

DUX4 drives global histone H3 hyperacetylation; and a new P300 inhibitor, iP300w, blocks DUX4-mediated transcription and toxicity.
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) results from mutations causing overexpression of the transcription factor, DUX4, which interacts with the histone acetyltransferases, EP300 and CBP. We describe the activity of a new spirocyclic EP300/CBP inhibitor, iP300w, which inhibits the cytotoxicity of the DUX4 protein and reverses the overexpression of most DUX4 target genes, in engineered cell lines and FSHD myoblasts, as well as in an FSHD animal model. In evaluating the effect of iP300w on global histone H3 acetylation, we discovered that DUX4 overexpression leads to a dramatic global increase in the total amount of acetylated histone H3. This unexpected effect requires the C-terminus of DUX4, is conserved with mouse Dux, and may facilitate zygotic genome activation. This global increase in histone H3 acetylation is reversed by iP300w, highlighting the central role of EP300 and CBP in the transcriptional mechanism underlying DUX4 cytotoxicity and the translational potential of blocking this interaction.
INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD), one of the most prevalent neuromuscular genetic disorders (1), is caused by loss of epigenetic repression of the D4Z4 macrosatellite repeat at 4qter (2–4). When this occurs together with a permissive allele of distal sequence, which provides a polyadenylation signal (5), the D4Z4 transcript is stabilized, and the DUX4 protein encoded by this transcript (6) is expressed. DUX4 is momentarily expressed in early cleavage–stage embryos at the onset of zygotic gene expression (7–9) but is not normally expressed in muscle. High levels of DUX4 expression lead to death of myoblasts (10, 11) and other cell types (10, 12), while low levels of expression inhibit myogenic differentiation (10, 13); thus, pathological mechanisms explaining muscle loss that involve both death of myofibers and impaired regeneration have been postulated. Mouse models, in which DUX4 is expressed in skeletal myofibers, show muscle pathologies (14–16). In FSHD-affected individuals, expression levels of DUX4 are extremely low, as monoclonal antibodies have to date failed to detect DUX4 by immunostaining of FSHD biopsies; however, the presence of DUX4 can be inferred by RNA sequencing (RNA-seq) of biopsies, where average levels of a panel of DUX4 target genes are elevated in FSHD biopsies compared to control biopsies (17).
Mechanistically, it has been shown that the DUX4 N terminus, containing two homeodomains, wraps around one complete turn of the DNA double helix (18), while the C terminus recruits the histone acetyltransferases (HATs) p300 and CBP (CREB binding protein), which results in local acetylation of H3K27 and expression of nearby target genes (19). The involvement of p300/CBP suggests a potential choke point at which DUX4 activity could be blocked if suitable inhibitors that block the DUX4-p300/CBP interaction or inhibit the acetyltransferase activity of p300 and CBP could be found. Although several compounds have been identified as HAT inhibitors, it has recently been shown that the general acceptance and spread of these inhibitors through the scientific literature was based on insufficient validation, and that as a group, they are nonselective assay interference compounds (20), meaning that they affect many pathways and are therefore unreliable as chemical probes. For example, the commonly used HAT inhibitor C646 (21) shows such high thiol reactivity that, besides affecting an array of unrelated targets, its acetyltransferase inhibitory activity can be severely diminished by its degradation in live cells (22), thus making it unsuitable even for in vitro work.
Next-generation specific p300 inhibitors are being developed (23). In this study, we investigate the activity of a new compound, iP300w, against the cytopathological activity of DUX4. In the course of evaluating iP300w, we found that DUX4 expression leads to an enormous global accumulation of acetylated histone H3, i.e., it does not redirect HAT activity such that it is lost from histones residing in some parts of the genome and gained by histones residing near DUX4 binding sites but changes the global balance of acetylated:nonacetylated histones. We show that iP300w reverses both this global effect and the specific ability of DUX4 to induce transcription of its target genes. Furthermore, through one or both mechanisms, iP300w protects cells from DUX4-mediated cytotoxicity.
RESULTS
A series of spirocyclic HAT inhibitors with selectivity for EP300 was recently described in patent WO2016044770 by AbbVie, one of which, A-485, has been shown to have antitumor potential in vitro and in vivo (23). Before the publication of A-485, we selected a different compound from the same series for synthesis, which we refer to as iP300 (Fig. 1A). This compound was prepared as a mixture of eight diastereomers. This mixture displayed an IC50 (median inhibitory concentration) of 33 nM versus p300 H3K9Ac in an HTRF (homogeneous time-resolved fluorescence) assay. The mixture was separated into two diastereomers (v and w; chirality not defined) by standard high-performance liquid chromatography. Testing of these two diastereomers showed that potent activity resided in only one of the two diastereomers, iP300w.
Fig. 1. iP300w diminishes DUX4-induced toxicity.
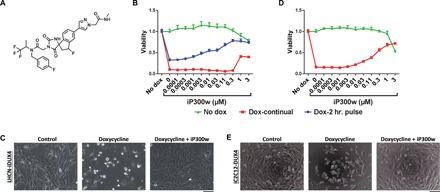
(A) Chemical structure of iP300w. (B) Cell viability assay [adenosine triphosphate (ATP) assay] on LHCN-iDUX4 myoblasts at 48 hours. Red series (Dox-continual): Cells were continually exposed to dox (200 ng/ml) for the full 48 hours and treated with serial dilutions of iP300w. Blue series (Dox-2-hr. pulse): Cells were induced for 2 hours with dox (200 ng/ml) and then washed, and medium was replaced with dox-free medium containing different concentrations of iP300w. Green series (Control): Noninduced LHCN-iDUX4 cells were treated only with different concentrations of iP300w. Data are presented as fold difference compared to the control untreated cells (n = 8). (C) Morphology of LHCN-iDUX4 cells after 48 hours with continual dox (200 ng/ml) induction and treatment with iP300w (1 μM). (D) ATP assay on iC2C12-DUX4 at 48 hours of induction with dox (500 ng/ml) and treatment with various concentrations of iP300w, as in (B). (E) Morphology of iC2C12-DUX4 cells at 48 hours of continual dox (500 ng/ml) induction and iP300w (1 μM) treatment. Concentrations of dox for both cell lines were determined to provide rapid cell death within 48 hours of induction.
