

Summary:
Enterococcal pheromone responsive conjugative plasmids like pCF10 promote horizontal spread of antibiotic resistance genes following induction of plasmid-containing cells by potential recipients. Transcription of conjugation genes from promoter PQ is inhibited by the master regulator PrgX, further repressed when PrgX is in complex with the inhibitory I peptide, and allowed when PrgX is in complex with the C inducing peptide. Single cell analysis has shown that heterogeneity in the pheromone response is prevalent. Here, we systematically varied levels of regulatory molecules to better understand why some individual cells have increased propensity for induction. In this study, PrgX was confirmed to repress PQ in the absence of exogenous peptides in vivo, but cells with increased levels of PrgX were shown to be more prone to induction. Further, ablation of endogenous I reduced PrgX levels, resulting in reduced basal repression and loss of inducibility. Reduction of both endogenous peptides by washing increased the inducibility of cells. Together, these results show that endogenous PrgX, C, and I levels can impact the induction potential of a cell and establish the importance of basal I for regulation. These results also suggest that PrgX/C complexes may directly activate prgQ transcription, contrary to a long-standing working model.
Keywords: antibiotic resistance, microscopy, resistance factors, plasmids, gene expression regulation, transcriptional activation
Graphical Abstract:
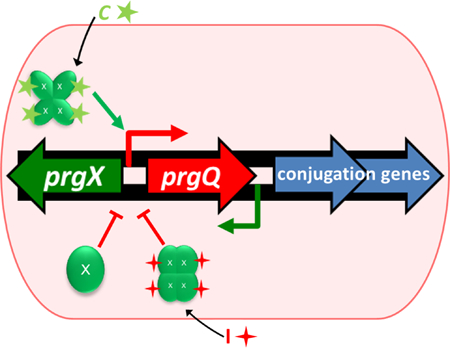
Abbreviated Summary:
Gene transfer by Enterococcus faecalis is controlled by a complex regulatory system. Recipients must signal donors for transfer to occur, and endogenous levels of negative regulators, PrgX and I keep donors poised to respond to recipient signals.
Introduction
Regulation of expression of the conjugative transfer genes on the enterococcal plasmid pCF10 is mediated by interactions between the master transcription factor PrgX, and two competing peptide pheromones cCF10 (C) and iCF10 (I). These interactions control the level from transcription initiation from PQ, the promoter for the prgQ operon, which encodes the pCF10 conjugation machinery (Fig. 1A and 1B). The understood role for the regulatory protein PrgX is as a repressor, either alone or as a complex with I. In complex with C, the PrgX structure is thought to change, de-stabilizing a critical interaction with a DNA operator binding site (Chen et al., 2017), and abolishing repression of transcription (Fig. 1B). This ultimately converts donor cells to an induced (conjugation-proficient) state. Following induction, shut-down is mediated by increased production of I. The prgX gene is transcribed from a promoter convergent with the opposing prgQ promoter (Bae et al., 2000). As a result of this genetic organization, additional layers of reciprocal regulation of the two operons are added by antisense RNA interactions and interference between RNA polymerase elongation complexes originating from the prgX and prgQ promoters (Chatterjee et al., 2011; Johnson et al., 2010). Previous work supports PrgX-mediated repression via two binding sites located in the prgQ promoter region, one with an 11 base pair palindromic sequence and another with weaker affinity and only half of the palindromic sequence (Fig. 1B); occupancy of the latter site by PrgX is required for repression of prgQ transcription (Bae et al., 2002). Further, analysis of dominant negative mutants of prgX indicated a crucial role for oligomerization of PrgX for negative regulation of pheromone-inducible conjugation (Bae & Dunny, 2001).
Fig. 1.
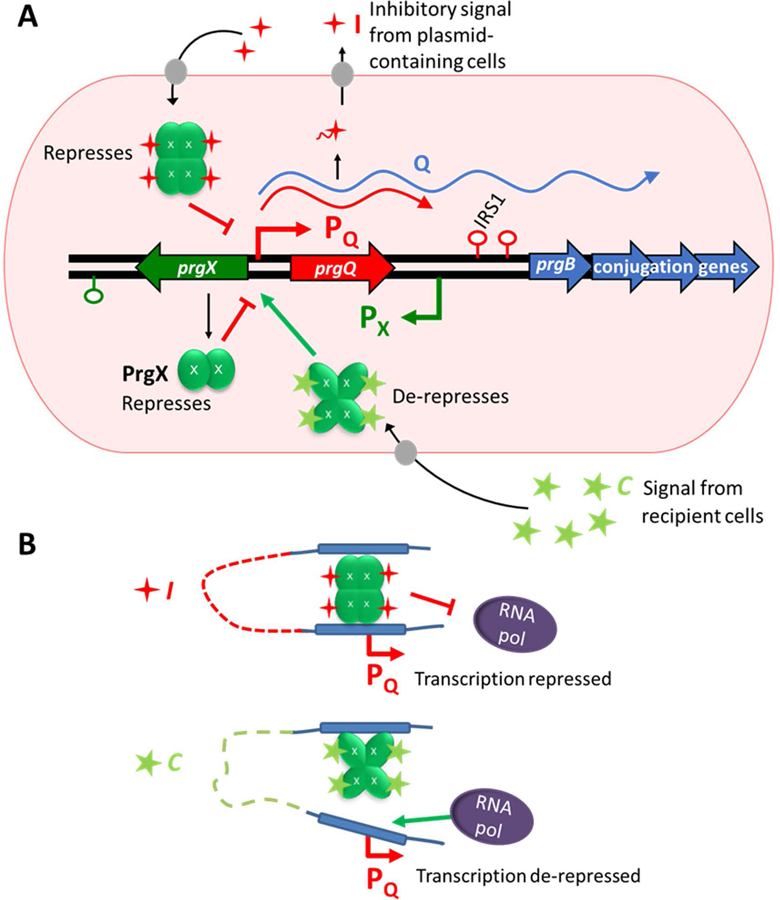
Models depicting pCF10 regulation. Peptides C and I are depicted as stars in green and red respectively. PrgX protein monomers are depicted as green ovals.
A. Regulatory region of pCF10 and overview of prgQ regulation. The I precursor is encoded by prgQ which is transcribed in un-induced cells with termination at IRS1. Mature I is produced via cleavage of immature I upon export from the cell. C is encoded on the chromosome of E. faecalis and produced via processing in a similar manner. The PrgX protein is encoded by pCF10 and regulates promoter PQ. Upon induction by PrgX-C complexes, transcription past the IRS1 terminator is induced and expression of the genes required for conjugation may occur.
B. Structural models of the PrgX binding region of pCF10 in the presence of I or C depicting predicted looped structures that prevent or allow transcription from promoter PQ. Figure based on model presented by Chen et al., 2017.
Models for the roles of PrgX in regulation were extensively refined based on genetic evidence (Bae & Dunny, 2001, Bae et al., 2002, Kozlowicz et al., 2004), but molecular studies have also enhanced our understanding of pCF10’s complex regulatory system. The molecular basis for control of induction by PrgX complexes was better revealed by the solved PrgX molecular structure with and without C and further analysis of predicted mutant structures (Kozlowicz et al., 2006; Shi et al., 2005). Although initial studies suggested that the PrgX oligomerization state in vivo was a dimer, subsequent work has revealed that in complex with C or I, PrgX forms a tetramer in both crystals (Shi et al., 2005) and in solution (Chen et al., 2017). However, apo-PrgX exists as a dimer in solution and its direct repressive effects can be observed via in vitro transcription experiments (Caserta et al., 2012). Overall, all current evidence supports PrgX’s ability to repress prgQ transcription as an apo-protein or in complex with I; in either case, the effective repressing complex is believed to contain a DNA loop stabilized by a PrgX tetramer with each dimer bound to an operator binding site (Fig. 1B, top). The prgQ upstream regulatory sequence features, PrgX oligomerization states, and peptide and DNA binding sites described above are represented in the context of our current models for PrgX-mediated regulation and binding (Fig. 1).
The response to C was assessed in single cells via labeling of induced transcripts and was shown to be very heterogeneous (Breuer et al., 2017). Not all cells exposed to similar conditions became induced and induced cells appeared to have a range of induction intensities. In light of these results, we sought to understand whether variation in the levels of individual components of this system might impact the inducibility of individual cells. In this study, we examined the effects of PrgX levels and endogenous C and I on the system, using both population-based assays and single cell analysis. Overall, we confirmed that PrgX impacts transcript initiation from promoter PQ which is essential for plasmid transfer, as expected from the working models (Fig. 1). It also follows that understanding variability in initiation of this response may reveal insights into how this system functions and what factors might push cells to be more or less likely to be induced and undergo conjugation. In this study, we found that in donor cells the endogenous levels of negative regulators PrgX and I impact inducibility, but in all conditions examined the system allows a subset of cells to be poised to respond to low levels of C. Results from recent and ongoing analysis of pheromone induction and plasmid transfer in the gastrointestinal tract of mice (Hirt et al., 2018) suggests that key features of the system, such as the inverse correlation between donor population density and inducibility, are relevant to the behavior of the system in the natural habitat of the organism. This system effectively prevents wasteful induction events to keep the fitness cost of plasmid maintenance low and allows for dissemination of the plasmid over time. Understanding the parameters that govern this process is important to understanding how to prevent gene transfer.
