

ABSTRACT
Cytosine arabinoside (Ara-c) is a pyrimidine anti-metabolite that is capable of interfering with cellular proliferation by inhibiting DNA synthesis. Each inhibitor of cyclin-dependent kinase 4 (INK4) family member has the ability to bind to cyclin-dependent kinase 4 (CDK4) and inhibit the formation of the cell cycle-dependent CDK4/cyclin D1 complex, subsequently leading to cell cycle arrest in the G1/S phase. In this study, the expression of INK4 family genes in kidney cancer and the impact of these genes on patient prognosis were examined. Additionally, the effects of INK4 family genes and Ara-c on cell proliferation and tumor formation and development were examined. Finally, a potential association between Ara-c-induced cell cycle arrest and INK4-associated gene expression was evaluated. An upregulation of INK4 family genes was found to be positively correlated with the prognosis of patients with kidney cancer. Both the INK4 family genes and Ara-c were shown to induce cell cycle arrest and inhibit tumor formation and development. Moreover, Ara-c-induced cell cycle arrest was found to be associated with an Ara-c-induced upregulation of INK4 family gene expression, which ultimately inhibited the formation of the CDK4/cyclin D1 complex. These findings suggested that an upregulation of INK4 family genes has a positive effect on kidney cancer prognosis and can inhibit the formation and development of tumors. Moreover, Ara-c was shown to promote the upregulation of INK4 family genes, at the same time, Ara-c could directly regulate the cell cycle-dependent genes CDK4 and cyclin D1 (CCND1), independent of the INK4 family genes.
KEYWORDS: INK4 family, Ara-c, cell proliferation, cell cycle, CDK4, Cyclin D1
1. Introduction
The INK4 (inhibitors of CDK4) family consists of four members, including cyclin-dependent kinase inhibitor 2A [CDKN2A (p16)], cyclin-dependent kinase inhibitor 2B [CDKN2B (p15)], cyclin-dependent kinase inhibitor 2C [CDKN2C (p18)], and cyclin-dependent kinase inhibitor 2D [CDKN2D (p14)], which are named based on their order of discovery [1]. Each INK4 protein has a functional domain consisting of 4–5 ankyrin repeats and can bind to CDK4 and inhibit the formation of the cell cycle-dependent CDK4/cyclin D1 complex [2]. Thus, the CDK4/cyclin D1 complex inhibits retinoblastoma protein phosphorylation (pRb), subsequently resulting in cell cycle arrest in the G1/S phase [3]. Furthermore, p16 (encoded by CDKN2A) forms a stable complex with CDK4 and acts as a negative cell cycle regulator via the classical CDKN2A-CDK4/cyclin D1-pRb pathway [4,5]. Additionally, the CDKN2A gene is able to respond rapidly to retinoblastoma (Rb) inactivation, Ras oncogene activation or cell death, and can be rapidly upregulated [6]; however, the specific mechanisms governing its response remains unclear. Unlike CDKN2A, CDKN2B expression is induced by transforming growth factor-β (TGF-β) via a complex of mothers against decapentaplegic homolog 2/3/4 (Smad2/3/4) and specificity protein 1 (Sp1) [7,8], with high TGF-β expression promoting p15 (encoded by CDKN2B) protein stability, which subsequently promotes CDK4/cyclin D1 binding [8,9]. Thus, CDKN2B expression not only promotes G1/S phase arrest, but also influences the G2/M phase [10,11] and is associated with cyclin D1, c-myc, and c-Ha-ras inhibition [10].
The third family member, p18 (encoded by CDKN2C), directly binds to CDK4 and prevents its binding to cyclin D1 [12]. Additionally, p18 can bind to CDK4/cyclin D1 to form a stable inactive ternary complex [13], with p18 binding distorting the ATP-binding site and altering the catalytic subunit, thus inhibiting kinase activity [13]. Furthermore, the cyclin D1 binding site can also be greatly distorted, thus reducing the binding of cyclin-dependent kinases (CDKs). Therefore, p18 can both prevent CDK4 from binding to cyclin D1 and inhibit the CDK4/cyclin D1 complex [12]. The final family member, p14 (encoded by CDKN2D), functions by inhibiting mouse double minute 2 homolog (Mdm2)-mediated p53 hydrolysis [14]. When the p14 N-terminus binds to the C-terminus of Mdm2, the Mdm2 nuclear shuttle is attenuated, and p53 cytoplasmic transport is blocked; thus, Mdm2-mediated p53 proteolysis in the cytoplasm is attenuated [15]. This gradual accumulation of p53 induces the expression of the pro-apoptotic gene BCL2 associated X (BAX, apoptosis regulator), which is within the WAF1/p21 pathway, and ultimately leads to cell cycle arrest and apoptosis [16,17]. Additionally, p14 competitively binds to CDK4, which inhibits the formation of the CDK4/cyclin D1 complex, resulting in cell cycle arrest in the G1/S phase [18].
Ara-c (cytosine arabinoside or cytarabine) was first synthesized in 1959 at the University of California, Berkeley. Normally, cytosine binds to another carbohydrate (deoxyribose) to form a component of DNA called deoxycytidine [19]. However, some organisms can combine arabinose with cytosine to form another compound (not a component of DNA) [20,21]. Furthermore, Ara-c is so similar to deoxycytidine that it can be substituted for it in human DNA, but structural differences prevent the DNA from replicating [21]. Thus, cytarabine is administered to kill cancer cells via this mechanism [22,23]. This drug is cell cycle-specific, and it predominantly targets S-phase proliferative cells and exerts a weak inhibitory effect on RNA and protein synthesis [22]. While some aspects of this drug have been characterized, the aim of this study was to determine if its actions are mediated through INK4-associated genes.
2. Materials and methods
2.1. Cells and treatment
The human renal cell line OSRC was supplied by the Cell Bank of Chinese Academy of Sciences. Prior to examination with quantitative real-time PCR (qRT-PCR), fresh cells were snap frozen in liquid nitrogen and stored at −80°C. Each sample was examined in triplicate. Cells were treated with Ara-c at a concentration of 50 µmol/L. Ara-c was purchased from Actavis Italy S.p.A. Upregulation or downregulation of the INK4 family genes and cell cycle-dependent genes CDK4 and cyclin D1, as well as related genes sequences and primer sequences are presented in the Supplement 1 and Supplement 2.
2.2. RNA extraction and cDNA synthesis
Total RNA was extracted using TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions, with RNA quality verified spectrophotometrically and an A260/A280 ratio of 1.8–2.0 considered optimal. Reverse transcription was performed using DNase- treated total RNA (2 µg), oligo (dT) primers, and PrimeScript™ Reverse Transcriptase (TaKaRa, Japan) according to the manufacturer’s instructions. The resultant cDNA was then diluted 1:10 prior to use.