iP300w inhibits DUX4-induced cytotoxicity
To test the effect of iP300w on cells expressing DUX4, we used LHCN-M2 immortalized human myoblasts (24) modified with a doxycycline (dox)–inducible DUX4 transgene (19). In the presence of dox, DUX4 is expressed and most cells die within 48 hours. Addition of iP300w to the medium enabled a dose-dependent rescue of this toxicity at 1 μM and above (Fig. 1, B and C). To exclude any effects of iP300w on transcription of DUX4 by the Tet-On system, we separated the window of dox treatment with that of iP300w treatment. In LHCN-iDUX4 cells, although less effectively than with continual dox, a short 2-hour pulse of dox also causes most cells to die within 48 hours (Fig. 1B, blue series). Therefore, we pulsed cells with dox for 2 hours to allow 2 hours of transcription of DUX4, washed to remove dox and stop further accumulation of DUX4 mRNA, and provided fresh medium with different doses of iP300w. Under these conditions also, iP300w provided substantial rescue (Fig. 1B, blue series), demonstrating that its inhibitory effects occur at some point after DUX4 transcription. To test whether cytoprotective effects were generalizable to other cell types, we tested iP300w against 293T-iDUX4 cells and found that it protected these cells in a similar manner (fig. S1). We also tested iP300w in murine cells: C2C12 myoblasts, which are known to express p300 (25), modified with a dox-inducible DUX4 transgene (10), were similarly protected (Fig. 1, D and E). In addition, we confirmed that dox does not have an effect on cell morphology or proliferation of the LHCN-rtTA and iC212 parent cell lines (fig. S2).
iP300w inhibits DUX4-mediated transcription
To investigate the effects of iP300w on DUX4 activity, we performed further dox pulse-chase experiments in which dox was applied for 2 hours, and iP300w was added after dox was removed, i.e., added to the chase medium (Fig. 2). In the absence of iP300w, DUX4 protein is visible within 1 hour, increases to 4 hours, and begins to decline by 8 hours. This suggests that both the DUX4 RNA and the protein are relatively short-lived (Fig. 2A). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) showed that DUX4 RNA declined to virtually undetectable levels by 8 hours and that iP300w had no effect on stability of the DUX4 message. Notably, in the presence of iP300w, the DUX4 protein was similar in abundance across the time series until the last time point (8 hours), when it was less abundant in the treated arm. Because values were similar before this point, this is likely a secondary effect, related to DUX4 toxicity reducing the efficiency of protein degradation in the untreated arm, although we cannot rule out the less likely possibility that EP300/CBP activity stabilizes the DUX4 protein. We then measured the activity of DUX4 target genes across this time series. Most target genes are increased by DUX4 already at the start of the chase period. After the addition of iP300w, their expression was severely blunted, while in the control arm, they continued to rise. Some DUX4 target genes were more dependent on DUX4-mediated EP300/CBP acetyltransferase activity (e.g., LEUTX) than others (e.g., ZSCAN4). Similar effects were observed in cells that were continually induced with dox for 8 hours (fig. S3). iP300w inactivation of DUX4-induced target genes was not cell line specific, as the same effect was observed in other myogenic (RD-iDUX4) and nonmyogenic (A204-iDUX4 and 293T-iDUX4) cell lines (fig. S4, A and B).
Fig. 2. iP300w disables transcription of DUX4-induced target genes.
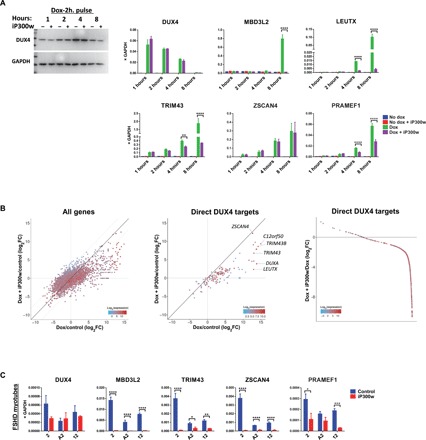
(A) Western blot for DUX4 and RT-qPCR for DUX4 target genes in LHCN-iDUX4 cells pulse induced for 2 hours with dox (200 ng/ml) and then treated with 0.1 μM iP300w. Protein and RNA samples were harvested at 1, 2, 4, and 8 hours of treatment with iP300w. Data represent means ± SEM; ****P < 0.0001, **P < 0.01, and *P < 0.05 by two-way analysis of variance (ANOVA) with Tukey’s post hoc test. Results are presented as fold difference compared to GAPDH (n = 3). (B) RNA-seq in LHCN-iDUX4 cells induced for 12 hours with dox (200 ng/ml) and treated with 0.25 μM iP300w. Left: Total gene expression given as log2 fold change of dox-treated versus control cells on the x axis and iP300w + dox–treated cells versus controls on the y axis. Middle: Same analysis showing only presumed DUX4 direct targets, defined as at least eight-fold up-regulated by 6 hours (from the Choi et al., 2016 dataset) and with a DUX4 chromatin immunoprecipitation sequencing (ChIP-seq) peak within 10 kb of the transcription start site. Right: Effect of iP300w on all strongly up-regulated DUX4 target genes (defined as all genes up-regulated by dox at least eight-fold in the current 12-hour dataset), organized by magnitude of the iP300w effect. The great majority of these are down-regulated by iP300w. In all panels, color indicates the level of gene expression. (C) RT-qPCR for DUX4 and DUX4 target genes in myotubes from three different FSHD myoblast clonal cell lines (2, A2, and 12) after 12 hours of treatment with 0.25 μM iP300w (n = 9). ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05 by two-way analysis of variance (ANOVA) with Tukey’s post hoc test.