Results
PrgX acts as a repressor in a concentration dependent fashion in vivo
To confirm the expected role of PrgX as a repressor of prgQ expression in vivo, strains and plasmids with varied intracellular concentrations of PrgX were utilized in the context of a β-galactosidase (LacZ) reporter of expression from the prgQ promoter (Fig. 2). Whereas PrgX is normally encoded by pCF10, in this experiment the strains and plasmids were constructed to produce varied levels of PrgX which were confirmed by western blots (Fig. 2A). Strain OG1Sp lacks prgX, while DM100–5 expresses a cloned prgX from its chromosome under control of the native promoter with an optimized ribosomal binding sequence (RBS). Into each of these strains, we inserted either p043lacZdx, which does not confer PrgX production due to a truncation of the 3’ end of the prgX gene, or pBK2 which contains a nearly identical segment of the pCF10 regulatory region, but with an intact prgX gene (Fig. 2B). In this fashion, we generated nearly identical strains containing the same set of PrgX operator binding sites, but where the intracellular concentrations of PrgX ranged from zero to very high levels as shown in the upper section of Fig. 2A.
Fig. 2.
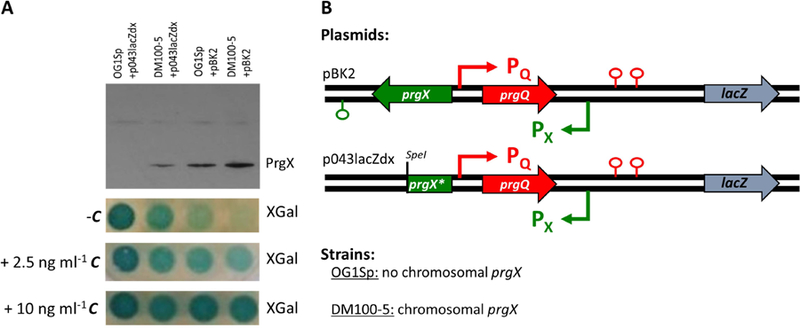
The repressing effect of PrgX can be observed in vivo in engineered strains expressing varied basal levels of PrgX.
A. Western blot and XGal spot plates. Top: Western blot for PrgX shows relative levels of PrgX expression in these strains. Bottom: XGal spot plates show development of blue relative to the level of expression of LacZ under control of the prgQ promoter from the pBK2 or p043lacZdx reporter constructs in strains OG1Sp and DM100–5. Plates contain 250 µg ml−1 XGal and varied levels of C, from 0– 10 ng ml−1.
B. Plasmids and strains used in this analysis. Maps show the prgX-prgQ regulatory region of reporter plasmids pBK2 and p043lacZdx. For p043lacZdx, the * indicates the deletion of the 3’ end of the prgX gene beginning at the SpeI site.
Spotting of normalized exponential phase cultures of these strains on plates containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (XGal) resulted in development of blue relative to the level of LacZ present (Fig. 2A). From this experiment, in the absence of C, we observed uncontrolled production of LacZ from strain OG1Sp+p043lacZdx which completely lacks PrgX. However, for strains with increasing levels of PrgX (DM100–5+p043lacZdx, OG1Sp+pBK2, and DM100–5+pBK2), production of LacZ in the absence of C was reduced in proportion to intracellular PrgX levels. With the addition of low levels of C to plates (2.5 ng ml−1), production of LacZ was induced in strains with higher levels of PrgX (OG1Sp+pBK2, and DM100–5+pBK2). With the addition of higher levels of C to plates (10 ng ml−1), production of LacZ was further induced to comparable levels in strains that produced PrgX (OG1Sp+pBK2 and DM100–5+pBK2). Strain DM100–5+p043lacZdx had very weak induction with high levels of C. In the absence of PrgX (strain OG1Sp+p043lacZdx), LacZ expression was not further increased by C.
These results confirm the role of PrgX as a repressor in the absence of C and demonstrate the role of C as an inducer in the presence of PrgX. It is interesting to note that LacZ expression from DM100–5+p043lacZdx did not appear to increase with the addition of 2.5 ng ml−1 C (relative to increases observed in strains with higher levels of PrgX- OG1Sp+pBK2 and DM100–5+pBK2). In the next section, we further explore the impact of PrgX levels on inducibility by quantitative mRNA analysis at the single cell level.
Cells with increased levels of PrgX are more inducible by low levels of exogenous C
To investigate the impact of PrgX levels on inducibility of pCF10, PrgX was expressed from the chromosome of OG1RF under control of either the prgX promoter with an optimized RBS (DM100–5) or a mutated RBS (DM101–12) and inducibility was assessed (Fig. 3). Strains DM100–5+pCF10, DM101–12+pCF10, and OG1RF+pCF10 were shown to have 3 distinct levels of PrgX by western blot to PrgX (Fig. 3A). These strains were grown to log phase and then induced by addition of 2.5 ng ml−1 C. Induction of prgB transcripts (Fig. 1), previously shown to be a reliable indicator of the conjugation induction state of pCF10 (Hirt et al., 2005), in individual cells was assessed 30 and 120 min after C addition by fluorescence in situ hybridization chain reaction (HCR) labeling and analysis. HCR analysis showed that all three strains had similar basal levels of expression and were induced and shut-down within the same time frame. However, strains with increased levels of PrgX (DM101–12+pCF10 and DM100–5+pCF10) were shown to have an increased percent of cells induced (or increased inducibility) compared to wild type (OG1RF+pCF10), (Fig. 3B). To illustrate how the distributions of the single cell labeling intensities compared in these strains, relative per cell average fluorescence intensities were plotted (Fig. 3C and Fig. S1). These show that the range of labeled cell intensities was the same for all three strains, even though the relative number of induced cells were much higher for the two strains with high basal levels of PrgX.
Fig. 3.
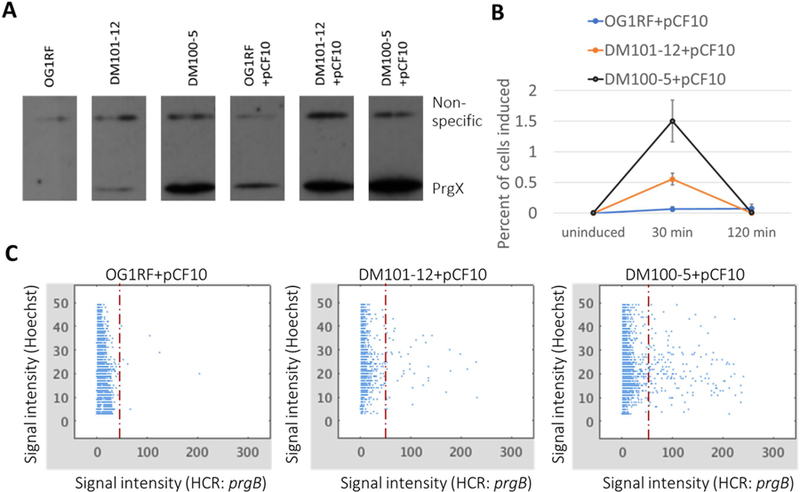
HCR labeling of prgB transcripts in induced cells shows that cells with increased levels of PrgX are more inducible by low levels of exogenous C.
A. Western blot for PrgX shows expected relative levels of PrgX expression in these strains with and without pCF10.
B. Cells with varied levels of PrgX expression were harvested for HCR labeling from uninduced populations and at 30 and 120 min after addition of 2.5 ng ml−1 C. Induced prgB transcripts were fluorescently labeled by HCR, cells were imaged, and fluorescence corresponding to HCR labeling was quantified for each cell. Cells were considered induced if HCR fluorescence exceeded a threshold. Results are the mean of 3 biological replicates with bars showing standard error. These plots were generated from the analyses of single cells, as depicted in C.
C. Single cell fluorescent intensity data from one replicate of the induction and HCR labeling experiment. Relative fluorescence of individual cells are plotted based on the per cell average fluorescence intensity of Hoechst:33342 (DNA label) and HCR: prgB (induced transcript label). The dashed line indicates the threshold value of 50, above which cells were considered to be induced (see Methods for further information).
Cells lacking endogenous I alone have decreased PrgX levels and are not inducible with low levels of exogenous C
All wild type E. faecalis cells produce peptide C endogenously and cells containing pCF10 produce both C and I endogenously. We sought to assess the effects of these endogenous peptides on the functioning of the pCF10 regulatory system. We accomplished this by again using β-galactosidase reporter construct pBK2, from which lacZ is expressed in lieu of the conjugation genes downstream of the prgQ promoter. Reporter plasmid pBK2idT is similar, but whereas pBK2 produces endogenous I, pBK2idT does not produce endogenous I due to deletion of one codon within the I coding sequence. To vary endogenous C production, we used wild type E. faecalis strain OG1Sp and strain JRC104, an OG1 derivative that does not produce endogenous C due to a nonsense point mutation in the ccfA gene. The resulting strains, genotypes, phenotypes, and numerical abbreviations used in the following figures (Figs. 4, 5, and 6) are 1: OG1RF+pBK2idT (C+ I−), 2: JRC104+pBK2 (C− I+), 3: OG1Sp+pBK2 (C+ I+), and 4: JRC104+pBK2idT (C− I−).
