

Abstract
The first catalytic enantioselective carbonyl (α-amino)allylations are described. Phthalimido-allene 1 and primary alcohols 2a-2z, 2a’−2c’ engage in hydrogen auto-transfer-mediated carbonyl reductive coupling by way of (α-amino)allyliridium-aldehyde pairs to form vicinal amino alcohols 3a-3z, 3a’−3c’ with high levels of regio-, anti-diastereo and enantioselectivity. Reaction progress kinetic analysis and KIE studies corroborate a catalytic cycle involving turn-over limiting alcohol dehydrogenation followed by rapid allene hydrometalation.
Graphical Abstract
Asymmetric carbonyl addition ranks foremost among methods used for the convergent construction of enantiomerically enriched alcohols.1 Data mining of patents from the pharmaceutical industry reveals that carbonyl addition (alongside Suzuki coupling) remains one of the most frequently utilized methods for C-C bond formation.2 The vast majority of carbonyl addition reactions rely on the use of preformed carbanions, which can be moisture sensitive, unsafe, and often require multi-step preparation and cryogenic conditions. Metal-catalyzed carbonyl reductive coupling of π-unsaturated pronucleophiles has emerged as an alternative to the use of stoichiometric carbanions.3 However, many of the terminal reductants utilized in such processes (e.g. Mn, Zn, Et3B, Et2Zn) are as problematic as the premetalated reagents they replace. Carbonyl reductive coupling via hydrogen auto-transfer does not require an exogenous reductant, as alcohol reactants serve dually as reductant and carbonyl proelectrophile.4
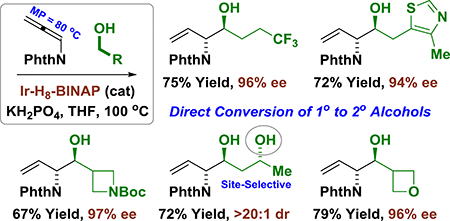
Based on this concept and motivated by the prevalence (>40%) of chiral amines (including vicinal amino alcohols) in FDA approved drugs,5a,b a catalytic enantioselective carbonyl (α-amino)allylation was sought.6,7,8 In 1993, Barrett reported a boron reagent for asymmetric carbonyl (α-amino)allylation.7 Remarkably, after more than 25 years, corresponding catalytic enantioselective processes have remained elusive, and the only related catalytic transformation to have appeared is the 2-azadiene-ketone reductive coupling reported by Malcolmson.9 Here, we disclose that phthalimido-allene 1, a tractable crystalline solid (M.P. = 79–81 °C), participates in catalytic enantioselective carbonyl reductive coupling via hydrogen auto-transfer to deliver vicinal amino alcohols with high levels of regio-, anti-diastereo- and enantiocontrol (Figure 1). This work represents a rare example of the use of allene pronucleophiles in enantioselective carbonyl reductive coupling.10
Figure 1.

Selected enantioselective methods for convergent construction of vicinal amino alcohols via classical and metal-catalyzed carbonyl addition.
