed that this signal was unlikely to be an artifact (see Materials and Methods). Thus, explicit consideration of haplotypes explains the lack of observed population structure when disregarding nonindependence of SNPs (Jeffares et al. 2015).
Fig. 1.

Distribution of Sp (red) and Sk (blue) ancestry blocks along the Schizosaccharomyces pombe genome. (a) Example of principal component analysis (PCA) of a representative genomic window in chromosome I (top) and the whole mitochondrial DNA (bottom). Samples fall into two major clades, Sp (red square) and Sk (blue square). The proportion of variance explained by PC1 and PC2 is indicated on the axis labels. Additional examples are found in supplementary figure 1 (Supplementary Material online). (b) Proportion of variance explained by PC1 (black line) and PC2 (gray line) for each genomic window along the genome. Centromeres are indicated with red bars. Note the drop in proximity to centromeres and telomeres where genotype quality is significantly reduced. (c) Heatmap for one representative of 57 near-clonal groups indicating ancestry along the genome (right panel). Samples are organized according to a hierarchical clustering, grouping samples based on ancestral block distribution (left dendrogram). Colors on the tips of the cladogram represent cluster membership by chromosome (see supplementary fig. 9, Supplementary Material online). Samples changing clustering group between chromosomes are shown in gray. (d) Estimate of Dxy between ancestral groups and genetic diversity (π) within the Sp (red) and Sk clade (blue) along the genome.
The strong signal from the PCA systematically differentiating groups along the genome was likewise reflected in population genetic summary statistics including Watterson’s theta (θ), pairwise nucleotide diversity (π), and Tajima’s D statistic (figs. 1d and2). Significant differences in these statistics (Kendall’s τ P-value ≤ 2.2·10−16) were also present in mitochondrial genetic variation (fig. 1a) and allowed polarizing the two groups across windows into a “low-diversity” group (red) and a “high-diversity” group (blue) (fig. 1a, supplementary fig. 5, Supplementary Material online). Genetic divergence between groups (Dxy) was 15 and 3 times higher than mean genetic diversity (π) within each group, respectively, and thus supports a period of independent evolution. Painting genomic windows by group membership revealed blocks of ancestry distributed in sample specific patterns along the genome (fig. 1c, supplementary fig. 6, Supplementary Material online). The sample corresponding to the reference genome isolated originally from Europe (Leupold’s 972; JB22) consisted almost exclusively of “red” ancestry (>96% red), whereas other samples were characterized near-exclusively by “blue” ancestry (>96% blue). The sample considered to be a different species from Asia, Schizosaccharomyces kambucha (JB1180; Singh and Klar 2002) had a large proportion of “blue” windows (>70% blue). Hereafter, we refer to the “red” and “blue” clade as Sp and Sk, for S. pombe and S. kambucha, respectively.
Fig. 2.

Population genetic summary statistics. (a) Proportion of Sp (red) and Sk (blue) ancestry across all 57 samples along the genome. (b) Tajima’s D statistic differentiated by Sp (red) and Sk (blue) ancestry and pooled across all samples irrespective of ancestry (gray line). Genomic regions previously identified under purifying selection (Fawcett et al. 2014) are shown with black triangles. Reported active meiotic drivers (Zanders et al. 2014; Hu et al. 2017; Nuckolls et al. 2017) are indicated by yellow triangles. The third panel shows the difference between ancestry specific Tajima's D statistic and the estimate from the pooled samples.
Next, we explored different historical processes that may underlie the deep divergence between these clades and their distribution along the genome. Using individual based forward simulations we contrasted a historical scenario of divergence in allopatry followed by extensive, recent hybridization with a scenario of low, but constant levels of gene flow during divergence. The empirical level of divergence between ancestral groups in combination with the observed distribution of recombinant block size along the genome could not be recovered under a demographic model of divergence with constant gene flow. The simulations much rather provide support for a scenario of divergence in isolation followed by a recent pulse of gene flow recovering both ancestral divergence and the observed introgression landscape (see Materials and Methods; supplementary figs. 7 and 8, Supplementary Material online).
The distribution of ancestry components was highly heterogeneous across the chromosome (fig. 2a). Most strains showed an excess of Sp ancestry in parts of chromosome I, whereas several regions of chromosome III had an excess of Sk ancestry. Grouping samples by the pattern of genomic ancestry across the genome revealed eight discrete clusters (fig. 1c). Consistent with independent and/or recent segregation of ancestral groups, cluster membership for several samples differed between chromosomes (fig. 1c) and genome components (supplementary figs. 9 and 10, Supplementary Material online). This was heterogeneity among genomic regions and chromosomes was reflected by low support in the relationship between the eight clusters.
Failing to incorporate this genome-wide variation of admixture proportions can mimic signatures of selection. For example, equal ancestry contributions for a certain genomic region will yield high positive values of both Tajima's D statistic (supplementary fig. 11, Supplementary Material online) and π and may be mistaken as evidence for balancing selection. Strong skew in ancestry proportions reduces both statistics to values of the prevailing ancestry and may appear as evidence for selective sweeps (fig. 2b). Taking ancestry into account, however, there was no clear signature of selection in either Sp or Sk genetic variation that could account for heterogeneity in the genetic composition of hybrids (supplementary fig. 11, Supplementary Material online). Signatures of selection identified previously (cf. Fawcett et al. 2014) are likely artifacts due to skewed ancestry proportions rather than events of positive or balancing selection in the ancestral populations.
Overall, our results provide strong evidence for the presence of at least two divergent ancestral populations: one genetically diverse group (Sk clade) and a less diverse group (Sp clade). We found a large range of ancestral admixture proportions between these two clades broadly clustering samples into eight weakly supported groups. These resemble clusters of strains previously identified by Structure and fineStructure without explicit modeling of ancestral admixture (Jeffares et al. 2015). Neglecting ancestry, Jeffares et al. (2015) argued that the shallow population structure likely results from extensive gene flow between clusters. Yet, considering the genome-wide distribution of discrete Sk and Sp ancestry, and lack of geographical structure, suggests that the eight clusters are derived from one or a few centers of ancient admixture (hybridization) without having to invoke subsequent or recent gene flow between them.
Age of Ancestral Lineages and Timing of Hybridization
To shed further light on the population history, we estimated the age of the parental lineages and the timing of initial hybridization. Calibrating mitochondrial divergence by known collection dates over the last 100 years, Jeffares et al. (2015) estimated that the time to the most recent common ancestor for all samples was around 2,300 years ago. Current overrepresentation of near-pure Sp and Sk in Europe or Africa/Asia, respectively, is consistent with an independent history of the parental lineages on different continents for the most part of the last millennia (supplementary fig. 10, Supplementary Material online). Yet, the variety of admixed genomes bears testimony to the fact that isolation has been disrupted by heterospecific gene flow. Using a theoretical model assuming secondary contact with subsequent hybridization (Janzen et al. 2018) as supported by the data, we estimated that hybridization occurred within the last around 20–60 sexual outcrossing generations depending on window size (fig. 3, supplementary figs. 12 and 13, Supplementary Material online). Considering intermittent generations of asexual reproduction, high rates of haploid selfing, and dormancy of spores (Farlow et al. 2015; Jeffares 2018) it is difficult to obtain a reliable estimate of time in years. Yet, the range of estimates of hybridization timing is consistent with hybridization induced by the onset of regular transcontinental human trade between Europe with Africa and Asia (∼14th century) and with the Americas (∼16th century), with fission yeast as a human commensal (Jeffares 2018). This fits with the observation that all current samples from the Americas were hybrids, whereas samples with the purest ancestry stem from Europe, Africa, and Asia. Moreover, negative genome-wide Tajima's D statistic estimates for both ancestral clades (mean ± SD for Sp: −0.8 ± 0.9 and Sk: −0.7 ± 0.6) signal a period of recent expansion.
Fig. 3.
