

Abstract
Sporadic colon cancer accounts for approximately 80% of colorectal cancer (CRC) with high incidence in Western societies strongly linked to long-term dietary patterns. A unique mouse model for sporadic CRC results from feeding a purified rodent Western-style diet (NWD1) recapitulating intake for the mouse of common nutrient risk factors each at its level consumed in higher risk Western populations. This causes sporadic large and small intestinal tumors in wild-type mice at an incidence and frequency similar to that in humans. NWD1 perturbs intestinal cell maturation and Wnt signaling throughout villi and colonic crypts and decreases mouse Lgr5hi intestinal stem cell contribution to homeostasis and tumor development. Here we establish that NWD1 transcriptionally reprograms Lgr5hi cells, and that nutrients are interactive in reprogramming. Furthermore, the DNA mismatch repair pathway is elevated in Lgr5hi cells by lower vitamin D3 and/or calcium in NWD1, paralleled by reduced accumulation of relevant somatic mutations detected by single-cell exome sequencing. In compensation, NWD1 also reprograms Bmi1+ cells to function and persist as stem-like cells in mucosal homeostasis and tumor development. The data establish the key role of the nutrient environment in defining the contribution of two different stem cell populations to both mucosal homeostasis and tumorigenesis. This raises important questions regarding impact of variable human diets on which and how stem cell populations function in the human mucosa and give rise to tumors. Moreover, major differences reported in turnover of human and mouse crypt base stem cells may be linked to their very different nutrient exposures.
A Western-style purified diet has profound and differential effects on Lgr5hi and Bmi1+ intestinal stem cells involving their transcriptional reprogramming and mutation accumulation. This results in reduced Lgr5hi cell and augmented Bmi1+ cell contribution to mucosal homeostasis and tumorigenesis.
Introduction
Sporadic colorectal cancer (CRC) is by far the most common form of the disease, accounting for approximately 80% of cases in Western high-risk societies. The incidence of sporadic CRC is tightly linked to long-term dietary patterns of the population (1,2). This can be modeled in the mouse by feeding NWD1, a purified rodent Western-style diet formulated to recapitulate intake levels for the mouse of common nutrients each at its level linked to higher CRC risk in the human (3–7). As a result, the diet is highly protumorigenic, accelerating and amplifying tumor phenotype in mouse genetic models, regardless of genetic etiology or aggressiveness (8–11). Most important, NWD1 fed to wild-type mice causes sporadic small and large intestinal tumors that reflect incidence, frequency and lag of human sporadic colon cancer (i.e. 25% of the mice develop one to two tumors over 2/3 of their lifespan) (7,12,13). Therefore, this is a unique mouse model of sporadic intestinal cancer. Thus, how NWD1 alters mucosal homeostasis and sporadic intestinal tumorigenesis provides fundamental insight into the etiology and mechanisms driving one of the most frequent cancers in human populations.
Field effects in a tissue are associated with probability of eventual tumor development (14). In the mucosa of NWD1 fed mice, there are multiple such field effects, including alterations in intestinal epithelial cell maturation; altered balance among expression of lineage-specific markers; ectopic expression of Paneth cell markers into the villi and colon; elevated Wnt signaling throughout small intestinal villi and colonic crypts (12,15). In mice maintained under standard conditions of mouse husbandry, Lgr5hi crypt base columnar (CBC) cells are the cycling stem cell population maintaining homeostasis and capable of initiating tumors (16). However, contrary to expectations, lineage tracing and tumorigenic potential of the Lgr5hi stem cells were reduced in NWD1 fed mice (17,18). An important contributor to this was lower vitamin D3 in the NWD1 because inactivation of the vitamin D receptor (Vdr) specifically in Lgr5+ CBC cells recapitulated the effects of feeding NWD1 on decreasing lineage tracing from this cell population (17,18). This conclusion is supported strongly and independently by the Lgr5hi cell stem cell signature, which showed expression of the Vdr is a robust marker of Lgr5hi cells, but is downregulated in their immediate Lgr5lower daughter cells that had lost capacity for self-renewal. This indicates a necessary role for Vdr signaling in Lgr5hi stem cell functions (19). The importance of this derives from the fact that studies of intestinal stem cells almost universally use mice fed chow diets. In mice fed these diets, the level of serum 1,25(OH)2 D are well above even the highest levels of the broad range that characterizes the human population (17,18). This raises the fundamental issue of which and how intestinal stem cells function under conditions that better mimic that of the human, especially those at higher risk for development of sporadic CRC.
Here we establish that feeding NWD1, in reducing stem cell functions of Lgr5hi CBC cells and their number, extensively reprograms transcription in these cells, and that nutrients are interactive in these effects. Among alterations induced by feeding the NWD1, levels of vitamin D3 and/or calcium have a major impact on the DNA mismatch repair pathway. Single-cell DNA whole exome sequencing showed there is a parallel altered accumulation of relevant mutations in Lgr5hi CBC cells. Moreover, in compensation for the reduction in Lgr5hi stem cell number and function, Bmi1+ cells are recruited by the NWD1, and persist, to function as stem-like cells in both homeostasis and tumorigenesis. It is clear that experimentally induced damage to Lgr5hi cells (e.g. radiation, chemical insult, diphtheria toxin) can recruit other cell populations to compensate (20). However, since diet is variable across the human population and changes daily for an individual, the major impact of the NWD1 on which and how cells function as stem cells in homeostasis and tumorigenesis has unique significance. This includes understanding the potential complexity of stem cell contribution to mucosal homeostasis in the human: why mouse and human crypt base stem cells exhibit very different kinetics in their turnover (21), and which cells may serve as the cell of origin for human sporadic CRC.
