

ABSTRACT
Human tumors exhibit plasticity and evolving capacity over time. It is difficult to study the mechanisms of how tumors change over time in human patients, in particular during the early stages when a few oncogenic cells are barely detectable. Here, we used a Drosophila tumor model caused by loss of scribble (scrib), a highly conserved apicobasal cell polarity gene, to investigate the spatial-temporal dynamics of early tumorigenesis events. The fly scrib mutant tumors have been successfully used to model many aspects of tumorigenesis processes. However, it is still unknown whether Drosophila scrib mutant tumors exhibit plasticity and evolvability along the temporal axis. We found that scrib mutant tumors displayed different growth rates and cell cycle profiles over time, indicative of a growth arrest-to-proliferation transition as the scrib mutant tumors progress. Longitudinal bulk and single-cell transcriptomic analysis of scrib mutant tumors revealed that the MAPK pathway, including JNK and ERK signaling activities, showed quantitative changes over time. We found that high JNK signaling activity caused G2/M cell cycle arrest in early scrib mutant tumors. In addition, JNK signaling activity displayed a radial polarity with the JNKhigh cells located at the periphery of scrib mutant tumors, providing an inherent mechanism that leads to an overall decrease in JNK signaling activity over time. We also found that ERK signaling activity, in contrast to JNK activity, increased over time and promoted growth in late-stage scrib mutant tumors. Furthermore, high JNK signaling activity repressed ERK signaling activity in early scrib mutant tumors. Together, these data demonstrate that dynamic MAPK signaling activity, fueled by intratumor heterogeneity derived from tissue topological differences, drives a growth arrest-to-proliferation transition in scrib mutant tumors.
This article has an associated First Person interview with the joint first authors of the paper.
KEY WORDS: Cell polarity, Drosophila tumor model, JNK, ERK
Summary: The authors provide evidence to show that a well-established Drosophila tumor model, caused by loss of apicobasal cell polarity, harbors a surprising degree of plasticity and evolvability along the temporal axis.
INTRODUCTION
Proteins essential for maintaining epithelial structures, such as cell polarity complexes, are involved in growth control (Bilder, 2004; Boggiano and Fehon, 2012; Sun and Irvine, 2016). For example, the basolateral Scribble complex, composed of Scribble (Scrib), Discs large (Dlg) and Lethal giant larvae [L(2)gl], was discovered as a group of ‘neoplastic tumor suppressor genes’ (nTSGs) in Drosophila (Bilder et al., 2000; Gateff, 1978; Woods and Bryant, 1991). Drosophila larvae homozygous mutants for any of the nTSGs grow into giant larvae with tumorous imaginal discs and optic lobes. These mutant tumors fail to differentiate and grow into masses that survive serial transplantations, induce cachexia and eventually kill the hosts (Figueroa-Clarevega and Bilder, 2015; Gateff, 1978). Studies of Drosophila nTSGs over decades have provided valuable insights into the mechanisms of growth control and tumorigenesis (Bilder, 2004; Gonzalez, 2013; Pastor-Pareja and Xu, 2013; Richardson and Portela, 2018; Sonoshita and Cagan, 2017). For example, analyses of nTSG mutant clonal growth have revealed the cooperative actions of multiple conserved signaling pathways during tumor development, cell competition-mediated tumor suppression mechanisms and tumor microenvironment influences (Brumby and Richardson, 2003; Chen et al., 2012; Cordero et al., 2010; Igaki et al., 2006; Katheder et al., 2017; Pagliarini and Xu, 2003; Vaughen and Igaki, 2016; Yamamoto et al., 2017). In particular, JNK and ERK signaling pathways, subgroups of the MAPK pathway (frequently deregulated in human cancers), have long been known as crucial for switching the growth outcome of scrib mutant cells when they are generated as mosaic loss-of-function clones surrounded by wild-type cells (Brumby and Richardson, 2003; Cordero et al., 2010; Igaki et al., 2006; Pagliarini and Xu, 2003; Uhlirova and Bohmann, 2006).
Interestingly, although Drosophila nTSG mutant tumors have successfully modeled many aspects of human epithelial cancers, it remains unclear whether and how the fast-growing fly nTSG mutant tumors change over time. The majority of mammalian tumors undergo evolution fueled by intratumor heterogeneity (McGranahan and Swanton, 2017). Many sources can contribute to intratumor heterogeneity. For example, individual tumor cells can acquire different types of mutations and grow into distinct subclones within the same tumor (de Bruin et al., 2014; Gerlinger et al., 2012). Moreover, most tumor cells co-exist with various immune and stromal cells, and different niches further contribute to intratumor heterogeneity (Jimenez-Sanchez et al., 2017). Whether the fly scrib mutant tumors exhibit some degree of intratumor heterogeneity is unclear.
Through quantitative analysis of Drosophila scrib mutant tumor growth, we found that over time scrib mutant tumors display different growth rates and cell cycle profiles. Moreover, quantitative changes in MAPK signaling activity underlie the change in growth rate. In particular, high JNK signaling activity is a primary cause of growth arrest in early scrib mutant tumors. We further found that JNK signaling activation is highly heterogeneous in scrib mutant tumors. JNK signaling activity displays a radial polarity, with JNKhigh cells located at the tumor periphery, which underlies a decrease in JNK signaling activity over time. Together, these data reveal that fly scrib mutant tumors are highly dynamic on the temporal axis.
RESULTS
The scrib mutant tumors display different growth rates and cell cycle profiles over time
To explore temporal changes in scrib mutant tumors, we monitored the growth of wing imaginal discs derived from eggs collected from a scrib1 allele over 3 h (Bilder et al., 2000) (Fig. 1A,B). We quantitatively measured the volumes of the scrib mutant tumors, which are known to grow over an extended larval period (Bunker et al., 2015; Vaccari and Bilder, 2005). On average, at 4 and 5 days after egg laying (AEL), the volumes of the scrib mutant tumors were around 15-30% of those of control imaginal discs raised under identical conditions (Fig. 1A,B). We calculated the growth rate (defined as the average volume change per day) in scrib mutant and wild-type imaginal discs based on the volume measurements in Fig. 1B and found that the growth rate of scrib mutant tumors increased over time and, starting from day 7, became comparable with that of the control group at 4 days AEL (Fig. 1C). Note that the scrib mutant tumors monitored in our experiments eventually grew to sizes consistent with previous reports (Bilder et al., 2000; Gateff, 1978).