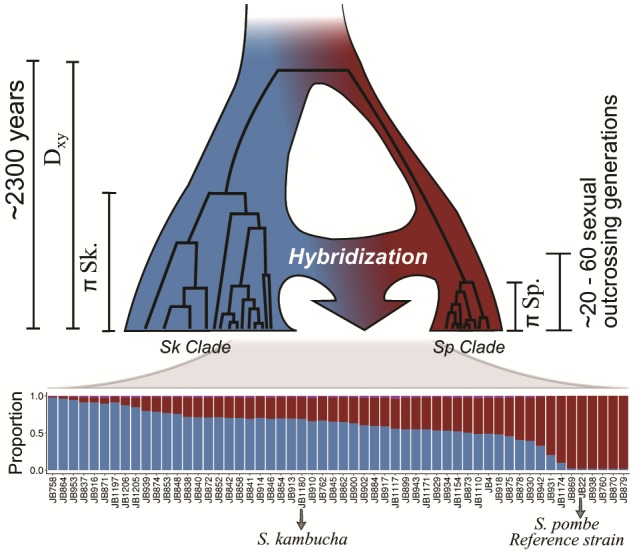
Inferred evolutionary history of contemporary Schizosaccharomyces pombe strains. An ancestral population diverged into two major clades, Sp (red) and Sk (blue), approximately 2,300 years ago (Jeffares et al. 2015). Recurrent hybridization upon secondary contact initiated around 20–60 sexual outcrossing generations ago resulted in the admixed genomes with a range of admixture proportions (bottom) prevailing today.
Heritable Phenotypic Variation and Reproductive Isolation Are Governed by Ancestry Components
Hybridization can lead to rapid evolution due to selection acting on the genetic and phenotypic variation emerging after admixture (Muller 1932; Fisher 1999; McDonald et al. 2016). We assessed the consequences of hybridization on phenotypic variation making use of a large data set including 228 quantitative traits collected from the strains under consideration here (Jeffares et al. 2015). Contrary to genetic clustering of hybrid genomes (fig. 1c), samples with similar ancestry proportions did not group in phenotypic space described by the first two PC-dimensions capturing 31% of the total variance across traits (fig. 4a). Moreover, phenotypic variation of hybrids exceeded variation of pure strains (>0.9 ancestry for Sp or Sk). This was supported by trait specific analyses. We divided samples into three discrete groups: pure Sp, pure Sk, and hybrids with a wide range of Sp admixture proportions (0.1–0.9). Sixty-three traits showed significant differences among groups (fig. 4b, supplementary fig. 14, Supplementary Material online). In the vast majority of cases (50 traits), hybrid phenotypes were indistinguishable from one of the parents, but differed from the other, suggesting dominance of one ancestral background. In seven traits, hybrid phenotypes were intermediate and differed from both parents. This is consistent with an additive or polygenic contribution of both ancestral backgrounds. For six traits, hybrids exceeded phenotypic values of both parents providing evidence for transgressive segregation. In all cases, the number of significantly differentiated traits was found to be higher than under the null model (mean number of significant traits after 10,000 permutations: dominant Sk 4 ± 2, dominant Sp 4 ± 2, transgressive 0 ± 0.3, intermediate 0 ± 0.1; supplementary fig. 15, Supplementary Material online). Jeffares et al. (2017) showed that for each trait the total proportion of phenotypic variance explained by the additive genetic variance component (used as an estimated of the narrow-sense heritability) ranged from 0% to around 90%. We found that across all 228 traits, considering Sp and Sk ancestry components across the 1,925 genomic windows explained an equivalent amount of phenotypic variance as all 172,935 SNPs segregating across all samples (both being highly correlated: fig. 4c and d;r = 0.82, P-value ≤ 2.2·10−16). Combinations of ancestral genetic variation appear to be the main determinants of heritable phenotypic variation with only little contribution from single-nucleotide mutations arising after admixture. In turn, this supports that the formation of hybrids is recent (see above), and few (adaptive) mutations have occurred after it.
Fig. 4.

Ancestry explains variation in phenotype and reproductive isolation. (a) PCA of normalized phenotypic variation across 228 traits. The proportion of variance explained by PC1 and PC2 is indicated on the axis labels. Admixed samples (dots) are color-coded by ancestry proportion (cf. fig. 3) ranging from pure Sp (red triangle) to pure Sk (blue triangle) ancestry. (b) Phenotypic distribution of example traits separated by the degree of admixture: admixed samples are shown in gray, pure ancestral Sp and Sk samples are shown in red and blue, respectively. The number of traits corresponding to a dominant, additive, and transgressive genetic architecture is indicated on the right hand side. (c) Comparison of heritability estimates of all 228 traits based on 172,935 SNPs (abscissa) and on 1,925 genomic windows polarized by ancestry (ordinate). Colors indicate statistical significance. NS: heritability values not significantly different from zero, AncHap: significant only using ancestral blocks, SNPs: significant only using SNPs, SB: significant in both analyses. Diagonal (slope = 1) added as reference. (d) Histogram of the difference between heritability estimates using SNPs and ancestry components for all 228 traits. (e) Correlation between the difference in ancestry proportions between two strains (cf. fig. 2) and spore viability of the cross. The red box highlights samples with low spore viability, but high genetic similarity.
Ancestry also explained most of the variation in postzygotic reproductive isolation between strains. Previous work revealed a negative correlation between spore viability and genome-wide SNP divergence between strains (Jeffares et al. 2015). The degree of similarity in genome-wide ancestry had the same effect: the more dissimilar two strains were in their ancestry, the lower the viability of the resulting spores (fig. 4e; Kendall correlation coefficients, τ= −0.30, T = 259, P-value = 6.66·10−3). This finding is consistent with reproductive isolation being governed by many, genome-wide incompatibilities between the Sp and Sk clade. Yet, in a number of cases spore survival was strongly reduced in strain combinations with near-identical ancestry. In these cases, reproductive isolation may be caused by few large-effect mutations, including structural genomic changes that arose after hybridization.
Structural Mutations Do Not Determine the Genome-Wide Distribution of Ancestry Blocks
Structural genomic changes (structural variants or SVs hereafter) are candidates for large-effect mutations governing phenotypic variation (Küpper et al. 2016; Jeffares et al. 2017), reproductive isolation (Hoffmann and Rieseberg 2008; Teresa Avelar et al. 2013), and heterospecific recombination (Ortiz-Barrientos et al. 2016). They may thus importantly contribute to shaping heterogeneity in the distribution of ancestry blocks observed along the genome (Poelstra et al. 2014; Jay et al. 2018) (fig. 2b). However, inference of SVs in natural strains of fission yeast has been primary based on short-read sequencing (Jeffares et al. 2017). SV calls from short-read sequencing data are known to differ strongly by bioinformatic pipeline, are prone to false positive inference and are limited in their ability to infer long-range SVs, in particular in repetitive regions of the genome (Jeffares et al. 2017).
To obtain a reliable and comprehensive account of SVs segregating across strains, and to test for a possible association of SVs with the skewed ancestry in the genome, we generated chromosome-level de novo genome assemblies for 17 of the most divergent samples using single-molecule real-time sequencing (mean sequence coverage 105×; supplementary table 7, Supplementary Material online). For the purpose of methodological comparison, we also generated de novo assemblies for a subset of eight strains (including the reference Leupold’s 972) based on nanopore sequencing (mean sequence coverage: 140×). SVs were called using a mixed approach combining alignment of de novo genomes and mapping of individual reads to the reference genome (Wood et al. 2002). Both approaches and technologies yielded highly comparable results (see Materials and Methods; supplementary figs. 16–19 and table 8, Supplementary Material online).