Materials and methods
Mice and diets
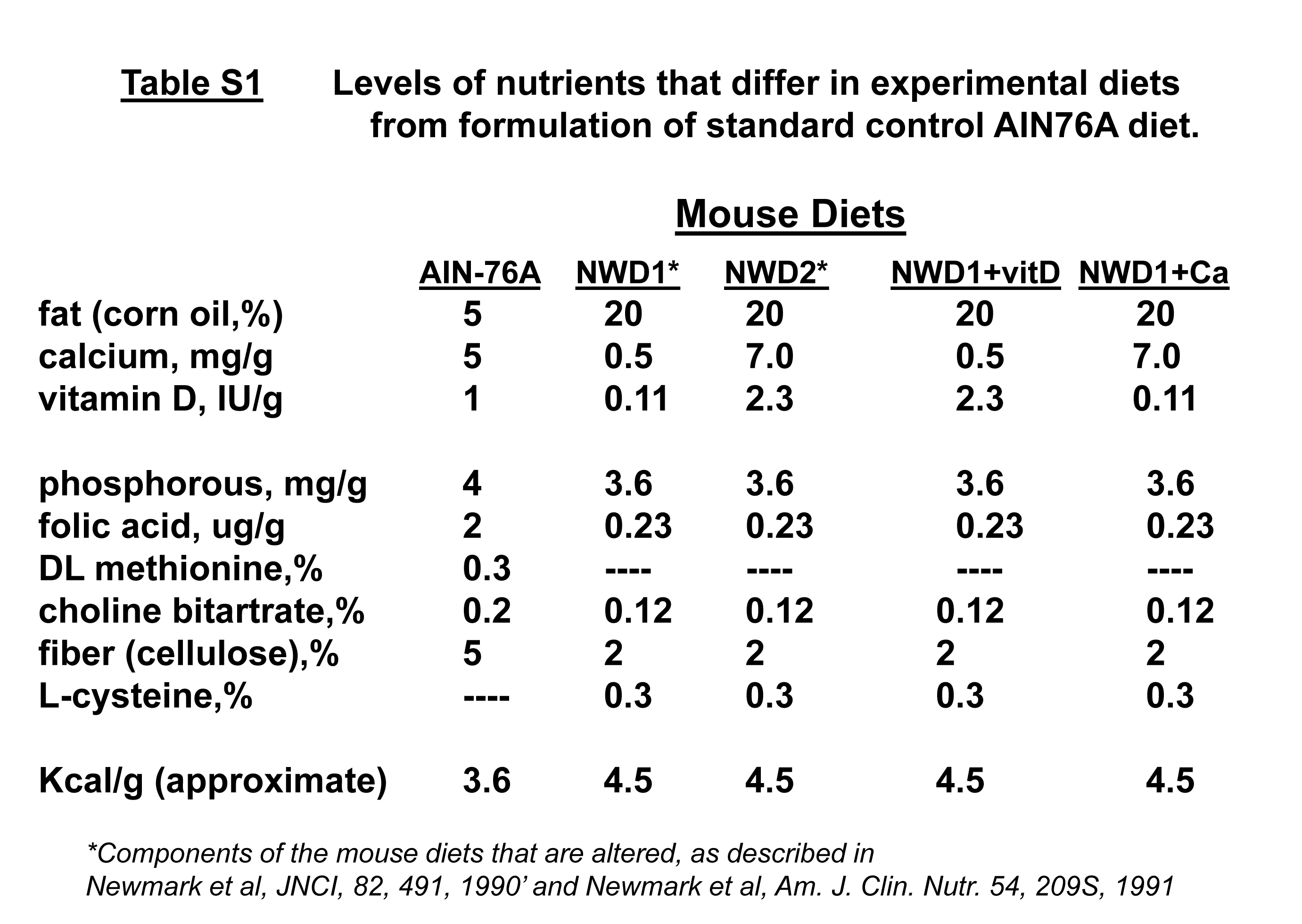
Mouse strains used were as follows: Lgr5tm1(cre/ERT2)Cle/J (Jax #008875, here in Lgr5EGFPcreERT2 (22)); Bmi1tm1(cre/ERT)Mrc/J (Jax #010531, here in Bmi1creERT2 (23)); Apc580S (Apcloxp/+ (24)); B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Jax #007914, here in Rosa26tom); Vdrloxp/loxp (25). All mice were on a C57BL/6 background. They were housed in a barrier facility of the Albert Einstein College of Medicine. Mice were fed mouse chow (PicoLab 5058) during strain maintenance and crosses. Experimental mice of correct genotype were weaned to purified diets at 3 weeks of age, maintained on these diets and sacrificed at 3 months of age. Diets (Supplementary Table S1, available at Carcinogenesis online) were from Research Diets (formulations at (http://www.researchdiets.com/)). Control purified diet was AIN76A (American Institute of Nutrition 76A). Changes to AIN76A formulation for NWD1 are reported (3,4,6) and Supplementary Table S1, available at Carcinogenesis online. This altered formulation for NWD1 was based on nutrient density to recapitulate intake levels of specific nutrients characteristic of populations in Western developed countries that have a high incidence of CRC. These changes include higher fat, lower vitamin D3, calcium, methyl donors and fiber in NWD1. In NWD2, vitamin D3 and calcium levels are increased back to higher levels, with levels of all other nutrients remaining as they are in NWD1 (Supplementary Table S1, available at Carcinogenesis online). Induction of creERT2 activity was by a 100 µl intraperitoneal injection of 1 mg freshly prepared Tamoxifen (TAM) in corn oil. All experiments were approved by the Albert Einstein College of Medicine IACUC.
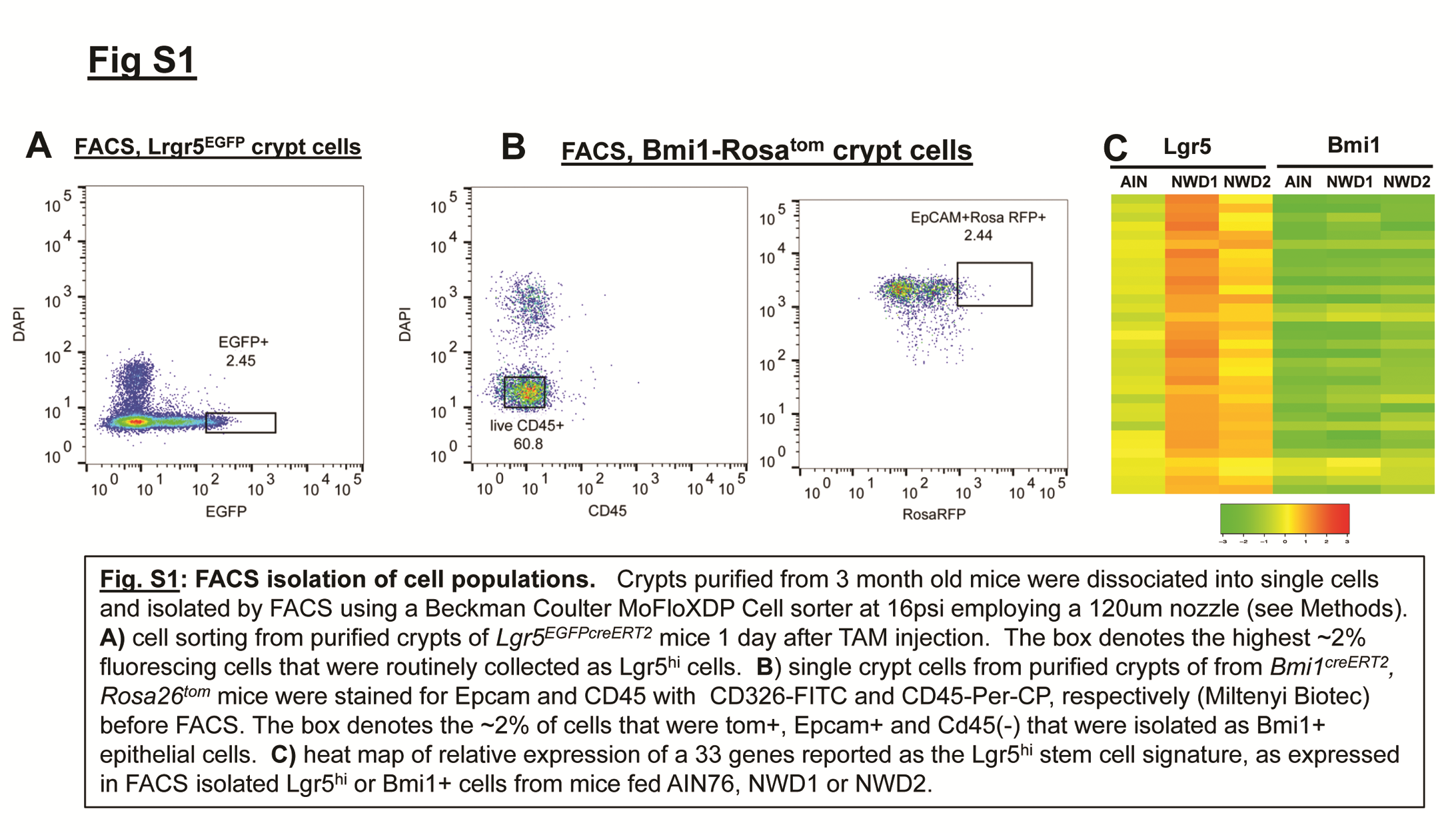
Crypt purification for stem cell isolation was as reported (26), with modifications to shorten the procedure (27). Isolated crypts were dispersed into single-cell suspensions and cells sorted with a Beckman Coulter MoFloXDP Cell sorter at 16 psi employing a 120 µm nozzle. Lgr5hi cells were the highest 2–3% of the total fluorescent cell population from Lgr5EGFP-creERT2 mice (Supplementary Figure S1, available at Carcinogenesis online). Bmi1creERT2 marked cells were those that exhibited high endogenous red fluorescence from BmicreERT2, Rosa26tom mice and that were also Epcam+ and CD45 (−) (Supplementary Figure S1, available at Carcinogenesis online). During sorting, single cells were collected individually into 192 microtubes and then frozen for DNA isolation and single-cell sequencing. The remaining cells were sorted and collected in bulk for RNA sequencing (RNAseq).
Marking of cells with Bmi1creERT2
Bmi1 creERT2 , Rosa26 tom mice were weaned to specific diets. After 3 months, the mice were given a single injection of Tamoxifen as mentioned previously and sacrificed at specified times thereafter. Dissected intestinal tissue was used for isolation of red fluorescent cells by fluorescence-activated cell sorting (FACS) or tissue was frozen in Optimal Cutttng Temperature compound (Tissue-Tek, Sakura Finetek, USA) compound for analysis of sections by fluorescence microscopy. Frozen sections were counter stained with 4′,6-diamidino-2-phenylindole for identification of nuclei before observation.
RNA sequencing
High-quality total RNAs (1–10 ng, RIN ≥ 9) from Lgr5hi and Bmi1creERT2 marked cells were prepared from cells isolated by FACS. The RNA was converted to cDNAs, using a SMART-Seq v4 ultra low input RNA kit (Clontech). Details of probe preparation, RNAseq and data analysis are in Supplementary Methods, available at Carcinogenesis online. The accession numbers for the Lgr5hi and the Bmi1+ cell RNAseq data are, respectively, GSE 102629 and 102630.
Single-cell exome sequencing