iP300w blocks up-regulation of most DUX4 target genes
We next performed RNA-seq on immortalized human myoblasts expressing DUX4 in the presence or absence of iP300w. At 12 hours after induction, we observed up-regulation of numerous DUX4 target genes, as well as many down-regulated genes, as recently described (26). However, in the presence of iP300w, the degree of up-regulation of most of the DUX4 targets was severely diminished (deviation of points from the diagonal to the right of center, Fig. 2B). This skewing was not present for most of the down-regulated genes, indicating that their down-regulation is not dependent on p300, i.e., for the most part, transcriptional down-regulation is not the secondary effect of overexpression of certain DUX4 target genes. This is consistent with the recent finding that DUX4-mediated down-regulation is not dependent on its C terminus (26).
iP300w erases the DUX4-mediated transcriptional fingerprint in FSHD myoblasts
We next tested the effect of inhibiting EP300/CBP activity in FSHD myotubes using cell lines from a panel of immortalized myoblasts carrying a contracted D4Z4 array with three repeats (27). When preformed myotubes from three independent lines were exposed to 0.25 μM iP300w for 12 hours, expression of DUX4 target genes was severely attenuated (Fig. 2C). Because endogenous DUX4 was expressed before adding iP300w, mRNA from target genes is already present at the time of iP300w addition; thus, this assay will vary from gene to gene, depending on the stability of the transcript. Notably however, ZSCAN4 was strongly inhibited, despite it being only weakly p300 dependent in the cells with inducible DUX4. Expression of DUX4 itself was unaffected in two of the three cell lines and only moderately affected in the other. Similar results were observed at the myoblast level (fig. S5A); however, effects were less pronounced, presumably because levels of DUX4 expression are lower in myoblasts than in myotubes. The effectiveness of iP300w on DUX4 activity was time and dose dependent (fig. S5, B and C).
iP300w impairs DUX4-mediated transcription in vivo in the iDUX4pA FSHD mouse model
The experiments performed in cell culture suggested that iP300w effectively negates the transcriptional activity of DUX4. To test the potential in vivo activity of iP300w, we used the iDUX4pA mouse model (14). Four-week-old female mice were given a 12-day treatment of a low dose of dox (5 mg/kg) with or without iP300w (0.3 mg/kg), delivered intraperitoneally. At this dose of dox, significant muscle loss was only observed in the quadriceps muscle, although the gastrocnemius + soleus trended toward a lower muscle mass; both effects were reversed by iP300w (Fig. 3A).Our previous work showed that DUX4 expression led to the accumulation of fibroadipogenic progenitors (FAPs) within skeletal muscle (14); therefore, we evaluated mononuclear cells from whole muscle digests for FAPs by fluorescence-activated cell sorting (FACS) for CD45neg CD31neg Itga7neg PDGFRα+ cells (28, 29). This revealed the expected significant increase in FAPs provoked by DUX4, but FAP numbers were significantly reduced in the presence of iP300w (Fig. 3, B and C). In addition to these phenotypic changes, we observed changes in gene expression of DUX4 targets (Myo1g and Wfdc3), as well as genes indicative of a fibrotic response (Collagens 1 and 3 and Tgfb1; Fig. 3D and fig. S6). These data show that the strategy of p300/CBP inhibition to inhibit the pathogenic changes caused by DUX4 has the potential to work in vivo.
Fig. 3. Effect of iP300w on DUX4-induced alterations in vivo.
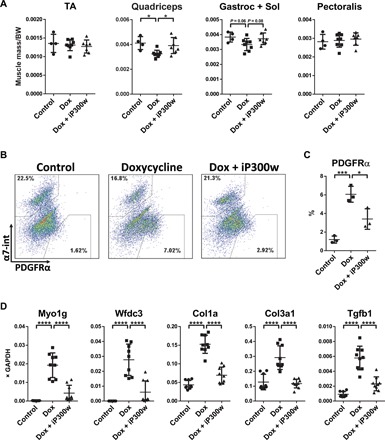
(A) Muscle mass of tibialis anterior (TA), quadriceps, gastrocnemius and soleus, and pectoralis, normalized to the body weight (BW) at day 12. iDUX4pA;HSA female mice were daily injected with dox (5 mg/kg, intraperitoneally) with or without iP300w (0.3 mg/kg) (n = 8). Wild-type (WT) siblings were used as a control (n = 4). (B) Representative FACS analyses for PDGFRα in pooled sample from quadriceps, gastrocnemius and soleus, pectoralis, and triceps at day 12. (C) Quantification of FACS analyses presented in (B). Data are presented as means ± SEM; *P < 0.05 and ***P < 0.001 by one-way ANOVA with Tukey’s post hoc test (n = 3). (D) RT-qPCR on RNA isolated from gastrocnemius at day 12. Note suppression of DUX4 target genes in the iP300w-treated group and reduction of expression of markers related to fibrosis. Data are presented as means ± SEM; ****P < 0.001 by one-way ANOVA with Tukey’s post hoc test. Results are presented as relative expression to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (n = 3).