After quality filtering, we retained a total of 832 variant calls including 563 insertions or deletions (indels), 118 inversions, 110 translocations, and 41 duplications. The 17 strains we examined with long-reads could be classified into six main karyotype arrangements (fig. 5a). The previously reported list of SVs of the same strains using short-reads consisted of only 52 SVs (Jeffares et al. 2017) of which only 8 were found to overlap with the 832 calls from long-read data. The vast majority of SVs were smaller than 10 kb (fig. 5b). The size distribution was dominated by elements of 6 and 0.5 kb in length corresponding to known transposable elements (TEs) and their flanking long terminal regions (LTRs), respectively (Kelly and Levin 2005). Only a small number of SVs corresponded to large-scale rearrangements (50 kb–2.2 Mb) including translocations between chromosomes (fig. 5a). A subset of these have been characterized previously as large-effect modifiers of recombination promoting reproductive isolation (Brown et al. 2011; Teresa Avelar et al. 2013; Jeffares et al. 2017).
Fig. 5.

Characterization of structural variation based on long-read, real-time sequencing. (a) Schematic representation of the three chromosomes in different strains displaying SVs larger than 10 kb relative to reference genome JB22 (left panel). Chromosome arms are differentiated by color; orientation is indicated with arrows relative to the reference; black bars represent centromeres. In the second panel, additional SVs, their type and ID of the corresponding strain are illustrated in brackets. (b) Size distribution of SVs below 10 kb. Colors indicate the type of SV. (c) Distribution of SV density along the genome. Black bars represent centromeres. (d) Two-dimensional, folded site frequency spectrum between inferred ancestral populations for all SVs, SNPs, and LTR INDELs. Numbers and colors show the percentage of the total number of variants in each category. Variants with low frequency in both populations are shown in the blue box. Variants highly differentiated between populations are show in red boxes with total in the upper right box. Fills with percentage lower than 0.01 are empty. Abbreviations: DEL, deletion; DUP, duplication; INS, insertion; INV, inversion.
Contrary to previous SV classification based on short-reads (Jeffares et al. 2017), SV density was not consistently increased in repetitive sequences such as centromeric and telomeric regions illustrating the difficulty of short-read data in resolving SVs in repetitive regions (fig. 5c). Instead, we found that the frequency of SVs was significantly elevated in close proximity to developmentally programed DNA double-strand breaks (DSB) associated with recombination initiation (Fowler et al. 2014). The proportion of SVs observed within [0, 0.5) kb and [0.5, 1) kb of DSB was increased by 46% (P-value < 1·10−4) and 67% (P-value < 1·10−4) relative to random expectations. On the contrary, regions more distant than 10 kb from DSB were relatively depleted of SVs (supplementary fig. 20, Supplementary Material online).
Next, we imputed the ancestry of SV alleles from SNPs surrounding SV break points. We calculated allele frequencies for SVs in both ancestral clades and constructed a folded 2D site frequency spectrum (fig. 5d). The majority of variants (66%) segregated at frequencies below 0.3 in both ancestral genetic backgrounds. Very few SVs were differentiated between ancestral populations (3% of variants with frequency higher than 0.9 in one population and below 0.1 in the other). This pattern contrasted with the reference spectrum derived from SNPs where the proportion of low frequency variants was similar at 60%, but genetic differentiation between populations was substantially higher (21% of SNP variants with frequency higher than 0.9 in one population and below 0.1 in the other). The difference was most pronounced for large SVs (larger than 10 kb) and TEs, for which we estimated allele frequencies for all 57 strains by means of polymerase chain reaction (PCR) and short-read data, respectively. For TE’s, 98% of the total 1,048 LTR variants segregated at frequencies below 0.3 in both ancestral populations without a single variant differentiating ancestral populations (fig. 5d). Large SVs likewise segregated at low frequencies, being present at most in two strains out of 57. This included the translocation reported for S. kambucha between chromosomes II and III (Zanders et al. 2014), which we found to be specific for that strain. Only the large inversion on chromosome I segregated at higher frequency being present in 5 strains out of 57, of which three were of pure Sp ancestry including the reference strain (supplementary table 10, Supplementary Material online). Additionally, SVs segregating at high frequency (>0.7) did not cluster in genomic regions with steep transitions between Sp and Sk ancestry (large changes in ancestral frequency in fig. 2a; P-value > 0.1; supplementary fig. 21, Supplementary Material online).
In summary, long-read sequencing provided a detailed account of species-wide diversity in structural genetic variation including over 800 high-quality variants ranging from small indels to large-scale interchromosomal rearrangements. SV calls showed substantial overlap among technologies (Pacific Biosciences, Nanopore) and approaches (de novo assembly vs. mapping), but less than 1% of this variation was inferred from short-read data. This finding admonishes to caution when interpreting SV calls from short-read data which are moreover sensitive to genotyping methods. In contrast to genome-wide SNPs, SVs segregated near-exclusively at low frequencies and were rarely differentiated by ancestral origin. This is consistent with stronger diversity-reducing purifying selection relative to SNPs. The fact that SVs, including large-scale rearrangements with known effects on recombination and reproductive isolation (Brown et al. 2011; Teresa Avelar et al. 2013; Zanders et al. 2014), were often unique to single strains precludes a role of SVs in shaping patterns of ancestral heterospecific recombination. Moreover, although being concentrated in proximity to DSB, possibly due to improper repair upon recombination (Currall et al. 2013), SVs were not significantly associated with steep transitions in ancestry blocks. Summarizing the evidence, SVs appear to have had limited influence in shaping genome-wide patterns of ancestral admixture. Although they may reduce fitness of hybrids between specific strains, SV cannot explain the prevalence of reproductive isolation as a function on ancestral similarity (fig. 4e).
Negative Epistasis and the Distribution of Ancestral Blocks
Alternatively, heterogeneity in the distribution and frequency of ancestry along the genome may result from negative epistatic interactions of incompatible genetic backgrounds (Schumer et al. 2016). An excess of homospecific combinations of physically distant loci can serve as an indication of epistatic selection against genetic incompatibilities which can be segregating at appreciable frequencies even within species (Corbett-Detig et al. 2013). We tested this hypothesis by measuring ancestry disequilibrium (AD) between all possible pairs of genomic windows within a chromosome. Specifically, we quantified linkage disequilibrium (LD) between windows dominated by alleles from the same ancestral group (>0.7) Sp–Sp or Sk–Sk (reflecting positive AD) and contrasted it to the degree of LD arising between heterospecific allele combinations Sp–Sk (negative AD) (supplementary fig. 22, Supplementary Material online). LD differed significantly between these two cases (fig. 6). Whereas negative AD decreased rapidly with genetic distance (R2 < 0.2 after 66, 19, and 21 kb, respectively for each chromosome) positive AD was higher in magnitude and extended over larger distances (R2 < 0.2 after 1.02, 0.54, and 0.18 Mb, respectively for each chromosome in Sk–Sk comparisons and 1.59, 1.12, and 0.32 Mb for Sp–Sp comparisons). This relationship remained significant after controlling for the potential effect of secondary population structure which can likewise increase apparent AD mimicking signatures of selection on epistatic genetic variation. Choosing only a single representative from each of the hybrid clusters (fig. 1c) LD remained larger for comparisons of genomic regions with the same ancestry. These results are overall consistent with a contribution of epistatic selection during the course of hybridization in shaping the ancestry composition of admixed genomes.
Fig. 6.

Decay in linkage disequilibrium (LD) with genetic distance. Relationship between LD (R2) and physical distance is depicted for each chromosome. Black points represent values for each window-pair comparison. Lines show nonlinear regression model based on Hill and Weir (1988) and Remington et al. (2001). LD estimates were divided into three categories representing comparison between windows of shared ancestry (Sp–Sp or Sk–Sk) reflecting positive ancestry disequilibrium (AD) or of opposite ancestry (Sk–Sp) reflecting negative AD.