Phthalimido allene 1 is readily prepared through base-catalyzed isomerization of commercially available N-propargyl phthalimide.11 Guided by seminal findings from our laboratory,12 it was posited that hydrogen transfer from primary alcohols to allenimide 1 would generate transient (phthalimido)allyliridium-aldehyde pairs that combine by way of closed six-centered transition structures to furnish anti-vicinal amino alcohols. The feasibility of this transformation was rendered uncertain by competing conventional transfer hydrogenation of allene 1 in response to the steric demand of the phthalimide moiety, which may retard the rate of aldehyde addition. An assay of diverse chiral ruthenium and iridium complexes was undertaken and a promising result was obtained using the cyclometallated π-allyliridium complex modified by 3-nitrobenzoic acid and (R)-SEGPHOS, Ir-I, which delivered the desired amino alcohol 3a in 10% yield and 40% ee with >20:1 anti-diastereoselectivity (Table 1). Enantioselectivity improved using the more Lewis acidic 4-cyano-3-nitro-C,O-benzoate, Ir-II, but the isolated yield of 3a remained modest due to low conversion. Similar trends were observed with the corresponding catalysts based on (R)-BINAP, Ir-III and Ir-IV, but with a small increase in enantioselectivity. A pronounced improvement in both conversion and enantioselectivity was observed upon use of Ir-V, which incorporates (R)-H8-BINAP.13 Use of the (R)-H8-BINAP iridium complex bound by 3,4-dinitro-C,O-benzoic acid, Ir-VI, provided still higher levels of enantioselectivity. Finally, introduction of mono-basic potassium phosphate led to higher conversion, allowing 3a to be formed in 80% yield, 96% ee with complete anti-diastereoselectivity (Table 1). As borne out by single crystal X-ray diffraction analysis of Ir-VI (see Supporting Information), the dihedral angle between the tetralin rings of (R)-H8-BINAP (ca. 86°) is significantly larger than the dihedral angle between the naphthalene rings of BINAP (ca. 75°) or SEGPHOS (ca. 72°),14 which may better accommodate the sterically demanding phthalimide moiety to facilitate alkoxide exchange at the metal center.
Table 1.
Selected optimization experiments in the enantioselective iridium-catalyzed (α-amino)allylation of phthalimido-allene 1 with alcohol 2a.a
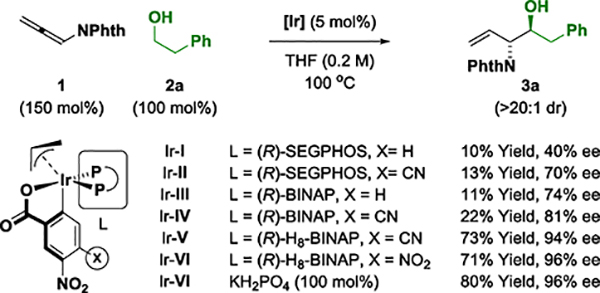 |
Yields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR of crude reaction mixtures. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for experimental details.
Reaction scope was evaluated by applying optimal conditions identified for the (α-amino)allylation of 2-phenylethanol 2a to diverse alcohols 2b-2z, 2a′−2c′ (Table 2). All vicinal amino alcohols 3a-3z, 3a′−3c′ were formed in good yield with excellent levels of diastereo- and enantioselectivity. The (α-amino)allylations of N-Boc-ethanolamine 2j, N-Boc-propanolamine 2k and trifluorobutanol 2m, which are commercially available, are significant as the corresponding aldehydes are not available for purchase and are relatively unstable. Modification of the heteroaryl-containing alcohols 2c-2i, 2t and 2u, which includes perphenazine 2g, an FDA approved drug, establishes the feasibility of utilizing this method for late-stage functionalization.15 Due to a pronounced kinetic bias for primary alcohol dehydrogenation,16 free secondary hydroxyl groups are tolerated, as illustrated in the site-selective formation of (R)-butane diol adducts 3b′ and 3c′, which occur with complete levels of catalyst-directed diastereoselectivity. Using this first generation catalytic system, benzylic alcohols are converted to the amino alcohols in high yield but lower enantioselectivities are observed. As demonstrated by the conversion of dehydro-2l to amino alcohol 3l, the reactions can also be conducted from the aldehyde oxidation level using 2-propanol as terminal reductant (eq. 1). Given the frequent appearance of morpholines as substructures in pharmaceutical ingredients,17 compound 3a was converted to the morpholine 5a (eq. 2).18 To further demonstrate utility of amino alcohols 3a-3z, 3a′−3c′, adduct 3m was subjected to alkene oxidative cleavage to provide the non-proteinogenic amino acid derivative 6m (eq. 3).19
Table 2.
