

Supplemental Digital Content is available in the text.
Keywords: Alzheimer disease, hypertension, inositol, risk factor, umbilical cord
Abstract
Transient hypertension is a risk factor for Alzheimer disease (AD), but the effects of this interaction on brain vasculature are understudied. Addressing vascular pathology is a promising avenue to potentiate the efficacy of treatments for AD. We used arterial spin labeling magnetic resonance imaging to longitudinally assess brain vascular function and immunohistopathology to examine cerebrovascular remodeling and amyloid load. Hypertension was induced for 1 month by administration of l-NG-nitroarginine-methyl-ester in TgF344-AD rats at the prodromal stage. Following hypertension, nontransgenic rats showed transient cerebrovascular changes, whereas TgF344-AD animals exhibited sustained alterations in cerebrovascular function. Human umbilical cord perivascular cells in combination with scyllo-inositol, an inhibitor of Aβ oligomerization, resulted in normalization of hippocampal vascular function and remodeling, in contrast to either treatment alone. Prodromal stage hypertension exacerbates latter AD pathology, and the combination of human umbilical cord perivascular cells with amyloid clearance promotes cerebrovascular functional recovery.
Alzheimer disease (AD) is a fatal disorder characterized by amnesia, progressive cognitive impairment, disorientation, behavioral disturbance, and loss of daily function. It afflicts over 50 million people worldwide.1 The hallmarks of AD include gradual accumulation of protein aggregates (Aβ and neurofibrillary tangles) and cerebrovascular amyloid, neuronal and cerebrovascular dysfunction, and eventually neuronal death.2 Sporadic AD constitutes over 95% of late-onset AD cases3 and is caused by an incompletely understood pathophysiological cascade triggered by environment and lifestyle.3 Risk factors account for as much as 35% of AD burden,4 with hypertension representing the major contributing factor towards development of AD.5 Epidemiological studies have found little to no reduction of dementia incidence following blood pressure (BP) normalization in late life, suggesting that even transient high BP irreversibly damages the brain.6,7 Although the incidence of severe hypertension in the senior population is limited through monitoring and pharmacological interventions,8 the prevalence of moderate hypertension in young adults is between 24% and 32%,9–11 and its incidence is rising.12 This is particularly alarming as even moderate hypertension may impair the vasculature9 by leading to loss of elastin, arteriosclerosis, arterial stiffening, and ultimately reducing cerebral blood flow.13
To date, disease-modifying approaches targeting Aß accumulation, cholinergic transmission, and inflammation have failed to deliver cognitive benefits even in mild-to-moderate AD patient population.14,15 Given the supra-additive effects of the interaction between amyloid and vascular pathologies16 and the high prevalence of vascular pathologies, there have been widespread calls for development of novel therapeutic approaches targeting the interaction between these 2 conditions.16,17 Due to the methodological complexity of these studies, no longitudinal investigations to date have assessed in situ brain physiological effects of combinatorial treatments in AD comorbid with transient hypertension. To fill this gap, we longitudinally examined cerebrovascular function in the TgF344-AD rat model made transiently hypertensive in the prodromal phase of amyloid pathology (Figure 1A). This AD model exhibits progressive deposition of amyloid-β peptides, tau hyperphosphorylation and tangles formation, and chronic neuroinflammation, starting after 6 months of age, followed by neuronal injury and loss starting between 9 and 16 months of age.18–20 The variability of antihypertensive approaches used in the clinic21 makes the direct preclinical model arduous. To model transient hypertension, we targeted NOS (nitric oxide synthase) in the endothelial cells (eNOS [endothelial NOS]) via the specific inhibitor l-NAME widely used in hypertension modeling.22,23 To ensure that the hypertension paradigm is transient and that the effects of l-NAME on NOS are no longer present, we measured NOS expression in vessel-enriched hippocampal tissue at end point. This approach has the advantage of increasing systemic BP24 without permanently impairing NOS activity, which recovers within 24 hours after the end of l-NAME administration.25 To promote cerebrovascular recovery,14 we combined amyloid clearance via scyllo-inositol (SI) treatment26 with first-trimester human umbilical cord perivascular cell (HUCPVC) treatment. The use of multipotent cell type was motivated by our earlier observations of fast proliferation and low immunogenicity of HUCPVCs27; their improved angiogenic potential and pericyte-like behavior28 and their promising effects on cognition and daily living activities in elderly vascular dementia patients.29 We allowed BP normalization (ie, 3 weeks after l-NAME cessation) before commencing treatment so as to examine the potential of treatments to mitigate the chronic effects of transient hypertension. We allowed 1 week for SI to reach stable concentration in the brain,30 before administering a single dose of HUCPVC and beginning immunosuppression with cyclosporine-A for 4 weeks to increase the survival of transplanted cells.31 Final imaging to evaluate cerebrovascular function was conducted 4 weeks after the HUCPVC administration so as to assess an extended-term outcome. We found that transient moderate hypertension in the prodromal stage of AD permanently compromised cerebrovascular function in the TgF344-AD animals, but not in their nontransgenic littermates, and that latter stage HUCPVCs treatment required concomitant amyloid clearance for resolution of the cerebrovascular deficits, suggesting synergistic effects may be obtained by combining cerebrovasculature-targeted treatments with amyloid clearance.
Figure 1.
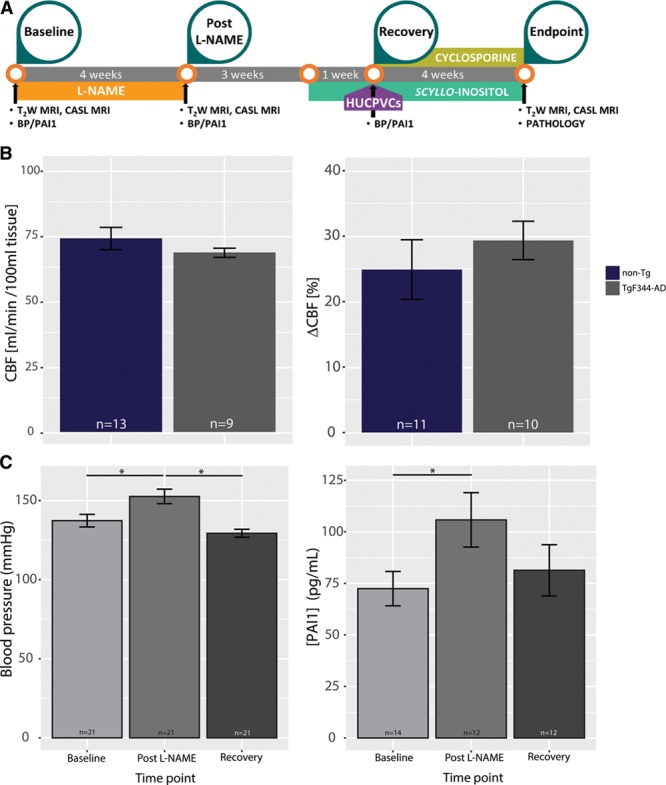
Experimental approach. A, Experimental timeline; (B) baseline hemodynamics. Resting hippocampal perfusion (left: 74.2±3.4 mL/[min·100 mL] tissue for the nontransgenic [non-Tg] vs 71.1±1.9 mL/[min·100 mL] tissue for the TgF344-AD, P=0.5) and cerebrovascular responsivity to hypercapnia (right: 24.8±4.5% for the non-Tg vs 29.3±2.9% tissue for the TgF344-AD, P=0.4) were indistinguishable between the 2 genotypes at baseline. C, N(G)-Nitro-l-arginine methyl ester (l-NAME) effect on blood pressure (BP) and PAI1 (plasminogen activator inhibitor-1). C, left, Blood pressure rose from baseline level of 137.3±3.9 to 152.7±4.5 mm Hg at the end of the l-NAME administration (P=0.02); the latter time point coincided with post–l-NAME time point of magnetic resonance imaging (MRI) experiments. One month after termination of l-NAME treatment (ie, at recovery time point) BP normalized to baseline levels (129.3±2.5 mm Hg, P=0.3 when compared with baseline, P=0.0002 when compared with post–l-NAME, 1-way ANOVA with post hoc Tukey honestly significant difference test). Right, PAI1 concentration rose from baseline level of 72.4±8.3–105.8±13.2 pg/mL at the end of the l-NAME administration (P=0.041); the latter time point coincided with post–l-NAME time point of MRI experiments. One month after termination of l-NAME treatment (ie, at recovery time point) PAI1 concentration normalized to baseline levels (81.3±12.4 pg/mL, P=0.56 when compared with baseline, P=0.5 when compared with post–l-NAME). CASL indicates continuous arterial spin labeling; CBF, cerebral blood flow; and HUCPVC, human umbilical cord perivascular cell.