Discussion
This study adds to the increasing evidence that hybridization plays an important role as a rapid, “creative” evolutionary force in natural populations (Seehausen 2004; Mallet 2007; Soltis and Soltis 2009; Abbott et al. 2013, 2016; Schumer et al. 2014; Pennisi 2016; Nieto Feliner et al. 2017). Recent heterospecific recombination between two ancestral S. pombe populations shuffled genetic variation of genomes that diverged since classical antiquity about 2,300 years ago. With the necessary caution, the timing of hybridization can be inferred to coincide with the onset of intensified transcontinental human trade suggesting an anthropogenic contribution. Several samples showed a similar distribution of ancestral blocks along the genome suggesting comparable evolutionary histories within eight discrete clusters. These clusters, in general showed weak geographical grouping, initially interpreted as evidence for reduced population structure with large recent world-wide gene flow (Jeffares et al. 2015). In contrast, the world-wide distribution of the two ancestral linages suggests rapid and recent global dispersion after hybridization followed by local differentiation. This study thus highlights the importance of taking genomic nonindependence into account. Allowing for the fact that genomes are mosaics reflecting different evolutionary histories can fundamentally alter inference on a species' evolutionary history.
Moreover, conceptualizing genetic variation as a function of ancestry blocks alternating along the genome changes the view on adaptation. Admixture is significantly faster than evolutionary change solely driven by mutation. Accordingly, phenotypic variation was near-exclusively explained by ancestry components with only little contribution from novel mutations. Importantly, admixture not only filled the phenotypic space between parental lineages, but also promoted transgressive segregation in several hybrids. This range of phenotypic outcomes opens the opportunity for hybrids to enter novel ecological niches (Nolte and Sheets 2005; Pfennig et al. 2016) and track rapid environmental changes (Eroukhmanoff et al. 2013).
Structural mutations have been described as prime candidates for rapid large-effect changes with implications on phenotypic variation, recombination, and reproductive isolation (Faria and Navarro 2010; Ortiz-Barrientos et al. 2016; Wellenreuther and Bernatchez 2018). This study contributes to this debate providing a detailed account of over 800 high-quality SVs identified across 17 chromosome-level de novo genomes sampled from the most divergent strains within the species. On the whole, SVs had little effect in our analysis. SVs segregated at low frequencies in both ancestral populations and, contrary to what has been suggested for specific genomic regions in other systems (Jay et al. 2018), they did not account for genome-wide heterogeneity in introgression among strains. Moreover, reproductive isolation was overall best predicted by the degree of shared ancestry with little contribution from SVs. Few crosses, however, showed strong reproductive isolation despite a high degree of shared ancestry (outliers in lower left corner of fig. 4e). In these cases, combining SVs from different strains into hybrid genomic backgrounds may have a significant impact. This is consistent with the observation that large, artificially generated rearrangements affect fitness (Teresa Avelar et al. 2013; Nieuwenhuis et al. 2018) and may promote reproductive isolation between specific S. pombe strains (Teresa Avelar et al. 2013; Zanders et al. 2014). Thus, reproductive isolation may arise by a combination of factors: negative epistasis between many loci with small to moderate effect differing in ancestry, and for specific strain combinations, single major effect (structural) mutations such as selfish elements or meiotic drivers (Zanders et al. 2014; Hu et al. 2017; Nuckolls et al. 2017). Functional work is needed to isolate these genetic elements.
Materials and Methods
Strains
This study is based on a global collection of S. pombe consisting of 161 world-wide distributed strains (see supplementary table 1, Supplementary Material online) described in Jeffares et al. (2015).
Inferring Ancestry Components
To characterize genetic variation across all strains, we made use of publically available data in variant call format (VCF) derived for all strains from Illumina sequencing with an average coverage of around 80× (Jeffares et al. 2015). The VCF file consists of 172,935 SNPs obtained after read mapping to the S. pombe 972 h− reference genome (ASM294v264) (Wood et al. 2002) and quality filtering (see supplementary table 1, Supplementary Material online, for additional information). We used a custom script in R 3.4.3 (R Core Team 2018) with the packages gdsfmt 1.14.1 and SNPRelate 1.12.2 (Zheng et al. 2017, 2012), to divide the VCF file into genomic windows of 200 SNPs with overlap of 100 SNPs. This resulted in 1,925 genomic windows of 1–89 kb in length (mean 13 kb). For each window, we performed PCAs using SNPRelate 1.12.2 (Zheng et al. 2017, 2012) (example in fig. 1a and supplementary fig. 1, Supplementary Material online). The proportion of variance explained by the major axis of variation (PC1) was consistently high and allowed separating strains into two genetic groups/clusters, Sp and Sk (see main text, fig. 1b). Using individual based forward simulations with SLiM 3.2.1 (Haller and Messer 2019), we found that this pattern cannot be obtained in the absence of population structure even for smaller window sizes including 5, 10, 20, 50, or 100 SNPs (supplementary fig. 23, Supplementary Material online).
We calculated population genetic parameters within clusters including pairwise nucleotide diversity (π) (Nei and Li 1979), Watterson theta (θw) (Watterson 1975), and Tajima’s D statistic (Tajima 1989), as well as the average number of pairwise differences between clusters (Dxy) (Nei and Li 1979) using custom scripts. Statistical significance of the difference in nucleotide diversity (π) between ancestral clades was inferred using Kendall’s τ as test statistic. Since values of adjacent windows are statistically nonindependent due to linkage, we randomly subsampled 200 windows along the genome with replacement. This was repeated a total of ten times for each test statistic, and we report the maximum P-value. Given the consistent difference between clusters (fig. 1 and supplementary figs. 2, 3, and 5, Supplementary Material online), normalized PC score could be used to attribute either Sp (low-diversity) or Sk (high-diversity) ancestry to each window (summary statistics for each window are given in supplementary table 2, Supplementary Material online). This was performed both for the subset of 57 samples (fig. 1c) and for all 161 samples (supplementary fig. 6, Supplementary Material online). Using different window sizes (150, 100, and 50 SNPs with overlap of 75, 50, and 25, respectively) yielded qualitatively the same results. Intermediate values in PC1 (between 0.25 and 0.75) were only observed in few, sequential windows where samples transitioned between clusters (supplementary fig. 3, Supplementary Material online). The only exception was sample JB1207, which we found to be diploid (for details see below).
Population Structure after Hybridization
To characterize the genome-wide distribution of ancestry components along the genome, we ran a hierarchical cluster analysis on the matrix containing ancestry information (Sp or Sk) for each window (columns) and strain (rows) using the R package Pvclust 2.0.0 (Suzuki and Shimodaira 2006). Pvclust includes a multiscale bootstrap resampling approach to calculate approximately unbiased probability values (P-values) for each cluster. We specified 1,000 bootstraps using the Ward method and a Euclidian-based dissimilarity matrix. The analysis was run both for the whole genome (fig. 1c) and by chromosome (fig. 1c, supplementary fig. 9, Supplementary Material online).
Phylogenetic Analysis of the Mitochondrial Genome
From the VCF file, we extracted mitochondrial variants for all 161 samples (Jeffares et al. 2015) and generated an alignment in .fasta format by substituting SNPs into the reference S. pombe 972 h− reference genome (ASM294v264) using the package vcf2fasta (https://github.com/JoseBlanca/vcf2fasta/, Accessed November 2015.). We excluded variants in mitochondrial regions with SVs inferred from long-reads. A maximum likelihood tree was calculated using RaxML (version 8.2.10-gcc-mpi) (Stamatakis 2014) with default parameters, GTRGAMMAI approximation, final optimization with GTR + GAMMA + I and 1,000 bootstraps. The final tree was visualized using FigTree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/, last accessed January 2019) (supplementary fig. 10, Supplementary Material online).