Single-cell whole genome amplification for exome sequencing, sequencing methodology and the rigorous data analysis pipeline are described in detail in Supplementary Methods, available at Carcinogenesis online. The accession number for the raw exome sequencing data is SRP106917.
Tumorigenesis assay
Mice were bred to be heterozygous for either the Lgr5EGFPcreERT2 or Bmi1creERT2 allele, and also heterozygous for the Apc580Sloxp allele. The mice were weaned to specific diets. After 3 months, each mouse received a single Tamoxifen injection as mentioned previously. This strategy of conditional inactivation of only one Apc allele at 3 months more closely reflects sporadic tumor development over time that takes place in the human rather than simultaneous genetic inactivation of both alleles. This permits tumors to develop with sequential inactivation of the second Apc allele, and other potential influences of the diet over a lengthy period of time. Therefore, after TAM injection, each mouse continued on its respective diet for an additional 6 months until sacrifice. At sacrifice, the intestine was dissected, visible tumors removed, fixed in formalin, paraffin embedded and hematoxylin/eosin stained sections examined to confirm histopathology. The remaining intestine was prepared as a Swiss roll, similarly fixed and embedded, and stained sections examined for histopathology. The total number of all tumors was recorded.
Statistical analyses
(i) Two-tailed Student’s t-test was used to compare mean values of data; (ii) gene set enrichment analysis (GSEA) for RNAseq data was done using Desktop GSEA(v3.0); (iii) mutational signature assay for single-cell sequencing was downloaded from https://www.mathworks.com/matlabcentral/fileexchange/38724-wtsi-mutational-signature-framework; (iv) Poisson regressions for tumor development were performed by R (function glm); and (v) quantitative RT-PCR analysis employed the comparative ΔΔCt method (the delta CT of each targeted gene and Gapdh). Statistical analysis among dietary groups was assessed by two-way ANOVA with two factors: dietary groups and target genes.
Results
Bmi1 marked cells respond to feeding NWD1
We established that feeding NWD1 decreased both the number of Lgr5hi cells and lineage tracing from the Lgr5hi cells at the bottom of the crypt (17,18). Feeding NWD1 also decreased the number of tumors that developed on targeted inactivation of an Apc allele in Lgr5+ cells (17,18). Since the mucosa is maintained, and NWD1 is protumorigenic, Bmi1+ cells were investigated as potentially mobilized in compensation. Bmi1creERT2, Rosa26tom mice fed purified diets from weaning (Supplementary Table S1, available at Carcinogenesis online) were injected with TAM at 3 months to visualize progeny of Bmi1+ cells. One day post-TAM, there were few red fluorescent epithelial cells in each crypt-villus. These could be identified as flat, broad bands localized by 4′,6-diamidino-2-phenylindole staining to the epithelial cell layer (Figure 1Aa, b, c; yellow arrows). These were readily distinguished from positive immune cells in the intra-villus regions derived from Bmi1 expressing bone marrow progenitors. As reported, Bmi1+ epithelial cells were not confined to the crypt base (19,23,28). At 3 and 5 days post-TAM, this was similar for mice fed AIN76A (Figure 1Aa, d, g). However, at 5 days, there was a small cluster of epithelial cells (Figure 1Ag) from a burst of division of a Bmi1+ cell, as reported for mice fed standard chow diets (19,28). In mice fed NWD1, by 3 day post-TAM, there were many more Bmi1+ cells and multiple clusters (Figure 1Ae). This was a 3-fold expansion of cells from the Bmi1+ compartment in NWD1 versus AIN76A fed mice (Figure 1B, P = 5 × 10−15). By 5 days, many crypt-villi in mice fed NWD1 were completely populated by Bmi1+ cells (Figure 1Ah). Feeding NWD2 (NWD1 + higher vitamin D3 and calcium, Supplementary Table S1, available at Carcinogenesis online) partially eliminated the mobilization of Bmi1+ cells caused feeding NWD1 (Figure 1Ac, f, i).