Methods
All experiments followed the Animal Research: Reporting of In Vivo Experiments guidelines and were approved by the Animal Care Committee of the Sunnybrook Research Institute. A total of 115 rats were used in this study, 41 non-Transgenic and 74 TgF344-AD. Animals were housed in pairs on a regular 12-hour light/dark cycle. All data processing was performed blinded to treatment group allocation. Exclusion criteria and group size are summarized in Table S1 in the online-only Data Supplement. Data and analytical methods will be made available upon request. Only procedures that differ from previously published methods are described here (details provided in the online-only Data Supplement).
Hypertension, BP and Plasminogen Activator Inhibitor-1 Measurement
One hundred two (34 non-Transgenic and 68 TgF344-AD, Table S1) 4-month old TgF344-AD rats and their nontransgenic littermates were administered l-NAME (Sigma-Aldrich, Oakville, ON) to induce transient hypertension in the prodromal phase of the AD via drinking water (males: 10 mg/kg per day, females: 7.5 mg/kg per day) for 30 days. We measured systolic BP using the CODA-HT2 tail-cuff system (Kent Scientific). PAI1 (plasminogen activator inhibitor-1) plasma levels, commonly investigated in patients,32,33 were measured using a commercially available kit (PAI1 ELISA Kit, ab201283) following manufacturer’s instructions.
Drug Treatment
SI (Transition Therapeutics, Inc) was given adlibitum in drinking water 10 mg/mL from 3 weeks post–l-NAME treatment until sacrifice.26 Cyclosporine treatment was initiated 4 weeks post–l-NAME cessation (SandImmune I.V.; Novartis Canada, Dorval, QC) at a dosage of 10 mg/kg per day via subcutaneous injection for 5 days, followed by delivery via drinking water beginning on the fifth day of injections and continuing until end point (at 10 mg/kg per day). The dosage in drinking water was calculated using average water consumption (males: 16 mL/day, females: 14 mL/day) according to weight (or average weight of group-housed animals).
HUCPVC Treatment
HUCPVC (CReATeIVF, Toronto) were harvested as in our previous work.27 HUCPVC (106 cells/0.5 cc) or saline (0.5 cc) was injected via tail vein in rats anesthetized with isoflurane (5% induction, 3% maintenance) on the third day of cyclosporine treatment.
Magnetic Resonance Imaging
Continuous arterial spin labeling magnetic resonance imaging was performed at baseline, post–l-NAME, and end point to measure cerebrovascular reactivity changes over time; continuous arterial spin labeling data were analyzed as described previously.34,35 Subject-specific hemodynamic response functions were produced by averaging the signal over the thalamic nuclei: this region was used to define reference hemodynamic response function as it spared from amyloid pathology at this stage of the disease (Figure S1).
Immunohistopathology
At the end point, the animals were euthanized, and their brains were extracted.17 Four hippocampus-containing coronal sections per animal, evenly spaced from −2.8 mm to −5.5 mm relative to bregma, were selected for immunostaining. Slices were stained for elastin/collagen IV and for Aβ load.19, 20
Statistical Analysis
Unless specified otherwise, linear mixed effects36 modeling was used in the statistical analysis of all normally distributed data, and results are expressed as mean±SEM.
Results
Baseline Hippocampal Hemodynamics Are not Different Between Genotypes
Four-month-old TgF344-AD rats and their nontransgenic littermates were administered l-NAME to induce transient hypertension in the prodromal phase of the AD. To ensure that overexpression of human APPswe and PS1ΔE9 in this model did not affect vascular function before overt disease onset, we examined cerebrovascular function using continuous arterial spin labeling magnetic resonance imaging. At 4 months of age, before the appearance of Aβ pathology, resting perfusion and responsivity to hypercapnia were indistinguishable between the nontransgenic and TgF344-AD animals (Figure 1B: resting perfusion: 74.2±3.4 mL/[min·100 mL] tissue for the nontransgenic versus 71.1±1.9 mL/[min·100 mL] for the TgF344-AD, P=0.5; blood flow increase to hypercapnia: 24.8±4.5% for the nontransgenic versus 29.3±2.9% for the TgF344-AD, P=0.4).
l-NAME Induces Mild-Moderate, Transient Hypertension in Both Genotypes
To model transient hypertension in the young adult, we targeted NOS in the eNOS via the specific inhibitor l-NAME.23 The transient hypertension terminology was motivated by the time-limited period of hypertension and the lack of blood pressure targeting treatment in the present study. Four-month-old TgF344-AD rats and their nontransgenic littermates were thus administered l-NAME for 1 month to induce transient hypertension in the prodromal phase of the AD. In the human population, BPs increases with age; and hypertension is defined as severe when BP is >20% higher than normal and mild-moderate when it is within 20% of the normal level.8 In our experiments, systolic BP rose 11±2% (from baseline level of 137.3±3.9 to 152.7±4.5 mm Hg post–l-NAME), therefore, falling in the middle of the mild-moderate interval.8 ANOVA with Tukey multiple comparisons of means did not indicate a significant difference between the BP at recovery when compared with that at baseline (P=0.3, n=21). The TgF344-AD rats and their nontransgenic littermates did not differ in either baseline BP or in post–l-NAME BP; however, female rats required a lower l-NAME dose to achieve similar increases in BP as the male rats (data not shown). Two-way ANOVA of BP data revealed no interaction between time and genotype and no effect of genotype (P=0.28 and P=0.14, respectively, Figure S2). In contrast, time had a significant effect on BP, which rose 12±2% at post–l-NAME time point (from 137.3±3.9 to 152.7±4.5, P=0.002) and then was no longer distinguishable from baseline level at the recovery time point (129.3±2.5, P=0.61, Figure 1C). This model hence recapitulated transient mild-moderate increase in BP in the young-adult population.
To evaluate whether this mild-moderate increase in BP was sufficient to exert the biological effects typical of hypertension in patient population,37 we measured PAI1 plasma levels 1 day before l-NAME administration (baseline), the day after the end of l-NAME treatment (post–l-NAME), and 1 month after the end of l-NAME administration (recovery). l-NAME administration resulted in a 47±6% increase in plasma level of PAI1 across all animals (PAI1, 72.4±8.3 pg/mL at baseline versus 105.8±13.2 pg/mL post–l-NAME, P=0.041), comparable to ≈30% plasma activity increase observed in patients.38 One month after termination of l-NAME treatment, PAI1 concentration returned to its baseline level (81.3±12.4 pg/mL, P=0.56, Figure 1C). To examine whether NOS expression levels were affected by the treatments, we measured eNOS, iNOS (inducible NOS), nNitric Oxide Synthases expression at end point: we found no significant differences in any NOS isoform levels between the treatment groups (Figure S3, ANOVA, F[3,68]=0.5, P=0.7).