Fig. 1.

The scrib mutant cells display different growth rates and cell cycle profiles over time. (A) Examples of a control imaginal disc collected at 5 days AEL and scrib1 mutant wing imaginal discs at 4-10 days AEL, stained for actin (red) and DNA (blue). Control genotype was FRT82B. Scale bar: 20 μm. (B) Quantification of volumes for control and scrib1 mutant wing imaginal discs over time. Control genotype was FRT82B raised under identical conditions. Control, 4 days n=20, 6±1×105 μm3, 5 days n=27, 1.8±0.4×106 μm3; scrib1 mutant, 4 days n=22, 2±1×105 μm3, 5 days n=30, 3±1×105 μm3, 6 days n=26, 8±3×105 μm3, 7 days n=29, 1.5±0.7×106 μm3, 8 days n=31, 3±2×106 μm3, 9 days n=24, 5±2×106 μm3, 10 days n=22, 6±2×106 μm3. Note that larvae from the control group become pupae at 5 days AEL. (C) Quantification of volume change per day for control and scrib1 mutant wing imaginal discs over time. Control genotype was FRT82B raised under identical conditions. (D-F) FACS analysis of cell cycle profiles of controls at 4 days AEL (D) and scrib1 mutant wing imaginal disc cells at 4 (E) and 8 (F) days AEL. Genotype of the control group was Ubi-GFP.E2f1 Ubi-mRFP1.CycB; FRT82B. Genotype of the scrib mutant group was Ubi-GFP.E2f1 Ubi-mRFP1.CycB; scrib1. At least 5000 cells were recorded for each cell group. Each experiment was replicated at least three times. (G) Average percentage of cells in G0/G1, S and G2/M phases. Error bars indicate standard deviation.
To test whether the growth rate change we observed was specific to the scrib1 allele, we also quantified the growth rate of tumors depleted of Scrib or Dlg protein at the posterior region of wing discs using engrailed-Gal4-mediated RNAi. At 4 and 5 days AEL, the volume of scrib RNAi and dlg RNAi tumors was much smaller than the size of the posterior region in control wing discs (Fig. S1). However, the anterior regions of the scrib RNAi, dlg RNAi and control imaginal discs had comparable volumes (Fig. S1), indicating that the growth arrest phenotype observed in early scrib tumors is likely to be inherent and independent of an overall delay in larval development. Interestingly, from 4 to 6 days AEL we detected an increase in phosphorylated histone H3-positive (PH3+) cell number in the posterior scrib RNAi and dlg RNAi tumors (Fig. S1), indicative of changing growth rates during tumor progression. The fly larvae harboring scrib RNAi or dlg RNAi imaginal discs turned into pupae by day 7 AEL, preventing further measurement of growth rates. Notably, the cell competition process mediated by Minute mutations and differential Myc levels does not cross compartment boundaries (Johnston, 2009; Morata and Ripoll, 1975; Simpson, 1979; Simpson and Morata, 1981). However, tumorigenic dCsk-deficient cells generated in the patched expression domain are known to undergo apoptosis at the anterior–posterior boundary (Vidal et al., 2006). It is therefore likely that the growth of posterior scrib RNAi and dlg RNAi tumors is largely independent of the influence of cell competition, except for cells at the anterior–posterior boundary.
It is possible to attribute the growth arrest phenotype of early scrib mutant tumors to increased apoptosis, defects in cellular growth or defective cell proliferation. However, we found that prevention of apoptosis by overexpressing p35 did not rescue the growth arrest of posterior scrib RNAi cells (Fig. S2). Moreover, although we were able to detect apoptotic cells in the posterior scrib RNAi region, a similar number of apoptotic cells could also be detected in the anterior control region (data not shown). Therefore, the growth arrest phenotype of early scrib mutant tumors is unlikely to be caused by increased apoptosis. The cell volume of individual scrib mutant cells was larger than that of the control wing disc cells (Fig. S3). Therefore, the growth arrest of early scrib mutant tumors is also unlikely to be caused by defects in the growth of individual cells.
We then performed flow cytometry analysis with wild-type imaginal discs and scrib mutant tumors expressing fluorescence ubiquitin cell cycle indicator (FUCCI) sensors to examine possible cell proliferation defects (Zielke et al., 2014). In wild-type discs at day 4 AEL, about 60% of cells were in the G0/G1 phase, 25% in the S phase and 15% in the G2/M phase (Fig. 1D,G), in agreement with previous reports (Milan et al., 1996). However, in scrib mutant tumors at 4 days AEL about 25% of cells were in the G0/G1 phase, 25% in the S phase and 50% in the G2/M phase (Fig. 1E,G). The results suggest that a population of cells in early scrib mutant tumors is arrested at the G2/M stage.
Drosophila larval brain neuroblasts are an excellent model for analyzing mitosis because of their accessibility for live imaging (Cabernard and Doe, 2013). We observed that scrib mutant neuroblasts frequently displayed a significantly prolonged entry into mitosis (Fig. S4), consistent with the G2/M arrest defects observed in early scrib mutant wing disc cells. Notably, by day 8 AEL, the cell cycle distribution of scrib mutant cells was comparable with that of day 4 wild-type imaginal discs (Fig. 1F,G). Therefore, the growth arrest phenotype in early scrib mutant tumors is caused by cell cycle defects, which can resolve over time.
The scrib mutant tumors from different time points display distinctive global transcriptomic signatures
Early and late scrib mutant tumors display different growth rates and cell cycle profiles, indicative of potential evolving capacity. We therefore performed bulk RNA sequencing (RNA-seq) for wild-type imaginal discs and scrib mutant tumors collected at different time points to gain a global view of transcriptomic changes associated with scrib tumor progression (Fig. 2). The biological repeats were well clustered per sample for each time point and the scrib mutant tumors formed a transition trajectory along time on the principal component analysis (PCA) plot (Fig. 2A). Both PCA and hierarchical clustering analyses suggested that the scrib mutant tumors could be grouped into three stages with distinctive transcriptomic signatures: an early stage at 4-6 days AEL, an intermediate stage at 8 days and a late stage at 10-14 days (Fig. 2A,B).