Figure 1.

Mobilization and persistence of Bmi1+ cells in mice fed different diets. (A) Red fluorescent intestinal epithelial cells derived from Bmi1+ cells in Bmi1creERT2, Rosa26tom mice fed different diets from weaning for 3 months, and injected with TAM 1, 3 or 5 days before sacrifice. Yellow arrows: examples of marked epithelial cells; yellow dotted boxes: examples of lineage tracing from BmicreERT2 marked cells. (B) Quantification of the number of marked Bmi1+ epithelial cells per crypt-villus in mice fed AIN76A or NWD1 from weaning for 3 months, and sacrificed 3 days following TAM injection: n = 77 and 67 crypt-villi scored for marked epithelial cells for mice fed AIN76A and NWD1 diets, respectively, for random fields of three mice in each dietary group. ****P = 5 × 10−15, Student’s t-test. (Ca, b) mice were fed AIN76A or NWD1 diet from weaning, injected with TAM at 3 months, and each mouse continued on its respective diet for an additional 33 days. (Cc, d) mice fed NWD1 from weaning, injected with TAM at 3 months, continued on NWD1 for 30 additional days and then switched to AIN76A for an additional 30 days (Cc) or continued on NWD1 for an additional 30 days (Cd). (D) Quantification of the % villi under each condition that exhibit lineage tracing of the Bmi1creERT2 marked cells. *P = 0.06; **P = 0.015, Student’s t-test.
Longer term activity of Bmi1+ derived cells marked by the pulse of TAM was determined. At 33 or 60 days after TAM injection at 3 months, all mice had labeled immune cells in the intra-villus regions (e.g., Figure 1Ca–d). This established efficacy of TAM in promoting recombination. Thirty-three days after TAM injection of mice fed AIN76A or NWD1, a similar percentage of villi in mice fed AIN76A or NWD1 exhibited regions of lineage tracing from the Bmi1+ targeted cells (15–20%; Figure 1Ca, b, D). This demonstrated persistence of stem/progenitor function of cells marked by the Bmi1-driven cre recombinase. However, when mice fed NWD1 were switched to AIN76A for an additional 30 days (i.e. 63 days total from TAM injection – 5 months overall from weaning), villi exhibiting lineage tracing decreased to 5%. But when the mice were continued to be fed NWD1 for the additional 30 days, villi with lineage tracing remained higher, at 21% (Figure 1Cc, D). The difference between the groups was highly significant (Figure 1D, P = 0.015). Thus, although the progenitor cells marked by the pulse of TAM turnover substantially, with presumably new cells recruited during the subsequent two months in mice fed NWD1, continuous exposure of the mice to NWD1 over this period leads to greater retention of the initially marked progenitors. When present, lineage tracing was seen either from the crypt base or in shorter bursts from further up in the villi (Figure 1Ca–d).
Following inactivation of a floxed Apc allele specifically in Lgr5+ cells there was greatly decreased tumor number in mice fed NWD1 relative to those fed AIN76A (Figure 2A, top; P = 0.001, Poisson distribution; data from ref. (17)). However, when the inactivating Apc mutation was targeted to Bmi1+ cells (i.e. Bmi1creERT2, Apcloxp/+ mice), the result was complementary. In mice fed NWD1, there was elevated tumor development compared with mice fed AIN76A or NWD2 mice (Figure 2B, bottom; P = 0.0005, Poisson distribution). The differential dietary effect on tumorigenesis in Lgr5+ compared with Bmi1+ targeted cells was highly significant (P = 10−6). Tumors arising from the Bmi1+ targeted cells were tubular and villous adenomas (Figure 2B). Thus, the ability of Lgr5+ or Bmi1+ cells to lineage trace under different dietary conditions was directly paralleled by their ability to cause tumors upon introduction of an initiating mutation.
Figure 2.

A) Complementary effects of diet on tumor development when targeting Lgr5hi versus Bmi1+ cells. (A) Lgr5EGFPcreERT2, Apcloxp/+ (top) or Bmi1creERT2, Apcloxp/+ mice (bottom) were fed different diets from weaning for 3 months, then received a single injection of TAM, following which each mouse continued on its respective diet for an additional 6 months (i.e., until 9 months of age) and then sacrificed. On dissection, visible tumors were scored, Swiss rolls of the small intestine prepared, fixed in formalin and paraffin embedded. Sections of these Swiss rolls were examined microscopically for scoring of smaller tumors. Total tumor number for each mouse is shown. For Lgr5 targeted cells (top, data from Peregrina et al. (17)), there were five mice in each dietary group; for Bmi1 targeted cells (bottom), there were 12, 11 and 8 mice in the AIN76A, NWD1 and NWD2 dietary groups, respectively. P = 0.001 and 0.0005, for differences in tumor number for Lgr5 and Bmi1 targeted cells, respectively, by Poisson distribution and application of a general linear model. For the different and complementary results of dietary effect on tumor number for the two different cell populations, P = 1.3 × 10−6. B) Example of tumor histopathology.
Diet profoundly altered RNA expression profile of Lgr5hi and Bmi1+ cells
Lgr5hi or Bmi1+ cells isolated by FACS (Supplementary Figure S1A and B; Supplementary Methods, available at Carcinogenesis online) of 3 month old mice fed different diets from weaning were analyzed by RNAseq. Thirty-three genes comprising a robust signature of the Lgr5hi cell population (19) were expressed under all dietary conditions, but each at considerably lower levels in Bmi1+ compared with Lgr5hi sorted cells (Supplementary Figure S1C, available at Carcinogenesis online). This documents distinctly altered gene expression profiles for cells of the two populations, consistent with their distinct distributions in the intestinal crypt-villus architecture.