DUX4 drives a global hyperacetylation of histone H3
Previous work on the global distribution of H3K27Ac in response to DUX4 led to the hypothesis that DUX4 recruited p300/CBP away from many of its normal sites of action for it to act in the vicinity of DUX4 (19). This was evident from the global altered pattern of H3K27Ac in LHCN-M2-iDUX4 human myoblasts. However, these chromatin immunoprecipitation sequencing (ChIP-seq) experiments are intrinsically normalized to total reads; thus, they cannot detect overall changes in global levels of H3 acetylation. In the process of evaluating activity of iP300w in DUX4-expressing cells, we evaluated overall levels of various histone acetylations in the presence of DUX4. This revealed an unexpected and remarkable increase in total levels of H3K27Ac and H3K18Ac within 12 hours of initiating DUX4 expression (Fig. 4A). DUX4 is expressed at extremely low levels in FSHD; therefore, it is of interest to evaluate cellular phenotypes at very low levels of expression. Although total levels of acetylation were not seen to rise within 12 hours when very low doses of dox were used, by 48 hours, a dose of dox as low as 3 ng/ml, at which DUX4 is virtually undetectable by Western blot, causes a large increase in H3K27Ac and H3K18Ac in human myoblasts (fig. S7A). The acetylation ability of DUX4 is mediated by its C terminus, i.e., the domain that interacts with p300/CBP (Fig. 4B and fig. S7B).We tested the effect of iP300w on these global acetylation level changes and found that this compound completely blocked the ability of DUX4 to induce the global increases in H3K27Ac and H3K18Ac described above (Fig. 4C and fig. S7C). It also reduced the basal level of H3 acetylation even in the absence of DUX4. Of note, it had no effect on the total amount of histone H3, nor did it affect acetylation of histone H4. The ability of DUX4 to induce global H3 acetylation was not cell type restricted. We observed similar effects in nonmyogenic 293T cells and in murine myoblast cells (iC2C12-DUX4, figs. S8 and S9A), where iP300w blocked DUX4-induced H3 acetylation and prevented the increase in expression of DUX4 target genes (fig. S9, A and B).
Fig. 4. DUX4 induces acetylation on H3 that is reversible by iP300w.
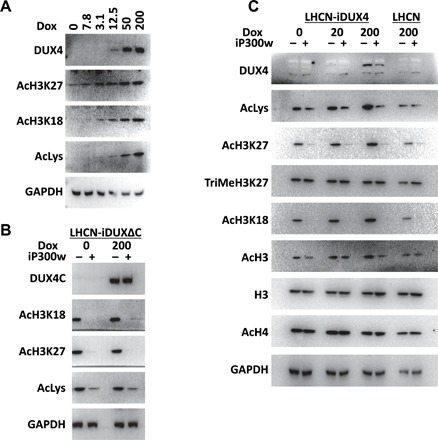
(A) Western blot analyses for acetylation of H3K27, H3K18, and total lysine in LHCN-iDUX4 cells induced for 12 hours with various concentrations of dox. Note that increased acetylation appears at levels of dox at which DUX4 is still not detectable. (B) Western blot analyses for the markers of H3 acetylation in LHCN-iDUX4ΔC cells continually induced with dox (200 ng/ml) and treated with 0.25 μM iP300w for 12 hours. DUX4ΔC is the C-terminal deletion construct of DUX4. (C) Western blot for acetylation and methylation of H3 and H4 in LHCN-iDUX4 cells that were continually induced with dox (20 and 200 ng/ml) for 12 hours. Cells were continually treated with 0.25 μM iP300w. Acetylation in LHCN cells was evaluated as an additional control.
H3 hyperacetylation is a property conserved with mouse Dux
Mouse Dux and human DUX4 have diverged substantially in protein sequence, with highest similarity in their homeodomains, as well as two short stretches of similarity within their C termini (30). These C-terminal similarities are within the previously defined 98–amino acid EP300/CBP-interacting domain of DUX4 (19), suggesting that these two proteins may engage similar transcriptional mechanisms. To test whether mouse Dux expression leads to a similar global increase in acetylated histone H3, we evaluated both human and mouse myoblasts modified with inducible mDux transgenes. We generated a human LHCN-M2 immortalized myoblast derivative expressing mDux using the same inducible lentivector strategy previously used to deliver DUX4 and made use of a previously described mouse cell line, iC2C12-mDux (31). In both cases, addition of dox led to a large increase in the total amount of acetylated (K27) H3, an increase that could be reversed by iP300w (Fig. 5A). iP300w also prevented mouse Dux from inducing its target genes in both human (Fig. 5B) and mouse (Fig. 5C) myoblasts.
Fig. 5. mDUX induces H3 acetylation that is p300 dependent.
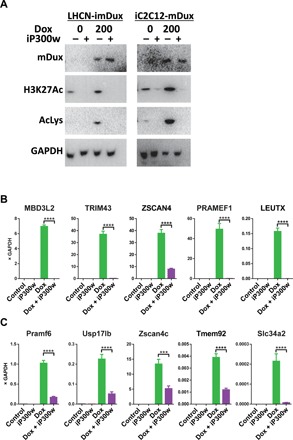
(A) Western blot analyses for H3K18 and H3K27 acetylation in LHCN-imDux and iC2C12-imDux cells continually induced with dox (200 ng/ml) and treated with 0.25 μM iP300w for 12 hours. (B) RT-qPCR for mDux target genes in LHCN-mDux after 12-hour induction with dox (200 ng/ml) and 0.25 μM iP300w. Data are presented as means ± SEM; ****P < 0.001 by one-way ANOVA with Tukey’s post hoc test. Results are presented as relative expression to GAPDH (n = 3). (C) RT-qPCR for mDux target genes in iC2C12-mDux cells after 12-hour induction with dox (200 ng/ml) and 0.25 μM iP300w (n = 3).