Nontransgenic Animals Showed Spontaneous Recovery of Cerebral Hemodynamics
Nontransgenic rats exhibited transient hypoperfusion, which was followed by spontaneous recovery: resting perfusion dropped from 74.2±3.4 mL/(min·100 mL) at baseline to 45.8±4.5 mL/(min·100 mL) post–l-NAME, P=0.0002 and then recovered to 57.6±11.5 mL/(min·100 mL) at end point, P=0.53 when compared with baseline (Wilcoxon rank-sum test with Bonferroni, Figure 2A). In parallel, responsivity to hypercapnia increased from 38.7±10.5% at baseline to 82.3±14.4% post–l-NAME (P=0.026) and then recovered to 64.9±21.6% at end point (P=0.28 when compared with baseline, Figure 2A). Reduced resting perfusion yet elevated cerebrovascular responsivity to hypercapnia is a pattern also observed in the subacute stage after cortical ischemia,34,35,39 where it has been shown to underlie metabolic dysfunction and to correlate to the extent of inflammation and neurovascular damage. At end point, nontransgenic animals showed resting perfusion and responsivity to hypercapnia similar to nontransgenic animals at baseline (resting perfusion: 93.1±17 versus 76.7±0.96 mL/[min·100 mL] tissue, P=0.12; hypercapnia challenges: 57.7±23.1 versus 37.8±11.1%, P=0.65). In light of this spontaneous resolution of l-NAME induced cerebrovascular deficits in nontransgenic animals and high attrition of these studies, the nontransgenic animals were not subsequently treated.
Figure 2.
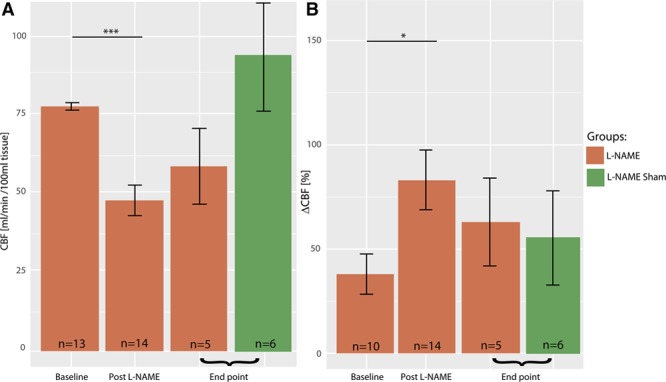
Cerebral hemodynamics in nontransgenic (non-Tg) animals. Non-Tg rats show a drop in the resting perfusion from 76.7±0.96 mL/(min·100 mL) tissue at baseline to 45.8±4.5 mL/(min·100 mL) post–l-NAME, which then recovers to 57.6±11.5 mL/(min·100 mL) at the end point (A, P=0.00015 and P=0.53 respectively, Wilcoxon rank-sum test with Bonferroni adjustment). Non-Tg rats’ responsivity to hypercapnia increases from 37.8±11.1% at baseline to 67.9±10.8% post–l-NAME and then recovers to 71.0±21.9% at the end point (B, P=0.0066 and P=0.21, respectively, Wilcoxon rank-sum test with Bonferroni adjustment). Sham l-NAME non-Tg animals showed resting perfusion and responsivity to hypercapnia similar to Sham administered non-Tg animals at baseline (resting perfusion: 93.1±17 vs 76.7±0.96 mL/min/100 mL tissue, P=0.12; hypercapnia challenges: 57.7±23.1 vs 37.8±11.1%, P=0.65). CBF indicates cerebral blood flow.
Cerebral Hemodynamics Alterations Persisted in Transgenic Animals
Rather than treating hypertension per se, our interventions were aimed at ameliorating lasting (ie, nonspontaneously recoverable) deficits in cerebrovascular function induced by transient hypertension. We, therefore, allowed 3 weeks after cessation of l-NAME for spontaneous recovery processes to play out before beginning SI treatment to lower Aβ levels. We subsequently allowed 1 week for SI to reach stable concentration within the brain,30 before beginning immunosuppression with cyclosporine-Aβ for 4 weeks to increase the survival of transplanted cells.31,40 Before administration of HUCPVCs, expression of characteristic mesenchymal stromal cell surface markers CD34, CD90, CD44, CD105, CD73, and the absence of nonmultipotent stem cell markers CD45, CD31, HLA-DPQR was confirmed using flow cytometric analyses of HUCPVCs (Figure 3). Final imaging session was conducted 4 weeks after the HUCPVC administration, that is, when the animals were 7 months old. Cerebral blood flow was assessed at rest to estimate steady-state tissue perfusion. Hippocampal hemodynamics in representative animals from each transgenic cohort are shown in Figure 4A: resting perfusion of a representative TgF344-AD rat at baseline (Ai) and post–l-NAME (Aii), as well as a vehicle-administered TgF344-AD rat (Aiii), a SI+HUCPVC treated TgF344-AD animal (Aiv) and Sham l-NAME animal (Av) at end point. Population analysis (Figure 4B) showed a significant drop, of 32.4±1.5%, in resting perfusion, from 73.8±1.6 mL/(min·100 mL) at baseline to 48±3 mL/(min·100 mL) post–l-NAME (P=0.00004). Resting perfusion in vehicle-administered TgF344-AD animals at the end point (47±1 mL/[min·100 mL] tissue) was still significantly reduced (by 33.8±1.8%) when compared with that at baseline (P=0.00015).
Figure 3.
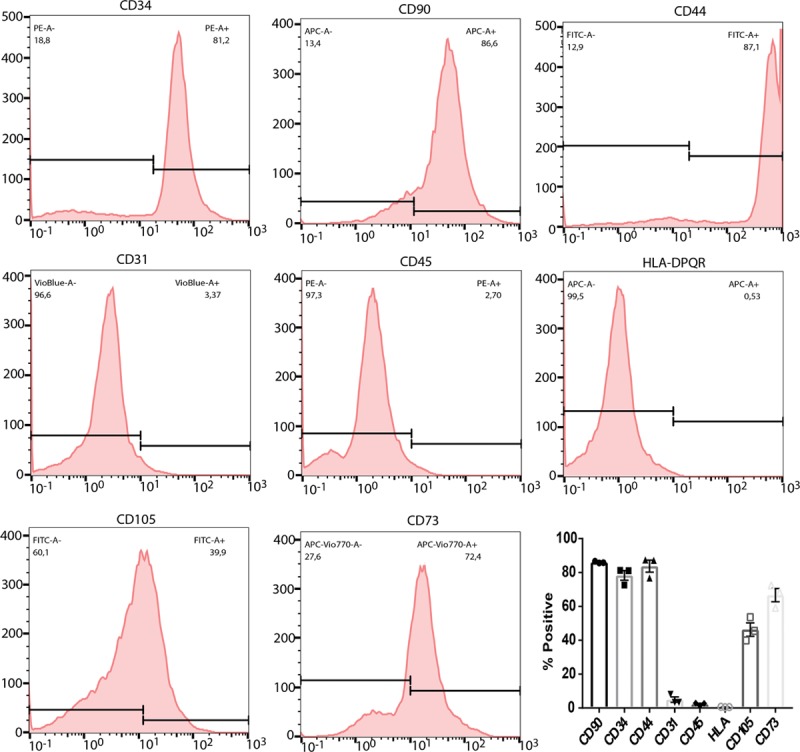
Human umbilical cord perivascular cell markers. Flow cytometry experiments targeting cell surface mesenchymal stromal cell markers show expression of CD90-PE, CD34-APC, CD44-FITC, CD105-FITC, CD73-PEVio770, and negative or low expression of nonmesenchymal markers CD45-PE, HLA-DPQR-APC, CD31-VioBlue. APC indicates activated protein C; FITC, fluorescein isothiocyanate; and PE, phycoerythrin.