Fig. 2.

The scrib mutant tumors from different stages display distinctive global transcriptomic signatures. (A) PCA of transcriptomes from control and scrib mutant wing imaginal discs collected at different ages. Control genotype was FRT82B. Three or four biological replicates were plotted for each time point. Low-expression genes were filtered using a cutoff of baseMean>100 calculated in DEseq2. A matrix of 6901 gene counts×34 samples was used to compute PCA after gene counts were transformed using the default DEseq2 rlogTransformation function. (B) Hierarchical clustering of transcriptomes from control and scrib mutant wing imaginal discs collected at different ages. Control genotype was FRT82B. Three or four biological replicates were plotted for each time point. Gene filtering criteria were the same as for A. A matrix of 6901 gene counts×34 samples was used for hierarchical clustering after gene counts were transformed to counts per million (cpm) value using the edgeR cpm function and scaled using the scale function in R. (C,D) KEGG pathway enrichment analysis for the top 10% of upregulated genes (C) and the bottom 10% of downregulated genes (D) over time with a cutoff of adjusted P-value <0.05. The pathway enrichment analysis was performed using enrichKEGG function in the clusterProfiler package.
We performed linear regression analysis for each gene in the scrib tumor time series and ranked the genes by the slope of the regression line (Fig. S5). For the 10% of genes ranked at the top and bottom, we performed pathway enrichment analysis with the KEGG pathway database (Kyoto Encyclopedia of Genes and Genomes, https://www.genome.jp/kegg/) (Fig. 2C,D) as well as the Drosophila GLAD database (Gene List Annotation for Drosophila, https://www.flyrnai.org/tools/glad/web/) (Hu et al., 2015) (Fig. S6). For example, the expression level of genes related to oxidative phosphorylation decreased over time and the expression level of glycolytic genes increased over time in the scrib mutant tumors, consistent with a glucose metabolism drift widely observed in tumors called the Warburg effect (Eichenlaub et al., 2018; Wang et al., 2016). Of the several conserved signaling pathways that show significant changes in scrib mutant tumors over time, the pathway enrichment analysis with both KEGG and GLAD databases pointed us to the MAPK signaling pathway (Fig. 2C; Fig. S6A). Notably, two of the three Drosophila MAPK pathway subgroups, the JNK and ERK pathways, have long been known as crucial for regulating the growth outcome of scrib mutant mosaic clones (Brumby and Richardson, 2003; Cordero et al., 2010; Igaki et al., 2006; Pagliarini and Xu, 2003; Uhlirova and Bohmann, 2006). Therefore, although the transcriptomic analysis suggested that scrib mutant tumors have extensive changes besides the MAPK pathway, including several metabolic pathways and other signaling pathways such as the Notch and JAK/STAT pathways, we first focused on examining whether JNK and ERK signaling activities change over time and whether these changes have functional implications regarding scrib tumor growth.
High JNK signaling activity inhibits proliferation in the early scrib mutant tumors
We first examined JNK signaling activity because the expression levels of two widely used JNK signaling target genes, Mmp1 and puckered (puc) (Igaki et al., 2006; Uhlirova and Bohmann, 2006), as measured in the bulk RNA-seq experiments (Fig. 2), decreased significantly in scrib mutant tumors over time (Fig. 3A). Although JNK signaling activity is elevated in scrib tumors of all stages compared with wild-type wing discs (Fig. 3A,B), in agreement with previous reports (Bunker et al., 2015; Igaki et al., 2006), we found that the protein levels of Mmp1 and TRE-DsRED, a JNK signaling reporter (Chatterjee and Bohmann, 2012), decreased significantly in scrib tumors at 8 days AEL compared with scrib tumors at 5 days AEL (Fig. 3B). These data suggest that JNK signaling activity decreases as the scrib tumors grow over time.
Fig. 3.

High JNK signaling activity causes growth arrest in early scrib mutant tumors. (A) Gene expression levels for puc and Mmp1, the JNK signaling activity reporter genes, as FPKM (fragments per kilobase million) values measured in the bulk RNA-seq experiments shown in Fig. 2A,B. Control genotype was FRT82B. Statistical analysis was performed using the one-way ANOVA test. (B) Western blot analysis of Mmp-1 and TRE-DsRed protein levels in control imaginal discs at 5 days AEL and scrib1 mutant wing imaginal discs at 5 and 8 days AEL. Control genotype was scrib1/TM6B. (C,D) Imaginal discs from dlgGH19 (C) and dlgGH19 Tak1DN (D) at 5 days AEL, stained for actin (red). Genotype for C was dlgGH19/Y; c855a-Gal4/+, n=19, 2.2±0.6×105 μm3. Genotype for D was dlgGH19/Y; UAS-Tak1DN/+; c855a-Gal4/+, n=30, 1.3±0.6×106 μm3. (E) Quantification of the tumor volumes. Statistical analysis was performed by unpaired t-test. (F,G) Imaginal disc from scrib1 (F) and scrib1 Tak1DN (G) at 5 days AEL, stained for actin (red). Genotype for F was c855a-Gal4 scrib1/scrib1, n=19, 8±3×105 μm3. Genotype for G was UAS-Tak1DN/+; c855a-Gal4 scrib1/scrib1, n=17, 1.4±0.9×106 μm3. (H) Quantification of the tumor volumes. Statistical analysis was performed by unpaired t-test. (I) t-SNE projection of scrib1 mutant cells at 4, 5, 8 and 14 days AEL, where individual cells are colored by the normalized expression level of Mmp1. The gene expression level normalization was performed using the default LogNormalize method in Seurat. (J,L) AEL scrib1 imaginal disc at 5 (J) and 8 (L) days AEL, stained for actin (blue) and PH3 (green). Genotype was TRE-dsRED/+; scrib1/scrib1. (K,M) Quantification of the PH3+ cell number per unit volume. 5 days TRE-DsRed–, n=20, 3.1±0.5/108 μm3; 5 days TRE-DsRed+, n=20, 10±2/107 μm3. 8 days TRE-DsRed–, n=15, 4±1/108 μm3; 8 days TRE-DsRed+, n=15, 5±2/107 μm3. Statistical analysis was performed by unpaired t-test. Scale bars: 10 μm.