DISCUSSION
These studies demonstrate the biological activity of the novel p300 (EP300/CBP) inhibitor iP300w, with significant advantages over many previously described HAT inhibitors (20). The activity of iP300w hints at the unique SAR (structure-activity relationship) in this series of compounds, which includes A-485. iP300w contains a spiroimidazolidine-2, 4-dione and an extra fluorinated stereogenic center relative to A-485, which features a spirooxazolidine-2,4-dione. The two compounds also differ in their nitrogen-containing substitution on the dihydroindene moiety. Nevertheless, the potent activity of iP300w validates its selection for resynthesis and use in the studies described here. Previous work showed that an interaction between DUX4 and EP300/CBP was essential to DUX4 activity (19) and motivated the testing of these novel P300 inhibitors in FSHD cell and animal models. Consistent with this rationale, we find that iP300w inhibits both DUX4 target gene induction and DUX4-induced cytotoxicity. In support of its utility, studies in cell lines demonstrated potent activity of iP300w at 1 μM, and studies in the iDUX4pA FSHD mouse model demonstrated inhibition of DUX4 target gene expression at 0.3 mg/kg per day.
In the course of investigating histone acetylation, we found that expression of both DUX4 and its homolog, mouse Dux, leads to global hyperacetylation of histone H3. Histone H3 acetylation is the most prominent lysine acetylation in these cells, occurring mainly at K27, as well as at K18 in the human system. Although DUX4 interacts with EP300 and CBP, simple overexpression of an interactor would not a priori be expected to increase the global activity of EP300/CBP, unless a large inactive fraction of these HATs existed. A model of histone modifications based on mass action of acetyltransferases envisages a limited pool of acetyltransferase activity competitively recruited to different target sites within the genome based on the abundance and affinity of the numerous transcription factors that interact with EP300/CBP. Thus, while the specific domains of enrichment of acetylated histone H3 across the genome may change with time related to changes in the constellation of competing transcription factors, the overall amount of acetylation would be predicted to be largely unchanged and linked to the abundance of EP300/CBP. The fact that this is not the case and that expression of DUX4 leads to grossly increased frequencies of acetylated histone H3 suggests that a large inactive store is present and that its activity is somehow released by DUX4, by either direct activation or abrogation of negative regulation. Given this marked global increased histone H3 acetylation, DUX4 therefore appears to be a powerful global epigenetic modulator. It is very interesting in this regard that DUX4 and mouse Dux are expressed at the onset of zygotic genome activation during mammalian embryogenesis (7, 9). Such a global increase in H3 acetylation could potentially facilitate zygotic genome activation and may therefore reflect the key conserved activity of DUX transcription factors expressed during early embryogenesis.
In contrast to the effect of DUX4 overexpression, the inhibitor iP300w drives the opposite response: a severe reduction in the levels of histone H3K18 and H3K27 acetylation. It also protects DUX4-expressing cells from the cytotoxicity caused by the DUX4 protein better than any of the compounds identified to date, from screening libraries representing more than 200,000 compounds (32, 33) as well as other approaches to inhibit DUX4-mediated toxicity (34). The previously published compound A-485 from the same chemical series has similar effects in our hands, although being slightly less potent than iP300w. This correlation of acetyltransferase activity and DUX4 toxicity solidifies the notion that DUX4 activity is mediated by EP300/CBP, previously established by showing that the C-terminal domain necessary for toxicity interacts with EP300 and CBP (19). In looking at the effect of iP300w on DUX4 target gene expression, it is clear that the vast majority of DUX4 targets show diminished expression; however, the level of suppression of transactivation shows some variance across DUX4 targets, and there are a few targets that are apparently insensitive, at least in the immortalized human myoblasts used for RNA-seq, for example, ZSCAN4. This may indicate that some targets, such as ZSCAN4, are alternatively regulated and may require another coactivator.
It is notable that iP300w suppressed the DUX4 fingerprint in FSHD patient myoblasts, in this case, including ZSCAN4. Together with the fact that iP300w treatment led to similar effects on DUX4 target gene expression in the iDUX4pA mouse model for FSHD, this suggests that targeting the EP300/CBP aspect of DUX4 function may be a productive approach to therapy for FSHD. Long-term treatment with this compound or other HAT inhibitors might be expected to have off-target effects; thus, risks would need to be carefully weighed against potential benefits for this specific class of inhibitors. However, given that DUX4 recruits EP300/CBP through its C terminus, compounds that disrupted the DUX4-EP300 or CBP interaction would be expected to have similar effects on DUX4 gene expression and cytotoxicity as iP300w, but without the off-target effects of blocking EP300/CBP activity. The results presented here provide the proof of principle for such an approach. Understanding the structural features of the DUX4 C terminus–EP300/CBP interaction would facilitate this approach and should therefore be a high priority.
METHODS
Cell culture
Immortalized human myoblast LHCN-M2 cell lines and FSHD clonal cell lines were cultured in proliferation media [F10 (HyClone) supplemented with 20% fetal bovine serum (FBS) (PeakSerum, Ps-FB3, lot 293Q16), 1× 2-mercaptoethanol (Gibco), 10−9 M dexamethasone (Sigma-Aldrich), basic fibroblast growth factor (10 ng/ml; PeproTech), GlutaMAX (Gibco), and penicillin/streptomycin (Gibco)] at 37°C in a 5% CO2 atmosphere. Myogenic differentiation was initiated on confluent cell cultures with differentiating media [Dulbecco’s modified Eagle’s medium (DMEM)/F12; Corning Cellgro], supplemented with 1× Insulin-Transferrin-Selenium (Gibco), 1× non-essential amino acids (Gibco), GlutaMAX, and penicillin/streptomycin. 293T, A204-iDUX4, RH30-iDUX4, and RD-iDUX4 cells were cultured in DMEM/F12 supplemented with 10% FBS and penicillin/streptomycin. iC2C12-DUX4 and iC2C12-mDUX cells were cultured in DMEM (HyClone) and 20% FBS.
