

Abstract
The endocannabinoid system (CS) has been implicated in the development of hepatic fibrosis such as schistosomiasis-associated liver fibrosis (SSLF). However, the mechanisms mediating the action of the CS in hepatic fibrosis are unclear. The present study hypothesized that Schistosoma J. infection upregulates cannabinoid receptor 1 (CB1) due to activation of NADPH oxidase leading to a fibrotic phenotype in hepatic stellate cells (HSCs). The SSLF model was developed by infecting mice with Schistosoma J. cercariae in the skin, and HSCs from control and infected mice were then isolated, cultured, and confirmed by analysis of HSC markers α-SMA and desmin. CB1 significantly increased in HSCs isolated from mice with SSLF, which was accompanied by a greater expression of fibrotic markers α-SMA, collagen I, and TIMP-1. CB1 upregulation and enhanced fibrotic changes were also observed in normal HSCs treated with soluble egg antigen (SEA) from Schistosoma J. Electron spin resonance (ESR) analysis further demonstrated that superoxide production was increased in infected HSCs or normal HSCs stimulated with SEA. Both Nox4 and Nox1 siRNA prevented SEA-induced upregulation of CB1, α-SMA, collagen I, and TIMP-1 by inhibition of production, while CB1 siRNA blocked SEA-induced fibrotic changes without effect on production in these HSCs. Taken together, these data suggest that the fibrotic activation of HSCs on Schistosoma J. infection or SEA stimulation is associated with NADPH oxidase-mediated redox regulation of CB1 expression, which may be a triggering mechanism for SSLF.
Keywords: Cannabinoid receptor-1, Hepatic stellate cell, NADPH oxidase, Schistosomiasis-associated liver fibrosis
Introduction
Schistosomiasis is a common parasitic trematode infection in humans that is prevalent in Africa, South America, and Asia [1,2], where more than 600 million people live in schistosomiasis transmission zones [3]. Infection with Schistosoma japonicum (Schistosoma J.) is a common cause for chronic liver and intestinal fibrosis, which occurs in approximately one million people who are infected with another 50 million at risk of this fibrosis in China alone [4]. The major pathology in schistosomiasis is the formation of egg granulomas in the liver or intestines, and the death of patients suffering from schistosomiasis is mainly due to hepatic fibrosis, termed schistosomiasis-associated liver fibrosis (SSLF) [5]. Many studies have shown that the granuloma inflammatory reactions and fibrosis in the liver continue aggressively even though efficacious schistosomicides are used for treatment [6–8]. So far, there has been little success in the development of antifibrotic drugs with low toxicity and high efficacy in the prevention or reversal of hepatic fibrosis during schistosomiasis. The major reason may be due to the poor understanding of the early mechanisms leading to the formation of egg granulomas in the liver, which is a major focus of the present study.
Liver fibrosis is characterized by two major events: activation and proliferation of hepatic stellate cells (HSCs) and an increase in extracellular matrix (ECM) deposition, particularly collagen type I [9]. It has been reported that the abnormal activation and consequent proliferation of HSCs along with disturbed ECM metabolism play critical roles in the initiation and development of liver fibrosis including SSLF [10,11]. Although some reports have indicated that Schistosoma J. eggs or their soluble egg antigens (SEA) activate HSCs and promote the formation of fibrotic phenotype in these cells, the mechanisms mediating this response of HSCs to Schistosoma J. eggs or SEA are still poorly understood. In this regard, recent studies have reported that the endocannabinoid system is a novel activating mechanism in the pathogenesis of liver fibrosis. In particular, the cannabinoid receptors 1 and 2 (CB1 and CB2) have been demonstrated to contribute to the regulation of pro- and antifibrogenic effects in the liver [12,13]. CB1 silencing by markedly ameliorated dimethylnitrosamine-induced hepatic fibrosis in rat HSCs and livers [14]. In our previous studies, CB1 was found to be increased in the area of the liver with Schistosoma J. egg granulomas from SSLF mice [15]. However, it remains unknown whether the endocannabinoid system is implicated in the activation of HSCs and consequent pathological fibrotic changes such as abnormal metabolism of ECM in these cells during Schistosoma J. infection or on SEA stimulation. The present study tried to answer this important question and explored the possible mechanisms mediating the action of cannabinoid receptors on the activation of HSCs and the formation of their fibrotic phenotypes. These mechanisms may involve the expression of CB1 and CB2 receptors as well as redox signaling, given that the generation of reactive oxygen species (ROS) has been reported to play a relevant role in liver fibrosis, especially those derived from NADPH oxidases (Noxs) [16–18].