Figure 4.
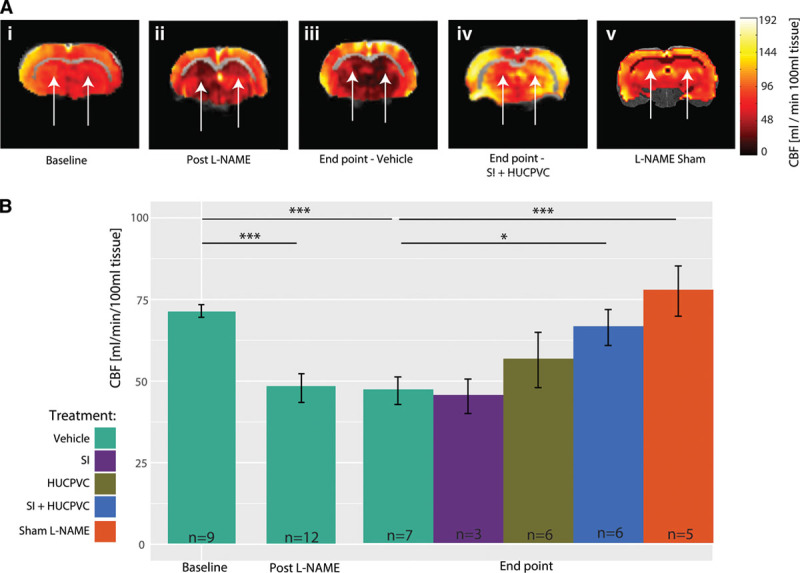
Resting perfusion in TgF344-AD rats. A, Representative continuous arterial spin labeling (CASL) cerebral blood flow (CBF) maps showing a drop in resting perfusion in the hippocampi (white arrows) of TgF344-AD rats from baseline, to post–l-NAME and the perfusion contrast between vehicle administration vs scyllo-inositol (SI) + human umbilical cord perivascular cell (HUCPVC) treatment at end point. B, Population analysis reveals a significant drop in resting perfusion from 73.8±1.6 at baseline to 48.3±3.8 mL/(min·100 mL) tissue post–l-NAME (P=8.52×10−6). Resting perfusion in vehicle administered animals at end point was still significantly reduced when compared with that at baseline (46.8±1.1 mL[min·100 mL] tissue, P=0.000235). SI-only and HUCPVC-only treated cohorts were indistinguishable from vehicle administered group (P=0.8 and P=0.14, respectively), whereas animals treated with a combination of SI and HUCPVC showed improvement in resting perfusion (63.9±1.9 mL/[min·100 mL] tissue, P=0.06 when compared with the vehicle-administered group). At end point, the TgF344-AD Sham l-NAME group showed higher resting perfusion than did the l-NAME and vehicle administered (77.7±7.7 vs 47±1 mL/[min·100 mL] tissue, P=0.000476).
When compared with vehicle-administered subjects at end point, neither SI-only nor HUCPVC-only treatments changed resting perfusion (SI-only: 45.3±5.2 mL/[min 100 mL] tissue, P=0.86; HUCPVC-only: 54.6±8.5 mL/[min·100 mL] tissue, P=0.13). In contrast, TgF344-AD rats treated with SI+HUCPVC showed 40±1% higher perfusion when compared with vehicle-administered animals at end point (SI+HUCPVC: 66±4 mL/[min·100 mL] tissue, P=0.014). At end point, the TgF344-AD Sham l-NAME group showed higher resting perfusion when compared with the l-NAME and vehicle-administered TgF344-AD rats (77.7±7.7 versus 47±1 mL/[min·100 mL] tissue, P=0.000476). Sustained hippocampal hypoperfusion reveals cerebral circulatory insufficiency that may destabilize neurons and synapses, trigger gradual neurodegeneration, and impair cognitive functioning.41 Cerebrovascular functioning was further assessed by vasodilatory challenges via elevating inspired CO2 tension. Figure 5A shows responses to hypercapnia of representative TgF344-AD rats at baseline, post–l-NAME, and at end point in a vehicle-administered TgF344-AD rat, in a SI+HUCPVC treated TgF344-AD and in a Sham l-NAME animal. Figure 5B presents the averaged time courses of the hippocampal perfusion responses to hypercapnia in the TgF344-AD animals. Population analysis (Figure 5C) revealed that hippocampal vascular responsivity to hypercapnia was increased by 217±10.1% (P=0.0004) from baseline (29±4%) to post–l-NAME (92±12%). The responsivity to hypercapnia remained elevated in vehicle-administered TgF344-AD rats at end point when compared with baseline (106.1±5.5% versus 29±4%, P=0.002). When compared with vehicle-administered animals, SI treated rats and HUCPVC treated rats did not show significantly different responses (SI-only: 111.5±27.7%, P=0.87 and HUCPVC-only: 88.9±25.9%, P=0.42, respectively). At end point, the TgF344-AD Sham l-NAME group showed reduced responses to hypercapnia (51.6±16.2% versus 106.1±5.5%, P=0.006) when compared with the l-NAME and vehicle-administered TgF344-AD rats. Such increased cerebrovascular responsivity to hypercapnia may underlie a dampened capacity of microvessels to resume resting tone post vasodilatation,42 potentially due to structural changes in the microvessels’ walls. In contrast to singly treated cohorts, rats that received a combined SI+HUCPVC treatment showed a significant reduction in their cerebral blood flow responses to hypercapnia when compared with vehicle-administered animals at end point (31.3±18.7%, P=0.009).
Figure 5.
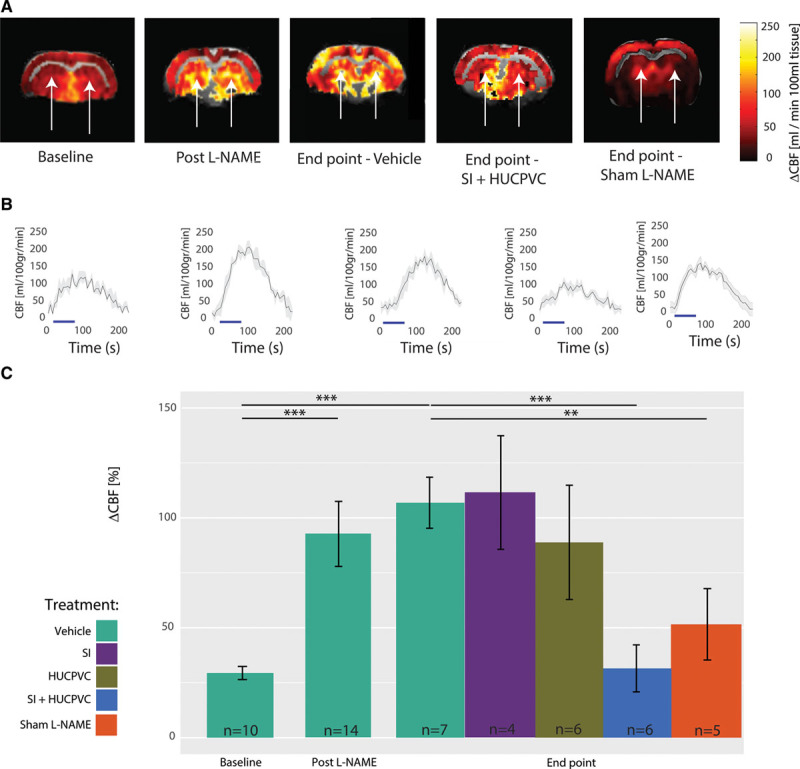
Cerebrovascular reactivity in the TgF344-AD rats. A, Representative experiments showing increased responsivity to hypercapnia in the hippocampus (white arrows) of TgF344-AD rats from baseline to post–l-NAME and at end point in a vehicle-administered animal but with relative resolution of this hyperreactivity in the scyllo-inositol (SI) + human umbilical cord perivascular cell (HUCPVC) treated rat. B, Averaged (black line and SD in gray) time courses of the responses over 4 hypercapnic challenges are shown at baseline, post–l-NAME and at end point for vehicle administered and SI+HUCPVC treated subject. Population analysis (C) reveals significantly increased (P=0.000105) responsivity to CO2 challenges from baseline (29.4±3.9%) to post–l-NAME (92.7±12.4%). The responsivity to hypercapnia remained increased at the end point in vehicle-administered rats when compared with baseline (106.1±5.5%, n=7, P=0.000381). When compared with vehicle-administered animals, SI-only treated rats did not show significant reduction in the cerebrovascular responses to hypercapnia (111.5±27.7%, P=0.87); similarly, HUCPVC-only treated rats showed no significant reduction in the cerebral blood flow (CBF) responses to hypercapnia (88.9±25.9%, P=0.42), whereas rats that received a combined treatment of SI+HUCPVC showed a pronounced reduction in their CBF responses to hypercapnia (31.2±18.4%, P=0.01). At end point, the TgF344-AD Sham l-NAME group showed reduced responses to hypercapnia than did the l-NAME and vehicle administered (51.6±16.2% vs 106.1±5.5%, P=0.006758).