Fig. 4.
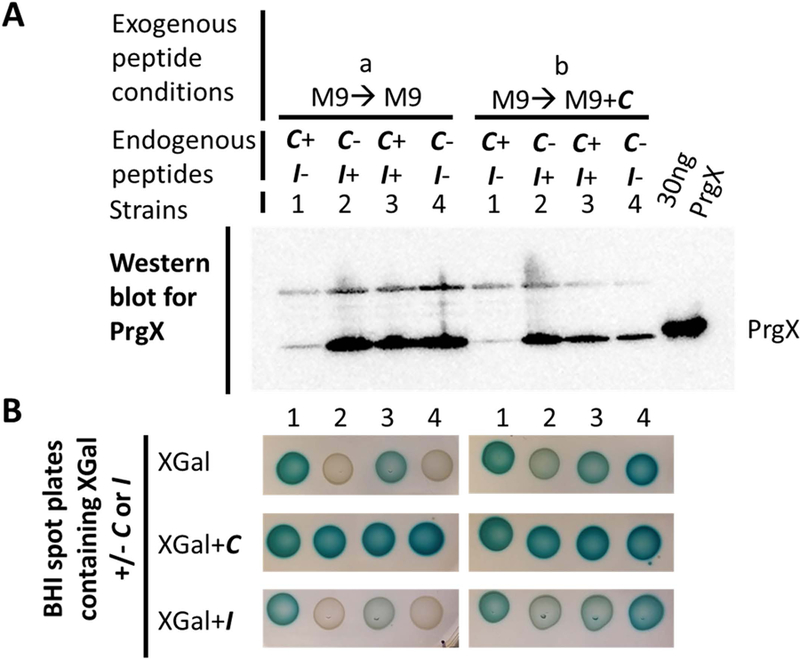
Cells lacking endogenous I alone (strain 1 (C+ I−)) have decreased PrgX levels and are less impacted by exogenous peptides C and I. Cultures were grown to log phase in M9 medium without C (left, condition “a”) or + 2.5 ng ml−1 C (right, condition “b”) and normalized. Strains are 1: OG1RF+pBK2idT (C+ I−), 2: JRC104+pBK2 (C− I+), 3: OG1Sp+pBK2 (WT-like, C+ I+), and 4: JRC104+pBK2idT (C− I−).
A. Levels of PrgX are compared by PrgX western blot. The far-right lane was loaded with 30 ng purified his tagged PrgX.
B. XGal spot plates show development of blue relative to the level of expression of LacZ under control of the prgQ promoter from the pBK2 or pBK2idT reporter constructs. Plates contain 250 µg ml−1 XGal and 2.5 ng ml−1 C (middle row) or 250 ng ml−1 I (bottom row).
Fig. 5.
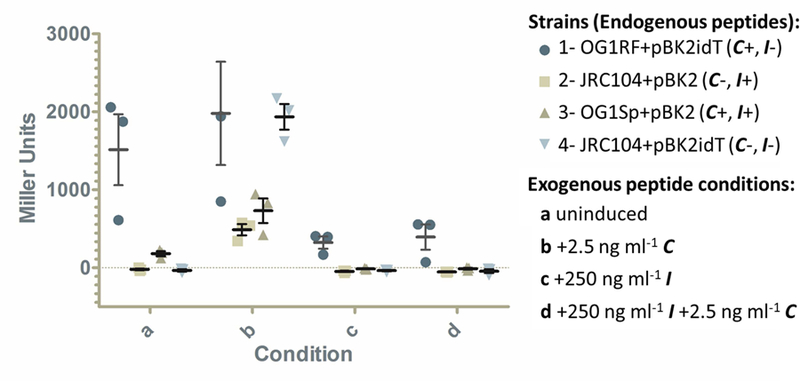
Cells lacking endogenous I (which have very low PrgX levels) do not have inducible expression with the addition of low levels of exogenous C. Cultures were grown overnight then diluted and grown to log phase in medium without C (uninduced, condition “a”) or with 2.5 ng ml−1 C (condition “b”). For conditions “c” and “d”, cultures were grown overnight in medium containing 250 ng ml−1 I, and then diluted and grown to log phase in medium containing 250 ng ml−1 I (condition “c”) or 250 ng ml−1 I and 2.5 ng ml−1 C (condition “d”). Strains include: 1: OG1RF+pBK2idT (C+ I−), 2: JRC104+pBK2 (C− I+), 3: OG1Sp+pBK2 (WT-like, C+ I+), and 4: JRC104+pBK2idT (C− I−). Cultures were harvested 2 hours after dilution and assayed for relative β-galactosidase activity which is reflected by Miller units and reflects production of the LacZ reporter. Results show 3 biological replicates with bars indicating standard error of the mean. The third replicate for strain 1 (C+ I−) condition “b” is not seen because it is too high (3145.5 Miller Units) to be on the y-axis. Pairwise comparisons to strain 3 (C+ I+) for each condition show the following comparisons with statistically significant differences where P<0.01: 1a vs 3a, 1b vs 3b, and 4b vs 3b. All other pairwise comparisons to strain 3 of the same condition are not statistically significant (P>0.05).
Fig. 6.
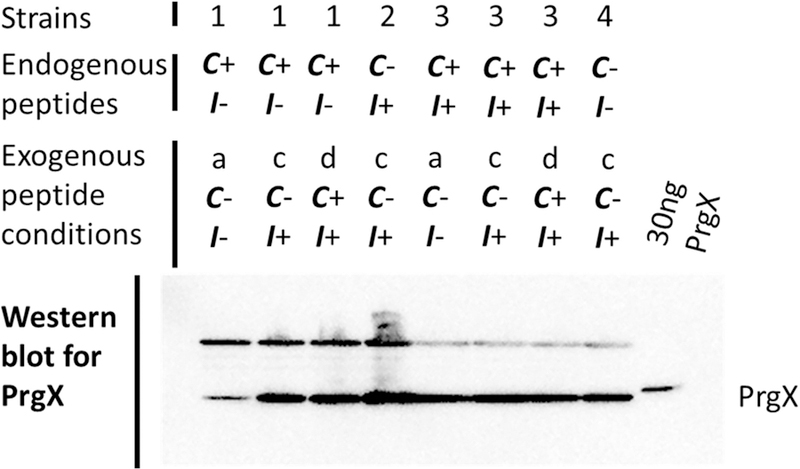
Incubation with exogenous I increases PrgX levels in C+, I− cells (strain 1). Numbers represent strains and letters represent culture conditions as follows. Cultures were grown overnight then diluted and grown to log phase in medium without C (uninduced, condition “a”). For conditions “c” and “d”, cultures were grown overnight in medium containing 250 ng ml−1 I, and then diluted and grown to log phase in medium containing 250 ng ml−1 I (condition “c”) or 250 ng ml−1 I and 2.5 ng ml−1 C (condition “d”). Strains include 1: OG1RF+pBK2idT (C+ I−), 2: JRC104+pBK2 (C− I+), 3: OG1Sp+pBK2 (WT-like, C+ I+), and 4: JRC104+pBK2idT (C− I−). Cultures were harvested 2 hours after dilution and normalized for comparison of PrgX levels by western blot. The far-right lane was loaded with 30 ng purified his tagged PrgX.
To determine whether changes to endogenous I and C impacted basal levels of PrgX, strains were grown to log phase in M9 medium without or with low levels of exogenous C, culture densities were normalized, and relative levels of PrgX were compared by PrgX western blot (Fig. 4A). Western blots on cells grown in the absence of exogenous C showed that PrgX levels were similar to wild type strain 3 (C+ I+) for strains 2 (C− I+) and 4 (C− I−). However, in strain 1 (C+ I−) PrgX protein levels were reduced relative to wild type. For cells grown to log phase in the presence of exogenous C, PrgX levels in strain 1 (C+ I−) remained reduced relative to wild type. In cultures of strains 2 (C− I+) and 4 (C− I−) grown with exogenous C, PrgX levels were again similar to wild type, but appeared reduced relative to the same strains grown in the absence of exogenous C. This slight reduction in PrgX expression is expected based on the effects of antisense RNA interactions reducing prgX transcript levels after induced expression from the prgQ promoter and interference between RNA polymerase elongation complexes originating from the prgX and prgQ promoters (Fig. 1A), (Chatterjee et al., 2011; Johnson et al., 2010).
The effective reduction in PrgX levels in strain 1 (C+ I−) (observed by western blot) allowed us to further examine the effects of basal PrgX levels on inducibility. We looked at inducibility of these strains in two ways: (1) by spotting of cultures on plates containing XGal +/− C or I and (2) by assaying for β-galactosidase activity via liquid β-gal assays (Figs. 4B and 5 respectively).