Diastereo- and enantioselective iridium-catalyzed hydrohydroxyalkylation of phthalimido-allene 1 with alcohols 2a-2z, 2a′−2c′ to form 1,2-amino alcohols 3a-3z, 3a′−3c′.a
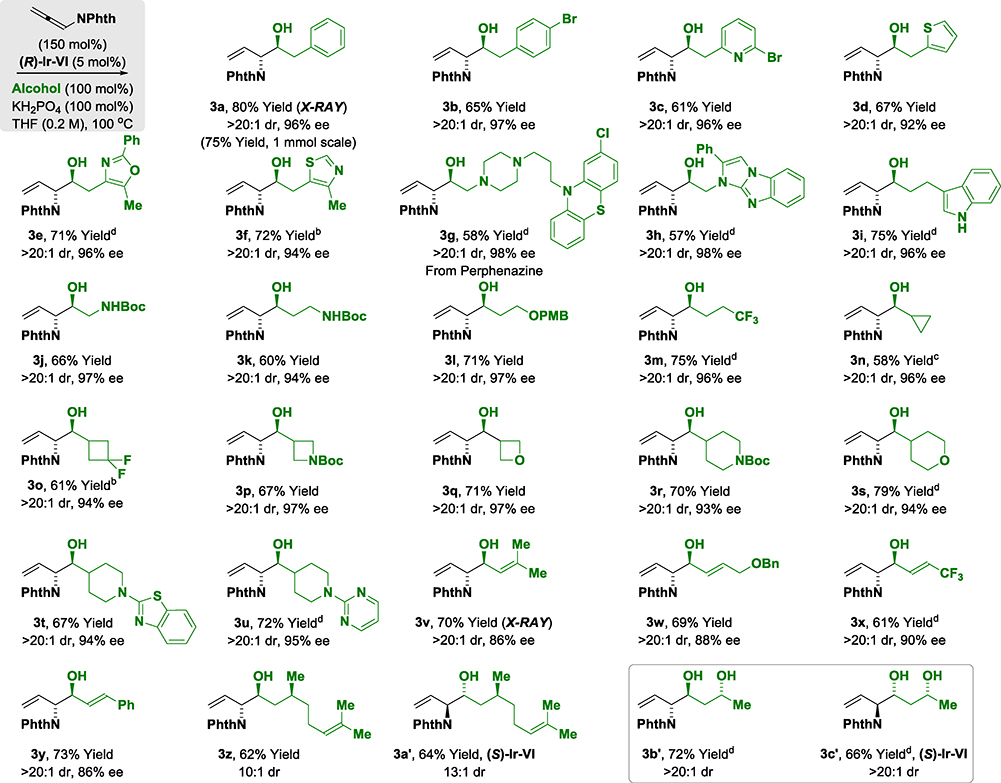 |
Yields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR of crude reaction mixtures. Enantioselectivities were determined by chiral stationary phase HPLC analysis. Standard conditions: 0.2 mmol scale, 48 h. See Supporting Information for experimental details. b72 h, cIr-V, dIr-VI (7.5 mol%).
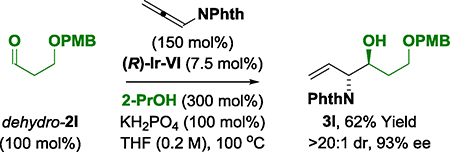 |
(eq. 1) |
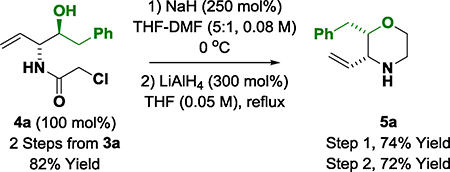 |
(eq. 2) |
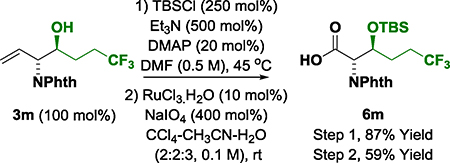 |
(eq. 3) |
Reaction progress kinetic analysis (RPKA) using the “different excess” protocol was used to gain mechanistic insight (Figure 2).20 The kinetic order of reactants varied over time; therefore, general trends were evaluated. Doubling the initial concentration of allene 1 slightly decreases the rate of product formation. This data suggests allene hydrometalation is rapid, allene 1 is not involved in the turnover-limiting step and, at higher concentrations, allene 1 inhibits the rate of product formation (Figure 2, left). Doubling the initial concentration of alcohol 2a results in a slight increase in the rate of product formation, signifying a positive order in alcohol 2a (Figure 2, middle). Increasing the loading of iridium catalyst, (R)-Ir-VI, results in a dramatic increase in the rate of product formation, demonstrating the reaction is positive order in catalyst (Figure 2, right). Separate experiments using the “same excess” protocol reveal significant catalyst deactivation that is contributed to by product inhibition.21 Additionally, introduction of aldehyde dehydro-2a (10 mol%) inhibits product formation, suggesting carbonyl addition may not be turn-over limiting.21
Figure 2.