Principal component analysis (PCA) of the RNAseq data of Lgr5hi cells from mice fed control AIN76A diet (gene list Supplementary Table S2, available at Carcinogenesis online) showed clustering of three independent biological replicates (Figure 3A). This differed substantially for mice fed NWD1, demonstrating a major dietary impact (Figure 3A). There is a key role for lower vitamin D3 and calcium in NWD1 in altering cell maturation, Wnt signaling, reduced lineage tracing from Lgr5hi stem cells, and eventual tumor development (12,15,17). Therefore, the impact of these nutrients on Lgr5hi cell expression signature was investigated. Feeding NWD2, elevating both vitamin D3 and calcium levels in NWD1 (Supplementary Table S1, available at Carcinogenesis online), substantially shifted clustering by PCA toward the AIN76A pattern for Lgr5hi cells (Figure 3A). Incomplete restoration to the pattern in mice fed AIN76A was expected since higher fat, and lower methyl donors and fiber in NWD1 relative to AIN76A were still present in NWD2 ((12) and Supplementary Table S1, available at Carcinogenesis online). Clustering was distinct for Lgr5hi cells from mice fed diets in which only vitamin D3 or only calcium were elevated in NWD1 (Figure 3A). Each of these was distinct from cells from NWD2 fed mice for which both nutrients were elevated, or when only vitamin D3 was lowered in AIN76A (Figure 3A). Furthermore, conditional Vdr inactivation targeted to Lgr5+ cells from mice fed AIN76A was distinct from lowering vitamin D3 in the diet (Figure 3A). Thus, there was profound sensitivity of Lgr5hi cells to the nutrient complement fed the mice and to Vdr-mediated signaling. Moreover, levels of different nutrients were interactive in Lgr5hi cell transcriptional reprogramming. RNAseq data for isolated Bmi1+ cells from AIN76A fed mice also clustered together, feeding NWD1 again shifted this clustering, and repletion of vitamin D3 and calcium by feeding NWD2 shifted clustering of Bmi1+ cells back toward the control pattern (Figure 3B). For all diets tested, Lgr5hi cell and Bmi1+ cell expression patterns were distinctly different (Figure 3B).
Figure 3.

Transcriptional profiles of Lgr5hi and Bmi1+ cells are profoundly influenced by diet. Lgr5EGFP,creERT2 or Bmi1creERT2, Rosa26tom mice were randomized to different diets at weaning, the Bmi1 group given a single injection of TAM at 3 months, then all mice sacrificed 2 days later and Lgr5hi or Bmi1creERT2 marked cells isolated by FACS from purified intestinal crypts (Supplementary Figure S1). RNAs from these cells were then isolated and used for RNAseq. n = 3 mice per group (data in Supplementary Table S2, diets defined in Supplementary Table S1). An additional group was RNAseq analysis for Lgr5hi cells from Lgr5EGFP,creERT2, Vdrloxp/loxp mice fed AIN76A control diet from weaning and given a single injection of Tamoxifen to genetically inactivate the Vdr gene in Lgr5hi cells 3 days before sacrifice at 3 months of age. PCA of RNAs from mice fed different diets are shown: (A) Lgr5hi cells; (B) Bmi1creERT2 marked cells. For comparison, in (B), PCA for Lgr5hi cells is shown to be distinct from Bmi1creERT2 marked cells for each of AIN76A, NWD1 and NWD2 diets.
Pathways altered by diet
GSEA identified 46 functional gene groups significantly altered in Lgr5hi cells from mice fed NWD1 compared with those fed AIN76A (Supplementary Table S3, available at Carcinogenesis online), emphasizing the major impact and complexity of nutrients in programming these cells. Of these, the DNA mismatch repair (MMR) pathway is fundamental in defining risk for both inherited and sporadic CRC (29,30). This functional group was significantly elevated in Lgr5hi cells from mice fed NWD1 compared with those fed AIN76A [Figure 4A; P = 0.003, false discovery rate (FDR) = 0.021, Supplementary Table S4, available at Carcinogenesis online]. Moreover, Msh2, rate limiting in MMR, was in the leading edge of the GSEA (Figure 4A, Supplementary Table S4, available at Carcinogenesis online). Lgr5hi cells isolated from mice fed NWD2 in which both vitamin D3 and calcium were elevated compared with NWD1 showed a significant reversal of this elevation (Figure 4B; P = 0.02; FDR = 0.06). This reversal was also present when either vitamin D3 (Figure 4C; P < 0.001, FDR = 0.004) or calcium (Figure 4D; P = 0.002, FDR = 0.005) were elevated in NWD1 individually. This indicates that MMR-upregulation is vitamin D/calcium dependent. The increased expression in Lgr5hi cells from NWD1 fed mice of five genes in the leading edge of the GSEA for MMR, and absence of this increase in Lgr5hi cells from mice fed NWD2, was confirmed by quantitative RT-PCR and was highly statistically significant (Supplementary Figure S2, available at Carcinogenesis online). Quantitative RT-PCR also confirmed that there were no significant changes in the expression of MMR genes in Bmi1+ cells as a function of diet (Supplementary Figure S2, available at Carcinogenesis online). Ingenuity pathway analysis, employing the RNAseq data, modeled regulation of Msh2, Exo1 and PolD1, key effector genes in MMR consistently upregulated by lower dietary vitamin D3 and/or calcium (Figure 4E). There are multiple paths by which the two nutrients alter the expression of these MMR target genes. These paths encompass genes at many nodes altered in expression by vitamin D3 and/or calcium (Figure 4E).
Figure 4.

Altered expression of the MMR repair pathway in Lgr5hi cells from mice fed different diets. GSEA of genes encoding enzymes of the MMR pathway in mice fed: (A) NWD1 versus AIN76A (P < 0.001, FDR = 0.05); (B) NWD2 versus NWD1 (P = 0.022, FDR = 0.061); (C) NWD1+ higher vitD versus NWD1 (P < 0.001, FDR = 0.004; (D) NWD1+higher Ca++ versus NWD1 (P = 0.002; FDR = 0.005). Diets are defined in Supplementary Table S1. (E) Ingenuity pathway analysis modeling of the complex regulation of Exo1, Msh2 and PolD1 in the MMR pathway by dietary vitamin D3 or calcium levels. Nodes in color are genes consistently altered in expression by dietary vitamin D3 and/or calcium levels in NWD1 (red, upregulation; green, downregulation; blue, gene not identified in the data set).