We also found that the growth arrest phenotype in early scrib or dlg tumors was effectively rescued through overexpression of a dominant-negative form of Tak1 (Tak1DN) or Basket (BskDN), which block JNK signaling activity (Fig. 3C-H; Fig. S7). Notably, overexpression of RasV12, NICD or YkiS168A (an active form of Yki), which are known to promote scrib clonal growth in a mosaic setting (Brumby and Richardson, 2003; Chen et al., 2012; Pagliarini and Xu, 2003), did not rescue the growth arrest phenotype in early scrib tumors (Fig. S8). Note that we used a C855a-Gal4 driver that expresses early in whole scrib or dlg tumors and does not cause lethality when RasV12 or NICD are co-expressed (Fig. S9). Together, the data suggest that high JNK activity is a potent inhibitor of tumor growth in early scrib tumors. This result is consistent with a recent study showing that injury-induced JNK activation in wing imaginal discs also leads to cell cycle arrest at G2 (Cosolo et al., 2019).
We next examined why JNK signaling activity decreases as the scrib tumors grow. From single-cell transcriptomic data generated from scrib mutant tumors, we found that expression of the JNK signaling reporter gene Mmp1 is highly heterogeneous (Fig. 3I). This data led us to re-examine the Mmp1 expression pattern; we found that Mmp1 showed strongest signals at the periphery of scrib mutant tumors (Movies 1, 2). To exclude the possibility that the Mmp1 staining pattern was an artifact from poor antibody penetrance, we examined the expression pattern of a JNK signaling reporter TRE-DsRED without using antibody staining. Similarly, we found that TRE-dsRED displayed a radial polarity, with the strongest fluorescence signals at the periphery of scrib mutant tumors (Fig. 3J-M). Moreover, single-cell transcriptomic analysis demonstrated that Mmp1 expression shows high correlation with a number of cytoskeletal genes, such as cindr (Table S1; Fig. S10). A protein-trap line of cindr also revealed a radial polarity in scrib mutant tumors (Movies 3, 4). Therefore, JNK signaling activation in homozygous scrib mutant tumors exhibited a radial polarity, with JNKhigh cells located at the surface of the scrib tumors. Because scrib mutant tumors grow in a compact manner, the surface-to-volume ratio drops as the tumor volumes increases, leading to a decrease in the percentage of JNKhigh G2/M-arrested cells over time.
ERK signaling activity increases over time and promotes tumor growth in late-stage scrib mutant tumors
The bulk scrib tumor time series transcriptomic data demonstrated that components of the ERK signaling pathway also showed significant changes (Fig. 4A). Moreover, when we ordered genes by the change in the percentage of cells expressing the specific gene in single-cell transcriptomic data, we noticed that kekkon1 (kek1), a well-established ERK signaling target (Ghiglione et al., 2003, 1999), was ranked at the top. Few kek1+ cells existed in early scrib tumors and the majority of cells were kek1+ in late scrib tumors (Fig. 4B; Fig. S11). Using a kek1-lacZ line (Musacchio and Perrimon, 1996), we confirmed that late scrib tumors indeed harbored many more kek1+ cells than early scrib tumors (Fig. 4C-E). Similarly, the expression of sprouty (sty) (Casci et al., 1999) and argos (aos) (Golembo et al., 1996), two other genes induced by ERK activity, also increased over time (Fig. 4A; Fig. S12). Moreover, the expression of vein (vn), an EGFR ligand promoting patterning and proliferation in wing imaginal discs (Schnepp et al., 1996), also increased over time (Fig. 4A; Fig. S12). Together, these data suggest that ERK activity increases over time as scrib mutant tumors grow.
Fig. 4.

ERK signaling activity increases over time and promotes growth of late-stage scrib mutant tumors. (A) Expression levels for aos, kek1, sty, the EGFR signaling activity reporter genes, and vn, an EGFR signaling ligand, in scrib mutant tumors normalized by the respective expression level in control imaginal discs at 5 days AEL using log2(FPKM fold change) values. Control genotype was FRT82B. (B) t-SNE projection of scrib1 mutant cells at 4, 5, 8 and 14 days AEL, where individual cells are colored by the normalized expression level of kek1. The gene expression level normalization was performed using the default LogNormalize method in Seurat. (C) Western blot analysis of β-galactosidase protein level in control imaginal discs at 5 days AEL and in kek1-lacZ; scrib1 mutant wing imaginal discs at 5 and 8 days AEL. Control genotype was kek1-lacZ/+; scrib1/TM6B. (D,E) kek1-lacZ; scrib1 mutant tumors at 5 (D) and 8 (E) days AEL, stained for β-galactosidase (green) and actin (red). (F,G) scrib RNAi (F) and scrib RNAi RasV12(G) imaginal discs at 8 days AEL, stained for actin (red). Genotype for F was c855a-Gal4/UAS-scrib RNAi, n=32, 8±3×106 μm3. Genotype for G was UAS-RasV12/+; c855a-Gal4/UAS-scrib RNAi, n=28, 1.6±0.7×107 μm3. (H) Quantification of the tumor volumes. (I,J) scrib1 (I) and scrib1 EGFRDN (J) imaginal discs at 8 days AEL, stained for actin (red). Genotype for (I) was c855a-Gal4 scrib1/scrib1, n=21, 4±2×106 μm3. Genotype for J was UAS-EGFRDN/+; c855a-Gal4 scrib1/ scrib1, n=31, 2±1×106 μm3. (K) Quantification of the tumor volumes. (L,M) dlgGH19 (L) and dlgGH19 EGFRDN (M) imaginal discs at 8 days AEL, stained for actin (red). Genotype for L was dlgGH19/Y; c855a-Gal4/+, n=21, 1.4±0.6×106 μm3. Genotype for M was dlgGH19/Y; UAS-EGFRDN/+; c855a-Gal4/ UAS-EGFRDN, n=18, 8±7×105 μm3. (N) Quantification of the tumor volumes. All statistical analyses were performed by unpaired t-test. Scale bars: 10 μm.