Product formation as monitored by 1H NMR analysis in reactions conducted using the “different excess” protocol: [Ir] = 0.01 M; [KH2PO4] = 0.2 M. (left) [2a]0 = 0.2 M, [1]0 = as noted; (middle) [1]0 = 0.3 M, [2a]0 = as noted. (right) Product formation varying catalyst loading reactions as monitored by NMR analysis: [1] = 0.3 M; [2a] = 0.2 M; [KH2PO4] = 0.2 M; [cat] = as noted.
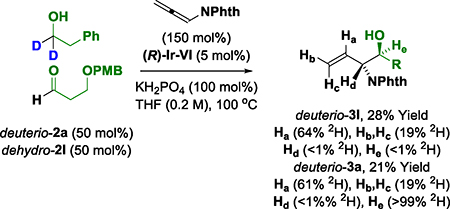
Deuterium labeling studies provide additional information on the reaction mechanism (eq. 4–6).22 Exposure of allene 1 to deuterio-2a under standard reaction conditions delivers deuterio-3a (eq. 4). Deuterium is completely retained at the carbinol position, suggesting deuterio-3a is inert with respect to dehydrogenation. Incorporation of deuterium at both the internal and terminal vinylic positions corroborates reversible allene hydrometalation with incomplete regioselectivity. In a competition kinetics experiment, allene 1 was exposed to equimolar quantities of alcohol 2a and deuterio-2a (eq. 5). The observed levels of deuterium incorporation at the carbinol position of deuterio-3a are consistent with a normal primary kinetic isotope effect (kH/kD ≈ 2.3). Evaluation of the initial rates for the reaction of both 2a and deuterio-2a also reveals a primary kinetic isotope effect (kH/kD ≈ 1.5) (Figure 3). Along with the reaction orders suggested from the RKPA experiments, this KIE data was consistent with two scenarios: (1) reversible alcohol dehydrogenation followed by rate-determining carbonyl addition, or (2) rate-determining alcohol dehydrogenation.22 To determine which of these processes is operative an additional experiment was undertaken (eq. 6). When pthalimido-allene 1 is exposed to equimolar quantities of deuterio-2a and dehydro-2l under standard conditions, hydrogen-deuterium exchange is not observed at the carbinol position of deuterio-3a and dehydro-3l, suggesting alcohol-aldehyde redox equilibration does not occur in advance of carbonyl addition. Hence, the collective data implicate turnover-limiting alcohol dehydrogenation followed by rapid allene hydrometalation.
Figure 3.

Initial rates study: [1]0 = 0.3 M; [2a]0 or [deuterio-2a]0 = 0.2 M; [cat] = 0.01 M.