2.3. Fluorescence-based qRT-PCR
Fluorescence-based qRT-PCR experiments were performed on a qRT-PCR detection system (CFX96; BIO-RAD, USA) using a SYBR Premix Ex Taq kit (TaKaRa, China) according to the manufacturers’ recommendations. All of the samples were normalized to β-actin, which served as an internal control. Transcriptional abundance was calculated based on the mean of three replicates.
2.4. Cell transfection
Cells were transfected using an empty vector or CDKN2A (CDKN2A-oe), CDKN2B (CDKN2B-oe), CDKN2C (CDKN2C-oe), CDKN2D (CDKN2D-oe), CDK4 (CDK4-oe), or cyclin D1 (cyclin D1-oe) overexpression vectors using PolyJet™ DNA In Vitro Transfection Reagent (SignaGen Laboratories, USA) according to the manufacture’s protocols (see Supplement 2).
2.5. Cell proliferation and cell cycle analysis
Cells were transfected as described above with a 70% transfection rate. Cells were then collected at 0 h, 24 h, 48 h, and 72 h post-transfection, and changes in cell numbers and in the cell cycle were detected by flow cytometry. To determine proliferation levels, a Thiazolyl Blue Tetrazolium Bromide (MTT) reduction assay was performed as previously described [24]. At 48 h post-transfection, cellular suspensions were prepared and transferred into a 96-well culturing plate, with 10 µl of MTT Reagent (Beyotime Biotechnology, China) added to each well. The samples were then incubated for 4 h and colorimetric changes were determined using a microtiter plate reader at an absorbance of 570 nm, with samples normalized to a blank control.
2.6. Co-immunoprecipitation
Cells were harvested by adding IP cell lysis buffer containing protease inhibitors at 4°C for 30 min. The samples were then centrifuged at 12,000 g for 30 min. A small amount of lysate was then analyzed via Western blot, and the remaining lysate was combined with antibodies (1 μg) and protein A/G-beads (10–50 μl). The immunoprecipitation experiments were then incubated overnight at 4°C with slow shaking. Next, the samples were centrifuged at 3,000 g for 5 min at 4°C, and the supernatants were removed. The A/G-beads were then washed three times with 1 ml lysis buffer. Finally, 2x SDS sample buffer was added to each sample and the samples were then placed in boiling water for 10 min. The obtained lysates were then examined via Western blot or by mass spectrometry. Specific information regarding the antibodies used in this study is presented in Supplement 3.
2.7. Immunofluorescence
Changes in the distributions of CDK4 and cyclin D1 were detected using an immunofluorescence assay. Samples were stained with primary rabbit anti-CDK4 and rat anti-cyclin D1 (Cell Signaling Technology, USA) antibodies, followed by the addition of fluorescently labeled secondary antibodies (Beyotime Biotechnology, China). The nuclei were stained with DAPI (Beyotime Biotechnology) and images were obtained using an Olympus TCS SP5 confocal microscope with a 40X/1.25 NA oil objective.
2.8. Gene expression analysis
To analyze the obtained transcriptional data in relation to the INK4 family, UALCAN (http://ualcan.path.uab.edu/), an interactive web resource used to analyze cancer transcriptome data, was utilized. UALCAN was developed using PERL-CGI, with high-quality graphics generated using JavaScript and CSS [25]. UALCAN was designed to provide the following outcomes: 1) provide easy access to publicly available cancer transcriptome data (TCGA and MET500 transcriptome sequencing); 2) allow users to identify biomarkers or perform in silico validation of potential genes of interest; 3) provide publication-quality graphs and plots depicting gene expression and patient survival information based on gene expression; 4) evaluate gene expression in molecular subtypes of breast and prostate cancers.
2.9. Immunohistochemistry
Renal cancer tissue sections were prepared by the Shanghai Chip Co. (China). The tumor area and the corresponding paracancerous areas were distinguished by performing hematoxylin and eosin (H&E) staining. Immunohistochemical procedure: 1. tissue chip dewaxing, if antigen repair is needed, can be carried out after this procedure. 2. use buffer to wash 3 min/2 times. 3. to reduce the nonspecific background staining caused by endogenous peroxidase, the chip was incubated in the Hydrogen Peroxide Block for 10–15 minutes. 4. use buffer to wash 5 min/2 times. 5. drops and Ultra V Block were incubated at room temperature for 5 minutes to block the non-specific background staining. 6. use buffer to wash 5 min/2 times. 7. drops and one anti-working fluid were incubated for 37 hours at 37 degrees. 8. use buffer to wash 5 min/2 times. 9. drops and Primary Antibody Enhancer were incubated for 20 minutes at room temperature. 10. use buffer to wash 5 min/2 times. 11. drops and HRP Polymer were incubated for 30 minutes at room temperature. 12. use buffer to wash 5 min/2 times. 13. add 1–2 drops of DAB Plus Chromogen to 1ml DAB Plus Substrate, mix well, add drops to slices, incubate for 3–15 minutes. 14. tap water is fully washed, dyed, dehydrated, transparent, sealed, and observed under the microscope.
2.10. Western blot
For western blot, tumour cells or specimens were homogenized with RIPA buffer (150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP40, 1 mM EDTA, 50 mM Tris (pH 8.0)). After boiling for 15min, supernatants were loaded onto SDS–PAGE gels. Nitrocellulose membranes were incubated with primary antibodies for immunoblotting. The protein bands were visualized with HRP-conjugated secondary antibodies.
2.11. Transwell invasion experiment
To examine the effects of CDK4 and CCND1 on cellular migration, a transwell cell migration assay was utilized. The transwell chamber (Corning, China) was placed in the culture plate, 1 × 104 cells were added to the upper chamber of the chamber, and fetal bovine serum was added to the lower chamber. After incubating for 24 h at 37°C in a 5% CO2 incubator, the remaining cells in the upper chamber were gently wiped off with a cotton swab. The cells migrated into the lower chamber were fixed with 4% paraformaldehyde for 30 min, and the cells were then stained with Giemsa cell staining solution for 30 min after dehydration with alcohol gradient. The number of randomly selected cells in 4 to 5 400X fields was measured under the microscope. This was repeated for at least three fields of view, and then statistics was calculated.
2.12. Nude mouse tumorigenicity assay
Tumor cells subcutaneously inoculated into nude mice were grown until reaching the logarithmic growth phase and a density of about 80%–90%. The obtained cellular suspension was placed on ice to reduce the cellular metabolism. After the cells were digested, they were immediately inoculated into the skin of nude mice(5–8 weeks old, 18–20 g). Blood-rich areas, such as the posterior part of the armpit and the middle and upper part of the groin, were chosen as injection sites.