Antibodies and Western blots
For Western blot analyses, proteins were separated on 10% SDS–polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membrane. Antibodies were diluted in 5% skim milk in TBST (tris-buffered saline with Tween 20) and incubated overnight at 4°C. Signal was visualized using Pierce ECL Western blotting substrate (Thermo Fisher Scientific).
Antibodies used in the study are as follows: rabbit anti-DUX4 (RD2-47c, 1:50, R&D Systems), GAPDH–horseradish peroxidase (HRP) (1:5000, 60004, Proteintech), mouse monoclonal acetylated histone H3 (D-4, 1:250, sc-518011, Santa Cruz Biotechnology), mouse monoclonal acetylated histone H4 (E-5, 1:250, sc-377520, Santa Cruz Biotechnology), mouse monoclonal histone H3 K9me3 (6F12-H4, 1:250, sc-130356, Santa Cruz Biotechnology), rabbit anti–histone H3 K18Ac [1:500, ab1191 (Abcam) and 9675T (Cell Signaling)], rabbit anti–histone H3 K27Ac (1:500, ab, Abcam), rabbit anti–acetylated lysine (1:500, 9441S, Cell Signaling), rabbit anti–histone H3 K9 (1:500, C5B11, Cell Signaling), anti–histone H3 (Poly6019, 1:500, 601901, BioLegend), and anti–rabbit-HRP (1:5000, 111-035-003, The Jackson Laboratory).
ATP assay
Cell lines were plated in a 96-well dish (2000 cells per well) and, the following day, induced with dox (200 ng/ml) with and without iP300w treatment. ATP assays were performed using CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. Luminescence was analyzed on the POLARstar Optima Microplate Reader (BMG Labtech, Offenburg, Germany).
Evaluation of effectiveness of iP300w in vivo
The iDUX4pA mouse model for FSHD was used for this study. Four-week-old female mice carrying both the iDUX4pA and HSA-rtTA transgenes were injected with dox (5 mg/kg intraperitoneally) in phosphate-buffered saline (PBS) for 12 days. Half of these mice further received intraperitoneal injections of iP300w (0.3 mg/kg; 0.5 μl from a 10 mM stock, diluted in 100 μl of PBS) per day, while the other half received PBS only. Untreated wild-type siblings were used for unaffected comparisons.
FACS analyses of FAPs from muscle
Analysis of muscle infiltration with FAPs was performed as previously explained (14). Briefly, pooled muscles (tibialis anterior, gastrocnemius, soleus, quadriceps, triceps, and pectoralis) were digested with collagenase type II and dispase. Mononuclear cells were stained with the following antibodies: phycoerythrin (PE)–Cy7–conjugated CD45 and CD31 (552848 and 561410, respectively; BD Biosciences), PE-conjugated PDGFRα (562776; BD Biosciences), and allophycocyanin (APC)–conjugated rat anti–integrin α7 (67-0010-05; Ablab), resuspended in PBS/1% FBS. Samples were run on a BD FACSAria instrument, and data were analyzed using FlowJo (BD Biosciences). All experiments were performed on at least three biological replicates.
Animal studies
Mice were maintained in specific pathogen–free conditions under a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee.
RNA isolation, quantitative real-time RT-PCR (RT-qPCR), and RNA-seq
RNA was extracted using an RNA extraction kit (Zymo), and complementary DNA (cDNA) was made using 0.75 μg of total RNA with oligo-dT primer and Verso cDNA Synthesis Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. qPCR was performed by using Premix Ex Taq (Probe qPCR, Takara) and commercially available probes: ZSCAN4, Hs00537549_m1; MBD3L2, HS00544743_m1; DUX4, Hs03037970_g1; GAPDH, Hs99999905_m1; TRIM43, Hs00299174_m1; LEUTX, Hs01028718_m1; PRAMEF1, Hs04401269_sH; CCNA1, Hs00171105_m1; CDK1A, Hs00355782_m1; EGR1, Hs00152928_m1; Gapdh, Mm99999915_g1; Myo1g, Mm00617991-m1; Wfdc3, Mm01243777_m1; Zscan4c, Mm02581232_m1; Slc34a2, Mm01215846_m1; Pramef6, Mm01728753_g1; Tmem92, Mm01299147_m1; Usp17lb, Mm04214102_s1 (Applied Biosystems). Endogenous DUX4 expression in FSHD myoblast cell lines was detected by FAM-labeled probe (TCTCTGTGCCCTTGTTCTTCCGTGAA) and primers PLH298-PAS-F: CCCAGGTACCAGCAGACC and PLH395-PAS-R: TCCAGGTTTGCCTAGACAGCGTC, which target the 3′ splice and polyadenylation site (14). Gene expression levels were normalized to that of GAPDH and analyzed with 7500 System Software using the ∆∆CT method (Applied Biosystems). RNA-seq libraries were prepared using 500 ng of total RNA and the KAPA mRNA Hyper Prep Kit following the manufacturer’s instructions. Fifty–base paired end sequencing was performed on an Illumina NovaSeq instrument at the University of Minnesota Genomics Center.
Bioinformatics
A total of 163,937,137 reads were generated for the samples (mean is 20.49 million reads, between 19.35 million and 21.76 million reads per sample). The FASTQ files were analyzed using a customized pipeline (gopher-pipelines; https://bitbucket.org/jgarbe/gopher-pipelines/overview) developed and maintained by the Minnesota Supercomputing Institute. The sequencing quality was checked using FastQC v0.11.5 (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads were discarded using Trimmomatic v0.33 (www.usadellab.org/cms/index.php?page=trimmomatic) (35). Reads passing quality checks were then aligned to GRCh38/hg38 reference genome using HISAT2 v2.0.2 (https://ccb.jhu.edu/software/hisat2/index.shtml) (36). The transcript abundances were then counted using subread v1.4.6 (http://subread.sourceforge.net/) (37). The reads were filtered to include genes with counts per million value over 5 in at least 20% of the samples for subsequent analysis. Sequencing data have been deposited in the Gene Expression Omnibus repository under accession code GSE130522.