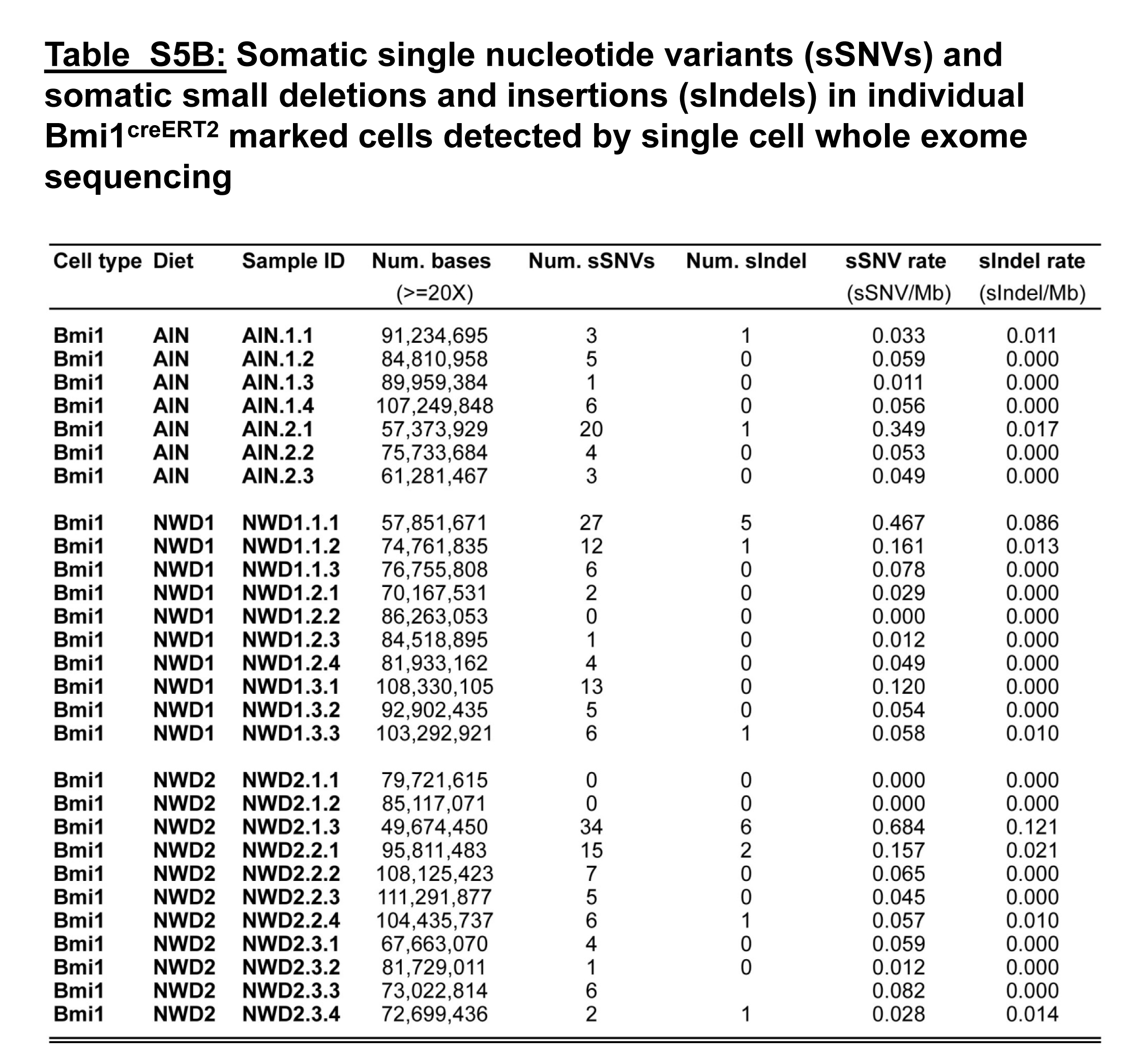
It was reported that differential MMR, but not differential rate of initial mutagenesis, is the principal determinant of altered mutation frequency in comparing tissues (30). Therefore, the impact of NWD1 on single-cell somatic mutations was determined by whole exome sequencing of genomes from single isolated Lgr5hi and Bmi1+ cells (methods, and the rigorous computational-bioinformatic pipeline, are described in detail in Supplemental Methods, available at Carcinogenesis online). Somatic single nucleotide variants (sSNVs; i.e. somatic point mutations) decreased 60% in Lgr5hi cells from mice fed NWD1 compared with cells from mice fed AIN76A, from 0.14 to 0.06 SNV/Mb, respectively (Figure 5A, Supplementary Table S5A, available at Carcinogenesis online; P = 0.01). This frequency is similar to that for mouse intestinal organoids (31). There was a similar decrease for small insertions and deletions (indels) in Lgr5hi cells from mice fed NWD1. However, indel frequency was low and the difference not significant (Supplementary Table S5A, available at Carcinogenesis online). Feeding NWD2 eliminated the decrease in mutations in parallel to lowering MMR gene expression and the pathway (Figure 5A). In contrast, there was no change in the single-cell point mutation frequency of Bmi1+ cells with diet (Figure 5A, Supplementary Table S5B, available at Carcinogenesis online), in parallel with the lack of dietary-induced change of the MMR pathway in these cells (Supplementary Figure S2, available at Carcinogenesis online; data not shown). Mutations for both cell populations were located in intergenic, exonic, intronic, 5′UTR and 3′UTR regions (Supplementary Table S6, available at Carcinogenesis online). There was no significant alteration in the distribution in relation to diet (Supplementary Table S6, available at Carcinogenesis online), consistent with mutations arising randomly in unselected mucosal stem cells.
Figure 5.

Mutation frequency, spectra and signature in Lgr5hi or Bmi1creERT2 marked cells from mice fed different diets. (A) SNVs (frequency per megabase, mean ± SEM) determined by single-cell sequencing of Lgr5hi or Bmi1 marked cells from mice fed AIN76A, NWD1 or NWD2 from weaning for 3 months (**P = 0.01, Student’s t-test). (B) Mutation spectra for Lgr5hi or Bmi1 marked cells after 3 months on diets. (C, a–d). The four most abundant mutation signatures identified in Lgr5hi or Bmi1 marked cells in mice fed any diet. (D) Relative abundance of each of the four mutational signatures in Lgr5hi or Bmi1 marked cells in mice fed each diet.
The single-cell mutation spectrum (base substitutions) and signature (base substitutions in the context of surrounding sequences (32)) in Lgr5hi and Bmi1+ cells were similar regardless of diet fed the mice (Figure 5B, C and D). For each diet, all identified sSNVs in both cell populations comprised 4 mutational signatures from the database of 30 mutational signatures defined for human tumors (http://cancer.sanger.ac.uk/cosmic/signatures) (32)—termed a, b, c, d (Figure 5C and D). The identities of the two major signatures were noteworthy: signature a resembled reported Sig18, identical to sSNVs in organoids derived from human intestinal, colon and liver stem cells (31); signature b resembled Sig23 and Sig6, signatures of replication error, that may reflect the prominent role of MMR (Figure 5) and/or altered replication of the Lgr5hi cells. Of the minor signatures, signature c resembled Sig2 characterized by C to T transition and C to G transversion at TCA and TCT sequences and may reflect off-target modification activity of APOBEC1/3 genes, which are transcribed in Lgr5hi and Bmi1+ cells (data not shown); signature d resembled Sig28, characterized by T to G transversion at CTT and TTT.