In the absence of exogenous peptides there was some basal lacZ expression for wild-type-like strain 3 (C+ I+) (Fig. 4B). This basal expression was completely obliterated in strains 2 (C− I+) and 4 (C− I−), which lack endogenous C. Strain 1 (C+ I−) had slightly increased basal β-galactosidase expression relative to strain 3 (C+ I+), probably resulting from the decreased level of PrgX repression in strain 1 (C+ I−) (Fig. 1A). As expected, spotting of cultures that had been grown in liquid medium in the presence of C on XGal plates without peptides resulted in some induction from strains 2 (C− I+) and 4 (C− I−) (Fig. 4B, top right). This inducibility was also observed by spotting cultures which had been grown in the presence or absence of C on XGal plates containing C (Fig. 4B, middle row), where strains 2 (C− I+) and 4 (C− I−) were both inducible to wild type-like strain 3 (C+ I+) levels. Strain 1 (C+ I−), which lacks endogenous I and had reduced PrgX levels, appeared to have slightly decreased levels of induction relative to strains 2, 3, and 4 (Fig. 4B, middle row), and the blue intensity appeared unchanged relative to the strain 1 spot on the XGal plate without C (Fig. 4B, top left). Further, when spotted on XGal plates containing I (Fig. 4B, bottom row), where strain 3 (C+ I+) appeared to have a slight decrease in inducibility (as compared to plates without peptide), strain 1 (C+ I−) activity was largely unchanged. Taken together, this result suggested that reduction of PrgX in cells via complete elimination of endogenous I made strain 1 cells less repressible by exogenous I and possibly less responsive to induction by exogenous C. It is also likely that induction of this strain was already at a maximum without exogenous C addition.
To confirm the observation that cells with reduced PrgX are less inducible by low levels of exogenous C and not repressible by exogenous I, liquid β-gal assays were performed following varied exposure to exogenous C and/ or I (Fig. 5). Specifically, cultures were grown overnight then diluted and grown to log phase in medium without C (uninduced, condition “a”) or with 2.5 ng ml−1 C (condition “b”). For conditions “c” and “d”, cultures were grown overnight in medium containing 250 ng ml−1 I, and then diluted and grown to log phase in medium containing 250 ng ml−1 I (condition “c”) or 250 ng ml−1 I and 2.5 ng ml−1 C (condition “d”). Cultures were harvested 2 hours after dilution and assayed for β-galactosidase activity. Results of β-gal assays on uninduced cells (condition “a”) were generally as expected: there was some basal induction of wild type-like strain 3 (C+ I+), strains 2 (C− I+) and 4 (C− I−) (which both lack endogenous C) completely lacked activity, and strain 1 (C+ I−) (which has reduced PrgX levels) had high basal levels of expression. β-gal assays on cells induced by 2.5 ng ml−1 (condition “b”) showed that strains 2 (C− I+), 3 (C+ I+), and 4 (C− I−) had strongly inducible expression. It is not clear whether expression of strain 1 (C+ I−) in condition “b” was increased relative to basal expression in condition “a” due to the high level of β-galactosidase activity and great deal of variability in both conditions. Interestingly, in condition “b”, strain 4 (C− I−) which lacks endogenous C and I appeared to be more inducible than wild type-like strain 3 (C+ I+) and this difference was statistically significant.
To look at whether reporter expression from these strains could be affected by exogenous I, cells were exposed to conditions “c” and “d” (Fig. 5). β-gal assays on cells grown in conditions “c” and “d” showed that I effectively reduces expression in all 4 strains when compared to strains grown in condition “b”. Although the reduction did not appear as complete in strain 1 (C+ I−), the differences between strain 1 (C+ I−) and strain 3 (C+ I+) in conditions “c” and “d” were not statistically significant. Additionally, the low level of C in condition “d” did not appear to have allowed for measurable induction by any of the 4 strains. Next, we sought to determine why reporter expression from strain 1 (C+ I−) now appeared to be repressible in conditions “c” and “d” (where it did not in the previous XGal spot plate experiments in Fig. 4B). A likely possibility was that extended incubation with exogenous I had effectively complemented I in this strain and allowed the strain to produce a more wild-type-like level of PrgX. Interestingly, for strain 4 (C− I−) without any endogenous peptides, the order of peptide addition likely determined the varied induction outcomes observed in the Fig. 4B and 5 experiments.
To investigate whether PrgX levels were increased in strain 1 (C+ I−) during extended incubation with exogenous I, western blots were performed to assess PrgX levels in these strains and with these varied treatment conditions (Fig. 6). The data showed that I was effectively complemented extracellularly in cells lacking endogenous I and it is hypothesized that this complementation allowed for the repressive effects of PrgX and I to be observed when cells were assessed for β-galactosidase activity in conditions “c” and “d”. Also, when these cells were subsequently exposed to exogenous C (condition “d”), expression remained shut-down (Fig. 5). Growth of strains 2 (C− I+), 3 (C+ I+), and 4 (C− I−) in the presence of I (condition “c”) did not appear to have impacted PrgX levels in these strains.
Washing cells (which reduces endogenous I and C) increases inducibility of cells exposed to low levels of exogenous C
Although cells permanently lacking endogenous I (strain 1, (C+ I−)) had high basal promoter PQ activity and decreased inducibility due to low endogenous levels of PrgX, we sought to determine whether temporary removal of endogenous peptides would alter inducibility over a short time frame. We tested shorter term peptide removal by washing cells in buffer before resuspending in growth medium and inducing with 2.5 ng ml−1 C (Fig. 7). The percent of cells induced were assessed by HCR labeling of induced prgB expression and analysis. Strains with varied levels of PrgX (due to additional chromosomal expression) were used to look at the effects of washing. Without washing, the results mirrored those shown in Fig. 3B; strains with increased levels of PrgX had increased inducibility relative to wild-type OG1RF+pCF10. The results of this experiment looking at fold changes in the percent of cells induced showed that washing drastically increases the frequency of induction within a population of cells, likely due to reduction of endogenous I, in strains with wild-type PrgX levels or with increased levels of PrgX (Fig. 7).
Fig. 7.
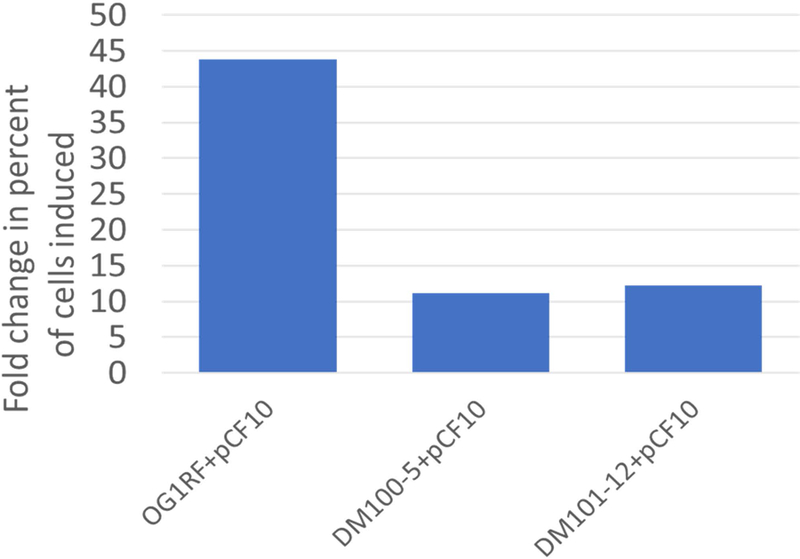
Washing increases the frequency of induction for cells exposed to low levels of exogenous C. Cells with varied levels of PrgX expression were washed in buffer before resuspending in medium containing 2.5 ng ml−1 C and compared to unwashed cells of the same strain to calculate fold change in percent of cells induced (y-axis). Cultures were harvested for HCR labeling 30 min after C addition. Induced prgB transcripts were fluorescently labeled by HCR, cells were imaged, and fluorescence corresponding to HCR labeling was quantified for each cell. Cells were considered induced if HCR fluorescence exceeded a threshold.
Notably, the distributions of induced prgB expression levels in individual cells (as reflected by HCR labeling intensity distributions) were similar for induced cells with or without washing (Fig. 8). This suggests that washing does not change the range of induced expression levels for individual cells, but rather increases frequency of induction across the population of cells. Thus, the overall range of prgB expression levels (reflected by HCR intensity values) remained the same. The drastic fold increase in induction observed for OG1RF+pCF10 relative to the other strains (Fig. 7) likely reflects the extreme rarity of induction in this strain without washing (shown in Figs. 3B, 3C, and 8A and B). Thus, although the washed strains with higher PrgX levels had much higher absolute numbers of highly induced cells (Fig. 8C and D), the fold change in induction relative to unwashed cells was lower than the fold change for OG1RF+pCF10 (Fig. 7).
Fig. 8.