Transgenic Animals Showed Improved Pathological Correlates of Vessel Function and Decreased Amyloid
Collagen and elastin are essential components of blood vessel walls. Elastin comprises 90% of elastic arterial fibers and imbues the vessels with the capacity to resume their resting tone after stretching or contracting. Collagen is a main protein of connective tissue. Changes in ratio between elastin and collagen have been shown an indicator of vascular degeneration and dysfunction.43 In light of the role of elastin in governing cerebrovascular elasticity,42 we examined the elastin/collagen IV ratio in hippocampal arterioles at end point. Figure 6 demonstrates that vehicle-administered TgF344-AD rats showed an arteriolar elastin/collagen IV ratio of 0.47±0.05, which was nondifferent from either the 0.51±0.01 ratio observed in SI-only treated rats (P=0.24) or the 0.46±0.03 ratio found in HUCPVC-only treated animals (P=0.82). Rats treated with SI+HUCPVC showed a 46±2% higher elastin/collagen IV ratio (0.67±0.03) when compared with that of vehicle-treated TgF344-AD animals (P=1×10−5). To demonstrate the efficacy of SI treatment on Aβ plaque reduction and to relate changes in cerebrovascular structure and function to amyloid pathology progression, we evaluated plaques coverage in the hippocampus of all groups at the end point. Quantitative histological analysis of hippocampal regions showed that Aβ plaques cover 1.1±0.2% of the hippocampi in vehicle-administered animals (Figure 6D, Figure S4). When compared with vehicle-administered animals, SI-only treated and SI+HUCPVC treated subjects show 67±6% and 275 75±7% less Aβ coverage, respectively (SI-only: 0.33±0.51%, P=0.0012; and SI+HUCPVC: 0.25±0.11%, P=0.0005 respectively), whereas HUCPVC-only treated subjects presented an Aβ load comparable to those of vehicle-administered animals (HUCPVC-only: 0.82±0.07%, P=0.14, Figure 6E, Figure S4), supporting the effectiveness of SI in clearing the amyloid. Interestingly, TgF344-AD animals that did not receive l-NAME showed Aβ plaques coverage comparable to that of l-NAME and vehicle-administered animals (0.97±0.01 versus 1.14±0.15, P=0.4) and higher Elastin/CollagenIV ratio than that of l-NAME and vehicle-administered animals (0.90±0.09 versus 0.46±0.04, P=0.00001).
Figure 6.
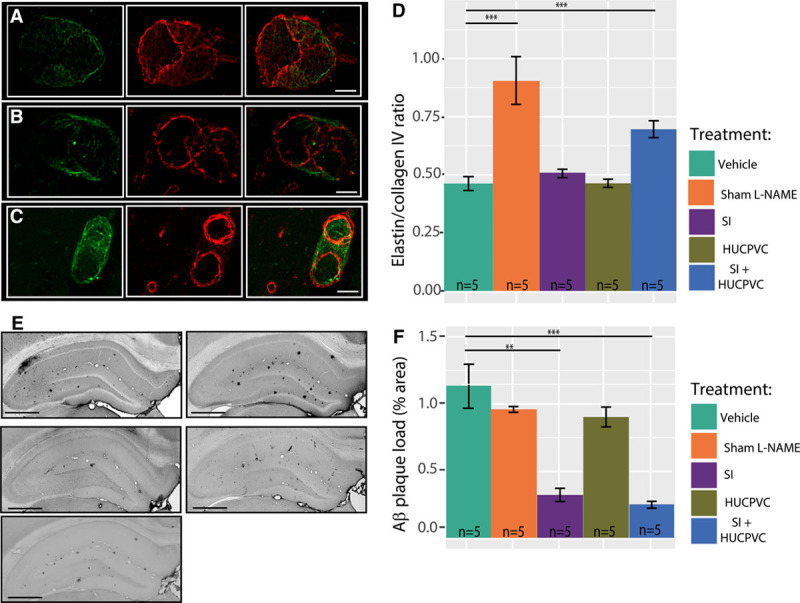
Cerebrovascular pathology and Aβ plaques in the TgF344-AD rats. Hippocampal arterioles stained for elastin (left) and collagen IV (middle), and overlay (right) in a representative vehicle-treated TgF344-AD rat (A), in an scyllo-inositol (SI) + human umbilical cord perivascular cell (HUCPVC) treated TgF344-AD rat (B) and in a TgF344-AD Sham l-NAME (C); scale bar 25 m. Population analysis (D) showed that combined treatment of SI+HUCPVC increased the elastin to collagen IV ratio. E, Representative 6F3D staining for Aβ plaques in the hippocampus of a vehicle-administered TgF344-AD rat (top left), SI-only treated TgF344-AD rat (top right), HUCPVC-only treated TgF344-AD rat (middle left), SI+HUCPVC treated TgF344-AD rat (middle right), and a TgF344-AD Sham l-NAME rat (bottom left). Scale bar: 0.8 mm. Population analysis (E) revealed a lower Aβ plaque load in SI-only and SI+HUCPVC treated TgF344-AD rats when compared with that of vehicle-administered and HUCPVC-only treated animals.
Discussion
This study was motivated by the observations that transient mild-moderate hypertension is becoming more common in young adults and that it may induce permanent cerebrovascular damage,9,10 thus exacerbating latter life AD progression. We provide the first data on in situ brain vascular dysfunction in AD-like pathology comorbid with mild-moderate hypertension showing how the interaction between the 2 pathologies results in cerebral hypoperfusion and exaggerated reactivity to hypercapnia, a pattern that has been shown to underlie metabolic dysfunction and to correlate with inflammation and neurovascular damage following ischemia. To achieve higher sensitivity in the detection of cerebrovascular abnormalities, we longitudinally examined in situ cerebrovascular dysfunction caused by the interaction between hypertension and AD. We further provide a proof-of-principle for the use of HUCPVCs in the presence of Aβ reduction therapeutic, SI, to drive cerebrovascular recovery.