2.13. Statistical analysis
All data are expressed as mean ± SD. In all relevant experiments, the t-test was used to evaluate the significance of the differences between two groups of data. The P value was bilateral and the value of P < 0.05 was statistically significant.
3. Results
3.1. INK4 gene family expression in OSRC cells
Transcriptional expression data were obtained and INK4 gene family expression was examined using the UALCAN database (Figure 1A). Within the tumor cells, the expression level of CDKN2A and CDKN2B was upregulated, while the expression level of CDKN2C and CDKN2D was downregulated. Furthermore, INK4 family genes expression was detected at different stages during kidney cancer (Figure 1B) and found to be upregulated during the late stage of tumor development. This finding suggests a possible feedback mechanism. Additionally, the relationship between INK4 gene family expression and the prognosis of patients with kidney cancer was also examined (Figure 1C), with some correlation with occurrence and development in kidney cancer patients discovered. These findings suggest that INK4 family members exert a degree of influence on tumor formation and development and are significantly positively correlated with prognosis.
Figure 1.
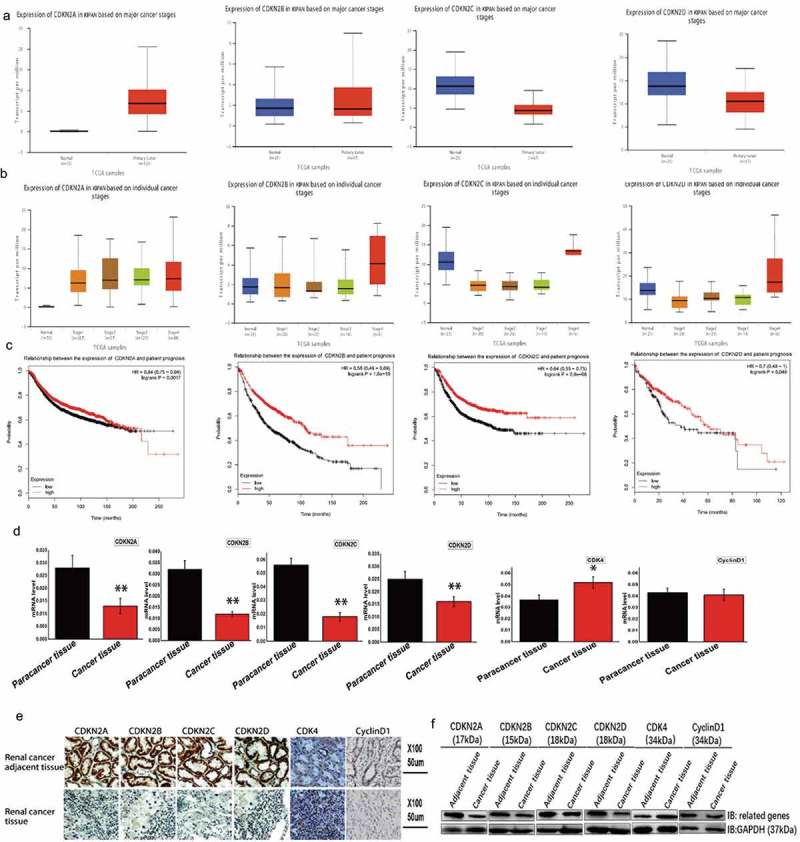
Analysis of the expression of INK4 family genes in kidney cancer.
(A) Transcriptional analysis using UALCAN showed that CDKN2A and CDKN2B were upregulated in the tumor tissues, while CDKN2C and CDKN2D were downregulated. INK4 family genes: CDKN2A (cyclin dependent kinase inhibitor 2A), CDKN2B (cyclin dependent kinase inhibitor 2B), CDKN2C (cyclin dependent kinase inhibitor 2C), and CDKN2D (cyclin dependent kinase inhibitor 2D). KIRC: kidney renal clear cell carcinoma. (B) Expression of INK4 family genes at different stages of kidney cancer. CDKN2A, CDKN2B, CDKN2C, and CDKN2D are highly expressed in the late stage of tumor development. (C) The examination of a potential correlation between INK4 family genes and the prognosis of patients with kidney cancer. Kaplan-Meier plots showing overall survival, with patients with expression levels above the median indicated in red and those below indicated in black. These plots revealed a correlation between these genes and the overall survival rate. Expressional changes in INK4 family genes in cancerous tissues and adjacent tissues from patient samples as detected by (D) fluorescence-based qRT-PCR, (E) immunohistochemistry, and (F) Western blot.
Moreover, expressional changes in INK4 family members were examined in cancer kidney tissues and adjacent tissues (Figure 1D) and showed that CDKN2A, CDKN2B, CDKN2C, and CDKN2D expression were upregulated in the adjacent tissues when compared to the corresponding cancer tissues. Furthermore, immunohistochemistry and Western blot analyses showed that all four genes were upregulated in the paracarcinoma tissues when compared to the corresponding cancer tissue (Figure 1E,F).
3.2. The INK4 family genes directly inhibited CDK4 expression and affected the formation of the cell cycle-dependent CDK4/cyclin D1 complex
To explore the mechanisms of four INK4 family members in renal cell carcinoma, these four genes were upregulated in OSRC cell line. To explore the effects of these four genes on the cell cycle-dependent CDK4/cyclin D1 complex, several expression vectors were developed and validated via qRT-PCR, with the expected overexpression in OSRC cells observed (Figure 2A). Next, changes in CDK4 and cyclin D1 expression were examined in OSRC cells overexpressing CDKN2A, CDKN2B, CDKN2C, or CDKN2D (Figure 2B,C) and showed that CDK4 expression was inhibited, but cyclin D1 expression was unaffected. These same results were also obtained at the protein level (Figure 2D,E). To further examine the relationship between these four genes and CDK4, immunofluorescence was used to determine their cellular locations (Figure 2F). The protein products of the four INK4 family members were found to colocalize with CDK4. Moreover, co-immunoprecipitation showed that the upregulation of these four proteins inhibited CDK4 expression and the formation of the CDK4/cyclin D1 complex; however, these four proteins did not affect cyclin D1 expression (Figure 2G).
Figure 2.