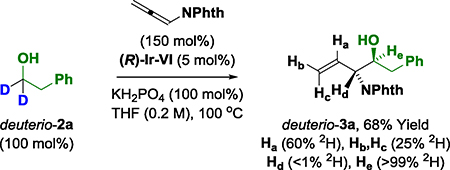 |
(eq. 4) |
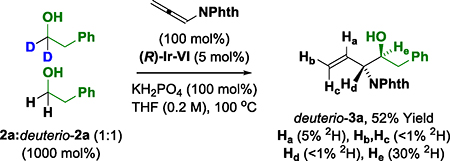 |
(eq. 5) |
 |
(eq. 6) |
Based on the kinetic and isotopic labeling studies, the indicated catalytic mechanism is proposed (Scheme 1). Entry into the catalytic cycle occurs through protonolysis of the allyliridium complex (R)-Ir-VI by the reactant alcohol. The resulting iridium alkoxide I undergoes irreversible dehydrogenation to form the iridium hydride II, which is rapidly consumed by reversible allene hydrometalation. Due to the steric demand of the phthalimide moiety, the (Z)-σ-(amino)allyliridium complex IIIa is anticipated to be the kinetic product of allene hydrometalation. Isomerization to the thermodynamically preferred (E)-σ-allyliridium complex IIIb, is followed by aldehyde coordination and carbonyl addition through a closed chair-like transition structure to form iridium(III) alkoxide IV. Exchange with the primary alcohol reactant releases product and regenerates iridium alkoxide I to close the catalytic cycle.
Scheme 1.

General catalytic mechanism as corroborated by kinetic and isotopic labeling studies.
In summary, we report a catalytic method for the direct conversion of primary alcohols to vicinal amino alcohols that occurs with high levels of regio-, anti-diastereo- and enantioselectivity. This hydrogen auto-transfer process exploits the tractable, crystalline phthalimido-allene 1 as pronucleophile and represents the first protocol for catalytic enantioselective carbonyl (α-amino)allylation. More broadly, this work contributes to an evolution from use of traditional carbonyl addition methods that exploit preformed carbanions to byproduct-free catalytic carbonyl reductive couplings, where alcohol proelectrophiles and π-unsaturated pronucleophiles combine by way of transient organometallics.4
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445, 1 S10 OD021508–01). We thank Professor Kami Hull for helpful discussions of the kinetic data.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available: Experimental procedures and spectral data. X-Ray diffraction data for compounds 3a, 3v and the (R)-H8-BINAP-modified iridium complex Ir-VI. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).For selected reviews on carbonyl addition chemistry, see:; (a) Noyori R; Kitamura M Enantioselective Addition of Organometallic Reagents to Carbonyl Compounds: Chirality Transfer, Multiplication and Amplification Angew. Chem. Int. Ed 1991, 30, 49. [Google Scholar]; (b) Soai K; Shibata T Alkylation of Carbonyl Groups In Comprehensive Asymmetric Catalysis I-III; Jacobsen EN, Pfaltz A, Yamamoto H, Eds.; Springer-Verlag Berlin Heidelberg: Germany, 1999; Vol. 2, pp 911. [Google Scholar]; (c) Pu L; Yu H-B Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds Chem. Rev 2001, 101, 757. [DOI] [PubMed] [Google Scholar]; (d) Trost BM; Weiss AH The Enantioselective Addition of Alkyne Nucleophiles to Carbonyl Groups Adv. Synth. Catal. 2009, 351, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Comprehensive Organic Synthesis, 2nd ed.; Knochel P; Molander GA, Eds.; Elsevier: Oxford, 2014; Vols. 1 and 2. [Google Scholar]
- (2).Schneider N; Lowe DM; Sayle RA; Tarselli MA; Landrum GA Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem. 2016, 59, 4385. [DOI] [PubMed] [Google Scholar]