The present study hypothesized that Schistosoma J. infection upregulates cannabinoid receptor 1 (CB1) by activation of NADPH oxidase to produce superoxide , thereby leading to a fibrotic phenotype of HSCs. To test this hypothesis, we first characterized the expression of CB1 and CB2 in HSCs from Schistosoma J.-infected mice as well as in HSCs exposed to SEA and then observed the fibrotic changes in HSCs. Next, we determined whether the activation of HSCs and the formation of a fibrotic phenotype are associated with altered expression of cannabinoid receptors. Finally, we explored whether CB1 is upregulated by Schistosoma J. infection or by SEA through redox signaling associated with the activation of NADPH oxidase in HSCs.
Materials and methods
Animals and reagents
Healthy 4- to 6-week-old male BALB/C mice were obtained from a Schistosomiasis Control Laboratory in Hubei province, China. The SSLF model was established by abdominal infection with Schistosoma J. cercariae according to the method used in a previous study [19]. In brief, mice in the model group were percutaneously infected with Schistosoma J. by placing a glass slide carrying 20±2 cercariae in nonchlorinated water in the abdomen for 15 min. Mice in the control group were treated with nonchlorinated water containing no cercariae. The control and infected mice were raised for 6 weeks under pathogen-free conditions with free access to food and water. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the Chinese Council on Animal Care. Liver samples taken from mice were fixed in 4% (v/v) paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for morphological examinations and other analyses. Schistosoma J. egg antigen [0.01 g/ml in PBS] was obtained from the Hubei Schistosomiasis Control Laboratory and diluted to the working concentration in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2% FBS before use.
Hepatic stellate cell isolation and culture
In different groups of control and infected mice, HSCs were prepared by the discontinuous density gradient centrifugation technique previously described by Schafer et al. [19] with minor modifications. Briefly, the liver was perfused through the hepatic portal vein with solution I (137 mM NaCl, 5.4 mM KCl, 0.6 mM NaH2PO4.2H2O, 0.8 mM Na2HPO4.12H2O, 10 mM Hepes, 0.5 mM EGTA, 4.2 mM NaHCO3, 5 mM glucose, 100 U/ml penicillium, and 100 U/ml streptomycin (Gibco, Grand Island, NY), pH 7.4) and solution II (137 mM NaCl, 5.4 mM KCl, 0.6 mM NaH2PO4.2H2O, 0.8 mM Na2HPO4.12H2O, 10 mM Hepes, 3.8 mM CaCl2.2H2O, 24.2 mM NaHCO3, 5 mM glucose, 600 mg/L collagenase IV (Gibco), 400 mg/L pronase (Sigma-Aldrich, St. Louis, MO), 20 mg/L DNAase (Gibco), pH 7.4). The digested liver tissues were removed and forced through a 200-gauge mesh. Parenchymal cells were separated by centrifugation at 20 g for 5 min. The supernatant was transferred to a 50-ml centrifuge tube and centrifuged at 500 g for 7 min. The pellet was resuspended in 15% OptiPrep (Sigma), 5 ml 11% OptiPrep, and 5 ml GBSS (120 mM NaCl, 5 mM KCl, 0.84 mM Na2HPO4.2H2O, 0.22 mM KH2PO4, 1.9 mM MgCl2.6H2O, 1.5 mM CaCl2.2H2O, 27 mM NaHCO3, 5 mM glucose, pH 7.4) were then layered on top of the 15% OptiPrep. With such gradient the sample was centrifuged at 1400 g for 17 min. The interface between the 11% OptiPrep and the GBSS was collected and washed twice with GBSS. The collected cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (Gibco). The cell viability, measured by a Trypan Blue exclusion assay, was approximately 90%.