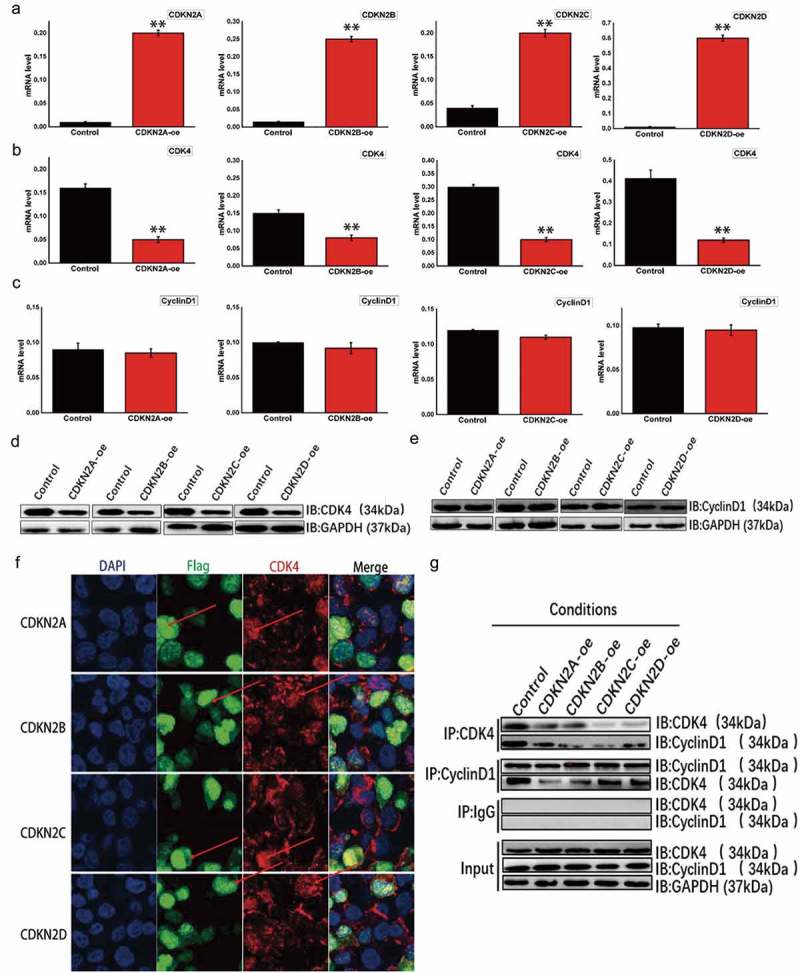
The INK4 family genes directly inhibited CDK4 expression and affected the formation of the cell cycle-dependent CDK4/cyclin D1 complex in OSRC cells.
(A) Expressional changes within the INK4 family genes were detected using fluorescence-based qRT-PCR and found to be upregulated. Samples included a control: blank load transfection group; CDKN2A-oe: CDKN2A overexpression; CDKN2B-oe: CDKN2B overexpression; CDKN2C-oe: CDKN2C overexpression; and CDKN2D-oe: CDKN2D overexpression. (B) Changes in CDK4 expression were detected by fluorescence-based qRT-PCR, with all four INK4 family genes were upregulated. (C) Expression changes in cyclin D1 as detected by fluorescence-based qRT-PCR, with all four INK4 family genes upregulated. (D) Expressional changes in CDK4 as detected by Western blot, with all four INK4 family genes upregulated. (E) Expressional changes in cyclin D1 as detected by Western blot, with all four INK4 family genes upregulated. (F) Regional expression of INK4 family genes and CDK4 visualized with immunofluorescence. (G) Co-immunoprecipitation to determine any associations between the INK4 family genes and the cell cycle-dependent CDK4/cyclin D1 complex.
3.3. The INK4 family genes regulated cell cycle and proliferation and interacted with CDK4 and cyclin D1
To further characterize the roles of CDKN2A, CDKN2B, CDKN2C, and CDKN2D, their impacts on cell cycle changes were detected in cells with altered gene expression. When these genes were overexpressed, the cell numbers in the G1 phase increased, while those in the S and G2 phases decreased (Figure 3A). Thus, this finding may explain the observed inhibition of cellular proliferation. Additionally, the effect of CDK4 or CCND1 overexpression was also examined, with CDK4 overexpression shown to promote the transition from G1 phase to S phase, while CCND1 overexpression had no apparent effect on cell cycle switching (Figure 3B). Furthermore, when both CDK4 and cyclin D1 were upregulated, the INK4 family genes had a weaker effect on cell cycle arrest (Figure 3C). Additionally, proliferation changes were observed at 0 h, 24 h, 36 h, and 48 h via flow cytometry and an MTT assay. When comparing these samples to the time 0 h control, INK4 gene family expression was lower overall relative to the control (Figure 3D), with the same result seen flow cytometry for cell number detection (Figure 3E). The results showed that most of the examined INK4 family genes inhibited proliferation, with the exception of CCND1 that had no obvious effect on cell proliferation (Figure 3F and G), and CDK4 effectively increased cell numbers. Moreover, when both CDK4 and CCND1 were simultaneously upregulated, the proliferation rate was effectively increased (Figure 3F,G). To examine the effects of these genes on cellular migration, a transwell cell migration assay was utilized (Figure 3H). The results showed that the INK4 family genes effectively inhibited tumor cells from migrating from the upper chamber to the lower chamber, while CDK4 did promote migration and cyclin D1 had no obvious effect. Furthermore, when both CDK4 and cyclin D1 were upregulated, a stronger promotion of migration was noted.
Figure 3.
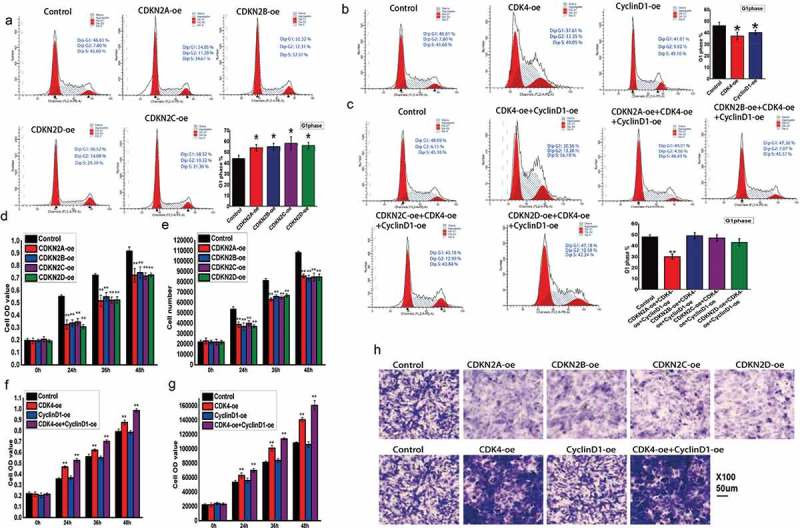
Regulation of cell cycle and cell proliferation by INK4 family genes via CDK4 and cyclin D1.