- (3).For a recent review on metal-catalyzed carbonyl reductive coupling, see: Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For selected reviews on carbonyl reductive coupling via hydrogen auto-transfer, see:; (a) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier Acc. Chem. Res 2017, 50, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed 2019, 58, In Press - 10.1002/anie.201905532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Ghislieri D; Turner NJ Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284. [Google Scholar]; (b) Breuer M; Ditrich K; Habicher T; Hauer B; Kesseler M; Stürmer R; Zelinski T Industrial Methods for the Production of Optically Active Intermediates. Angew. Chem., Int. Ed 2004, 43, 788. [DOI] [PubMed] [Google Scholar]; (c) Sehl T; Maugeri Z; Rother D Multi-step Synthesis Strategies towards 1,2-Amino Alcohols with Special Emphasis on Phenylpropanolamine. J. Mol. Catal. B: Enzy. 2015, 114, 65 and references cited therein. [Google Scholar]
- (6).For racemic vicinal amino alcohols via metal-catalyzed carbonyl (α-amino)allylation, see:; (a) Skucas E; Zbieg JR; Krische MJ anti-Aminoallylation of Aldehydes via Ruthenium Catalyzed Transfer Hydrogenative Coupling of Sulfonamido-Allenes: 1,2-Aminoalcohols. J. Am. Chem. Soc 2009, 131, 5054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR; McInturff EL; Krische MJ Allenamide Hydro-Hydroxyalkylation: 1,2-Aminoalcohols via Ruthenium Catalyzed Carbonyl anti-Aminoallylation. Org. Lett. 2010, 12, 2514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang W Chen W; Xiao H; Krische MJ Carbonyl anti-(α-Amino)allylation via Ruthenium Catalyzed Hydrogen Auto-Transfer: Use of an Acetylenic Pyrrole as an Allylmetal Pronucleophile. Org. Lett 2017, 19, 4876. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schwartz JL; Schäfers F; Tlahuext-Aca A; Lückemeier, L.; Glorius, F. Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc 2018, 140, 12705. [DOI] [PubMed] [Google Scholar]
- (7).(a) Barrett AGM; Seefeld MA B- [(E) - 3- (Diphenylamino) allyl] diisopinocampheylborane: An Excellent Reagent for The Stereoselective Synthesis of anti- β- Diphenylamino Alcohols. J. Chem. Soc., Chem. Commun 1993, 339. [Google Scholar]; (b) Barrett AGM; Seefeld MA The Use of B- [(E) - 3- (Diphenylamino) allyl] diisopinocampheylborane as A Reagent for The Stereoselective Synthesis of anti- β- Diphenylamino Alcohols and trans- 1- Diphenylamino- 2- (1- hydroxylalkyl) cyclopropanes. Tetrahedron 1993, 49, 7857. [Google Scholar]; (c) Barrett AGM; Seefeld MA; Williams DJ Convenient Asymmetric Synthesis of anti- β- Amino Alcohols: An X- Ray Crystallographic Study of (4R) - 2, 2- Dimethyl- 4- [(2S) - (diphenylmethyleneamino) - (1S) - hydroxy- 3- buten- 1- yl] - 1, 3- dioxolane. J. Chem. Soc., Chem. Commun 1994, 1053. [Google Scholar]; (d) Barrett AGM; Seefeld MA; White AJP; Williams DJ Convenient Asymmetric Syntheses of anti- β- Amino Alcohols. J. Org. Chem 1996, 61, 2677. [DOI] [PubMed] [Google Scholar]
- (8).For selected reviews on enantioselective carbonyl allylation, see:; (a) Ramachandran PV Pinane-based versatile “allyl” boranes. Aldrichim. Acta 2002, 35, 23. [Google Scholar]; (b) Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763. [DOI] [PubMed] [Google Scholar]; (c) Yu C-M; Youn J; Jung H-K Regulation of Stereoselectivity and Reactivity in the Inter- and Intramolecular Allylic Transfer Reactions. Bull. Korean Chem. Soc 2006, 27, 463. [Google Scholar]; (d) Marek I; Sklute G Creation of Quaternary Stereocenters in Carbonylallylation Reactions. Chem. Commun 2007, 1683. [DOI] [PubMed] [Google Scholar]; (e) Hall DG Lewis and Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds: From Discovery to Mechanism and Applications. Synlett 2007, 11, 1644. [Google Scholar]; (f) Hargaden GC; Guiry PJ The Development of the Asymmetric Nozaki–Hiyama–Kishi Reaction. Adv. Synth. Catal 2007, 349, 2407. [Google Scholar]; (g) Lachance H; Hall DG Allylboration of Carbonyl Compounds. Org. React. 