Time of Hybridization
Previous work (Jeffares et al. 2015) has shown that the time to the most recent common ancestor for 161 samples dates back to around 2,300 years ago. This defines the maximum boundary for the time of hybridization. We used the theoretical model by Janzen et al. (2018) to infer the age of the initial hybridization event. The model predicts the number of ancestry blocks and junctions present in a hybrid individual as a function of time and effective population size (Ne). First, we obtained an estimate of Ne using the multiple sequential Markovian coalescent (MSMC). We constructed artificial diploid genomes from strains with consistent clustering by ancestry (fig. 1c) and estimated change in Ne as function across time using MSMC 2-2.0.0 (Schiffels and Durbin 2014). In total we took four samples per group and produced diploid genomes in all possible six pairs for each group, except for one cluster that had only two samples (JB1205 and JB1206). Bootstraps were produced for each analysis, subsampling 25 genomic fragments per chromosome of 200 kb each. Resulting effective population size and time was scaled using the reported mutation rate of 2⋅10−10 mutations site−1 generation−1 (Farlow et al. 2015). Although it is difficult to be certain of the number of independent hybridization events, it is interesting to see that some clusters show similar demographic histories (supplementary fig. 24, Supplementary Material online). Regardless of the demographic history in each cluster, long-term Ne as estimated by the harmonic mean ranged between 1⋅105 and 1⋅109. Ne of the near-pure ancestral Sp and Sk cluster was 7⋅105 and 9⋅106, respectively. These estimates of Ne are consistent with previous reports of 1⋅107 (Farlow et al. 2015).
We then used a customized R script with the ancestral component matrix to estimate the number of ancestry blocks (Sp or Sk clade) (supplementary fig. 12, Supplementary Material online). We used the R script from Janzen et al. (2018), and ran the model in each sample and chromosome using: Ne = 1⋅106, r = number of genomic windows per chromosome, h0 = 0.298 (mean heterogenicity (h0) was estimated from the ancestral haplotype matrix) and c = 7.1, 5.6, and 4.1 respectively for chromosomes I, II, and III (values taken from Munz et al. [1989]) (supplementary fig. 13, Supplementary Material online). Given the large Ne, no changes in mean heterogenicity is expected over time after hybridization due to drift (the proportion of ancestral haplotypes Sp and Sk in hybrids, estimated as 2pq, where p and q are the proportion of each ancestral clade in hybrids). Accordingly, results did not change within the range of the large Ne values. For this analysis, samples with proportion of admixture lower than 0.1 were excluded. The analysis was repeated with different windows sizes (200, 100, and 50 SNPs per window).
Demographic Validation
To differentiate between possible historical scenarios underlying the observed pattern of ancestral divergence and introgression we performed individual based forward simulations using SLiM 3.2.1 (Haller and Messer 2019). These simulations were not intended to trace the “real” demographic history including estimates of split time, changes in effective population size, population structure after hybridization, or migration rates between subpopulations. Much rather, we contrasted two extremes of a continuum of possible scenarios: (1) a scenario of ancestral divergence in isolation follwed by a short, recent period of gene flow and (2) a scenario of divergence with continuous gene flow throughout the course of evolution. We asked the question which of the simulated scenarios may better recover the empirically observed deep divergence of ancestry components and their distribution in blocks along the genome. We parametrized the simulations with estimates from the literature including 1⋅106 for Ne, a mutation rate of 2⋅10−10 site−1 generation−1 (Farlow et al. 2015), an average recombination rate of 1⋅106 site−1 generation−1 (Munz et al. 1989), and a cloning rate of 0.95 generation−1 (equivalent to around 1 sexual cycle every 20 asexual generations). In order to reduce the computational effort, all parameters were scaled relative to an Ne of 1,000 as suggested in the SLiM manual. Simulations were divided in two parts: (1) simulations of single 15 kb genomic windows corresponding to ∼200 SNPs per window in the empirical data; (2) whole chromosome simulations with a fragment of 3 Mb corresponding to the size of S. pombe chromosome III. In both cases, we simulated divergence of two populations that were connected by different levels of symmetrical gene flow ranging from migration rates of 0 (complete isolation) to 1⋅10−4 (equivalent to 100 migrants per generation in a population of 1⋅106). To mimic the empirical difference in genetic diversity between populations, effective population sizes of the populations were set to 7⋅105 and 3⋅105, respectively. For the first part (single windows), a total of 100 replicate simulations were run for 30⋅Ne generations. Every 2 Ne generations 50 individuals were randomly sampled in total from both populations. At each time point, samples were processed in the same way as the empirical data per window estimating the percentage of variance explained by PC1, and Dxy between subgroups as inferred by normalized PC1 scores (supplementary fig. 7, Supplementary Material online). For the second part (whole chromosome), a total of 100 replicate simulations were run for 15⋅Ne generations after which a total of 50 genomes were randomly sampled from both populations. Simulated data were divided into genomic windows of 200 SNPs and processed in the same way as the empirical data (supplementary fig. 8, Supplementary Material online) for which the mean proportion of variance explained by PC1 was 64% and mean divergence between subpopulations (Dxy) was 0.0058. These values were only obtained in the simulations of a single genomic window under migration rates below 2⋅10−5 (equivalent to 20 migrants per generation in a population of 1⋅106 individuals). However, this migration rate was not enough to produce recombinant blocks in whole chromosome simulations. Blocks were only observed when migration rate exceeded 1⋅10−4 (100 migrants per generation). Yet, with this level of gene flow the proportion of variance explained by PC1 (around 25% in all simulations) and Dxy (below 0.0025 in all simulations) were significantly lower.
Phenotypic Variation and Reproductive Isolation
We sourced phenotypic data of 229 phenotypic measurements in the 161 strains including amino acid quantification on liquid chromatography (aaconc), growth and stress on solid media (smgrowth), cell growth parameters and kinetics in liquid media (lmgrowth), and cell morphology (shape1 and shape2) from Jeffares et al. (2015). Data on reproductive isolation measured as the percentage of viable spores in pairs of crosses were compiled from Jeffares et al. (2015) and Marsellach X (2019). A summary of all phenotypic measurements and reproductive data is provided in supplementary tables 4 and 5 (Supplementary Material online), respectively.
First, we normalized each phenotypic trait y using rank-based transformation with the relationship normal.y = qnorm (rank (y)/(1 + length (y))). We then conducted PCA on normalized values of all phenotypic traits using the R package missMDA 1.12 (Josse and Husson 2016). We estimated the number of dimensions for the PCA by cross-validation, testing up to 30 PC components and imputing missing values. In addition to PCA, decomposing variance across all traits, we examined the effect of admixture on each trait separately. Samples were divided into three discrete categories of admixture: two groups including samples with low admixture proportions (proportion of Sp or Sk clades higher than 0.9), and one for hybrid samples (proportion of Sp or Sk clades between 0.1 and 0.9). Significant differences in phenotypic distributions between groups were tested using Tukey–Kramer HSD as implemented in Stats 3.4.2 (Team 2014). Supplementary figure 14 (Supplementary Material online) shows the distribution of phenotypic values by admixture category for each trait. The number of traits with significant differences among groups was contrasted to values obtained by randomizing admixture categories without replacement (permutations of the Sp, Sk, or hybrid category). Observed values were contrasted with the distribution of the expected number of significant traits after running 10,000 independent permutations (supplementary fig. 15, Supplementary Material online).
Heritability
Heritability was estimated for all normalized traits using LDAK 5.94 (Speed et al. 2012), calculating independent kinship matrices derived from: (1) all SNPs and (2) ancestral haplotypes. Both SNPs and haplotype data were binary encoded (0 or 1). Jeffares et al. (2015) showed that heritability estimates between normalized and raw values are highly correlated (r = 0.69, P-value ≤ 2.2·10−16). Heritability estimated with SNP values were strongly correlated with those from ancestral haplotypes (r = 0.82, P-value ≤ 2.2·10−16). Heritability estimates and standard deviation for each trait for both SNPs and ancestral haplotypes are detailed in supplementary table 6 (Supplementary Material online).