ERK signaling activity is required for both growth and patterning in wing imaginal discs (Diaz-Benjumea and Hafen, 1994; Prober and Edgar, 2000; Zecca and Struhl, 2002a,b). Here, we focused on examining whether increasing ERK activity over time influences scrib tumor growth. Interestingly, although overexpression of RasV12 did not rescue early scrib mutant tumors from growth arrest (Fig. S8), it significantly increased the size of scrib mutant tumors at later stages (Fig. 4F-H). Conversely, blocking ERK signaling activity through expression of a dominant-negative form of EGFR (Buff et al., 1998) (Fig. 4I-N) and ERK and Ras RNAi constructs (Fig. S13) led to a reduction in scrib and dlg mutant tumor sizes at later stages. These data suggest that an increase in ERK signaling activity promotes scrib tumor growth at later stages.
Interestingly, ERK and JNK pathways have extensive cross-talks in scrib mutant tumors, as indicated by the high correlation of a number of ERK pathway components with Mmp1 expression at the single-cell level (Table S1; Fig. S14). Moreover, when we blocked JNK signaling activity in early scrib mutant tumors, we observed an increase in kek1+ cell number (Movies 5, 6; Fig. S15) in addition to an increase in tumor size (Fig. 3C-H), indicating that high JNK signaling activity possibly represses ERK activity in early scrib mutants. However, the molecular mechanism mediating JNK repression of ERK activity remains unclear.
DISCUSSION
Here we have demonstrated that dynamic changes in JNK and ERK activities underlie a transition from a growth arrest state to a proliferation state over time in Drosophila scrib mutant tumors.
We found that JNK signaling activation exhibited a radial polarity, with JNKhigh cells located at the surface of homozygous scrib mutant tumors. We do not yet know the underlying reason for the heterogeneous JNK activation pattern. One possible reason is that the active JNK ligand Eiger might be mostly provided from external sources. This might involve hemocytes (Cordero et al., 2010) or the fat body, as recently shown for another nTSG mutant alg3 (de Vreede et al., 2018). Another possible input is potential mechanical stresses that the tumor surface cells experience because JNK signaling is activated at leading-edge cells, which assemble supracellular actin cables and experience high actomyosin-dependent tensile force during dorsal closure (Martin-Blanco et al., 1998).
JNK activation induced by injury in the wing imaginal discs also leads to cell cycle arrest at G2, which can be restored by overexpression of Cdc25/String (Cosolo et al., 2019). We found that expression of Cdc25/string(stg) and Cks30A anti-correlates with Mmp1 in the single-cell data. Moreover, we could not rescue growth arrest in early scrib mutant tumors by overexpression of String alone (data not shown), indicating that the cell cycle defects in early scrib mutant tumors are probably mediated by multiple factors downstream of high JNK signaling activity.
Clonal scrib mutant cells are eliminated through cell competition when they are surrounded by wild-type neighbors (Brumby and Richardson, 2003; Vaughen and Igaki, 2016; Yamamoto et al., 2017). It is noteworthy that clonal scrib mutant cells are likely to be in a different state from growth-arrested homozygous scrib mutant cells. Studies have shown that overexpression of RasV12, NICD and p35 can effectively block the clonal scrib mutant cells from undergoing apoptosis induced by cell competition. In our study, we found that overexpression of RasV12, NICD and p35 had little effect in relieving the early scrib mutant tumors from growth arrest. It is likely that interactions between clonal scrib mutant cells and wild-type cells during cell competition induce an additional layer of complexity into determination of clonal scrib mutant cell state, as the effects of JNK activation are known to be highly context dependent (Pinal et al., 2019).
In human solid tumors, the tumor margin and core were also shown to experience different immune environments (Kather et al., 2018). Our study highlights that tissue topological factors (peripheral versus center) can be an inherent source of diversity in cell populations in growing tumors and that dynamic signaling rewiring during the processes of early tumorigenesis does not necessarily require the generation of de novo mutations or new cell clones.
MATERIALS AND METHODS
Fly stocks
The fly strains used in this study were scrib1 FRT82B/TM6B (Bilder et al., 2000), dlgGH19 FRT19A/FM7c (a kind gift from the Schupbach laboratory, Princeton University; a C-to-T mutation led to a stop codon at Q42), UAS-scrib RNAi on the second chromosome (Bloomington/BL38199), UAS-scrib RNAi on the third chromosome (BL35748), UAS-dlg RNAi on the third chromosome (BL35772), UAS-Ras85D RNAi (BL34619), UAS-ERK RNAi(BL34855), y[1] v[1]; P{y[+t7.7]=CaryP}attP2 (BL36303, the third chromosome TRiP line background strain), y[1] v[1]; P{y[+t7.7]=CaryP}attP40 (BL36304, the second chromosome TRiP line background strain), engrailed-Gal4 UAS-GFP (BL25752), engrailed-Gal4 (BL30564), UAS-p35 (Hay et al., 1994) (BL6298), Fly-FUCCI (BL55098), worGal4, UAS-cherry::Jupiter, Sqh::GFP (Cabernard and Doe, 2013), c855a-Gal4 (Hrdlicka et al., 2002) (BL6990), UAS-RasV12 (Karim and Rubin, 1998), UAS-YkiS168A (Oh and Irvine, 2009) (BL28818), UAS-NICD (Rebay et al., 1993), UAS-BskDN (Igaki et al., 2006) (a kind gift from Jose C. Pastor-Pareja School of Life Sciences, Tsinghua University, China), UAS-Tak1DN (BL58811), TRE-dsRED (Chatterjee and Bohmann, 2012), kek1-lacZ (Ghiglione et al., 2003) and UAS-EGFRDN (Buff et al., 1998) (BL5364), cindrCA06686 (BL50802).