For SEA stimulation, the HSCs were seeded onto 6-well plates (Greiner Bio-One, Monroe, NC) at a density of about 1 × 106 cells per well and under serum-starved conditions. Then, Schistosoma J. soluble egg antigens (SEA, 50 mg/ml) or vehicle without SEA were added to different wells with HSCs. After a 24-h treatment, HSCs were collected for corresponding experiments as described below.
Masson staining
Masson staining was performed using standard methods as described previously [20] to observe collagen deposition in liver tissue slides. The data were represented by the area percentage of each slide positive for the blue stain, which was calculated in Image Pro Plus 6.0 software.
Real-time reverse transcription–polymerase chain reaction (RT-PCR)
Total RNA from HSCs was isolated using TriZol reagent (Life Technologies, Grand Island, NY) according to the protocol described by the manufacturer. Isolated RNA samples were analyzed by measurement of optic absorbance at 260 and 280 nm in a spectrophotometer, with the A260/A280 ratio ranging from 1.8 to 2.0, indicating a high purity of RNA. The concentrations of RNA were calculated by the absorbance at 260 nm. Aliquots of total RNA (1 μg) from each sample were reverse-transcribed into cDNA according to the instructions of the first-strand cDNA synthesis kit of the manufacturer (BioRad, Hercules, CA) Equal amounts of the reverse transcriptional products were subjected to PCR amplification using SYBR Green as a fluorescent indicator on a CFX Connect Real-Time PCR Detection System (BioRad). The mRNA levels of target genes were normalized to the β-actin mRNA levels. The primers used in this study were synthesized by Eurofins MWG Operon (Huntsville, AL) and the sequences were α-SMA sense 5′-CCCAGACATCAGGGAGTAATGG-3′, α-SMA antisense 5′-TCTATCGGATACTTCAGCGTCA-3′; desmin sense, 5′-GTGGATGCAGCCA CTCTAGC-3′, desmin antisense 5′-TTAGCCGCGATGGTCTCATAC-3′; CB1 sense 5′-AAGTCGATCTTAGACGGCCTT-3′, CB1 antisense 5′-TCCTAATTTGGATGCCATGTCTC-3′; CB2 sense 5′-ACGGTGGCTTGGAGTTCAAC-3′, CB2 antisense 5′-GCCGGGAGGACAGGATAAT-3′; β-actin sense 5′-TCGCTGCGCTGGTCGTC-3′, antisense 5′-GGCCTCGTCACCCACATAGGA-3′ (all primer refer to http://pga.mgh.harvard.edu/primerbank).
Western blotting
After various treatment, HSCs were washed twice with PBS and scraped in a lysis buffer (20 mM Tris-HCl, 250 mM sucrose, pH 7.2) containing 1X protease inhibitor mixture (Roche, Indianapolis, IN). Proteins were denatured with SDS buffer and boiled for 5 min. Samples were run by SDS-PAGE, transferred onto a PVDF membrane, and blocked using 5% nonfat dry milk (NFDM) in Tris-buffered saline with 0.1% Tween 20 (TBST). Then, the membranes were probed with the following primary antibodies overnight at 4 °C: anti-α-SMA (1:1000, Abcam, Cambridge, MA), anti-desmin (1:500, BD Pharmingen, San Jose, CA), anti-CB1 (1:1000, Thermo, Waltham, MA), anti-CB2 (1:1000, Abcam), anti-TIMP-1 (1:500, Santa Cruz, Dallas, TX), anti-collagen I (1:1000, Calbiochem, Billerica, MA), anti-Nox1 (1:500, Abcam), anti-Nox4 (1:500, Santa Cruz), anti-Nrf2 (1:1000, Santa Cruz), and anti-β-actin (1:10000, Santa Cruz). After washing with TBST three times, membranes were incubated with the following secondary antibodies for 1 h at room temperature, respectively with corresponding goat anti-rabbit-HRP or rabbit anti-mouse-HRP (1:10000, Santa Cruz) based on animals used for raising primary antibody. After washing for the final three times, bands were detected by the addition of a chemiluminescent substrate and visualized on Kodak Omat X-ray film.