Identification of Ploidy Changes
Schizosaccharomyces pombe is generally considered haploid under natural conditions. Yet, for two samples ancestry components did not separate on the principle component axis 1 (see above) for much of the genome. Instead, these samples were intermediate in PC1 score. A possible explanation is diploidization of the two ancestral genomes. To establish the potential ploidy of samples, we called variants for all 161 samples using the Illumina data from Jeffares et al. (2015). Cleaned reads were mapped with BWA (version 0.7.17-r1188) in default settings and variants were called using samtools and bcftools (version 1.8). After filtering reads with a QUAL score >25, the number of heterozygous sites per base per 20 kb window were calculated. Additionally the nuclear content (C) as measured by Jeffares et al. (2015) (Supplementary Table S4 in Jeffares et al. [2015]) were used to verify increased ploidy. Two samples showed high heterozygosity along the genome (JB1169 and JB1207) of which JB1207 for which data were available also showed a high C-value, suggesting that these samples are diploid (supplementary figs. 4 and 25, Supplementary Material online). In JB1207, heterozygosity varied along the genome, with regions of high- and low-diversity. Sample JB1110 showed genomic content similar to JB1207, but did not show heterozygosity levels above that of haploid strains, suggesting the increase in genome content occurred by autopolyploidization.
High-Weight Genomic DNA Extraction and Whole-Genome Sequencing
To obtain high-weight gDNA for long-read sequencing, we grew strains from single colonies and cultured them in 200 ml liquid Edinburgh Minimal Medium (EMM) at 32 °C shaking at 150 r.p.m. overnight. Standard media and growth conditions were used throughout this work (Hagan et al. 2016) with minor modifications: We used standard liquid EMM (Per liter: Potassium Hydrogen Phthalate 3.0 g, Na HPO4⋅2H2O 2.76 g, NH4Cl 5.0 g, d-glucose 20 g, MgCl2⋅6H2O 1.05 g, CaCl2⋅2H2O 14.7 mg, KCl 1 g, Na2SO4 40 mg, Vitamin Stock ×1,000 1.0 ml, Mineral Stock ×10,000 0.1 ml, supplemented with 100 mg l−1 adenine and 225 mg l−1 leucine) for the asexual growth. DNA extraction was performed with Genomic Tip 500/G or 100/G kits (Qiagen) following the manufacturer’s instruction, but using Lallzyme MMX for lysis (Flor-Parra et al. 2014, doi: 10.1002/yea.2994). For each sample, 20 kb libraries were produced that were sequenced on one SMRT cell per library using the Pacific Biosciences RSII Technology Platform (PacBio, CA). For a subset of eight samples, additional sequencing was performed using Oxford Nanopore (MinION). Sequencing was performed at SciLifeLab, Uppsala, Gene Center LMU, Munich and the Genomics & Bioinformatics Laboratory, University of York. We obtained on average 80× (SMRT) and 140× (nanopore) coverage for the nuclear genome for each sample (summary in supplementary table 7, Supplementary Material online).
Additionally, 2.5 µg of the same DNA was delivered to the SNP&SEQ Technology Platform at the Uppsala Biomedical Centre (BMC), for Illumina sequencing. Libraries were prepared using the TruSeq PCRfree DNA library preparation kit (Illumina Inc.). Sequencing was performed on all samples pooled into a single lane, with cluster generation and 150 cycles paired-end sequencing on the HiSeqX system with v2.5 sequencing chemistry (Illumina Inc.). These data were used for draft genome polishing (see below).
De Novo Assembly of Single-Molecule Read Data
De novo genomes were assembled with Canu 1.5 (Koren et al. 2017) using default parameters. BridgeMapper from the SMRT 2.3.0 package was used to polish and subsequently assess the quality of genome assembly. Draft genomes were additionally polishing using short Illumina reads, running four rounds of read mapping to the draft genome with BWA 0.7.15 and polishing with Pilon 1.22 (Walker et al. 2014). Summary statistics of the final assembled genomes are found in supplementary table 7 (Supplementary Material online). De novo genomes were aligned to the reference genome using MUMmer 3.23 (Kurtz et al. 2004). Contigs were classified by reference chromosome to which they showed the highest degree of complementarity. We used customized Python scripts to identify and trim mitochondrial genomes.
Structural Variant Detection
SVs were identified by a combination of a de novo and mapping approach. De novo genomes were aligned to the reference genome using MUMmer, and SVs were called using the function show-diff and the package SVMU 0.2beta (Khost et al. 2017). Then, raw long-reads were mapped to the reference genome with NGMLR and genotypes were called using the package Sniffles (Sedlazeck et al. 2018). We implemented a new function within Sniffles “forced genotypes,” which calls SVs by validating the mapping calls from an existing list of breaking points or SVs. This reports the read support per variant even down to a single read. We forced genotypes using the list of de novo breaking points to generate a multisample VCF file. SVs were merged using the package SURVIVOR (Jeffares et al. 2017) option merge with a threshold of 1 kbp and requiring the same type. In total, it resulted in a list of 1,498 SVs with 892 in common between the mapping and de-novo approaches (supplementary fig. 16, Supplementary Material online).
Within the 892 common variants we compared the accuracy of genotyping for each sample by comparing genotypes obtained from de novo genomes and by mapping reads the reference genome. Additionally, we compared genotypes in samples sequenced with both PacBio and MinION. In total we sequenced eight samples with both technologies. We found high consistency for variants called with both sequencing technologies and observed that allele frequencies were highly correlated (r = 0.98, P-value ≤ 2.2·10−16) (supplementary figs. 16–19, Supplementary Material online).
Only common SVs between the mapping and de-novo approach were considered, and variants with consistency below 50% were removed. We manually checked large SVs (larger than 10 kb) by comparing the list of SVs with the alignment of the de novo genomes to the reference genome from MUMmer. This resulted in a final data set including 832 SVs (supplementary table 8, Supplementary Material online).
Distribution of SVs around Developmentally Programed DNA DSB
We tested the association between DSB and SVs by comparing the physical genomic coordinates of the final list of SVs with DSB locations accessed from Fowler et al. (2014). Maintaining the same number of SVs per chromosome, we used a customized R script to randomize SV coordinates and measure the distances to the closest DSB. We counted the number of SVs present within different intervals of physical genetic distance ([0, 500), [500, 1000), [1000, 2000), [2000, 4000), [4000, 10000), [10000, 20000), [20000, 30000) bp). Empirically observed values were contrasted with the randomized distribution after running 10,000 independent permutations. P-values of differences between randomization and observed values were obtained from the fraction of expected values higher than the observed value from the original data (supplementary fig. 20, Supplementary Material online).
PCR Validation of Large SVs
To validate the frequency of large inversions and rearrangements observed from long-read data, we performed PCR verification over the breakpoints in the 57 nonclone samples. PCR was performed for both sides of the breakpoints, with a combination of one primer “outside” of the inversion and both primers “inside” the inversion (supplementary fig. 26, Supplementary Material online). PCR was performed on DNA using standard Taq polymerase, with annealing temperature at 59 °C. The primers used, the coordinates in the reference and the expected amplicon length are given in supplementary table 9 (Supplementary Material online).
Distribution of SVs in Ancestral Population—2D Folded Site Frequency Spectrum
We used the location of break points of SVs to identify whether a variant was located in the Sp or Sk genetic background in each sample. Ancestral haplotypes are difficult to infer in telomeric and centromeric regions given the low confidence in SNP calling in those regions, resulting in low percentage of variance explained by PC1. Thus SVs with break points in those regions were excluded from this analysis (19 SVs). SVs were grouped by ancestral group and allele frequencies were calculated for each ancestral population. We used these frequencies to build a 2D folded site frequency spectrum (2dSFS). In order to compare this 2dSFS, we repeated the analysis using SNP data from all 57 samples. Considering that the majority of identified SVs with long-reads were TEs, we also made use of LRT insertion–deletion polymorphism (indels) inferred from short-reads. For this additional data we produced a similar folded 2dSFS. LTR indel data were taken from Jeffares et al. (2015) and are listed in supplementary table 11 (Supplementary Material online).