2008, 73, 1. [Google Scholar]; (h) Yus M; González-Gómez JC; Foubelo F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774. [DOI] [PubMed] [Google Scholar]; (i) Yus M; Gonzalez-Gomez JC; Foubelo F Diastereoselective Allylation of Carbonyl Compounds and Imines: Application to the Synthesis of Natural Products. Chem. Rev 2013, 113, 5595. [DOI] [PubMed] [Google Scholar]; (j) Huo H-X; Duvall JR; Huang M-Y; Hong R Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronates. Org. Chem. Front 2014, 1, 303. [Google Scholar]; (k) Kumar P; Tripathi D; Sharma BM; Dwivedi N Transition Metal Catalysis - A Unique Road Map in the Stereoselective Synthesis of 1,3-Polyols. Org. Biomol. Chem 2017, 15, 733. [DOI] [PubMed] [Google Scholar]; (l) Spielmann K; Niel G; de Figueiredo RM; Campagne JM Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev 2018, 47, 1159. [DOI] [PubMed] [Google Scholar]
- (9).(a) Li K; Shao X; Tseng L; Malcolmson SJ 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao X; Li K; Malcolmson S Enantioselective Synthesis of anti-1,2-Diamines by Cu-Catalyzed Reductive Couplings of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc 2018, 140, 7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Only five catalytic enantioselective allene-carbonyl reductive couplings have been reported, see:; (a) Han SB; Kim IS; Han H; Krische MJ Enantioselective Carbonyl Reverse Prenylation from the Alcohol or Aldehyde Oxidation Level Employing 1,1-Dimethylallene as the Prenyl Donor. J. Am. Chem. Soc 2009, 131, 6916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MJ Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C-H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc. 2017, 139, 8114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tsai EY; Liu RY; Yang Y; Buchwald SL A Regio- and Enantioselective CuH- Catalyzed Ketone Allylation with Terminal Allenes. J. Am. Chem. Soc 2018, 140, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schwartz LA; Holmes M; Brito GA; Gonç TP; Richardson J; Ruble JC; Huang K-W; Krische MJ Cyclometallated Iridium-PhanePhos Complexes Are Active Catalysts in Enantioselective Allene-Fluoral Reductive Coupling and Related Alcohol-Mediated Carbonyl Additions that Form Acyclic Quaternary Carbon Stereocenters. J. Am. Chem. Soc. 2019, 141, 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu RY; Zhou Y; Yang Y; Buchwald SL Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct. J. Am. Chem. Soc 2019, 141, 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Fenandez I; Monterde MI; Plumet J On the Base-Induced Isomerization of Cyclic Propargylamides to Cyclic Allenamides. Tetrahedron Lett. 2005, 46, 6029. [Google Scholar]; (b) Lindsay VNG; Fiset D; Gritsch PJ; Azzi S; Charette AB Stereoselective Rh2(S-IBAZ)4-Catalyzed Cyclopropanation of Alkenes, Alkynes, and Allenes: Asymmetric Synthesis of Diacceptor Cyclopropylphosphonates and Alkylidenecyclopropanes. J. Am. Chem. Soc 2013, 135, 1463. [DOI] [PubMed] [Google Scholar]
- (12).Bower JF; Skucas E; Patman RL; Krische MJ Catalytic C-C Coupling via Transfer Hydrogenation: Reverse Prenylation, Crotylation and Allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc 2007, 129, 15134. [DOI] [PubMed] [Google Scholar]
- (13).(a) Zhang X; Mashima K; Koyano K; Sayo N; Kumobayashi H; Akutagawa S; Takaya H Synthesis of Partially Hydrogenated BINAP Variants. Tetrahedron Lett. 1991, 32, 7283. [Google Scholar]; (b) Zhang X; Taketomi T; Yoshizumi T; Kumobayashi H; Akutagawa S; Mashima K; Takaya H Asymmetric Hydrogenation of Cycloalkanones Catalyzed by BINAP-Ir(I)-Aminophosphine Systems. J. Am. Chem. Soc 1993, 115, 3318. [Google Scholar]; (c) Zhang X; Mashima K; Koyano K; Sayo N; Kumobayashi H; Akutagawa S; Takaya H Synthesis of Partially Hydrogenated 2,2′-Bis(diphenylphosphenyl)-1,1′-binaphthyl (BINAP) Ligands and Their Application to Catalytic Asymmetric Hydrogenation. J. Chem. Soc. Perkin Trans 1 1994, 2309. [Google Scholar]
- (14).For X-ray diffraction data on analogous π-allyliridium-C,O-benzoate complexes modified by (R)-SEGPHOS and (R)-BINAP, see CCDC929090 and CCDC712857, respectively.