(A) Flow cytometry analysis examining cell cycle changes after altering INK4 family gene expression. (B) Flow cytometry analysis examining cell cycle changes after altering CDK4 or cyclin D1 expression. CDK4-oe: CDK4 overexpression and cyclin D1-oe: cyclin D1 overexpression. (C) Flow cytometry analysis examining cell cycle changes after altering INK4 family gene expression, and CDK4 and cyclin D1 expression. (D) MTT reduction assay evaluating proliferation at 0 h, 24 h, 36 h, and 48 h after altering INK4 family gene expression. (E) Flow cytometry analysis examining cell proliferation at 0 h, 24 h, 36 h, and 48 h after altering INK4 family expression. (F) MTT reduction assay evaluating proliferation at 0 h, 24 h, 36 h, and 48 h after altering CDK4 and cyclin D1 expression. (G) Flow cytometry analysis examining cell proliferation at 0 h, 24 h, 36 h, and 48 h after altering CDK4 and cyclin D1 expression. (H) Transwell cell migration assay examining the effects of INK4 family genes and CDK4 and cyclin D1 on tumor cell migration.
3.4. INK4 family genes inhibited the formation and development of kidney cancer
The INK4 family genes, including CDKN2A, CDKN2B, CDKN2C, and CDKN2D, were found to be upregulated in the OSRC kidney cancer cell line. To observe their behavior in vivo, cells were grown to the log phase and injected subcutaneously into nude mice, with the tumor formation and volume observed after eight weeks (Figure 4A,B). The results showed that when the four genes were upregulated, the tumor volume and mass were smaller than that of the control (Figure 4C and D). These findings show that the overexpression of these INK4 family genes can inhibit tumor growth. Furthermore, while the overexpression of these genes was found to have no effect on cyclin D1 expression (Figure 4E), but CDK4 expression was inhibited (Figure 4F), thereby affecting the tumor mass.
Figure 4.
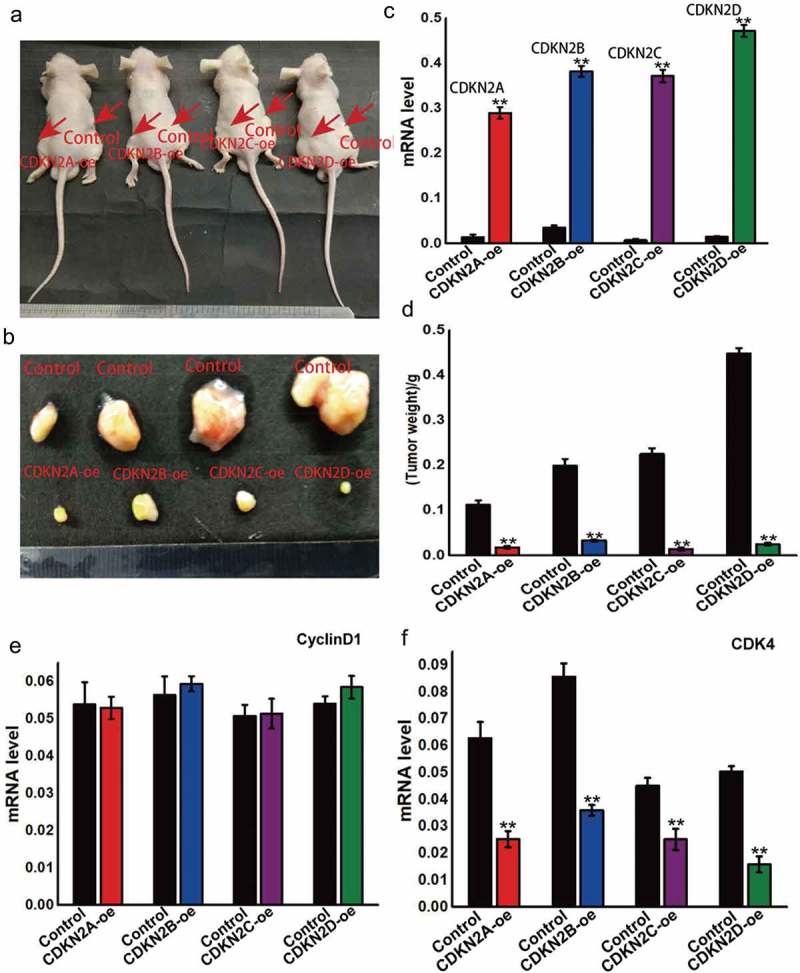
INK4 family genes can inhibit the formation and development of kidney cancer.
(A) The INK4 family genes, including CDKN2A, CDKN2B, CDKN2C, and CDKN2D, were upregulated in OSRC cells, with these cells injected subcutaneously into nude mice for in vivo experimentation. (B) After eight weeks later, the subcutaneous tumor was removed and the volume was measured. (C) INK4 family gene expression in subcutaneous tumors. (D) The weight was also measured after eight weeks. (E) Cyclin D1 expression in subcutaneous tumors with altered INK4 family gene expression. (F) CDK4 expression in subcutaneous tumors with altered INK4 family gene expression.
3.5. Ara-c modulated the cell cycle through the regulation of INK4 family genes and ultimately affected tumor formation and development
Ara-c, a commonly used chemotherapy drug, inhibits cell proliferation by inducing cell cycle arrest. However, the mechanism by which Ara-c regulates cell cycle requires further elucidation. To further examine the actions of Ara-c, cell cycle changes were examined at 0 h, 24 h, 36 h, and 48 h post Ara-c treatment and illustrated that G1 arrest occurs at all of the time points relative to the 0 h control (Figure 5A). Moreover, in addition to inducing cell cycle arrest, Ara-c also inhibited cell proliferation (Figure 5B and C). To further characterize the mechanisms of Ara-c in vivo, tumor cells were subcutaneously implanted in nude mice that were then intravenously treated with Ara-c for 10 d, 20 d, 30 d, or 40 d. The results showed that Ara-c treatment reduces tumor volume and mass and reduces the growth rate relative to the control group (Figure 5D,E). Additionally, Ara-c was found to promote expression of INK4 family genes and inhibit CDK4 and cyclin D1 expression within the tumor samples (Figure 5F–L). These findings collectively suggest that Ara-c inhibits the expression of the cell cycle-dependent CDK4/cyclin D1 complex by promoting expression of INK4 family genes, but further examination is required to confirm this assertion.
Figure 5.