Decay in LD
To contrast LD between alleles from alternative ancestral groups, we calculated LD between all described genomic windows within chromosomes (supplementary fig. 22, Supplementary Material online). For this analysis only hybrid samples were considered (strains with admixture proportion higher than 0.1). For each pair of windows, we polarized windows by ancestry (at a threshold of >0.7) and calculated standardized LD as the squared Pearson's correlation coefficient (R2) (Hill and Robertson 1968; Weir 1979). This measurement takes into consideration difference in allele frequencies. The expected value of R2 (E(R2)) can be approximated by Hill and Weir (1988):
where C corresponds to the product between the genetic distance (bp) and the population recombination rate (ρ) in n number of haplotypes sampled. The population recombination rate was calculated as: , where c is the recombination fraction between sites and Ne is the effective population size. We fitted a nonlinear model to obtain least squares estimates of ρ using a customized R script. The decay of LD with physical distance can be described with this model (Remington et al. 2001). LD values were grouped in three categories: (1) comparison between windows with high proportion (Sp > 0.7) of Sp ancestral group (Sp–Sp); (2) high proportion (Sk > 0.7) of Sk ancestral group (Sk–Sk); and (3) high proportion of opposite ancestral groups (Sp–Sk). (1) and (2) represent cases of positive AD, (3) will be denoted as negative AD. In order to reduce bias given the potential for secondary population structure (clusters in fig. 1c), the analysis was repeated using one random sample per cluster, resulting in similar general conclusions (supplementary fig. 27, Supplementary Material online).
Data Availability
Nanopore, single-molecule real-time sequencing data and de-novo genomes are available at NCBI Sequence Read Archive, BioProject ID PRJNA527756.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Fidel Botero-Castro, Ana Catalán, Sebastian Höhna, Ulrich Knief, Claire Peart, Joshua Peñalba, Ricardo Pereira, Matthias Weissensteiner, (LMU Munich) and S. Lorena Ament-Velásquez (Uppsala University) for providing valuable intellectual input on the various analyses, sharing scripts, and commenting on previous versions of this manuscript. We are further indebted to Bernadette Weissensteiner for extensive help with laboratory work and Saurabh Pophaly for bioinformatics support (LMU Munich). We further acknowledge support for data generation from the National Genomics Infrastructure, Uppsala, Sweden; the Gene Center, Munich, Germany; Sally James and Peter Ashton from the Bioscience Technology Facility, Department of Biology, University of York, United Kingdom and James Chong, Department of Biology, University of York, United Kingdom. The computational infrastructure was provided by the UPPMAX Next-Generation Sequencing Cluster and Storage (UPPNEX) project funded by the Knut and Alice Wallenberg Foundation and the Swedish National Infrastructure for Computing and the York Advanced Research Computing Cluster (YARCC), University of York, United Kingdom. This study was funded by LMU Munich to J.B.W.W. and NHGRI (UM1 HG008898) to F.J.S.
References
- Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R, et al. 2013. Hybridization and speciation. J Evol Biol. 262:229–246. [DOI] [PubMed] [Google Scholar]
- Abbott RJ, Barton NH, Good JM.. 2016. Genomics of hybridization and its evolutionary consequences. Mol Ecol. 2511:2325–2332. [DOI] [PubMed] [Google Scholar]
- Brown WRA, Liti G, Rosa C, James S, Roberts I, Robert V, Jolly N, Tang W, Baumann P, Green C, et al. 2011. A geographically diverse collection of Schizosaccharomyces pombe isolates shows limited phenotypic variation but extensive karyotypic diversity. G3 1:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Zhou J, Clark AG, Hartl DL, Ayroles JF.. 2013. Genetic incompatibilities are widespread within species. Nature 5047478:135–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currall BB, Chiangmai C, Talkowski ME, Morton CC.. 2013. Mechanisms for structural variation in the human genome. Curr Genet Med Rep. 12:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling TE, Smith GR, Brown WM.. 1989. Reproductive isolation and introgression between Notropis cornutus and Notropis chrysocephalus (family Cyprinidae): comparison of morphology, allozymes, and mitochondrial DNA. Evolution 433:620–634. [DOI] [PubMed] [Google Scholar]
- Eroukhmanoff F, Hermansen JS, Bailey RI, Sæther SA, Sætre G-P.. 2013. Local adaptation within a hybrid species. Heredity 1114:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria R, Navarro A.. 2010. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends Ecol Evol. 2511:660–669. [DOI] [PubMed] [Google Scholar]
- Farlow A, Long H, Arnoux S, Sung W, Doak TG, Nordborg M, Lynch M.. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 2012:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JA, Iida T, Takuno S, Sugino RP, Kado T, Kugou K, Mura S, Kobayashi T, Ohta K, Nakayama J, et al. 2014. Population genomics of the fission yeast Schizosaccharomyces pombe. PLoS One 98:e104241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1999. The genetical theory of natural selection: a complete variorum edition. Oxford University Press:Oxford. [Google Scholar]
- Flor-Parra I, Zhurinsky J, Bernal, M, Gallardo P and Daga RR. 2014. A Lallzyme MMX–based rapid method for fission yeast protoplast preparation. Yeast, 31: 61–66. doi:10.1002/yea.2994 [DOI] [PubMed] [Google Scholar]
- Fowler KR, Sasaki M, Milman N, Keeney S, Smith GR.. 2014. Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 2410:1650–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan IM, Carr AM, Grallert A, Nurse P.. 2016. Fission yeast: a laboratory manual. Cold Spring Harbor Laboratory Press. [Google Scholar]
- Haller BC, Messer PW.. 2019. SLiM 3: forward genetic simulations beyond the Wright–Fisher model. Mol Biol Evol. 363:632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A.. 1968. Linkage disequilibrium in finite populations. Theor Appl Genet. 386:226–231. [DOI] [PubMed] [Google Scholar]
- Hill WG, Weir BS.. 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol. 331:54–78. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Wood V, Fantes PA.. 2015. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics 2012:403–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AA, Rieseberg LH.. 2008. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol Syst. 391:21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Jiang Z-D, Suo F, Zheng J-X, He W-Z, Du L-L.. 2017. A large gene family in fission yeast encodes spore killers that subvert Mendel’s law. eLife 6:e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen T, Nolte AW, Traulsen A.. 2018. The breakdown of genomic ancestry blocks in hybrid lineages given a finite number of recombination sites: breakdown of ancestry blocks after hybridization. Evolution 724:735–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay P, Whibley A, Frézal L, Rodríguez de Cara MÁ, Nowell RW, Mallet J, Dasmahapatra KK, Joron M.. 2018. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr Biol. 2811:1839–1845. [DOI] [PubMed] [Google Scholar]
- Jeffares DC. 2018. The natural diversity and ecology of fission yeast. Yeast 353:253–260. [DOI] [PubMed] [Google Scholar]
- Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bähler J, Sedlazeck FJ.. 2017. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 8:14061.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares DC, Rallis C, Rieux A, Speed D, Převorovský M, Mourier T, Marsellach FX, Iqbal Z, Lau W, Cheng TMK, et al. 2015. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat Genet. 473:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josse J, Husson F.. 2016. missMDA: a package for handling missing values in multivariate data analysis. J Stat Softw. 70(1):1–31. [Google Scholar]
- Kelly FD, Levin HL.. 2005. The evolution of transposons in Schizosaccharomyces pombe. Cytogenet Genome Res. 110(1–4):566–574. [DOI] [PubMed] [Google Scholar]
- Khost DE, Eickbush DG, Larracuente AM.. 2017. Single-molecule sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Genome Res. 275:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrat’eva VI, Naumov GI.. 2001. The phenomenon of spore killing in Schizosaccharomyces pombe hybrids. Dokl Biol Sci. 379:385–388. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM.. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 275:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nat Genet. 481:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL.. 2004. Versatile and open software for comparing large genomes. Genome Biol. 52:R12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2017. Rapid hybrid speciation in Darwin’s finches. Science 359(6372): 224–228. [DOI] [PubMed] [Google Scholar]