Modeling AD and Hypertension
Transgenic mouse models of AD present limited (if any) neuronal loss or tau pathology, which are recognized as key determinants of AD progression in patients.44 We, therefore, undertook treatment evaluations in TgF344-AD rats, which exhibit progressive amyloidosis, tauopathy (starting from 9 months of age), neuronal loss, and hippocampal dysfunction dependent cognitive deficits starting at 7 to 9 months of age18–20 and have a typical lifespan of 24 months in our colony. Although l-NAME is frequently used to model hypertension,24 like any mono-factorial preclinical model of disease, l-NAME model of hypertension reduces multifactorial causes of a complex pathology to a single factor. In particular, l-NAME is a selective NOS inhibitor, and the decreased level of NO during l-NAME administration could be seen as confounding.22 However, NOS modulation has been reported in hypertensive patients,45 and previous reports show that normal NOS activity resumes within 24 to 48 hours of l-NAME administration cessation.24 Furthermore, the l-NAME model induces a sudden increase in BP: these kinetics thus fail to recapitulate the gradual increase in BP observed in primary hypertension. The most common alternatives to l-NAME for induced hypertension include the activation of the renin-angiotensin-aldosterone system (renin-angiotensin-aldosterone system–like Ang II [angiotensin II] infusion model) and surgical models as the renovascular and renoprival models. Although these can induce higher increase in BP than the one we observed, they also introduce other confounding factors, namely cardiac and renal injuries, which are typically absent in transient middle-age hypertension.46 l-NAME has been hypothesized to increase Aβ production23; however, the ≈1% Aβ coverage of the hippocampus that we observed at end point (ie, at 7 months of age) in the current work is the same we observed in a previous study in this AD model in the absence of hypertension19 and lower than the ≈2% observed by others in TgF344-AD rats at 10 months of age.47
Hypertension and AD Interaction in the TgF344-AD Rats
In patients, hypertension-induced cerebrovascular impairments manifest as resting hypoperfusion and structural degeneration of vascular wall and elastin loss, culminating in neuronal death and cognitive impairments.13 Elastin is responsible for the extensibility of arterial walls,42 and its reduction may increase the time needed for vessels to return to their baseline diameter following a dilatory stimulus, resulting in exaggerated responses to hypercapnia. Immediately after mild-moderate hypertension period, TgF344-AD rats and their nontransgenic littermates showed resting hippocampal hypoperfusion and elevated hippocampal vascular reactivity to hypercapnia. The persistence of this phenotype in TgF344-AD rats—but not in nontransgenic rats—is not due to vascular amyloid deposition as TgF344-AD rats do not show vascular amyloid in the hippocampus at 7 months of age (data not shown); however, we cannot rule out the contribution of soluble oligomeric Aβ. Indeed, nontransgenic animals show spontaneous recovery from mild-moderate hypertension-induced hypoperfusion and cerebrovascular hyperreactivity to hypercapnia. Furthermore, untreated 7-month old TgF344-AD rats show hippocampal cerebrovascular phenotype distinct from that presently observed postvehicle administration. Both TgF344-AD and non-Tg animals which did not experience transient hypertension showed resting perfusion and responsivity to hypercapnia indistinguishable from baseline levels. Altogether, this study indicates it is the co-occurrence between transient hypertension and AD-like pathology that drives permanent cerebrovascular damage.
Effectiveness of Combinatorial Treatment With HUCPVC and SI
Multipotent stem cells have been shown to partially rescue cognitive impairments in transgenic mouse models of progressive amyloidosis.48 As multipotent stem cell homing to the central nervous system is low (data not shown and Uccelli et al49), it has been hypothesized that their mechanisms of action rely on the modulation of peripheral immune system49,50 or perivascular macrophages51; however, more studies are needed to elucidate this mechanism. In light of the mesodermal origin of vasculature,52 we examined the regenerative effects of HUCPVCs on arterioles, which are damaged by transient hypertension.53 Four weeks after SI-only or SI+HUCPVC treatment, hippocampal amyloid plaque load was reduced, as expected; while HUCPVC-only treated rats showed amyloid load comparable to that of vehicle-administered animals. Moreover, the elastin/collagen IV ratio in HUCPVC-only cohort was comparable to that of vehicle-administered animals, suggesting that HUCPVC treatment alone could not promote vascular remodeling in the presence of Aβ toxicity. In contrast, SI+HUCPVC treated animals showed an increase in the elastin/collagen IV ratio, indicating effective cerebrovascular remodeling occurs following HUCPVC treatment in the absence of toxic Aβ peptides. In line with this, HUCPVC treatment alone failed to rescue resting perfusion and had limited effects on cerebrovascular hyperreactivity. Only the combination of SI and HUCPVC normalized resting perfusion and hippocampal vascular response to hypercapnia, suggesting that amyloid clearance acts as a permissive factor, necessary to enable HUCPVC-driven cerebrovascular recovery. At the end point of this study, the rats were 7 months old, that is, at an age when this model of AD shows pronounced cortical vascular deficits,19 but cognitive deficits are only starting.18 Extended treatment period and periodic cognitive assessments will be conducted in future work to determine whether the observed Aβ clearance, vascular wall remodeling, and improvement in hippocampal vascular function decelerate subsequent cognitive decline in combinatorially treated animals. Furthermore, studies examining the molecular pathways responsible for cerebrovascular damage from transient moderate hypertension and its interaction with AD pathology are currently underway.
Perspectives
This study constitutes the proof-of-principle that the combination of Aβ clearance and HUCPVC transplantation drives the rescue of hippocampal vascular impairment caused by moderate transient hypertension in the TgF344-AD rat model. The combinatorial treatment with HUCPVC and amyloid clearance thus constitutes a promising therapeutic strategy in AD comorbid with hypertension.
Acknowledgments
We thank Andree Gauthier-Fisher, Shlomit Kenigsberg, Leila Maghen, and Daniella R. Morrone for technical support.
Source of Funding
This work was funded by the W. Garfield Weston Foundation and from Canada Institute of Health and Research and National Institutes of Health (grant number R01AG057665-2).
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
These authors contributed equally to this work.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/HYPERTENSIONAHA.119.13187.
Novelty and Significance
What Is New?
We describe the dysfunction of brain vasculature caused by transient hypertension and amyloid pathology in the prodromal phase of Alzheimer disease.
We provided 4 treatments: human umbilical cord perivascular cells, scyllo-inositol (an agent that reduces the formation of amyloid plaques), a combination of human umbilical cord perivascular cells and scyllo-inositol, and a control group administered with vehicle.
Only the group receiving the combinatorial treatment shows significant recovery of brain vascular function.
What Is Relevant?
We show that transient hypertension has deleterious effects on brain vasculature and that these effects are long-lasting in the presence of amyloid pathology.
Summary
Transient hypertension in the young-adult age can cause long-lasting damage in brain vasculature and accelerate the progression of Alzheimer disease;
treatments aimed to restore brain vasculature function have greater chances of success in an amyloid-free environment.