Immunohistochemistry
About 50 embryos collected within 3 h were put into an individual vial of fly food to avoid crowding and the larvae raised in an incubator at 25°C for appropriate lengths of time before dissection. Imaginal discs were fixed and stained according to standard protocols. The primary antibodies used were mouse anti-phospho-Histone3 (1:1000; Cell Signaling Technology), mouse anti-Mmp 1(1:30; a mixture of 5H7B11, 3A6B4 and 3B8D12, DSHB) and mouse anti-beta-galactosidase (1: 25, 40-1a, DHSB). The secondary antibodies conjugated with various Alexa Fluor dyes (Thermo Fisher Scientific) were used at 1:500. Phalloidin conjugated with Alexa Fluor dyes (1:1000, Thermo Fisher Scientific) and Hoechst 33342 (1:10,000, Thermo Fisher Scientific) were used to stain F-actin and DNA, respectively. All images were acquired on a Leica TCS SP8 confocal microscope.
Western blotting
About 30 larvae were dissected in PBS. Cell lysates were homogenized in 1× RIPA (Millipore) with protease inhibitors (Roche). The primary antibodies used were mouse anti-MMP1 (1:100; a mixture of 5H7B11, 3A6B4 and 3B8D12, DSHB), rabbit anti-DsRed (1:1000; Takara) mouse anti-beta-galactosidase (1:250; 40-1a, DHSB) and mouse anti-alpha-tubulin (1:5000; 12G10, DSHB).
Image processing and data analysis
Images were taking as z-stacks with a step size of 1 μm. Tissue volume was measured with Measure Stack plugin in Fiji using the actin-staining channel; the wand tool was used to select regions of interest, where manual adjustment of tolerance value and recognition of edges were made for each slice. PH3+ cell number was calculated with the Cell Counter plugin in Fiji.
Fluorescence-activated cell-sorting
Wing imaginal discs were dissociated in 0.25% trypsin-EDTA solution at 37°C for 10 min and stained with Hoechst at 37°C for 45 min. Cells were sorted with BDFACSAria IIIu and the data analyzed with FlowJo.
Drosophila neuroblast live imaging
Female virgins of hsFLP; worGal4, Sqh::GFP,UASCherry::jupiter; FRT82B/TM6B were crossed with males of scribFRT82B/TM6B. Progeny were heat-shocked at 38°C (in the water bath) for 1 h and subsequently raised at 25°C until imaging. Larvae (5 days AEL) were then dissected and imaged according to standard protocols (Cabernard and Doe, 2013). For wild-type controls, larvae expressing wt; worGal4,Sqh::GFP,UASCherry::jupiter; Dr/TM6B were dissected and imaged with the same laser setting as that of the scrib mutant neuroblasts.
Bulk RNA-seq and data analysis
Total RNA was extracted from control and scrib1 wing imaginal discs with the RNeasy Mini Kit (Qiagen). Construction of cDNA libraries and 150 bp paired-end sequencing on Illumina HiSeq platform were performed by Novogene. Cleaned raw reads obtained from Novogene were mapped to the reference genome (BDGP6) using STAR (2.5.4a) (Dobin et al., 2013). Default parameters were used except for the max intron size, which was set at 100,000. Counts were generated by featureCounts available in Subread package (1.32.2) with the default setting (Liao et al., 2013). PCA analysis was performed in DESeq2 (1.22.2) after low expression genes were filtered (baseMean>100) (Love et al., 2014). Count normalization to counts per million (cpm) was performed using edgeR (3.24.3) before hierarchical clustering (hclust function in R) (Robinson et al., 2010). Linear regression was performed using the lm function in R (3.5.2). Pathway enrichment analysis was performed using the default setting of clusterProfiler (3.10.1) (Yu et al., 2012). The GEO accession number for these data is GSE130243.
10x Genomics single cell RNA-seq data analysis
Single-cell RNA-seq data from staged scrib1 wing imaginal discs were generated in another study (M.D., Y.W., L.Z., Yang Yang, S.H., J.W., Hao Ge, Toyotaka Ishibashi and Y.Y., unpublished; GEO accession number GSE130566). Raw data mapping and primary analysis was performed in the Cell Ranger pipeline. The t-SNE plot for marker gene expression pattern was plotted using Seurat (v2.3.4) using default settings (Butler et al., 2018). To calculate the correlation coefficients for any two genes x and y in single cells, the normalized expression levels of genes x and y form two numeric vectors that can be used to compute the correlation coefficient (r) using Pearson’s formula:
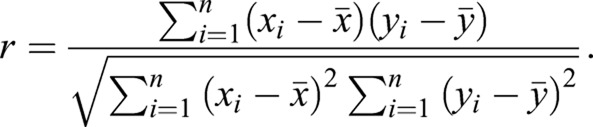 |
Supplementary Material
Acknowledgements
We thank Dr Chris Doe and Dr Jose C. Pastor-Pareja, Bloomington Stock Center and Developmental Biology Hybridoma Bank for providing fly stocks and reagents. We thank Dr Ting Xie for providing the 10× single-cell RNA-seq protocol. We thank Dr Zilong Wen and Dr Mingjie Zhang for sharing their confocal microscope. We thank Dr Gang Wang and Dr Zhenguo Wu for their help with FACS experiments. We thank Dr Chris Doe and Dr Trudi Schupbach for helpful comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Methodology: T.J., L.Z., M.D., S.H., J.W.; Software: M.D.; Formal analysis: T.J., L.Z., M.D., Y.Y.; Investigation: T.J., L.Z., Y.W., T.T.P., A.A.S., V.S., C.C.; Data curation: M.D.; Visualization: T.J., M.D.; Supervision: Y.Y.; Project administration: Y.Y.; Funding acquisition: Y.Y.
Funding
This work was supported by grants to Y.Y. from the Research Grants Council of the Hong Kong Special Administrative Region (Research Grants Council, University Grants Committee; GRF16103815, 16104018, 16150016, AoE/M-09/12) and the Shenzhen Science and Technology Innovation Commission (JCYJ20170306161537148, JCYJ20180223181229868), and grants to J.W. from the Hong Kong Research Grants Council (N_HKUST606/17, 26102719, C6002-17GF, C7065-18GF) and Innovation and Technology Commission - Hong Kong (ITCPD/17-9, ITS/480/18FP).