Differential gene expression (DGE) analysis was performed in R v3.4.3 using edgeR package (https://bioconductor.org/packages/release/bioc/html/edgeR.html) (38). The DGE genes were visualized using ggplot2 package.
Statistics
GraphPad Prism software was used for statistical analyses of the data, except where indicated. Differences between groups were evaluated by one- or two-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests. Differences were considered significant at P values of 0.05 or lower.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from the FSH Society (FSHS-22017-05) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR055685). Author contributions: Investigation: D.B., M.T.d.S., S.T.S., E.T.E., E.A.T., and Z.C.; analysis: D.B. and M.K. bionformatics analysis: C.Y.; chemical synthesis and medicinal chemistry analysis: M.A.W. and A.J.; research design and supervision and manuscript preparation: M.K. and D.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Materials reported in this study can be provided by M.K. pending scientific review and a completed material transfer agreement. Requests for materials should be submitted to kyba@umn.edu. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw7781/DC1
Fig. S1. iP300w protects 293T cells from DUX4-induced toxicity.
Fig. S2. Doxycycline effect on morphology and viability of parent cell lines.
Fig. S3. iP300w inactivates induction of DUX4 target genes.
Fig. S4. iP300w inactivates induction of DUX4 target genes in different cell types.
Fig. S5. Dose- and time-dependent DUX4 inactivation by iP300w in FSHD myoblasts.
Fig. S6. Effect of doxycycline on profibrotic gene expression in wild-type mice.
Fig. S7. Low levels of DUX4 induce H3 acetylation through p300 in human myoblasts.
Fig. S8. DUX4 induces H3 acetylation in nonmyogenic cells.
Fig. S9. Low levels of DUX4 induce H3 acetylation through p300 in mouse myoblasts.
REFERENCES AND NOTES
- 1.Deenen J. C. W., Arnts H., van der Maarel S. M., Padberg G. W., Verschuuren J. J. G. M., Bakker E., Weinreich S. S., Verbeek A. L. M., van Engelen B. G. M., Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 83, 1056–1059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Overveld P. G. M., Lemmers R. J. F. L., Sandkuijl L. A., Enthoven L., Winokur S. T., Bakels F., Padberg G. W., van Ommen G.-J. B., Frants R. R., van der Maarel S. M., Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 35, 315–317 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Van Deutekom J. C. T., Wljmenga C., Van Tlenhoven E. A. E., Gruter A.-M., Hewitt J. E., Padberg G. W., van Ommen G.-J. B., Hofker M. H., Fronts R. R., FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 2, 2037–2042 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Lemmers R. J. L. F., Tawil R., Petek L. M., Balog J., Block G. J., Santen G. W. E., Amell A. M., van der Vliet P. J., Almomani R., Straasheijm K. R., Krom Y. D., Klooster R., Sun Y., den Dunnen J. T., Helmer Q., Donlin-Smith C. M., Padberg G. W., van Engelen B. G. M., de Greef J. C., Aartsma-Rus A. M., Frants R. R., de Visser M., Desnuelle C., Sacconi S., Filippova G. N., Bakker B., Bamshad M. J., Tapscott S. J., Miller D. G., van der Maarel S. M., Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemmers R. J. L. F., van der Vliet P. J., Klooster R., Sacconi S., Camano P., Dauwerse J. G., Snider L., Straasheijm K. R., van Ommen G. J., Padberg G. W., Miller D. G., Tapscott S. J., Tawil R., Frants R. R., van der Maarel S. M., A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329, 1650–1653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabriëls J., Beckers M. C., Ding H., De Vriese A., Plaisance S., van der Maarel S. M., Padberg G. W., Frants R. R., Hewitt J. E., Collen D., Belayew A., Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236, 25–32 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Hendrickson P. G., Doráis J. A., Grow E. J., Whiddon J. L., Lim J.-W., Wike C. L., Weaver B. D., Pflueger C., Emery B. R., Wilcox A. L., Nix D. A., Peterson C. M., Tapscott S. J., Carrell D. T., Cairns B. R., Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiddon J. L., Langford A. T., Wong C.-J., Zhong J. W., Tapscott S. J., Conservation and innovation in the DUX4-family gene network. Nat. Genet. 49, 935–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Iaco A., Planet E., Coluccio A., Verp S., Duc J., Trono D., DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 49, 941–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosnakovski D., Xu Z., Gang E. J., Galindo C. L., Liu M., Simsek T., Garner H. R., Agha-Mohammadi S., Tassin A., Coppée F., Belayew A., Perlingeiro R. R., Kyba M., An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 27, 2766–2779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rickard A. M., Petek L. M., Miller D. G., Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 24, 5901–5914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowaljow V., Marcowycz A., Ansseau E., Conde C. B., Sauvage S., Mattéotti C., Arias C., Corona E. D., Nunez N. G., Leo O., Wattiez R., Figlewicz D., Laoudj-Chenivesse D., Belayew A., Coppée F., Rosa A. L., The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 17, 611–623 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Bosnakovski D., Toso E. A., Hartweck L. M., Magli A., Lee H. A., Thompson E. R., Dandapat A., Perlingeiro R. C. R., Kyba M., The DUX4 homeodomains mediate inhibition of myogenesis and are functionally exchangeable with the Pax7 homeodomain. J. Cell Sci. 130, 3685–3697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosnakovski D., Chan S. S. K., Recht O. O., Hartweck L. M., Gustafson C. J., Athman L. L., Lowe D. A., Kyba M., Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 8, 550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones T., Jones