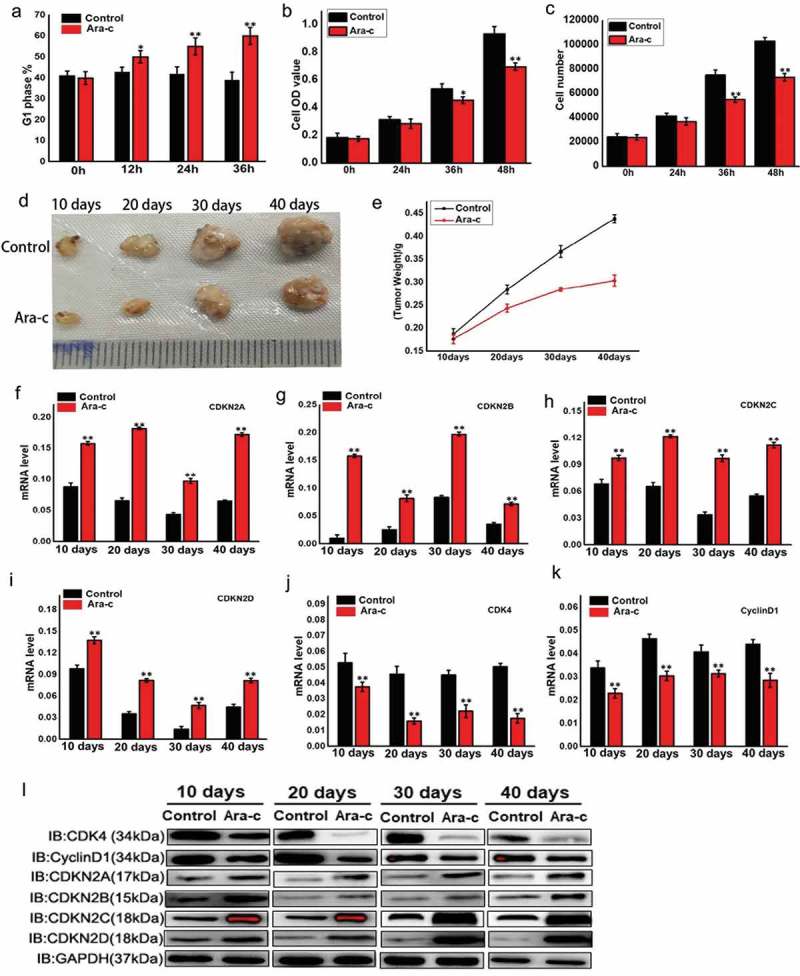
Ara-c regulates the cell cycle by modulating INK4 family genes and ultimately affects tumor formation and development.
(A) Flow cytometry analysis evaluating cell cycle changes after treatment with Ara-c at 0 h, 24 h, 36h, or 48h. Ara-c: cytosine β-D-arabinofuranoside, cytosine arabinoside, 4-Amino-1-beta-D-arabinofuranosyl-2(1H)-pyrimidinone, or arabinocytidine. (B) MTT reduction assay examining proliferation in OSRC cells treated with Ara-c for 0 h, 24 h, 36 h, or 48 h. (C) Flow cytometry analysis evaluating cell proliferation in OSRC treated with Ara-c for 0 h, 24 h, 36 h, or 48 h. (D) Tumor volume measurements for the extracted subcutaneous tumors. Tumor cells were injected subcutaneously into nude mice, while control mice were injected intraperitoneally with saline. The treatment group was injected with the same volume of Ara-c for 10 d, 20 d, 30 d, or 40 d. (E) Tumor weight measurements for the extracted subcutaneous tumors. Control mice were intraperitoneally injected with normal saline, and the treatment group was injected with the same volume of Ara-c for 10 d, 20 d, 30 d, or 40 d. (F-I) INK4 family genes expression levels in the extracted subcutaneous tumors. Control nude mice were intraperitoneally injected with saline, while the treatment group was injected with the same volume of Ara-c for 10 d, 20 d, 30 d, or 40 d. (J-K) CDK4 and cyclin D1 expression levels in the extracted subcutaneous tumors. Control nude mice were intraperitoneally injected with saline, while the treatment group was injected with the same volume of Ara-c for 10 d, 20 d, 30 d, or 40 d. (L) CDK4 and cyclin D1 protein expression levels in the extracted subcutaneous tumors. Control nude mice were intraperitoneally injected with normal saline, while the treatment group was injected with the same volume of Ara-c for 10 d, 20 d, 30 d, or 40 d.
3.6. Ara-c does not rely on INK4 family genes to directly regulate the expression of CDK4 and cyclin D1.
INK4 family, including CDKN2A, CDKN2B, CDKN2C, and CDKN2D was downregulated in the renal cancer cell line OSRC, and changes in the mRNA and protein levels of these genes and corresponding protein products were detected (Figure 6A–B). The INK4 family-related genes were successfully downregulated, and cells were treated with Ara-c for 48 h to detect the expression changes of cell cycle-dependent genes CDK4 and cyclin D1 (Figure 6C–F). When the INK4 family genes are downregulated, Ara-c can still inhibit the expression of cell cycle-related genes CDK4 and cyclin D1. Combined with the above experimental results (Figure 4–6), it was concluded that Ara-c can promote the expression of INK4 family genes and inhibit the cell cycle-dependent gene CDK4. It is worth noting that Ara-c can also directly inhibit the expression of the cell cycle-dependent genes CDK4 and cyclin D1, without the relying on the INK4 family genes.
Figure 6.
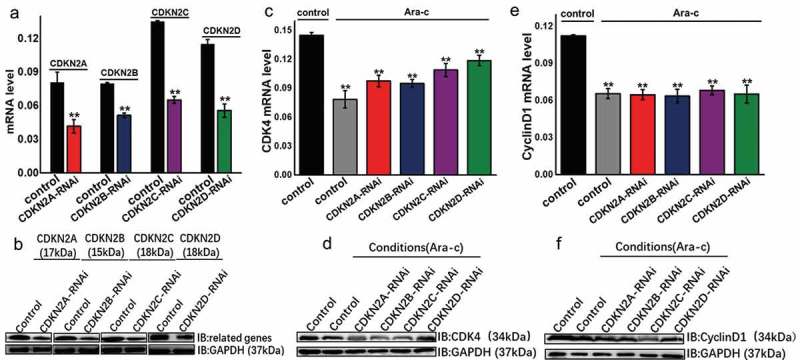
Ara-c does not rely on INK4 family genes to directly regulate cell cycle dependent gene CDK4 and cyclin D1 expression.
(A) CDKN2A, CDKN2B, CDKN2C, CDKN2D were downregulated in OSRC cells, and mRNA expression changes of these genes were examined. (B) CDKN2A, CDKN2B, CDKN2C, CDKN2D were downregulated in OSRC cells, and the expression changes in the proteins encoded by these genes were examined. (C) The cells were treated with Ara-c while down-regulating the expression of the INK4 family genes, and the mRNA expression of CDK4 was detected. (D) The cells were treated with Ara-c while downregulating the expression of the INK4 family gene, and the protein level changes of CDK4 was detected. (E) The cells were treated with Ara-c while downregulating the expression of the INK4 family gene, and the mRNA expression of cyclin D1 was detected. (F) The cells were treated with Ara-c while downregulating the expression of the INK4 family gene, and the protein level changes of cyclin D1 was detected.