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.040147.supplemental
References
- Bilder D. (2004). Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18, 1909-1925. 10.1101/gad.1211604 [DOI] [PubMed] [Google Scholar]
- Bilder D., Li M. and Perrimon N. (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289, 113-116. 10.1126/science.289.5476.113 [DOI] [PubMed] [Google Scholar]
- Boggiano J. C. and Fehon R. G. (2012). Growth control by committee: intercellular junctions, cell polarity, and the cytoskeleton regulate Hippo signaling. Dev. Cell 22, 695-702. 10.1016/j.devcel.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby A. M. and Richardson H. E. (2003). scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769-5779. 10.1093/emboj/cdg548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buff E., Carmena A., Gisselbrecht S., Jimenez F. and Michelson A. M. (1998). Signalling by the Drosophila epidermal growth factor receptor is required for the specification and diversification of embryonic muscle progenitors. Development 125, 2075-2086. [DOI] [PubMed] [Google Scholar]
- Bunker B. D., Nellimoottil T. T., Boileau R. M., Classen A. K. and Bilder D. (2015). The transcriptional response to tumorigenic polarity loss in Drosophila. Elife 4, e03189 10.7554/eLife.03189.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E. and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411-420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabernard C. and Doe C. Q. (2013). Live imaging of neuroblast lineages within intact larval brains in Drosophila. Cold Spring Harb. Protoc. 2013, 970-977. 10.1101/pdb.prot078162 [DOI] [PubMed] [Google Scholar]
- Casci T., Vinós J. and Freeman M. (1999). Sprouty, an intracellular inhibitor of Ras signaling. Cell 96, 655-665. 10.1016/S0092-8674(00)80576-0 [DOI] [PubMed] [Google Scholar]
- Chatterjee N. and Bohmann D. (2012). A versatile PhiC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS ONE 7, e34063 10.1371/journal.pone.0034063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-L., Schroeder M. C., Kango-Singh M., Tao C. and Halder G. (2012). Tumor suppression by cell competition through regulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA 109, 484-489. 10.1073/pnas.1113882109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero J. B., Macagno J. P., Stefanatos R. K., Strathdee K. E., Cagan R. L. and Vidal M. (2010). Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell 18, 999-1011. 10.1016/j.devcel.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosolo A., Jaiswal J., Csordas G., Grass I., Uhlirova M. and Classen A. K. (2019). JNK-dependent cell cycle stalling in G2 promotes survival and senescence-like phenotypes in tissue stress. Elife 8, e41036 10.7554/eLife.41036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin E. C., McGranahan N., Mitter R., Salm M., Wedge D. C., Yates L., Jamal-Hanjani M., Shafi S., Murugaesu N., Rowan A. J. et al. (2014). Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251-256. 10.1126/science.1253462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vreede G., Morrison H. A., Houser A. M., Boileau R. M., Andersen D., Colombani J. and Bilder D. (2018). A Drosophila tumor suppressor gene prevents tonic TNF signaling through receptor N-glycosylation. Dev. Cell 45, 595-605.e594. 10.1016/j.devcel.2018.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Benjumea F. J. and Hafen E. (1994). The sevenless signalling cassette mediates Drosophila EGF receptor function during epidermal development. Development 120, 569-578. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenlaub T., Villadsen R., Freitas F. C. P., Andrejeva D., Aldana B. I., Nguyen H. T., Petersen O. W., Gorodkin J., Herranz H. and Cohen S. M. (2018). Warburg effect metabolism drives neoplasia in a Drosophila genetic model of epithelial cancer. Curr. Biol. 28, 3220-3228.e3226. 10.1016/j.cub.2018.08.035 [DOI] [PubMed] [Google Scholar]
- Figueroa-Clarevega A. and Bilder D. (2015). Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 33, 47-55. 10.1016/j.devcel.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateff E. (1978). Malignant neoplasms of genetic origin in Drosophila melanogaster. Science 200, 1448-1459. 10.1126/science.96525 [DOI] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A. J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A. et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883-892. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglione C., Carraway K. L. III, Amundadottir L. T., Boswell R. E., Perrimon N. and Duffy J. B. (1999). The transmembrane molecule kekkon 1 acts in a feedback loop to negatively regulate the activity of the Drosophila EGF receptor during oogenesis. Cell 96, 847-856. 10.1016/S0092-8674(00)80594-2 [DOI] [PubMed] [Google Scholar]
- Ghiglione C., Amundadottir L., Andresdottir M., Bilder D., Diamonti J. A., Noselli S., Perrimon N. and Carraway I. K. (2003). Mechanism of inhibition of the Drosophila and mammalian EGF receptors by the transmembrane protein Kekkon 1. Development 130, 4483-4493. 10.1242/dev.00617 [DOI] [PubMed] [Google Scholar]
- Golembo M., Schweitzer R., Freeman M. and Shilo B. Z. (1996). Argos transcription is induced by the Drosophila EGF receptor pathway to form an inhibitory feedback loop. Development 122, 223-230. [DOI] [PubMed] [Google Scholar]
- Gonzalez C. (2013). Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13, 172-183. 10.1038/nrc3461 [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wolff T. and Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121-2129. [DOI] [PubMed] [Google Scholar]
- Hrdlicka L., Gibson M., Kiger A., Micchelli C., Schober M., Schöck F. and Perrimon N. (2002). Analysis of twenty-four Gal4 lines in Drosophila melanogaster. Genesis 34, 51-57. 10.1002/gene.10125 [DOI] [PubMed] [Google Scholar]
- Hu Y., Comjean A., Perkins L. A., Perrimon N. and Mohr S. E. (2015). GLAD: an online database of gene list annotation for Drosophila. J. Genomics 3, 75-81. 10.7150/jgen.12863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Pagliarini R. A. and Xu T. (2006). Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 16, 1139-1146. 10.1016/j.cub.2006.04.042 [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez A., Memon D., Pourpe S., Veeraraghavan H., Li Y., Vargas H. A., Gill M. B., Park K. J., Zivanovic O., Konner J. et al. (2017). Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 170, 927-938.e920. 10.1016/j.cell.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston L. A. (2009). Competitive interactions between cells: death, growth, and geography. Science 324, 1679-1682. 10.1126/science.1163862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F. D. and Rubin G. M. (1998). Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125, 1-9. [DOI] [PubMed] [Google Scholar]