- Leducq J-B, Nielly-Thibault L, Charron G, Eberlein C, Verta J-P, Samani P, Sylvester K, Hittinger CT, Bell G, Landry CR.. 2016. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat Microbiol. 1:15003.. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol Evol (Amst) 205:229–237. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2007. Hybrid speciation. Nature 4467133:279–283. [DOI] [PubMed] [Google Scholar]
- Marsellach X. 2019. A non-Lamarckian model for the inheritance of the epigenetically-coded phenotypic characteristics: a new paradigm for Genetics, Genomics and, above all, Ageing studies. bioRxiv 492280; doi: https://doi.org/10.1101/492280 [Google Scholar]
- McDonald MJ, Rice DP, Desai MM.. 2016. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 5317593:233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JI, Marques DA, Mwaiko S, Wagner CE, Excoffier L, Seehausen O.. 2017. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun. 8:14363.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1932. Some genetic aspects of sex. Am Nat. 66703:118–138. [Google Scholar]
- Munz P, Wolf K, Kohli J, Leupold U.. 1989. Genetics overview In: Nasim AM, Young P, Johnson BF, eds. Molecular biology of the fission yeast. p. 1–30. Academic Press Inc.: California. [Google Scholar]
- Naumov GI, Kondratieva VI, Naumova ES.. 2015. Hybrid sterility of the yeast Schizosaccharomyces pombe: genetic genus and many species in statu nascendi? Microbiology 842:159–169. [PubMed] [Google Scholar]
- Nei M, Li WH.. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 7610:5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto Feliner G, Álvarez I, Fuertes-Aguilar J, Heuertz M, Marques I, Moharrek F, Piñeiro R, Riina R, Rosselló JA, Soltis PS, et al. 2017. Is homoploid hybrid speciation that rare? An empiricist’s view. Heredity 1186:513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis BPS, Tusso S, Bjerling P, Stångberg J, Wolf JBW, Immler S.. 2018. Repeated evolution of self-compatibility for reproductive assurance. Nat Commun. 9(1):1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte AW, Sheets HD.. 2005. Shape based assignment tests suggest transgressive phenotypes in natural sculpin hybrids (Teleostei, Scorpaeniformes, Cottidae). Front Zool. 21:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuckolls NL, Bravo Núñez MA, Eickbush MT, Young JM, Lange JJ, Yu JS, Smith GR, Jaspersen SL, Malik HS, Zanders SE.. 2017. wtf genes are prolific dual poison-antidote meiotic drivers. eLife 6:e26033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Barrientos D, Engelstädter J, Rieseberg LH.. 2016. Recombination rate evolution and the origin of species. Trends Ecol Evol. 313:226–236. [DOI] [PubMed] [Google Scholar]
- Payseur BA, Rieseberg LH.. 2016. A genomic perspective on hybridization and speciation. Mol Ecol 25(11):2337–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E. 2016. A shortcut to a species. Science 3546314:818. [DOI] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, et al. 2018. Genome evolution across 1, 011 Saccharomyces cerevisiae isolates. Nature 5567701:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfennig KS, Kelly AL, Pierce AA.. 2016. Hybridization as a facilitator of species range expansion. Proc R Soc B. 2831839:20161329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelstra JW, Vijay N, Bossu C, Lantz H, Ryll B, Müller I, Baglione V, Unneberg P, Wikelski M, Grabherr M, et al. 2014. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 3446190:1410–1414. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. [Google Scholar]
- Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES.. 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci U S A. 9820:11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runemark A, Trier CN, Eroukhmanoff F, Hermansen JS, Matschiner M, Ravinet M, Elgvin TO, Sætre G.. 2018. Variation and constraints in hybrid genome formation. Nat Ecol Evol. 23:549–556. [DOI] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D.. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 5077492:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S, Durbin R.. 2014. Inferring human population size and separation history from multiple genome sequences. Nat Genet. 468:919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Cui R, Powell DL, Rosenthal GG, Andolfatto P.. 2016. Ancient hybridization and genomic stabilization in a swordtail fish. Mol Ecol. 2511:2661–2679. [DOI] [PubMed] [Google Scholar]
- Schumer M, Rosenthal GG, Andolfatto P.. 2014. How common is homoploid hybrid speciation? Perspective. Evolution 686:1553–1560. [DOI] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC.. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 156:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehausen O. 2004. Hybridization and adaptive radiation. Trends Ecol Evol. 194:198–207. [DOI] [PubMed] [Google Scholar]
- Singh G, Klar AJ.. 2002. The 2.1-kb inverted repeat DNA sequences flank the mat2, 3 silent region in two species of Schizosaccharomyces and are involved in epigenetic silencing in Schizosaccharomyces pombe. Genetics 162:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE.. 2009. The role of hybridization in plant speciation. Annu Rev Plant Biol. 601:561–588. [DOI] [PubMed] [Google Scholar]
- Speed D, Hemani G, Johnson MR, Balding DJ.. 2012. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet. 916:1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenkamp ET, Wingfield MJ, McTaggart AR, Wingfield BD.. 2018. Fungal species and their boundaries matter—definitions, mechanisms and practical implications. Fungal Biol Rev. 32:104–116. [Google Scholar]
- Stukenbrock EH. 2016. The role of hybridization in the evolution and emergence of new fungal plant pathogens. Phytopathology 1062:104–112. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H.. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2212:1540–1542. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DJ, Hebert PD.. 1993. Habitat-dependent hybrid parentage and differential introgression between neighboringly sympatric Daphnia species. Proc Natl Acad Sci U S A. 9015:7079–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R.C., 2014. R: A language and environment for statistical computing. [Google Scholar]
- Teresa Avelar A, Perfeito L, Gordo I, Godinho FM.. 2013. Genome architecture is a selectable trait that can be maintained by antagonistic pleiotropy. Nat Commun. 4:2235. [DOI] [PubMed] [Google Scholar]
- Turner LM, Harr B.. 2014. Genome-wide mapping in a house mouse hybrid zone reveals hybrid sterility loci and Dobzhansky–Muller interactions. eLife 3, e02504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford AD, Streisfeld MA, Lowry DB, Friedman J.. 2015. Genomic studies on the nature of species: adaptation and speciation in Mimulus. Mol Ecol. 2411:2601–2609. [DOI] [PubMed] [Google Scholar]
- Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, Wolf JBW.. 2016. Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun. 7, 13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 911:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 72:256–276. [DOI] [PubMed] [Google Scholar]
- Weir BS. 1979. Inferences about linkage disequilibrium. Biometrics 351:235–254. [PubMed] [Google Scholar]
- Wellenreuther M, Bernatchez L.. 2018. Eco-evolutionary genomics of chromosomal inversions. Trends Ecol Evol. 336:427–440. [DOI] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream M-A, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. 2002. Erratum: the genome sequence of Schizosaccharomyces pombe. Nature 4156874:871–880. [DOI] [PubMed] [Google Scholar]
- Zanders SE, Eickbush MT, Yu JS, Kang J-W, Fowler KR, Smith GR, Malik HS.. 2014. Genome rearrangements and pervasive meiotic drive cause hybrid infertility in fission yeast. eLife 3, e02630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Gogarten SM, Lawrence M, Stilp A, Conomos MP, Weir BS, Laurie C, Levine D.. 2017. SeqArray—a storage-efficient high-performance data format for WGS variant calls. Bioinformatics 3315:2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS.. 2012. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2824:3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Nanopore, single-molecule real-time sequencing data and de-novo genomes are available at NCBI Sequence Read Archive, BioProject ID PRJNA527756.