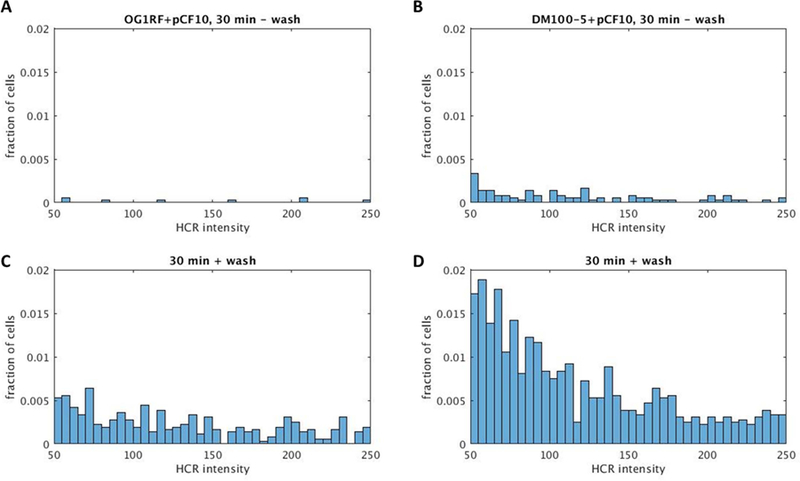
Distributions of induced prgB expression levels are similar for induced cells with or without washing. HCR labeling intensity histograms show the fraction of cells with different levels of HCR labeled prgB expressed upon pCF10 induction in OG1RF (A and C) or DM100–5 (B and D) 30 minutes after addition of 2.5 ng ml−1 C. HCR intensity reflects quantified fluorescent intensity of HCR labeled prgB transcripts in individual cells.
A and B: sample data for unwashed cells.
C and D: sample data for washed cells.
The results of this washing experiment intuitively make sense since washing likely reduces the concentration of the inhibitory I peptide, making induction more likely. Yet, at first glance, these results may seem to contradict the OG1RF+pBK2idT (strain 1 (C+ I−)) induction phenotype where this strain lacking endogenous I was less inducible than cells with endogenous I (Fig. 4). However, the experiments depicted in Figs. 7 and 8 are different in that they look at the effects of short-term reduction in endogenous peptides by washing and not the effect of permanent endogenous I eradication. In the case of short-term peptide reduction, (through washing), PrgX levels likely remain unaltered due to the stability of the protein (Bae et al., 2000).
Discussion
Insights from combined approaches
Although the pCF10-encoded pheromone response has been characterized extensively at the population and single-cell levels, the effects of varying the endogenous levels of the 3 primary regulators, C, I, and PrgX, on the response has never been determined. The combination of single-cell and population-averaging approaches used in this analysis of the impact of endogenous PrgX, C, and I levels has yielded several new insights. HCR analysis of induction in single cells allowed us to gauge the range of inducibility in cultures of responder cells. In parallel, the lacZ reporter construct allowed us to functionally assess the level of basal and induced expression via XGal spot plates and quantitative β-Gal assays at a population level. Lastly, quantitative western blot analysis of PrgX in cell lysates allowed us to assess PrgX levels to confirm PrgX levels in our engineered constructs and ask whether PrgX levels had been impacted by other changes to endogenous peptide concentrations in the system.
However, it is also apparent that each of these methods of analysis has its own advantages and shortcomings. While together the data overwhelmingly support the conclusions discussed below, there are some inconsistencies that might be explained by how the analysis approaches differ. For example, in comparison of the quantitative liquid β-gal assay (Fig. 5) and XGal spot plate (Fig. 4B) results, one may notice that for quantitative assay strain 1 (C+ I−), OG1RF+pBK2idT appears to have substantially increased basal expression relative to wild-type like strain 3 (C+ I+) that is much less apparent on the plate-based assay. This is likely due to differences between how these assays are run and how LacZ activity is detected. For the plate assay, cultures are grown and normalized before being spotted on plates containing XGal. Plates are incubated for a defined amount of time before being imaged. The resulting development of blue is relative to the amount of extracellular LacZ produced by the strain but is also determined by the amount of XGal in that portion of the plate and the incubation time. For the liquid assay, cultures are also grown and normalized, but cells are lysed and total LacZ activity is assessed at a single timepoint by reaction with ONPG. For expression of lacZ from this pCF10-like reporter system, the temporal effects of induction could also yield slightly different results with analysis of LacZ activity over time in the plate assay versus at a single timepoint in the liquid assay. Regardless, both of these methods are in agreement in that they show that for strain 1 (C+ I−), basal expression was high, and expression was not further increased by exogenous C.
Overall strain 1 (C+ I−), OG1RF+pBK2idT was shown to have decreased PrgX levels. Based on our model for pCF10 and our observation that increased levels of PrgX led to increased inducibility, we hypothesize that decreased PrgX levels in this strain may explain the loss of inducibility of strain 1 (C+ I−) observed in Figs. 4B and 5. However, in further consideration of the effect of endogenous I loss, when we look at the results of the wash experiment, there appears to be an inconsistency. Washing cells, which effectively removes endogenous peptides, leads to a significant increase in inducibility (Fig. 7). For unwashed strain 1 (C+ I−) there was a loss of inducibility (Fig. 5). It is likely that this difference is due to the time period for which endogenous peptides were altered. For strain 1 (C+ I−), endogenous I was permanently removed and the effects of this were allowed to compound over time. As a result, the levels of PrgX in strain 1 decreased and in turn, inducibility was lost. For the wash experiment, endogenous peptides were removed for a short period, effectively increasing the chance that PrgX produced by cells would remain without peptides until exposure to exogenous peptides. As a result of washing, cells became more inducible.
Other differences in the results of HCR and LacZ reporter experiments may be observed. HCR allows detection of very rare events that may not be detectable by population averaging approaches. It is likely that more common phenotypes obfuscate rare phenotypes when an average is measured or observed. For these reasons, complementary analyses using both single cell and population-level approaches are valuable. Analysis by HCR made it possible to detect changes in inducibility and compare the behavior of induced cells to determine that the timing and range of expression in the responding cells had remained similar.
Mechanistic implications
Work presented here expands our understanding of the impact of the levels of endogenous PrgX, C, and I on the inducibility of single cells. Previous studies of the system have not examined the effects of systematic variation of the basal levels of these regulators on behavior of the system. Specifically, analysis of induction in the absence of peptides demonstrates the in vivo role of the master regulator PrgX. Alone, PrgX represses in a concentration dependent manner. However, analysis of inducibility with increased levels of PrgX or with varied levels of endogenous peptide reveals that the level of PrgX can impact the tendency of cells to be induced. Taken together, these results show that with low levels of exogenous C, cells with increased endogenous PrgX levels have increased inducibility and cells with decreased endogenous PrgX levels have decreased inducibility. This suggests that the understood role of PrgX solely as a repressor alone or in complex with I may need to be altered (Fig. 1). PrgX binding with C was known to relieve repression of expression from promoter PQ allowing for induction, but these results suggest that PrgX-C complexes may actually activate transcription initiation. Promoter activation could occur via polymerase recruitment or through alteration of the DNA structure in such a way as to promote more transcription or give more access to helicase. These possibilities could be tested through in vivo and in vitro transcript quantification and foot-printing experiments to determine the direct impact of PrgX, PrgX-C, and PrgX-I complexes on expression.
Summary and conclusions
Given our understanding of the regulatory circuits that govern induced expression from the pCF10 regulatory region (Dunny & Berntsson, 2016; Dunny, 2013), the results from the experiments in which endogenous I and/ or C were eliminated make sense. In this work, we observed that loss of endogenous basal I in this system leads to increased basal expression of the prgQ operon, and an inability to have expression induced further. We reason that loss of endogenous I led to increased antisense interactions by prgQ transcripts that decreased productive prgX mRNA, which reduced PrgX levels, and resulted in a loss of repression in this strain. As expected based on our model of pCF10 regulation, extracellular complementation of I led to increased production of PrgX in the strain lacking endogenous I (Fig. 6), which can explain the restoration of repression of induction in this strain (Fig. 5). Examination of the effects of cell washing further demonstrated that removal of endogenous peptides could make cells more inducible. Basal levels of I are produced from short prgQ transcripts in cells containing the pCF10 plasmid and must be maintained to allow for adequate levels of PrgX in the system. These experiments demonstrate the importance of endogenous I in cells for facilitation of a rapid and sensitive induction response upon signaling by potential recipients with C.
Together, these results suggest that small stochastic differences in the levels of these individual regulatory components (PrgX, I, or C) in individual cells could lead to different propensities for induction in individual cells and contribute toward the heterogeneity observed in the response. Unfortunately, it is not currently possible to measure the single-cell intracellular concentrations of peptides like C and I. Further, we would predict that experimentally washing cells recapitulates a natural scenario where an individual donor cell (or small population of donors) enters the gastrointestinal tract of a new host already colonized by recipients. These donors would be highly inducible by the C produced by the plasmid-free residents in this environment and the spread of the plasmid in the new host would be favored. Conversely, when the resident population already contains a high donor population, the presence of I due to basal levels of prgQ expression would keep PrgX levels high, allow repression of promoter PQ to be maintained, and prevent wasteful induction events. However, the presence of a very small number of highly induced cells (detected by HCR) under conditions where the overall level of induction is low may extend the opportunity for dissemination of the plasmid. This could be significant over evolutionary time scales, without a high fitness cost for the donor population.