References
- 1.GBD 2016 Dementia Collaborators. Global, regional, and national burden of A lzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:88–106. doi: 10.1016/S1474-4422(18)30403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Lista S. Alzheimer disease: from inherited to sporadic AD-crossing the biomarker bridge. Nat Rev Neurol. 2012;8:598–600. doi: 10.1038/nrneurol.2012.202. doi: 10.1038/nrneurol.2012.202. [DOI] [PubMed] [Google Scholar]
- 4.Orgeta V, Mukadam N, Sommerlad A, Livingston G. The lancet commission on dementia prevention, intervention, and care: a call for action. Irish J Psychol Med. 2018;36:85–88. doi: 10.1017/ipm.2018.4. doi: 10.1017/ipm.2018.4. [DOI] [PubMed] [Google Scholar]
- 5.Gorelick PB. Risk factors for vascular dementia and A lzheimer disease. Stroke. 2004;35(11 suppl 1):2620–2622. doi: 10.1161/01.STR.0000143318.70292.47. doi: 10.1161/01.STR.0000143318.70292.47. [DOI] [PubMed] [Google Scholar]
- 6.in’t Veld BA, Ruitenberg A, Hofman A, Stricker BH, Breteler MM. Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol Aging. 2001;22:407–412. doi: 10.1016/s0197-4580(00)00241-4. [DOI] [PubMed] [Google Scholar]
- 7.Yasar S, Xia J, Yao W, Furberg CD, Xue QL, Mercado CI, Fitzpatrick AL, Fried LP, Kawas CH, Sink KM, Williamson JD, DeKosky ST, Carlson MC Ginkgo Evaluation of Memory (GEM) Study Investigators. Antihypertensive drugs decrease risk of A lzheimer disease: ginkgo evaluation of Memory Study. Neurology. 2013;81:896–903. doi: 10.1212/WNL.0b013e3182a35228. doi: 10.1212/WNL.0b013e3182a35228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reboussin DM, Allen NB, Griswold ME, Guallar E, Hong Y, Lackland DT, Miller EPR, III, Polonsky T, Thompson-Paul AM, Vupputuri S. Systematic review for the 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2018;138:e595–e616. doi: 10.1161/CIR.0000000000000601. doi: 10.1161/CIR.0000000000000601. [DOI] [PubMed] [Google Scholar]
- 9.Muntner P, Carey RM, Gidding S, Jones DW, Taler SJ, Wright JT, Jr, Whelton PK. Potential US population impact of the 2017 ACC/AHA high blood pressure guideline. Circulation. 2018;137:109–118. doi: 10.1161/CIRCULATIONAHA.117.032582. doi: 10.1161/CIRCULATIONAHA.117.032582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muntner P, Whelton PK, Woodward M, Carey RM. A comparison of the 2017 American College of Cardiology/American Heart Association blood pressure guideline and the 2017 American Diabetes Association diabetes and hypertension position statement for U.S. adults with diabetes. Diabetes Care. 2018;41:2322–2329. doi: 10.2337/dc18-1307. doi: 10.2337/dc18-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin EJ, Virani SS, Callaway CW, et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. doi: 10.1161/CIR.0000000000000558. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 12.De Venecia T, Lu M, Figueredo VM. Hypertension in young adults. Postgrad Med. 2016;128:201–207. doi: 10.1080/00325481.2016.1147927. doi: 10.1080/00325481.2016.1147927. [DOI] [PubMed] [Google Scholar]
- 13.Iadecola C, Yaffe K, Biller J, Bratzke LC, Faraci FM, Gorelick PB, Gulati M, Kamel H, Knopman DS, Launer LJ, Saczynski JS, Seshadri S, Zeki Al Hazzouri A American Heart Association Council on Hypertension; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Quality of Care and Outcomes Research; and Stroke Council. Impact of hypertension on cognitive function: a scientific statement from the American Heart Association. Hypertension. 2016;68:e67–e94. doi: 10.1161/HYP.0000000000000053. doi: 10.1161/HYP.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jan AT, Azam M, Rahman S, Almigeiti AMS, Choi DH, Lee EJ, Haq QMR, Choi I. Perspective insights into disease progression, diagnostics, and therapeutic approaches in A lzheimer’s disease: a judicious update. Front Aging Neurosci. 2017;9:356. doi: 10.3389/fnagi.2017.00356. doi: 10.3389/fnagi.2017.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, Sur C, Mukai Y, Voss T, Furtek C, Mahoney E, Harper Mozley L, Vandenberghe R, Mo Y, Michelson D. Randomized trial of verubecestat for mild-to- moderate A lzheimer’s disease. N Engl J Med. 2018;378:1691–1703. doi: 10.1056/NEJMoa1706441. doi: 10.1056/NEJMoa1706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koncz R, Sachdev PS. Are the brain’s vascular and alzheimer pathologies additive or interactive? Curr Opin Psychiatry. 2018;31:147–152. doi: 10.1097/YCO.0000000000000395. doi: 10.1097/YCO.0000000000000395. [DOI] [PubMed] [Google Scholar]
- 17.Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including A lzheimer’s disease. Alzheimers Dement. 2015;11:710–717. doi: 10.1016/j.jalz.2014.10.008. doi: 10.1016/j.jalz.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen RM, Rezai-Zadeh K, Weitz TM, et al. A transgenic alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013;33:6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013. doi: 10.1523/JNEUROSCI.3672-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joo IL, Lai AY, Bazzigaluppi P, Koletar MM, Dorr A, Brown ME, Thomason LA, Sled JG, McLaurin J, Stefanovic B. Early neurovascular dysfunction in a transgenic rat model of A lzheimer’s disease. Sci Rep. 2017;7:46427. doi: 10.1038/srep46427. doi: 10.1038/srep46427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazzigaluppi P, Beckett TL, Koletar MM, Lai AY, Joo IL, Brown ME, Carlen PL, McLaurin J, Stefanovic B. Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of A lzheimer’s disease. J Neurochem. 2018;144:669–679. doi: 10.1111/jnc.14136. doi: 10.1111/jnc.14136. [DOI] [PubMed] [Google Scholar]
- 21.Williams B. Drug treatment of hypertension. BMJ. 2003;326:61–62. doi: 10.1136/bmj.326.7380.61. doi: 10.1136/bmj.326.7380.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leong XF, Ng CY, Jaarin K. Animal models in cardiovascular research: hypertension and atherosclerosis. Biomed Res Int. 2015;2015:528757. doi: 10.1155/2015/528757. doi: 10.1155/2015/528757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katusic ZS, Austin SA. Neurovascular protective function of endothelial nitric oxide □ - recent advances. Circ J. 2016;80:1499–1503. doi: 10.1253/circj.CJ-16-0423. doi: 10.1253/circj.CJ-16-0423. [DOI] [PubMed] [Google Scholar]
- 24.Pechánová O, Dobesová Z, Cejka J, Kunes J, Zicha J. Vasoactive systems in L-NAME hypertension: the role of inducible nitric oxide synthase. J Hypertens. 2004;22:167–173. doi: 10.1097/00004872-200401000-00026. [DOI] [PubMed] [Google Scholar]
- 25.Salter M, Duffy C, Garthwaite J, Strijbos PJ. Substantial regional and hemispheric differences in brain nitric oxide synthase (NOS) inhibition following intracerebroventricular administration of N omega-nitro-L-arginine (L-NA) and its methyl ester (L-NAME). Neuropharmacology. 1995;34:639–649. doi: 10.1016/0028-3908(95)00036-6. doi: 10.1016/0028-3908(95)00036-6. [DOI] [PubMed] [Google Scholar]