Immunofluorescent staining
Immunofluorescent staining of α-SMA was used to identify the isolated HSCs. Briefly, cells on 8-well chamber slides were fixed with 4% paraformaldehyde and then incubated with a blocking solution of 0.1% Triton X-100 in 5% bovine serum albumin (BSA). This was followed by incubation with anti-α-SMA (1:200, Abcam) or anti-Nrf2 (1:100, Santa Cruz) primary antibodies at 4 1C overnight. After washing with phosphate-buffered saline containing 0.1% Tween 20 (PBST) three times, the slides were incubated with Alexa-488 or Alexa555 secondary antibodies (Life Technologies) for 1 h at 37 °C and then mounted in Vectorshield mounting medium with DAPI (Vector Laboratories Inc.). Staining was visualized through sequential scanning on an Olympus laser scanning confocal microscope (Fluoview FV1000, Olympus, Japan) or fluorescent microscopy (BX41TF, Olympus, Japan).
ESR detection of
For detection of NADPH oxidase-dependent production, protein was gently isolated from 1 × 106 cells and resuspended with modified Krebs–4-(2-hydroxyethyl)- 1-piperazineethanesulfonic acid buffer containing deferoximine (100 μM, Sigma) and diethyldithiocarbamate (5 μM; Sigma). A spin trap, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, Noxygen, Elzach, Germany) (1 mM final concentration), was then added to the mixture in the presence or absence of manganese-dependent superoxide dismutase (SOD, 200 U/ml; Sigma). The mixtures were loaded into glass capillaries and immediately analyzed for superoxide formation kinetics for 10 min in a Miniscope MS200 ESR spectrometer (Magnettech, Berlin, Germany). The ESR was set according to the following parameters: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; 4096 points of resolution; and receiver gain, 50 for cells. The ESR signal strength was recorded in arbitrary units and the final results were expressed as the fold changes of the treatment group compared to the control as recently described [21].
RNA interference
Nox1 and Nox4 small interfering RNAs (siRNAs) were purchased from Invitrogen (Valencia, CA). The DNA target sequences for the siRNAs are as follows: Nox1– 5′-AAGGCCGTGAGTTTCTACCT-3′, Nox4– 5′-AAGCGCTGGGAGACTTTCTA-3′. CB1 siRNA and Nrf2 siRNA were purchased from Santa Cruz (CB1 siRNA sc-142443; Nrf2 siRNA sc-37049). The scrambled siRNA (5′-AATTCTCCGAACGTGTCACGT-3′) has been confirmed as nonsilencing double-stranded RNA and was used as a control in the present study. In these experiments, siRNA transfection was performed according to the manufacturer’s instruction of BioRad’s SiLentFect lipid reagent (BioRad). Protein expression of each target was examined by Western blot to assess transfection efficiency of each siRNA.
Statistical analysis
The results of multiple observations were presented as the means ± SE of at least 3 independent experiments. The Student test was used to evaluate the differences in the ratio of collagen deposited area against the total between infected and normal mouse livers. One-way ANOVA was used to evaluate all other differences among multiple groups of data. The P values in the description or figures of results were two-sided and P<0.05 was considered to be statistically significant. All statistical analyses were performed using SigmaStat 11.0 (San Jose, CA).
Results
Mouse model of SSLF
After 6 weeks of infection with Schistosoma J. cercariae, the mouse liver exhibited collagen deposition at the periphery of the eosinophilic granuloma while Schistosoma J. eggs remained in the hepatic portal vein. These characteristics of SSLF were not observed in the livers of control mice (Fig. 1A). Acidophilic necrosis was also observed in Schistosoma J.-infected livers (Supplementary Fig. S1). These results suggest the successful establishment of the SSLF model in Schistosoma J.-infected mice. Quantification of the area percentage positive for collagen staining in the liver revealed that significantly more collagen deposited in the liver from Schistosoma J.-infected mice compared to that from control mice, which was associated with an upregulation of CB1 expression (Fig. 1B). Further, these Schistosoma J.-induced increases in collagen deposition and CB1 expression were markedly reduced in mice pretreated with a NADPH oxidase inhibitor diphenyleneiodonium (DPI) (purchased from Sigma).
Fig. 1.