Experimental Procedures
Bacterial growth
The E. faecalis strains and plasmids used in this chapter are listed in Table 1.
Table 1.
Strains and plasmids used in this study.
| Strains | Description | Reference |
|---|---|---|
| OG1 | Reference strain | (Gold et al., 1975) |
| OG1RF | OG1 derivative with Rifampicin and Fusidic acid resistance | (Bourgogne et al., 2008; Dunny et al., 1978) |
| OG1Sp | OG1 derivative with Spectinomycin resistance | (Kristich et al., 2007) |
| JRC104 | OG1RF derivative that does not produce the cCF10 peptide pheromone due to a nonsense point mutation in the ccfA gene | (Kristich et al., 2007) |
| DM100–5 | OG1RF derivative with chromosomally integrated expression of PrgX from a promoter with an optimized Ribosomal Binding Site | (Fixen et al., 2007) |
| DM101–12 | OG1RF derivative (100–5) with chromosomally integrated expression of PrgX from a promoter with a mutated Ribosomal Binding Site | This study |
| Plasmids | Description | Reference |
| pCF10 | Native E. faecalis conjugative plasmid encoding for Tetracycline resistance and inducible expression of genes for conjugation machinery. | (Dunny et al., 1981; Hirt et al., 2005; Ruhfel et al., 1993) |
| p043lacZdx | Contains pCF10 regulatory region with a lacZ fusion downstream of IRS1. It could have inducible expression of lacZ in lieu of genes encoding conjugation machinery, but due to a deletion in the prgX gene sequence from the SpeI site to the 3’ end and therefore lack of PrgX protein, it has constitutive lacZ expression. It allows for iCF10 production and encodes for Erythromycin resistance. | (Kozlowicz et al., 2004) |
| pBK2 | Contains pCF10 regulatory region with a lacZ fusion downstream of IRS1, thus it has inducible expression of lacZ in lieu of genes encoding conjugation machinery, allows for iCF10 production, encodes for Chloramphenicol resistance. | (Shokeen et al., 2010) |
| pBK2idT | Derivative of the pBK2, contains pCF10 regulatory region, has inducible expression of lacZ in lieu of genes encoding conjugation machinery, does not produce functional iCF10 due to deletion of 3 nucleotides from prgQ leading to production of an inactive iCF10-like peptide which cannot bind PrgX, encodes for Chloramphenicol resistance. | (Borrero et al., 2015; Chatterjee et al., 2013) |
E. faecalis strains were grown statically at 37ºC in M9 medium containing 3 g l−1 yeast extract, 10 g l−1 casamino acids, 36 g l−1 glucose, 0.12 g l−1 MgSO4, and 0.011 g l−1 CaCl2. Antibiotics, when used for selection, were at following concentrations: tetracycline (Tet), 10 μg ml−1; chloramphenicol (Cm), 20 μg ml−1; erythromycin (Erm), 20 μg ml−1; spectinomycin (Spec), 1000 μg ml−1; rifampicin (Rif), 200 μg ml−1; and fusidic acid (Fus), 25 μg ml−1. Antibiotics were generally used to check for expected antibiotic resistances and not during experimental growth.
pCF10 was transferred between strains by conjugation and where necessary plasmids p043lacZdx, pBK2, and pBK2idT were transferred into strains by electroporation. Strains were plated on Brain Heart Infusion medium with 15 g l−1 agar (BHI-agar) containing antibiotics and/ or 250 ug ml−1 XGal (5-Bromo-4-Chloro-3-Indolyl β-D-Galactopyranoside) for selection and phenotypic confirmations.
Peptides iCF10 (I) and cCF10 (C) were kept in DMF stock solutions at 50 mg ml−1 and freshly diluted to their final concentration upon each use.
Strain construction
Strain DM101–12 was engineered from strain OG1RF to express prgX from the chromosome via cloning into the upp locus. This is analogous to how prgX was cloned for strain DM100–5 (Fixen et al., 2007). For DM101–12, the gene encoding prgX was inserted with a mutated ribosomal binding site (the sequence from the ribosome binding site to the prgX start codon is AGGAGC-9 bases-ATG). For DM100–5, the sequence from the ribosome binding site to the prgX start codon is AGGAGG-9 bases-ATG (Fixen et al., 2007).
Western blots
For the western blots in Figs. 2A and 3A, cultures were grown in M9 medium overnight, diluted 1:10 in fresh M9, and incubated for 2 hours. Culture densities were normalized to an OD600 of 1 in KPBS and cells from 1 ml of culture were pelleted to be processed for western blot. PrgX antibody raised in rabbit (Bae et al., 2000) was diluted 1:300 for use (in 10% milk in PBST, further described below). Western blots were done according to the protocol below.
For the western blots in Figs. 4A and 6, cultures were grown according to four conditions (referred to as “a”, “b”, “c”, and “d”) and used for analysis by western blots, XGal spot plates, and liquid β-galactosidase assays. Specifically, cultures were grown overnight in M9 medium then diluted and grown to log phase in medium without C (uninduced, condition “a”) or with 2.5 ng ml−1 C (condition “b”). For conditions “c” and “d”, cultures were grown overnight in medium containing 250 ng ml−1 I, and then diluted and grown to log phase in medium containing 250 ng ml−1 I (condition “c”) or 250 ng ml−1 I and 2.5 ng ml−1 C (condition “d”). For all four conditions, cultures were harvested 2 hours after dilution, normalized to an OD600 of 1 in KPBS and cells from 1 ml of culture were pelleted to be processed for western blot. PrgX antibody raised in rabbit was freshly absorbed (described below) before dilution 1:150 for use (in 10% milk in PBST, further described below). Again, these western blots were done according to the protocol below.
Western blots were done based on previously published methods (Bae et al., 2000; Towbin & Gordon, 1984) with specific methods variations described below. Cells (1 OD600 worth) were pelleted via centrifugation and resuspended in 15 mg ml−1 lysozyme in TES buffer comprised of 10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, and 25% sucrose. Resuspended cells were incubated in a 37°C water bath for 15 minutes to allow for peptidoglycan degradation and the resulting protoplasts were pelleted via centrifugation. Pellets were resuspended in 50 µl 1× SDS-PAGE loading dye comprised of 31.3 mM Tris, pH 6.8, 20% sodium dodecyl sulfate (SDS), 5% glycerol, 0.125% bromophenol blue, and 5% β-Mercaptoethanol (βME) and vortexed to ensure resuspension and kept on ice until boiling. His-tagged purified PrgX protein dimer samples were thawed and diluted to a concentration of 10,000 ng ml−1 using a buffer comprised of 20 mM Tris and 300 mM NaCl. Toward the goal of loading 30 ng protein on the gel, 6 µl of 10,000 ng ml−1 protein was mixed with 4 µl ddH2O and 10 µl 2x SDS-PAGE loading dye. This was also kept on ice until boiling. Samples were boiled for 3–5 minutes before 10 µl of each sample was loaded into separate lanes of a 12% SDS-PAGE gel (further described below). 5 µl of the Precision Plus Protein Western C blotting standard (BioRad) was also loaded into one lane. Before loading, the gel running apparatus was filled with 1× Tank Buffer pH 8.3 comprised of 25 mM Tris, 192 mM glycine, and 0.1% SDS. The gel was run at 395 Amp and 100 Volts until the loading dye was out of the stacking gel, and then at 150 Volts until the loading dye had reached the bottom of the gel.
To make the SDS-PAGE gel, a Hoefer gel casting device, Illumina plates, glass slide, and 1.5 mm spacers were used. Gel casting components were washed with 1× RBS (Sigma-Aldrich) and rinsed with dH2O. The resolving gel was made by combining 1.5 M Tris, pH 8.8, dH2O, 30% acrylamide/ 0.8 bisacrylamide, 10% SDS, 10% ammonium persulfate (APS), and TEMED. The resolving gel was pipetted into the casting device and isopropanol was used to cover the gel during polymerization. After 45 minutes, the stacking gel was made by combining 0.5 M Tris pH 6.8, dH2O, 30% acrylamide/ 0.8 bisacrylamide, 10% SDS, 10% ammonium persulfate (APS), and TEMED. After dumping out the isopropanol and rinsing with dH2O, the stacking gel was pipetted on top of the resolving gel and combs were inserted in the gel. Polymerization was allowed for 15 minutes before the combs were removed, wells were washed with Tank Buffer, and the gel was loaded and run as described above.
After running, the gel was prepared for protein transfer to a Whatman Protran BA 83 0.2 µm nitrocellulose membrane (GE Healthcare Life Sciences). The gel, a piece of nitrocellulose, transfer sponges, and two Whatman papers were soaked/ pre-wet in Towbin buffer for 15 minutes. Towbin buffer was comprised of 25 mM Tris, 192 mM glycine, and 20% methanol. The gel was subsequently sandwiched in a transfer cassette between sponges, Whatman paper, and the nitrocellulose membrane cut to the size of the gel. The transfer cassette was inserted in a transfer apparatus filled with Towbin buffer and run at 90 Volts, 0.3 Amp, and 300 Watts, for 70 minutes with the current flowing toward the nitrocellulose membrane.