- 26.McLaurin J, Kierstead ME, Brown ME, et al. Cyclohexanehexol inhibitors of abeta aggregation prevent and reverse alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–808. doi: 10.1038/nm1423. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 27.Hong SH, Maghen L, Kenigsberg S, Teichert AM, Rammeloo AW, Shlush E, Szaraz P, Pereira S, Lulat A, Xiao R, Yie SM, Gauthier-Fisher A, Librach CL. Ontogeny of human umbilical cord perivascular cells: molecular and fate potential changes during gestation. Stem Cells Dev. 2013;22:2425–2439. doi: 10.1089/scd.2012.0552. doi: 10.1089/scd.2012.0552. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal F, Szaraz P, Librach M, Gauthier-Fisher A, Librach CL. Angiogenic potency evaluation of cell therapy candidates by a novel application of the in vitro aortic ring assay. Stem Cell Res Ther. 2017;8:184. doi: 10.1186/s13287-017-0631-1. doi: 10.1186/s13287-017-0631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He Y, Jin X, Wang J, Meng M, Hou Z, Tian W, Li Y, Wang W, Wei Y, Wang Y, Meng H, Lu X, Chen Z, Fu L. Umbilical cord-derived mesenchymal stem cell transplantation for treating elderly vascular dementia. Cell Tissue Bank. 2017;18:53–59. doi: 10.1007/s10561-017-9609-6. doi: 10.1007/s10561-017-9609-6. [DOI] [PubMed] [Google Scholar]
- 30.Fenili D, Brown M, Rappaport R, McLaurin J. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J Mol Med (Berl) 2007;85:603–611. doi: 10.1007/s00109-007-0156-7. doi: 10.1007/s00109-007-0156-7. [DOI] [PubMed] [Google Scholar]
- 31.Jensen MB, Krishnaney-Davison R, Cohen LK, Zhang SC. Injected versus oral cyclosporine for human neural progenitor grafting in rats. J Stem Cell Res Ther. 2012;23(suppl 10):91–105. doi: 10.4172/2157-7633.S10-003. doi: 10.4172/2157-7633.S10-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poli KA, Tofler GH, Larson MG, Evans JC, Sutherland PA, Lipinska I, Mittleman MA, Muller JE, D’Agostino RB, Wilson PW, Levy D. Association of blood pressure with fibrinolytic potential in the framingham offspring population. Circulation. 2000;101:264–269. doi: 10.1161/01.cir.101.3.264. doi: 10.1161/01.cir.101.3.264. [DOI] [PubMed] [Google Scholar]
- 33.Coban E, Ozdogan M. The plasma levels of plasminogen activator inhibitor-1 in subjects with white coat hypertension. Int J Clin Pract. 2004;58:541–544. doi: 10.1111/j.1368-5031.2004.00119.x. [DOI] [PubMed] [Google Scholar]
- 34.Bazzigaluppi P, Adams C, Koletar MM, Dorr A, Pikula A, Carlen PL, Stefanovic B. Oophorectomy reduces estradiol levels and long- term spontaneous neurovascular recovery in a female rat model of focal ischemic stroke. Front Mol Neurosci. 2018;11:338. doi: 10.3389/fnmol.2018.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bazzigaluppi P, Lake EM, Beckett TL, Koletar MM, Weisspapir I, Heinen S, Mester J, Lai A, Janik R, Dorr A, McLaurin J, Stanisz GJ, Carlen PL, Stefanovic B. Imaging the effects of β- hydroxybutyrate on peri- infarct neurovascular function and metabolism. Stroke. 2018;49:2173–2181. doi: 10.1161/STROKEAHA.118.020586. [DOI] [PubMed] [Google Scholar]
- 36.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 37.Cigolini M, Targher G, Seidell JC, Tonoli M, Schiavon R, Agostino G, De Sandre G. Relationships of blood pressure to fibrinolysis: influence of anthropometry, metabolic profile and behavioural variables. J Hypertens. 1995;13:659–666. doi: 10.1097/00004872-199506000-00013. [DOI] [PubMed] [Google Scholar]
- 38.Jeng JR. Association of PAI-1 gene promoter 4g/5g polymorphism with plasma PAI-1 activity in Chinese patients with and without hypertension. Am J Hypertens. 2003;16:290–296. doi: 10.1016/s0895-7061(03)00004-9. doi: 10.1016/s0895-7061(03)00004-9. [DOI] [PubMed] [Google Scholar]
- 39.Lake EMR, Bazzigaluppi P, Mester J, Thomason LAM, Janik R, Brown M, McLaurin J, Carlen PL, Corbett D, Stanisz GJ, Stefanovic B. Neurovascular unit remodelling in the subacute stage of stroke recovery. Neuroimage. 2017;146:869–882. doi: 10.1016/j.neuroimage.2016.09.016. doi: 10.1016/j.neuroimage.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Savitz SI. Introduction to cellular therapy: the next frontier for stroke therapeutics. Stroke. 2009;40(3 suppl):S141–S142. doi: 10.1161/STROKEAHA.108.535864. doi: 10.1161/STROKEAHA.108.535864. [DOI] [PubMed] [Google Scholar]
- 41.de la Torre J C. Critical threshold cerebral hypoperfusion causes A lzheimer’s disease? Acta Neuropathol. 1999;98:1–8. doi: 10.1007/s004010051044. [DOI] [PubMed] [Google Scholar]
- 42.Xu J, Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta. 2014;1842:2106–2119. doi: 10.1016/j.bbadis.2014.07.008. doi: 10.1016/j.bbadis.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu P, Sen U, Tyagi N, Tyagi SC. Blood flow interplays with elastin: collagen and MMP: TIMP ratios to maintain healthy vascular structure and function. Vasc Health Risk Manag. 2010;6:215–228. doi: 10.2147/vhrm.s9472. doi: 10.2147/vhrm.s9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in A lzheimer’s disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 45.Moss MB, Brunini TM, Soares De Moura R, Novaes Malagris LE, Roberts NB, Ellory JC, Mann GE, Mendes Ribeiro AC. Diminished L-arginine bioavailability in hypertension. Clin Sci (Lond) 2004;107:391–397. doi: 10.1042/CS20030412. doi: 10.1042/CS20030412. [DOI] [PubMed] [Google Scholar]
- 46.Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C, Coffman TM. Animal models of hypertension: a scientific statement from the American Heart Association. Hypertension. 2019;73:e87–e120. doi: 10.1161/HYP.0000000000000090. doi: 10.1161/HYP.0000000000000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anckaerts C, Blockx I, Summer P, Michael J, Hamaide J, Kreutzer C, Boutin H, Couillard-Després S, Verhoye M, Van der Linden A. Early functional connectivity deficits and progressive microstructural alterations in the TgF344-AD rat model of A lzheimer’s disease: a longitudinal MRI Study. Neurobiol Dis. 2019;124:93–107. doi: 10.1016/j.nbd.2018.11.010. doi: 10.1016/j.nbd.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Duncan T, Valenzuela M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther. 2017;8:111. doi: 10.1186/s13287-017-0567-5. doi: 10.1186/s13287-017-0567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uccelli A, Benvenuto F, Laroni A, Giunti D. Neuroprotective features of mesenchymal stem cells. Best Pract Res Clin Haematol. 2011;24:59–64. doi: 10.1016/j.beha.2011.01.004. doi: 10.1016/j.beha.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Hsuan YC, Lin CH, Chang CP, Lin MT. Mesenchymal stem cell-based treatments for stroke, neural trauma, and heat stroke. Brain Behav. 2016;6:e00526. doi: 10.1002/brb3.526. doi: 10.1002/brb3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, Younkin L, Younkin S, Zhou P, Carlson G, Anrather J, Iadecola C. Brain perivascular macrophages initiate the neurovascular dysfunction of alzheimer Aβ peptides. Circ Res. 2017;121:258–269. doi: 10.1161/CIRCRESAHA.117.311054. doi: 10.1161/CIRCRESAHA.117.311054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noden DM. Embryonic origins and assembly of blood vessels. Am Rev Respir Dis. 1989;140:1097–1103. doi: 10.1164/ajrccm/140.4.1097. doi: 10.1164/ajrccm/140.4.1097. [DOI] [PubMed] [Google Scholar]
- 53.Kontos HA, Wei EP, Dietrich WD, Navari RM, Povlishock JT, Ghatak NR, Ellis EF, Patterson JL., Jr. Mechanism of cerebral arteriolar abnormalities after acute hypertension. Am J Physiol. 1981;240:H511–H527. doi: 10.1152/ajpheart.1981.240.4.H511. doi: 10.1152/ajpheart.1981.240.4.H511. [DOI] [PubMed] [Google Scholar]