3.7. Ara-c affected mouse growth and development by regulating cell cycle-related genes
To further examine the impact of Ara-c on growth and development, mice were injected intraperitoneally with 2.5 mg/kg of cytarabine for 5 d, 10 d, 15 d, or 20 d, with the control group injected with an equal volume of saline. Body weight measurements were taken at various times, and a gradual weight loss was noted in the treatment group over time when compared to the control group (Figure 7A). At 20 d post-injection, the mice were dissected and the kidneys, livers, and lungs were measured separately and found to have a much lower weight than the control samples (Figure 7B–D). To further substantiate these findings, fluorescence-based qRT-PCR was utilized to examine the expression levels of the four INK4 family genes and CDK4 and cyclin D1 in the Ara-c-treated samples from various organs (Figure 7E–F). In the Ara-c treatment group, the expression of the four INK4 family genes was induced, while CDK4 and cyclin D1 expression were inhibited. These findings suggest that Ara-c can inhibit the development and growth of various organs in mice.
Figure 7.
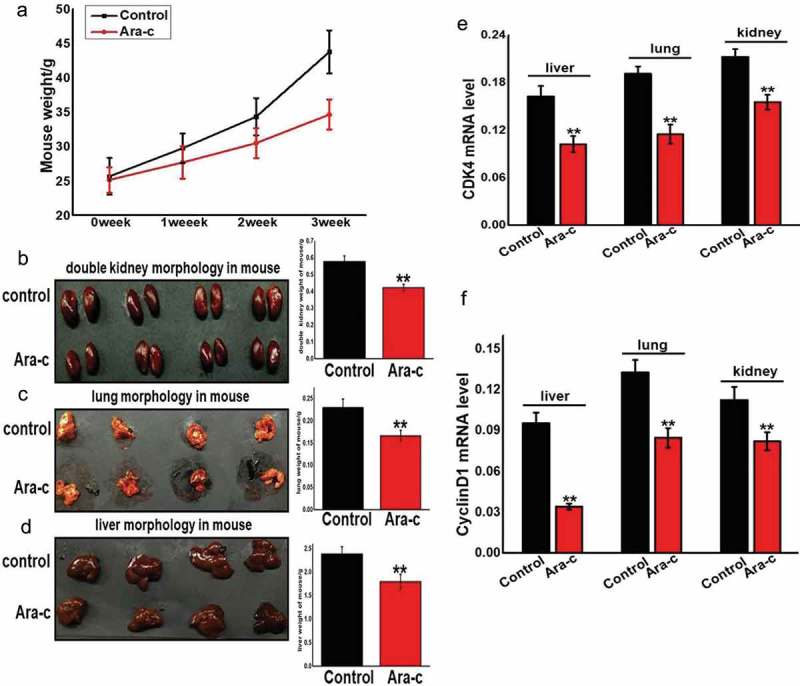
Ara-c affected mouse growth and development by regulating INK4 family genes and the cell cycle-dependent CDK4/cyclin D1 complex.
(A) Body weight changes were measured during different injection periods with Ara-c or saline. (B-D) Organ weights were measured during injection of Ara-c or saline for three weeks. (E-F) The expression of CDK4 and cyclin D1 in organs during injection of Ara-c or saline for three weeks.
4. Discussion
The findings presented herein demonstrate that the four examined INK4 family genes are able to inhibit the formation of the cell cycle-dependent CDK4/cyclin D1 complex, thereby leading to cell cycle arrest in the G1/S phase and inhibiting cell proliferation. While these genes did ultimately inhibit tumor formation and development, they did not affect cyclin D1 expression.
The results of this study further showed that the commonly used chemotherapeutic drug Ara-c induces cell cycle arrest and inhibits cell proliferation by modulating the four examined INK4 family genes. Moreover, Ara-c was seen to not only regulate these four genes, but also to inhibit the cyclin D1 expression, which resulted in the inhibition cellular proliferation (Figure 8). This indicates that Ara-C can directly affect the expression of cyclin D1 to a certain extent without depending on the INK4 family, thereby inhibiting the proliferation of kidney cancer cells. While the findings herein provide further understanding into the mechanism of action of Ara-c, they also speak to its complexity. However, further studies are needed in order to elucidate whether these two mechanisms are combined or one is the main one and the other is the auxiliary.
Figure 8.
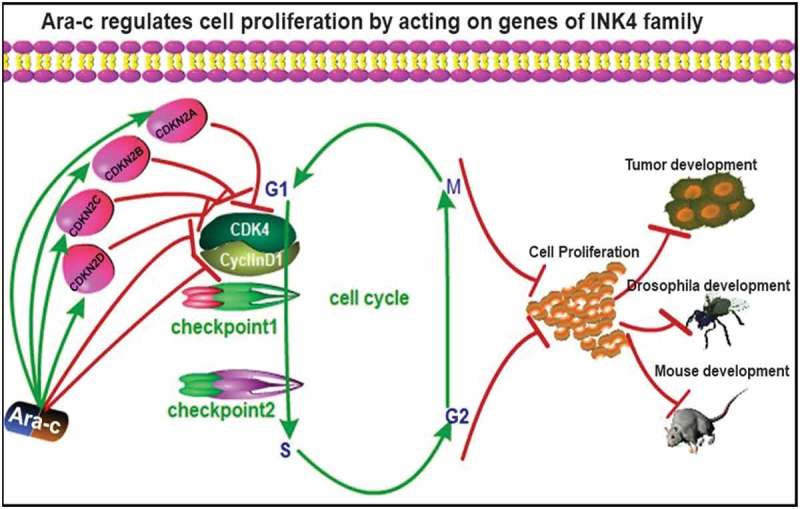
Mechanism map showing that Ara-c inhibited the formation and development of kidney cancer by regulating INK4 family gene expression.
Furthermore, while Ara-c serves as an important cancer treatment, it is undeniable that it has certain side effects. Herein, mice that received long-term Ara-c injections were found to exhibit maldevelopment accompanied by kidney inflammation (Figures 6 and 7), indicating that the drug has some toxicity. Therefore, these findings should be considered when utilizing this drug for treatment and an effort to reduce the side effects should be a primary focus in clinical research.
Funding Statement
The research was supported by the National Natural Science Foundation of China (No. 31830094, No. 31472153), the Hi-Tech Research and Development 863 Program of China Grant (No. 2013AA102507), the Fundamental Research Funds for the Central Universities in China (No. XDJK2019D009) and Funds of China Agriculture Research System (No. CARS-18-ZJ0102).
Acknowledgments
We would like to thank Dr. Jianfei Zhang for consulting on the manuscript, Dr. Yuyun Wu for providing the cell lines, and Dr. Songyuan Wu and Shubo Liang for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Statement of Ethics
Clinical data have been approved by Army Medical University. All animal experiments were approved by Army Medical University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Supplemental material
Supplemental data for this article can be accessed here.