P. L., A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLOS ONE 13, e0192657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giesige C. R., Wallace L. M., Heller K. N., Eidahl J. O., Saad N. Y., Fowler A. M., Pyne N. K., Al-Kharsan M., Rashnonejad A., Chermahini G. A., Domire J. S., Mukweyi D., Garwick-Coppens S. E., Guckes S. M., McLaughlin K. J., Meyer K., Rodino-Klapac L. R., Harper S. Q., AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight 3, 123538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Z., Snider L., Balog J., Lemmers R. J. L. F., Van Der Maarel S. M., Tawil R., Tapscott S. J., DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 23, 5342–5352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J. K., Bosnakovski D., Toso E. A., Dinh T., Banerjee S., Bohl T. E., Shi K., Orellana K., Kyba M., Aihara H., Crystal structure of the double homeodomain of DUX4 in complex with DNA. Cell Rep. 25, 2955–2962.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi S. H., Gearhart M. D., Cui Z., Bosnakovski D., Kim M., Schennum N., Kyba M., DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 44, 5161–5173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlin J. L., Nelson K. M., Strasser J. M., Barsyte-Lovejoy D., Szewczyk M. M., Organ S., Cuellar M., Singh G., Shrimp J. H., Nguyen N., Meier J. L., Arrowsmith C. H., Brown P. J., Baell J. B., Walters M. A., Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat. Commun. 8, 1527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers E. M., Yan G., Mukherjee C., Orry A., Wang L., Holbert M. A., Crump N. T., Hazzalin C. A., Liszczak G., Yuan H., Larocca C., Saldanha S. A., Abagyan R., Sun Y., Meyers D. J., Marmorstein R., Mahadevan L. C., Alani R. M., Cole P. A., Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 17, 471–482 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shrimp J. H., Sorum A. W., Garlick J. M., Guasch L., Nicklaus M. C., Meier J. L., Characterizing the covalent targets of a small molecule inhibitor of the lysine acetyltransferase P300. ACS Med. Chem. Lett. 7, 151–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasko L. M., Jakob C. G., Edalji R. P., Qiu W., Montgomery D., Digiammarino E. L., Hansen T. M., Risi R. M., Frey R., Manaves V., Shaw B., Algire M., Hessler P., Lam L. T., Uziel T., Faivre E., Ferguson D., Buchanan F. G., Martin R. L., Torrent M., Chiang G. G., Karukurichi K., Langston J. W., Weinert B. T., Choudhary C., de Vries P., Van Drie J. H., McElligott D., Kesicki E., Marmorstein R., Sun C., Cole P. A., Rosenberg S. H., Michaelides M. R., Lai A., Bromberg K. D., Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu C. H., Mouly V., Cooper R. N., Mamchaoui K., Bigot A., Shay J. W., Di Santo J. P., Butler-Browne G. S., Wright W. E., Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: Consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 6, 515–523 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Chen M. J., Yokomizo T., Zeigler B. M., Dzierzak E., Speck N. A., Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457, 887–891 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosnakovski D., Gearhart M. D., Toso E. A., Ener E. T., Choi S. H., Kyba M., Low level DUX4 expression disrupts myogenesis through deregulation of myogenic gene expression. Sci. Rep. 8, 16957 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krom Y. D., Dumonceaux J., Mamchaoui K., den Hamer B., Mariot V., Negroni E., Geng L. N., Martin N., Tawil R., Tapscott S. J., van Engelen B. G., Mouly V., Butler-Browne G. S., van der Maarel S. M., Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: A cellular model for FSHD. Am. J. Pathol. 181, 1387–1401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joe A. W. B., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M. A., Rossi F. M. V., Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uezumi A., Fukada S.-i., Yamamoto N., Takeda S., Tsuchida K., Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12, 143–152 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Clapp J., Mitchell L. M., Bolland D. J., Fantes J., Corcoran A. E., Scotting P. J., Armour J. A. L., Hewitt J. E., Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 81, 264–279 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosnakovski D., Daughters R. S., Xu Z., Slack J. M. W., Kyba M., Biphasic myopathic phenotype of mouse DUX, an ORF within conserved FSHD-related repeats. PLOS ONE 4, e7003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosnakovski D., Choi S. H., Strasser J. M., Toso E. A., Walters M. A., Kyba M., High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet. Muscle 4, 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi S. H., Bosnakovski D., Strasser J. M., Toso E. A., Walters M. A., Kyba M., Transcriptional Inhibitors Identified in a 160,000-Compound Small-Molecule DUX4 Viability Screen. J. Biomol. Screen. 21, 680–688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamel J., Tawil R., Facioscapulohumeral muscular dystrophy: Update on pathogenesis and future treatments. Neurotherapeutics 15, 863–871 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw7781/DC1
Fig. S1. iP300w protects 293T cells from DUX4-induced toxicity.
Fig. S2. Doxycycline effect on morphology and viability of parent cell lines.
Fig. S3. iP300w inactivates induction of DUX4 target genes.
Fig. S4. iP300w inactivates induction of DUX4 target genes in different cell types.
Fig. S5. Dose- and time-dependent DUX4 inactivation by iP300w in FSHD myoblasts.
Fig. S6. Effect of doxycycline on profibrotic gene expression in wild-type mice.
Fig. S7. Low levels of DUX4 induce H3 acetylation through p300 in human myoblasts.
Fig. S8. DUX4 induces H3 acetylation in nonmyogenic cells.
Fig. S9. Low levels of DUX4 induce H3 acetylation through p300 in mouse myoblasts.