- Katheder N. S., Khezri R., O'Farrell F., Schultz S. W., Jain A., Rahman M. M., Schink K. O., Theodossiou T. A., Johansen T., Juhasz G. et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature 541, 417-420. 10.1038/nature20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather J. N., Suarez-Carmona M., Charoentong P., Weis C. A., Hirsch D., Bankhead P., Horning M., Ferber D., Kel I., Herpel E. et al. (2018). Topography of cancer-associated immune cells in human solid tumors. Elife 7, e36967 10.7554/eLife.36967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G. K. and Shi W. (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 10.1093/nar/gkt214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E., Gampel A., Ring J., Virdee K., Kirov N., Tolkovsky A. M. and Martinez-Arias A. (1998). puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557-570. 10.1101/gad.12.4.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N. and Swanton C. (2017). Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613-628. 10.1016/j.cell.2017.01.018 [DOI] [PubMed] [Google Scholar]
- Milan M., Campuzano S. and Garcia-Bellido A. (1996). Cell cycling and patterned cell proliferation in the wing primordium of Drosophila. Proc. Natl. Acad. Sci. USA 93, 640-645. 10.1073/pnas.93.2.640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morata G. and Ripoll P. (1975). Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42, 211-221. 10.1016/0012-1606(75)90330-9 [DOI] [PubMed] [Google Scholar]
- Musacchio M. and Perrimon N. (1996). The Drosophila kekkon genes: novel members of both the leucine-rich repeat and immunoglobulin superfamilies expressed in the CNS. Dev. Biol. 178, 63-76. 10.1006/dbio.1996.0198 [DOI] [PubMed] [Google Scholar]
- Oh H. and Irvine K. D. (2009). In vivo analysis of Yorkie phosphorylation sites. Oncogene 28, 1916-1927. 10.1038/onc.2009.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini R. A. and Xu T. (2003). A genetic screen in Drosophila for metastatic behavior. Science 302, 1227-1231. 10.1126/science.1088474 [DOI] [PubMed] [Google Scholar]
- Pastor-Pareja J. C. and Xu T. (2013). Dissecting social cell biology and tumors using Drosophila genetics. Annu. Rev. Genet. 47, 51-74. 10.1146/annurev-genet-110711-155414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinal N., Calleja M. and Morata G. (2019). Pro-apoptotic and pro-proliferation functions of the JNK pathway of Drosophila: roles in cell competition, tumorigenesis and regeneration. Open Biol. 9, 180256 10.1098/rsob.180256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober D. A. and Edgar B. A. (2000). Ras1 promotes cellular growth in the Drosophila wing. Cell 100, 435-446. 10.1016/S0092-8674(00)80679-0 [DOI] [PubMed] [Google Scholar]
- Rebay I., Fehon R. G. and Artavanis-Tsakonas S. (1993). Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell 74, 319-329. 10.1016/0092-8674(93)90423-N [DOI] [PubMed] [Google Scholar]
- Richardson H. E. and Portela M. (2018). Modelling cooperative tumorigenesis in Drosophila. Biomed. Res. Int. 2018, 4258387 10.1155/2018/4258387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. and Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp B., Grumbling G., Donaldson T. and Simcox A. (1996). Vein is a novel component in the Drosophila epidermal growth factor receptor pathway with similarity to the neuregulins. Genes Dev. 10, 2302-2313. 10.1101/gad.10.18.2302 [DOI] [PubMed] [Google Scholar]
- Simpson P. (1979). Parameters of cell competition in the compartments of the wing disc of Drosophila. Dev. Biol. 69, 182-193. 10.1016/0012-1606(79)90284-7 [DOI] [PubMed] [Google Scholar]
- Simpson P. and Morata G. (1981). Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev. Biol. 85, 299-308. 10.1016/0012-1606(81)90261-X [DOI] [PubMed] [Google Scholar]
- Sonoshita M. and Cagan R. L. (2017). Modeling human cancers in Drosophila. Curr. Top. Dev. Biol. 121, 287-309. 10.1016/bs.ctdb.2016.07.008 [DOI] [PubMed] [Google Scholar]
- Sun S. and Irvine K. D. (2016). Cellular organization and cytoskeletal regulation of the hippo signaling network. Trends Cell Biol. 26, 694-704. 10.1016/j.tcb.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlirova M. and Bohmann D. (2006). JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 25, 5294-5304. 10.1038/sj.emboj.7601401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari T. and Bilder D. (2005). The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell 9, 687-698. 10.1016/j.devcel.2005.09.019 [DOI] [PubMed] [Google Scholar]
- Vaughen J. and Igaki T. (2016). Slit-robo repulsive signaling extrudes tumorigenic cells from epithelia. Dev. Cell 39, 683-695. 10.1016/j.devcel.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M., Larson D. E. and Cagan R. L. (2006). Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell 10, 33-44. 10.1016/j.devcel.2005.11.007 [DOI] [PubMed] [Google Scholar]
- Wang C. W., Purkayastha A., Jones K. T., Thaker S. K. and Banerjee U. (2016). In vivo genetic dissection of tumor growth and the Warburg effect. Elife 5, e18126 10.7554/eLife.18126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods D. F. and Bryant P. J. (1991). The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell 66, 451-464. 10.1016/0092-8674(81)90009-X [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Ohsawa S., Kunimasa K. and Igaki T. (2017). The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition. Nature 542, 246-250. 10.1038/nature21033 [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y. and He Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284-287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca M. and Struhl G. (2002a). Control of growth and patterning of the Drosophila wing imaginal disc by EGFR-mediated signaling. Development 129, 1369-1376. [DOI] [PubMed] [Google Scholar]
- Zecca M. and Struhl G. (2002b). Subdivision of the Drosophila wing imaginal disc by EGFR-mediated signaling. Development 129, 1357-1368. [DOI] [PubMed] [Google Scholar]
- Zielke N., Korzelius J., van Straaten M., Bender K., Schuhknecht G. F. P., Dutta D., Xiang J. and Edgar B. A. (2014). Fly-FUCCI: a versatile tool for studying cell proliferation in complex tissues. Cell Rep. 7, 588-598. 10.1016/j.celrep.2014.03.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.