Discussion
Feeding NWD1 to wild-type C57Bl/6 mice is of high relevance to the etiology of sporadic colon cancer, the most common form of the disease in human high-risk populations. Here we establish that feeding NWD1 rather than control diet redefines the contribution of two different stem cell populations to both mucosal homeostasis and tumorigenesis. As previously established, Lgr5hi cell contribution is reduced (17,18), whereas cells derived from the Bmi1+ compartment are substantially increased. This is accompanied by a profound and complex transcriptional reprogramming of both cell populations, with clear interactions of nutrients in the reprogramming. This encompasses a highly significant increased expression of the MMR pathway in Lgr5hi cells. There is also decreased mutation accumulation in the Lgr5hi cells determined by single-cell exome sequencing. This includes a mutation signature characteristic of replicative damage of human tumors that is reversed by elevating vitamin D3 and/or calcium in the NWD1. In compensation, the increased contribution of Bmi1+ derived cells to homeostasis and tumorigenesis was highly significant and persisted for months when the mice were continuously fed NWD1, but diminished when switched back to control AIN76A. Therefore, the plasticity of cells from different compartments in the intestinal mucosa to function as stem or progenitor cells when Lgr5hi cells are acutely damaged by experimental insults (20,28,33) is also a response to intake levels of common nutritional components similar to those in the Western-style diet that establishes higher risk for CRC in both the mouse and human.
The parallel between potentially altered capacity for DNA MMR in Lgr5hi cells and reduction of mutations in these cells is striking. However, changes in replication frequency or metabolic processes that generate mutagenic intermediates are also factors that could contribute to the decreased accumulation of mutations in these CBC stem cells. Regardless of the mechanism, the combination of fewer Lgr5hi cells (17,18) and decreased number of mutations per cell reduces the mutational burden that the Lgr5hi population presents to the mucosa in NWD1 fed mice. In contrast, there are no changes in mutation frequency per cell in cells derived from the Bmi1+ population, but the vastly increased contribution of Bmi1+ cells to mucosal maintenance translates to a much greater mutational burden from this compartment. Thus, both increased mutational burden from the Bmi1+ cells and the ability of these cells to function as stem/progenitor cells may contribute to the increased tumorigenic potential from the Bmi1+ compartment, whereas the opposite contributes to reduction of tumorigenic potential of the Lgr5hi cells (17,18).
Bmi1+ cells are fundamental in tumorigenesis. Germ-line or intestinal epithelial cell targeted inactivation of Bmi1 reduces mutant Apc initiated tumorigenesis (34); in humans, Bmi1 expression is a marker of colon tumorigenesis and progression (35,36). However, cells marked by cre activation in Bmi1creERT2 mice (28,33) are heterogeneous. These mobilized Bmi1+ cells may be derived from dedifferentiation of a sub-population of maturing epithelial cells (37,38). The Bmi1+ cell population that responds to decreased function of Lgrhi cells may overlap with other intestinal cell populations also recruited by damage to Lgr5hi cells (38–42), but this is not clear. Further investigations are aimed at determining the identity of the sub-population of Bmi1+ cells that may underlie the response of this compartment to feeding NWD1.
It has been emphasized that when necessary, multiple quiescent cell populations can be mobilized to ‘seamlessly’ take over maintenance of the intestinal mucosa (20). Signals recruiting other cell populations when Lgr5hi cells are damaged are not understood (43). However, it is noteworthy that NWD1 elevates Wnt signaling throughout villi and crypts of the small and large intestine (12,15), which can mediate reversion of differentiated cells to a progenitor state. Moreover, elevated Wnt signaling in the mucosa is also initiated by lower dietary vitamin D3 similar to levels in NWD1, or Vdr knockout in mucosal epithelial cells (44–46).
Experimentally induced acute damage to Lgr5hi cells recruits other cell populations to function as intestinal stem cells. However, there may be unique importance of the major impact of nutrient intake on also generating this response. Diet is highly variable in human populations and changes daily for an individual. Therefore, the contribution of different intestinal stem cell populations to human mucosal homeostasis may be more variable and in flux than currently appreciated. Moreover, under different nutritional conditions, different stem cell populations may be the cell of origin of human tumors. Furthermore, as discussed earlier, the probability of tumor development may depend on relative mutational burdens and/or functions of different stem cell populations established by the nutritional environment.
Finally, common control rodent diets are formulated for the maximal health and longevity of the mouse. However, human diets, as reflected by NWD1, are distinctly unhealthy and are a major driver of risk for sporadic tumors (1,47,48). For example, the very high level of vitamin D3 in control rodent diets generates mouse serum 1,25(OH)2D levels considerably exceeding even the highest levels of the range for the US population (discussed in refs (17,18)). This is fundamental since our data (17,18) and the Lgr5hi CBC cell signature (19) establish a key role for vitamin D3 signaling through the Vdr in supporting Lgr5hi stem cell function. In this regard, it was recently reported that turnover of stem cells at the crypt base and time to reach crypt clonality is orders of magnitude slower in the human than in the mouse colon (i.e. years versus weeks (21)). It was speculated that this may be due to environmental differences between human and mouse (49). The prolonged kinetics in the human may, therefore, reflect constraints on Lgr5hi CBC cells by the lower vitamin D3 in the human diet, or other dietary components, differentially present in the rodent and human diets, instead recruiting other cells, such as Bmi1+, to maintain homeostasis.
In conclusion, functions of at least two intestinal stem/progenitor cell populations in maintaining intestinal homeostasis and in causing tumors were profoundly influenced by a nutrient environment highly relevant to a diet strongly linked to human sporadic colon cancer. Therefore, to more accurately model the etiology and pathogenesis of human tumor development, and response to chemo-preventive and therapeutic interventions, it would be advantageous to mimic nutrient environments that are produced by the human diet (50).
Funding
National Cancer Institute, National Institutes of Health (R01CA174432, R01CA229216, R01214625 and P30-013330); American Institute for Cancer Research (314707); New York State Stem Cell Science (C029154).
Author contributions
Experimental design: W.L., K.P., M.H., L.H.A.; data generation: W.L., K.P., M.H., J.M., S.M., L.H.A.; data analysis: W.L., S.E.Z., K.P., M.H., J.M., J.Z., S.M., Z.Z., Y.C., Q.Y., L.H.A.; manuscript preparation: W.L., L.H.A.