After gel transfer, the gel was stained with GelCode Blue Stain Reagent (ThermoFisher) according to manufacture instructions and the nitrocellulose membrane was stained with 0.1% Ponceau S in 5% acetic acid. Ponceau S staining allowed observation of equal loading and transfer across the samples. Ponceau S de-staining was done with an aqueous solution of 0.1 M NaOH and subsequent rinsing in dH2O. The membrane was then transferred to 10% milk in PBST which was comprised of 10 mM KPBS pH 7.4 and 0.1% Tween 20 for blocking. Blocking was done gently rocking overnight at 4°C. After blocking, primary antibody to PrgX was diluted in PBST with 10% milk (details for each individual blot were described above) and membranes were incubated at room temperature, rocking with primary antibody for 1 hour. Next, the membrane was gently washed three times with PBST for 5 minutes each at room temperature. Horse Radish Peroxidase (HRP)- Goat Anti-rabbit IgG (H+L) secondary antibody (Invitrogen) was diluted 1:5000 in PBST with 10% milk and incubated for 1 hour at room temperature with the membrane with rocking. Finally, the membrane was again washed three times with PBST for 5 minutes each at room temperature.
To develop the western blots, SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher) was used according to manufacturer instructions. Blots were imaged on a FluorChem FC3 chemi-imager (proteinsimple) and/ or on x-ray film with varied exposure times from 30 seconds to overnight. Figs. 2A and 3A were imaged using x-ray film and Figs. 4A and 6 were imaged on the FluorChem FC3 chemi-imager.
For antibody absorption, (for the primary antibody to PrgX used in the blots shown in Figs. 4A and 6), 2 ml E. faecalis cell lysate was mixed with 150 µl of PrgX primary antibody-containing rabbit serum. This mix was inverted constantly overnight at 4°C and then centrifuged at 13,000 xg for 15 minutes. Aliquots of the resulting cell-free supernatant were stored at −20°C and used diluted 1:150 in PBST with 10% milk.
XGal (5-Bromo-4-Chloro-3-Indolyl β-D-Galactopyranoside) spot plates
In these spot plate experiments, β-galactosidase enzyme expression level is reflected by formation of blue 5,5’-dibromo-4,4’-dichloro-indigo product in the XGal spot plate assays and such activity is generally equated with inducibility in the summary of these results. For all spot plate experiments (Figs. 2A and 4B), 3 replicates were performed, and images shown are representative of the results.
For spot plates in Fig. 2A, cultures were grown in M9 medium overnight, diluted 1:10 in fresh M9, and incubated for 2 hours. Culture densities were normalized to an OD600 of 1 in KPBS and 5 µl of culture were spotted on BHI-agar plates containing 250 ug ml−1 XGal and varied concentrations of C (from 0 to 10 ng ml−1). Spots were dried, inverted, incubated for 16 h at 37°C before imaging.
For spot plates in Fig. 4B, the cultures used were the same as those used in the western blot and β-galactosidase assays presented in Figs. 4A and 5. These cultures were grown according to growth conditions “a” and “b” referenced above. Specifically, cultures were grown overnight in M9 medium then diluted and grown to log phase in medium without C (uninduced, condition “a”) or with 2.5 ng ml−1 C (condition “b”). Cultures were harvested 2 hours after dilution, normalized to an OD600 of 1 in KPBS, and 5 µl of culture were spotted on BHI-agar plates containing 250 ug ml−1 XGal and 2.5 ng ml−1 C or 250 ng ml−1 I. Spots were dried, inverted, incubated for 16 h at 37°C before imaging.
Beta-galactosidase assays
For the quantitative liquid β-galactosidase (β-gal) assays in Fig. 5, the cultures used were the same as those used in the western blots and XGal spot plate assays presented in Figs. 4A, 4B, and 6 or cultures grown as biological replicates according to the same conditions. All β-gal assays were performed with 3 biological replicates. Again, these 4 growth conditions were referenced as conditions “a”, “b”, “c”, and “d”. For conditions “a” and “b”, cultures were grown overnight in M9 medium then diluted and grown to log phase in medium without C (uninduced, condition “a”) or with 2.5 ng ml−1 C (condition “b”). For conditions “c” and “d”, cultures were grown overnight in medium containing 250 ng ml−1 I, and then diluted and grown to log phase in medium containing 250 ng ml−1 I (condition “c”) or 250 ng ml−1 I and 2.5 ng ml−1 C (condition “d”). For all four conditions, cultures were harvested 2 hours after dilution and a portion of the culture was used in the assay.
The liquid β-gal assays were done based on previous methods (Hedberg et al., 1996; Miller, 1972), but specifically, 1 ml of harvested culture was pelleted by centrifugation and resuspended in 1 ml 28°C Z buffer comprised of 60 mM Na2HPO47H2O, 40 mM NaH2PO4H2O, 10 mM KCl, 1 mM MgSO47H2O, and 50 mM βME. The OD600 of these resuspended cultures was determined. For each biological replicate of the β-gal assays for each sample, 2 technical replicates were set-up. To lyse cells, 40 µl toluene was added to 100 µl of culture in Z buffer, immediately vortexed for 10 seconds, and then incubated at 28°C for at least 10 minutes. Subsequently, 200 µl Z buffer containing 0.8 mg ortho-Nitrophenol-β-galactoside (ONPG) was added, tubes were vortexed, and incubations were continued at 28°C until yellow color had developed and this incubation time (tmin) was recorded. The tmin ranged from 5–15 minutes. Reactions were stopped via addition of 500 µl 1 M Na2CO3. Tubes were centrifuged for 15 minutes at 4°C and 13,000 xg and the resulting supernatant OD420 and OD550 were measured. Miller Unit values reflect amount of β-galactosidase activity (and therefore LacZ levels in cultures) and were calculated based on formation of colorimetric product ortho-nitrophenol by OD420. This calculation (formula shown below) takes into account enzyme assay run time (tmin), reaction volume (V), culture density (OD600), and light scattering by cell debris (OD550). The graph in Fig. 5 was made using GraphPad Prism (version 5).
Hybridization Chain Reaction (HCR) analysis
For the HCR experiment in Figs. 3B and 3C, overnight cultures were sub-cultured 1:10 in M9 medium, grown to early exponential phase (≈2 h to OD600 ≈ 1.2), induced with 0 or 2.5 ng ml−1 C, and harvested at varied times (0, 30, or 120 minutes) post C addition. Cells were fixed by mixing cultures 1:1 with 8% PFA (4% final concentration) and overnight incubation (>20 h) at 4°C before resuspension in KPBS with trace RNaseOUT (Invitrogen).
For the washing experiment in Figs. 7 and 8, overnight cultures were similarly sub-cultured 1:10 in M9 medium and grown to early exponential phase (≈2 h to OD600 ≈ 1.2). Cells were washed prior to resuspension with 2.5 ng ml−1 C for induction and harvest at 30 minutes post C addition. Cells were fixed by the same methods as above.
HCR labeling generally followed published methods (Breuer et al., 2017). Briefly, cells were permeabilized and HCR labeling and counterstaining was done on cells in suspension. HCR hybridization probes were designed to an early induced gene from pCF10 (prgB). Nucleic acid probes and hairpin amplifiers used in this study were obtained from Molecular Instruments (www.molecularinstruments.org) and used published probes to prgB transcripts (Breuer et al., 2017). The B2H1 and B2H2 amplifiers conjugated to Alexa Fluor 488 were used to detect prgB transcripts.
After cell labeling by HCR and counterstaining by Hoechst 33342, cells were mounted (Breuer et al., 2017). Images were acquired using a Zeiss Axio Observer.Z1 confocal microscope equipped with an LSM 800-based Airyscan super-resolution detector system (Zeiss). Confocal images were acquired through a 63× 1.40- numerical aperture (NA) objective (Zeiss) as z stacks at 0.15- μm intervals and using Zen software (version 2.1, Zeiss) these images were deconvolved and flattened using a maximum intensity projection with Ortho Display. From here, images were analyzed by high-throughput Matlab (version 2015b, Mathworks) analysis described below.