- (15).For selected reviews on late-stage functionalization of complex small molecules, see:; (a) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug- Like Molecules. Chem. Soc. Rev. 2016, 45, 546. [DOI] [PubMed] [Google Scholar]; (b) Shugrue CR; Miller SJ Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev. 2017, 117, 11894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blakemore DC; Castro L; Churcher I; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem. 2018, 10, 383. [DOI] [PubMed] [Google Scholar]; (d) Dominguez-Huerta A; Dai X-J; Zhou F; Querard P; Qiu Z; Ung S; Liu W; Li J; Li C-J Exploration of New Reaction Tools for Late-Stage Functionalization of Complex Chemicals. Can. J. Chem 2019, 97, 67. [Google Scholar]
- (16).Although primary alcohol dehydrogenation is more endothermic than secondary alcohol dehydrogenation, primary alcohol dehydrogenation is kinetically preferred due to the steric demand of the catalyst. Site-selective methods for the C-C coupling of diols and higher polyols are summarized in reference 4a.
- (17).For selected reviews highlighting the recurrence of N-heterocycles among FDA approved drugs, see:; (a) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (18).The synthesis of morpholine 5a was conducted in analogy to the following procedures:; (a) Alexander R; Balasundaram A; Batchelor M; Brookings D; Crépy K; Crabbe T; Deltent M-F; Driessens F; Gill A; Harris S; Hutchinson G; Kulisa C; Merriman M; Mistry P; Partone T; Turnera J; Whitcombe I; Wrighte, S. 4-(1,3-Thiazol-2-yl)morpholine Derivatives as Inhibitors of Phosphoinositide 3-Kinase. Bioorg. Med. Chem. Lett 2008, 18, 4316. [DOI] [PubMed] [Google Scholar]; (b) Hu P; Hu J; Jiao J; Tong X Amine-Promoted Asymmetric (4+2) Annulations for the Enantioselective Synthesis of Tetrahydropyridines: A Traceless and Recoverable Auxiliary Strategy. Angew. Chem. Int. Ed 2013, 52, 5319. [DOI] [PubMed] [Google Scholar]
- (19).Chen YK; Lurain AE; Walsh PJ A General, Highly Enantioselective Method for the Synthesis of d and l α-Amino Acids and Allylic Amines. J. Am. Chem. Soc 2002, 124, 12225. [DOI] [PubMed] [Google Scholar]
- (20).For a review on reaction progress kinetic analysis, see: Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem., Int. Ed 2005, 44, 4302. [DOI] [PubMed] [Google Scholar]
- (21).This experiment is described in the Supporting Information.
- (22).An authoritative treatment of the execution and interpretation of kinetic isotope effect experiments in reactions involving C-H bond cleavage is reported. Our catalytic mechanism appears consistent with reaction coordinate 1, Figure 1: Simmons, E. M.; Hartwig, J. F. On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed 2012, 51, 3066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.