Conflict of Interest Statement: None declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Glossary
Abbreviations
- CBC
crypt base columnar
- CRC
colorectal cancer
- FACS
fluorescence-activated cell sorting
- FDR
false discovery rate
- GSEA
gene set enrichment analysis
- MMR
mismatch repair
- PCA
principal component analysis
- RNAseq
RNA sequencing
- sSNV
somatic single nucleotide variants
- TAM
tamoxifen
References
- 1. Chan A.T., et al. (2010) Primary prevention of colorectal cancer. Gastroenterology, 138, 2029–2043.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giovannucci E., et al. (1992) Relationship of diet to risk of colorectal adenoma in men. J. Natl Cancer Inst., 84, 91–98. [DOI] [PubMed] [Google Scholar]
- 3. Newmark H.L. (1987) Nutrient density: an important and useful tool for laboratory animal studies. Carcinogenesis, 8, 871–873. [DOI] [PubMed] [Google Scholar]
- 4. Newmark H.L., et al. (1990) Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J. Natl Cancer Inst., 82, 491–496. [DOI] [PubMed] [Google Scholar]
- 5. Newmark H.L., et al. (1991) Colonic hyperproliferation induced in rats and mice by nutritional-stress diets containing four components of a human Western-style diet (series 2). Am. J. Clin. Nutr., 54(1 Suppl), 209S–214S. [DOI] [PubMed] [Google Scholar]
- 6. Newmark H.L., et al. (2009) Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis, 30, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newmark H.L., et al. (2001) A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis, 22, 1871–1875. [DOI] [PubMed] [Google Scholar]
- 8. Yang W.C., et al. (2001) Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res., 61, 565–569. [PubMed] [Google Scholar]
- 9. Yang W., et al. (2003) Targeted inactivation of p27kip1 is sufficient for large and small intestinal tumorigenesis in the mouse, which can be augmented by a Western-style high-risk diet. Cancer Res., 63, 4990–4996. [PubMed] [Google Scholar]
- 10. Yang W., et al. (2005) Inactivation of p21WAF1/cip1 enhances intestinal tumor formation in Muc2−/− mice. Am. J. Pathol., 166, 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang K., et al. (2008) Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res., 68, 7313–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang K., et al. (2008) Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res., 68, 7803–7810. [DOI] [PubMed] [Google Scholar]
- 13. Aslam M.N., et al. (2010) A mineral-rich red algae extract inhibits polyp formation and inflammation in the gastrointestinal tract of mice on a high-fat diet. Integr. Cancer Ther., 9, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lochhead P., et al. (2015) Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod. Pathol., 28, 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang D., et al. (2011) Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc. Natl Acad. Sci. USA, 108, 10272–10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barker N., et al. (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 17. Peregrina K., et al. (2015) Vitamin D is a determinant of mouse intestinal Lgr5 stem cell functions. Carcinogenesis, 36, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Augenlicht L.H. (2017) Environmental impact on intestinal stem cell functions in mucosal homeostasis and tumorigenesis. J. Cell. Biochem., 118, 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muñoz J., et al. (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J., 31, 3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mills J.C., et al. (2015) Reserve stem cells: differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci. Signal., 8, re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicholson A.M., et al. (2018) Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell, 22, 909–918.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barker N., et al. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457, 608–611. [DOI] [PubMed] [Google Scholar]
- 23. Sangiorgi E., et al. (2008) Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet., 40, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shibata H., et al. (1997) Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science, 278, 120–123. [DOI] [PubMed] [Google Scholar]
- 25. Van Cromphaut S.J., et al. (2001) Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc. Natl Acad. Sci. USA, 98, 13324–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato T., et al. (2013) Primary mouse small intestinal epithelial cell cultures. Methods Mol. Biol., 945, 319–328. [DOI] [PubMed] [Google Scholar]
- 27. Bas T., et al. (2014) Real time analysis of metabolic profile in ex-vivo mouse intestinal crypt organoid cultures. J. Vis. Exp., 93, e52026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tian H., et al. (2011) A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature, 478, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lynch H.T., et al. (2015) Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer, 15, 181–194. [DOI] [PubMed] [Google Scholar]
- 30. Supek F., et al. (2015) Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature, 521, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blokzijl F., et al. (2016) Tissue-specific mutation accumulation in human adult stem cells during life. Nature, 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexandrov L.B., et al. ; Australian Pancreatic Cancer Genome Initiative ; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yan K.S., et al. (2012) The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl Acad. Sci. USA, 109, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maynard M.A., et al. (2014) Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene, 33, 3742–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li D.W., et al. (2010) Expression level of Bmi-1 oncoprotein is associated with progression and prognosis in colon cancer. J. Cancer Res. Clin. Oncol., 136, 997–1006. [DOI] [PubMed] [Google Scholar]
- 36. Tateishi K., et al. (2006) Dysregulated expression of stem cell factor Bmi1 in precancerous lesions of the gastrointestinal tract. Clin. Cancer Res., 12, 6960–6966. [DOI] [PubMed] [Google Scholar]
- 37. Schwitalla S., et al. (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell, 152, 25–38. [DOI] [PubMed] [Google Scholar]
- 38. Tetteh P.W., et al. (2016) Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell, 18, 203–213. [DOI] [PubMed] [Google Scholar]
- 39. Powell A.E., et al. (2012) The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell, 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westphalen C.B., et al. (2014) Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest., 124, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Asfaha S., et al. (2015) Krt19(+)/Lgr5(−) cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell, 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buczacki S.J., et al. (2013) Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature, 495, 65–69. [DOI] [PubMed] [Google Scholar]
- 43. Jadhav U., et al. (2017) Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell, 21, 65–77.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gröschel C., et al. (2016) Effect of 1,25-dihydroxyvitamin D3 on the Wnt pathway in non-malignant colonic cells. J. Steroid Biochem. Mol. Biol., 155(Pt B), 224–230. [DOI] [PubMed] [Google Scholar]
- 45. Larriba M.J., et al. (2013) Vitamin D is a multilevel repressor of Wnt/b-catenin signaling in cancer cells. Cancers (Basel)., 5, 1242–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Larriba M.J., et al. (2011) Vitamin D receptor deficiency enhances Wnt/β-catenin signaling and tumor burden in colon cancer. PLoS One, 6, e23524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giovannucci E. (1999) Nutritional factors in human cancers. Adv. Exp. Med. Biol., 472, 29–42. [DOI] [PubMed] [Google Scholar]
- 48. McCullough M.L., et al. (2019) Circulating vitamin D and colorectal cancer risk: an international pooling project of 17 cohorts. J. Natl. Cancer Inst., 111, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hodder M.C., et al. (2018) Intestinal stem cell dynamics: a story of mice and humans. Cell Stem Cell, 22, 785–787. [DOI] [PubMed] [Google Scholar]
- 50. Augenlicht L. (2014) Hidden effects of mouse chow. Science, 346, 710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.