For HCR image analysis, the blue fluorescence channel (the reference channel corresponding to Hoechst fluorescence) was used as a proxy to define the pixel locations of individual cells and co-localized fluorescent overlap from HCR labeled transcripts was quantified (Figs. 3B, 3C, 7, and 8). Images corresponding to each fluorescent channel (that had been deconvolved and then flattened by maximum intensity projection and subjected to background subtraction) were imported into Matlab (version 2015b, Mathworks) in 16 bit TIFF format and analyzed as done previously (Breuer et al., 2017). To identify cell positions, the blue fluorescence (reference channel) images were binarized using Otsu’s method (Otsu, 1979) for thresholding. The internal Regionprops function was used to identify and characterize the sizes and pixel locations of objects in the image. Objects between 3 and 50 pixels in size were analyzed as cells in subsequent analysis. The PixelList property from the Regionprops function (called on the blue reference channel) was used to define the pixels corresponding to each cell. Using these coordinates, the HCR intensity value corresponding to each cell was calculated by taking the mean intensity of the pixels corresponding to each cell. The percentages of cells induced were determined based on a threshold HCR intensity of 50, which was chosen based on expression in the uninduced population and represents a conservative cut-off for induction (which likely mis-categorized some induced cells as uninduced). Graphs were created using Matlab and Excel (Microsoft Office, Version 1812). The image analysis script and supporting functions used in this study can be accessed at: https://github.com/dunnylabumn/Ef_HCR_ImageAnalysis
Supplementary Material
Acknowledgements:
We thank Arpan Bandyopadhyay for valuable discussions and help building the model figures and Aaron Barnes for microscopy assistance. These studies were supported by NIH grant R35GM118079 (G.M.D.). R.J.B.E. was supported in part by predoctoral traineeships under T32-GM008347 and T90-DE0227232. Computational resources were provided by the Minnesota Supercomputing Institute (MSI) at the University of Minnesota (http://www.msi.umn.edu). The data that support the findings of this study are available from the corresponding author upon reasonable request.
Footnotes
The authors state no conflicts of interest.
References:
- Bae T, & Dunny GM (2001). Dominant-negative mutants of prgX: Evidence for a role for PrgX dimerization in negative regulation of pheromone-inducible conjugation. Molecular Microbiology, 39(5), 1307–1320. 10.1046/j.1365-2958.2001.02319.x [DOI] [PubMed] [Google Scholar]
- Bae T, Clerc-Bardin S, & Dunny GM (2000). Analysis of expression of prgX, a key negative regulator of the transfer of the Enterococcus faecalis pheromone-inducible plasmid pCF10. Journal of Molecular Biology, 297(4), 861–875. 10.1006/jmbi.2000.3628 [DOI] [PubMed] [Google Scholar]
- Bae T, Kozlowicz B, & Dunny GM (2002). Two targets in pCF10 DNA for PrgX binding: Their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation. Journal of Molecular Biology, 315(5), 995–1007. 10.1006/jmbi.2001.5294 [DOI] [PubMed] [Google Scholar]
- Borrero J, Chen Y, Dunny GM, & Kaznessis YN (2015). Modified lactic acid bacteria detect and inhibit multiresistant enterococci. ACS Synthetic Biology, 4(3), 299–306. 10.1021/sb500090b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgogne A, Garsin DA, Qin X, Singh KV, Sillanpaa J, Yerrapragada S, et al. (2008). Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol, 9(7), R110 10.1186/gb-2008-9-7-r110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer RJ, Bandyopadhyay A, O’Brien SA, Barnes AMT, Hunter RC, Hu WS, & Dunny GM (2017). Stochasticity in the enterococcal sex pheromone response revealed by quantitative analysis of transcription in single cells. PLoS Genetics, 13(7). 10.1371/journal.pgen.1006878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta E, Haemig HAH, Manias DA, Tomsic J, Grundy FJ, Henkin TM, & Dunny GM (2012). In vivo and in vitro analyses of regulation of the pheromone-responsive prgQ promoter by the PrgX pheromone receptor protein. Journal of Bacteriology, 194(13), 3386–3394. 10.1128/JB.00364-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Johnson CM, Shu C-C, Kaznessis YN, Ramkrishna D, Dunny GM, & Hu W-S (2011). Convergent transcription confers a bistable switch in Enterococcus faecalis conjugation. Proceedings of the National Academy of Sciences, 108(23), 9721–9726. 10.1073/pnas.1101569108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Cook L, Shu C-C, Chen Y, Manias DA, Ramkrishna D, et al. (2013). Antagonistic self-sensing and mate-sensing signaling controls antibiotic-resistance transfer. Proceedings of the National Academy of Sciences, 110(17), 7086–7090. 10.1073/pnas.1212256110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bandyopadhyay A, Kozlowicz BK, Haemig HAH, Tai A, Hu W-SS, & Dunny GM (2017). Mechanisms of peptide sex pheromone regulation of conjugation in Enterococcus faecalis. MicrobiologyOpen, 6(4). 10.1002/mbo3.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny G, Funk C, & Adsit J (1981). Direct stimulation of the transfer of antibiotic resistance by sex pheromones in Streptococcus faecalis. Plasmid, 6(3), 270–278. 10.1016/0147-619X(81)90035-4 [DOI] [PubMed] [Google Scholar]
- Dunny GM (2013). Enterococcal sex pheromones: signaling, social behavior, and evolution. Annual Review of Genetics, 47(November), 457–82. 10.1146/annurev-genet-111212-133449 [DOI] [PubMed] [Google Scholar]
- Dunny GM, & Berntsson RPA (2016). Enterococcal sex pheromones: Evolutionary pathways to complex, two-signal systems. Journal of Bacteriology, 198(11), 1556–1562. 10.1128/JB.00128-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM, Brown BL, & Clewell DB (1978). Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proceedings of the National Academy of Sciences of the United States of America, 75(7), 3479–3483. 10.1073/pnas.75.7.3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fixen KR, Chandler JR, Le T, Kozlowicz BK, Manias DA, & Dunny GM (2007). Analysis of the amino acid sequence specificity determinants of the enterococcal cCF10 sex pheromone in interactions with the pheromone-sensing machinery. Journal of Bacteriology, 189, 1399–1406. 10.1128/JB.01226-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold OG, Jordan HV, & van Houte J (1975). The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Archives of Oral Biology, 20(7). 10.1016/0003-9969(75)90236-8 [DOI] [PubMed] [Google Scholar]
- Hedberg PJ, Leonard BAB, Ruhfel RE, & Dunny GM (1996). Identification and characterization of the genes of Enterococcus faecalis plasmid pCF10 involved in replication and in negative control of pheromone-inducible conjugation. Plasmid, 35(1), 46–57. 10.1006/plas.1996.0005 [DOI] [PubMed] [Google Scholar]
- Hirt H, Manias DA, Bryan EM, Marklund JK, Staddon JH, Paustian ML, et al. (2005). Characterization of the pheromone response of the Enterococcus faecalis conjugative plasmid pCF10: Complete sequence and comparative analysis of the transcriptional and phenotypic responses of pCF10-containing cells to pheromone induction. Journal of Bacteriology, 187(3), 1044–1054. 10.1128/JB.187.3.1044-1054.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt H, Greenwood-Quaintance KE, Karau MJ, Till LM, Kashyap PC, Patel R, & Dunny GM (2018). Enterococcus faecalis sex pheromone cCF10 enhances conjugative plasmid transfer in vivo. mBio, 9(1). 10.1128/mBio.00037-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Manias DA, Haemig HA, Shokeen S, Weaver KE, Henkin TM, & Dunny GM (2010). Direct evidence for control of the pheromone-inducible prgQ operon of Enterococcus faecalis plasmid pCF10 by a countertranscript-driven attenuation mechanism. Journal of Bacteriology, 192(6 PG-1634–42), 1634–1642. 10.1128/JB.01525-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowicz BK, Bae T, & Dunny GM (2004). Enterococcus faecalis pheromone-responsive protein PrgX: Genetic separation of positive autoregulatory functions from those involved in negative regulation of conjugative plasmid transfer. Molecular Microbiology, 54, 520–532. 10.1111/j.1365-2958.2004.04286.x [DOI] [PubMed] [Google Scholar]
- Kozlowicz BK, Shi K, Gu ZY, Ohlendorf DH, Earhart CA, & Dunny GM (2006). Molecular basis for control of conjugation by bacterial pheromone and inhibitor peptides. Molecular Microbiology, 62(4), 958–969. 10.1111/j.1365-2958.2006.05434.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristich CJ, Chandler JR, & Dunny GM (2007). Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid, 57(2), 131–144. 10.1016/j.plasmid.2006.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J (1972). Experiments in Molecular Genetics. Cold Spring Harbor protocols https://doi.org/papers2://publication/uuid/28F839D6-A272-4D50-9114-D2769DE72B38
- Otsu N (1979). A threshold selection method from gray-level histograms. IEEE Transactions on Systems, Man, and Cybernetics, 9(1), 62–66. 10.1109/TSMC.1979.4310076 [DOI] [Google Scholar]
- Ruhfel RE, Manias DA, & Dunny GM (1993). Cloning and characterization of a region of the Enterococcus faecalis conjugative plasmid, pCF10, encoding a sex pheromone-binding function. Journal of Bacteriology, 175(16), 5253–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, Brown CK, Gu Z-Y, Kozlowicz BK, Dunny GM, Ohlendorf DH, & Earhart CA (2005). Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proceedings of the National Academy of Sciences, 102(51), 18596–18601. 10.1073/pnas.0506163102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokeen S, Johnson CM, Greenfield TJ, Manias DA, Dunny GM, & Weaver KE (2010). Structural analysis of the Anti-Q-Qs interaction: RNA-mediated regulation of E. faecalis plasmid pCF10 conjugation. Plasmid, 64(1), 26–35. 10.1016/j.plasmid.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, & Gordon J (1984). Immunoblotting and dot immunobinding - Current status and outlook. Journal of Immunological Methods, 72(2), 313–340. 10.1016/0022-1759(84)90001-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.