Abbreviations
- Ara-c
Cytosine arabinoside
- BAX
BCL2 associated X
- CCND1
Cyclin D1
- CDK4
Cyclin-dependent kinase 4
- CDKN2A
Cyclin-dependent kinase inhibitor 2A
- CDKN2B
Cyclin-dependent kinase inhibitor 2B
- CDKN2C
Cyclin-dependent kinase inhibitor 2C
- CDKN2D
Cyclin-dependent kinase inhibitor 2D
- CDKs
Cyclin-dependent kinases
- INK4
Inhibitors of cyclin-dependent kinase 4
- Mdm2
Mouse double minute 2 homolog
- MTT
Thiazolyl Blue Tetrazolium Bromide
- pRb
Retinoblastoma protein phosphorylation
- Rb
Retinoblastoma
- Smad2/3/4
Mothers against decapentaplegic homolog 2/3/4
- Sp1
Specificity protein 1
- TGF-β
Transforming growth factor-β
References
- [1].Carnero A, Hannon GJ.. The INK4 family of CDK inhibitors. Curr Top Microbiol Immunol. 1998;227:43–55. [DOI] [PubMed] [Google Scholar]
- [2].Canepa ET, Scassa ME, Ceruti JM, et al. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59:419–426. [DOI] [PubMed] [Google Scholar]
- [3].Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia. 1998;12:845–859. [DOI] [PubMed] [Google Scholar]
- [4].Rodriguez-Diez E, Quereda V, Bellutti F, et al. Cdk4 and Cdk6 cooperate in counteracting the INK4 family of inhibitors during murine leukemogenesis. Blood. 2014;124:2380–2390. [DOI] [PubMed] [Google Scholar]
- [5].Lu F, Xu H, Wang Q, et al. Inhibition of enhancer of zeste homolog 2 increases the expression of p16 and suppresses the proliferation and migration of ovarian carcinoma cells in vitro and in vivo. Oncol Lett. 2018;15:3233–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang S, Ye H, Guo W, et al. CDK4/6 inhibitor suppresses gastric cancer with CDKN2A mutation. Int J Clin Exp Med. 2015;8:11692–11700. [PMC free article] [PubMed] [Google Scholar]
- [7].Kumar R, Smeds J, Berggren P, et al. A single nucleotide polymorphism in the 3ʹuntranslated region of the CDKN2A gene is common in sporadic primary melanomas but mutations in the CDKN2B, CDKN2C, CDK4 and p53 genes are rare. Int J Cancer. 2001;95:388–393. [DOI] [PubMed] [Google Scholar]
- [8].Louis‐Brennetot C, Coindre JM, Ferreira C, et al. The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well‐differentiated and dedifferentiated liposarcoma oncogenesis. An analysis of 104 tumors. Genes Chromosomes Cancer. 2011;50:896–907. [DOI] [PubMed] [Google Scholar]
- [9].Jafri M, Wake NC, Ascher DB, et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell Carcinoma. Cancer Discov. 2015;5:723–729. [DOI] [PubMed] [Google Scholar]
- [10].Ansems M, Sondergaard JN, Sieuwerts AM, et al. DC-SCRIPT is a novel regulator of the tumor suppressor gene CDKN2B and induces cell cycle arrest in ERalpha-positive breast cancer cells. Breast Cancer Res Treat. 2015;149:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Soto JL, Cabrera CM, Serrano S, et al. Mutation analysis of genes that control the G1/S cell cycle in melanoma: TP53, CDKN1A, CDKN2A, and CDKN2B. BMC Cancer. 2005;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhu S, Cao J, Sun H, et al. p18 inhibits reprogramming through inactivation of Cdk4/6. Sci Rep. 2016;6:31085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hirai H, Roussel MF, Kato JY, et al. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weber JD, Jeffers JR, Rehg JE, et al. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14:2358–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nishizawa H, Ota K, Dohi Y, et al. Bach1-mediated suppression of p53 is inhibited by p19(ARF) independently of MDM2. Cancer Sci. 2012;103:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yue Z, Rong J, Ping W, et al. Gene expression of the p16(INK4a)-Rb and p19(Arf)-p53-p21(Cip/Waf1) signaling pathways in the regulation of hematopoietic stem cell aging by ginsenoside Rg1. Genet Mol Res: GMR. 2014;13:10086–10096. [DOI] [PubMed] [Google Scholar]
- [17].Laine H, Doetzlhofer A, Mantela J, et al. p19(Ink4d) and p21(Cip1) collaborate to maintain the postmitotic state of auditory hair cells, their codeletion leading to DNA damage and p53-mediated apoptosis. J Neurosci. 2007;27:1434–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Watanabe Y, Watanabe T, Kitagawa M, et al. pRb phosphorylation is regulated differentially by cyclin-dependent kinase (Cdk) 2 and Cdk4 in retinoic acid-induced neuronal differentiation of P19 cells. Brain Res. 1999;842:342–350. [DOI] [PubMed] [Google Scholar]
- [19].Hui KP, Sit WH, Wan JM. Induction of S phase cell arrest and caspase activation by polysaccharide peptide isolated from Coriolus versicolor enhanced the cell cycle dependent activity and apoptotic cell death of doxorubicin and etoposide, but not cytarabine in HL-60 cells. Oncol Rep. 2005;14:145–155. [PubMed] [Google Scholar]
- [20].Wanda PE, Walker MM. Hemoglobin induction by Ara-C in human erythroleukemic cells (K562) is cell-cycle dependent. Leuk Res. 1989;13:683–688. [DOI] [PubMed] [Google Scholar]
- [21].Kitagawa J, Hara T, Tsurumi H, et al. Cell cycle-dependent priming action of granulocyte colony-stimulating factor (G-CSF) enhances in vitro apoptosis induction by cytarabine and etoposide in leukemia cell lines. J Clin Exp Hematop. 2010;50:99–105. [DOI] [PubMed] [Google Scholar]
- [22].Adema AD, Laan AC, Myhren F, et al. Cell cycle effects of fatty acid derivatives of cytarabine, CP-4055, and of gemcitabine, CP-4126, as basis for the interaction with oxaliplatin and docetaxel. Int J Oncol. 2010;36:285–294. [PubMed] [Google Scholar]
- [23].Wang YT, Yuan B, Chen HD, et al. Acquired resistance of phosphatase and tensin homolog‐deficient cells to poly(ADP‐ribose) polymerase inhibitor and Ara‐C mediated by 53BP1 loss and SAMHD1 overexpression. Cancer Sci. 2018;109:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aziz DM. Assessment of bovine sperm viability by MTT reduction assay. Anim Reprod Sci. 2006;92:1–8. [DOI] [PubMed] [Google Scholar]
- [25].Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: A portal for facilitating Tumor Subgroup Gene expression and survival analyses. Neoplasia. 2017;19:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.