Pathological changes and CB1 expression in livers from mice infected with Schistosoma J. for 6 weeks. (A) Representative images of Masson-stained liver tissue from normal mice with minor fiber deposition around the vein (Ctrl), from infected mice showing the formation of liver fibrosis (Infected), or from infected mice treated with diphenyleneiodonium (DPI) (ip, 1 mg/kg/day). Arrows indicate different pathological changes (100 × magnification). Summarized data from three control mice, six infected mice, or six infected mice with DPI showing the area percentage of positive stain (blue color) for collagen fiber deposition in the liver tissue sections. (B) Western blot gels representing the expression of CB1 in liver homogenates from three control mice, four infected mice, or four infected mice with DPI. The bar summarized densitometric analysis from Western blot gel documents of CB1 and β-actin. *P<0.05 vs control group; # P<0.05 vs infected group.
Identification of isolated HSCs
To confirm the identity of isolated and cultured mouse HSCs, the expression of α-SMA and desmin as classic markers for this type of cell was analyzed. As shown in Fig. 2A, both α-SMA and desmin were markedly increased at the mRNA level in infected HSCs compared to those from uninfected mice. Similarly displayed in Fig. 2B, Western blot analysis demonstrated that the infected HSCs had significantly elevated α-SMA and desmin proteins. The expression patterns of α-SMA and desmin in HSCs were similar to those in mouse podocytes and coronary arterial smooth muscle cells (Supplementary Fig. 2). Utilizing confocal microscopy, immunocytochemical detections further confirmed an increase in the expression of α-SMA in infected HSCs (Fig. 2C). Together, these results suggest that primary HSCs, which are enriched with α-SMA and desmin, were successfully isolated from normal and Schistosoma J.-infected mice and that Schistosoma J. infection elevates the levels of both markers in HSCs.
Fig. 2.

Characterization of hepatic stellate cells (HSCs) from mice with and without Schistosoma J. infection. (A) Summarized relative mRNA levels of α-SMA and desmin in normal and infected HSCs (n=6). (B) Western blot gels representing the upregulation of α-SMA and desmin in infected HSCs when compared to normal HSCs. The bar summarized densitometric analysis from Western blot gel documents of α-SMA and desmin (n=6). (C) Confocal images demonstrating increased staining of α-SMA protein in infected HSCs. *P<0.05 vs Normal HSCs.
Effect of in vivo chronic Schistosoma J. infection on the expression of cannabinoid receptors and the fibrogenic phenotype in HSCs
By real-time RT-PCR, the mRNA level of CB1, but not CB2, was increased in HSCs isolated from chronic Schistosoma J.-infected mice compared to HSCs isolated from uninfected mice (Fig. 3A). Western blot analysis demonstrated the same pattern, where infected HSCs showed increased protein expression of CB1 compared to normal HSCs, without significant changes in CB2 expression (Fig. 3B). Furthermore, we observed an upregulation of collagen I and TIMP-1 expression in infected HSCs (Fig. 3C), indicative of a Schistosoma J. infection-induced fibrogenic phenotype. These results tell us that chronic Schistosoma J. infection primarily increases the expression of CB1, rather than CB2, which may induce changes in HSC fibrogenic phenotype.
Fig. 3.

Effects of chronic Schistosoma J. infection or acute Schistosoma J. soluble egg antigen (SEA) treatment on the expression of CB1 and CB2 receptors and of fibrotic markers in HSCs. (A) Relative mRNA levels of CB1 and CB2 in normal control (Ctrl) and infected HSCs (n=4). (B) Western blot gels and summarized densitometric analysis representing the difference in expression of CB1 and CB2 receptors between normal and infected HSCs (n=6). (C) Western blot gels and summarized data showing the expression of collagen I and TIMP-1 expression in normal and infected HSCs (n=6). D. Relative mRNA levels of CB1 and CB2 in SEA-treated HSCs (n=6). E. Western blot gels and summarized data representing the effect of SEA on CB1 and CB2 receptors in normal and SEA-treated HSCs (n=6). (F) Western blot gels and summarized data showing the expression of collagen I and TIMP-1 expression in normal and SEA-treated HSCs (n=6). *P<0.05 vs Ctrl.
Effect of Schistosoma J. egg antigen on the expression of cannabinoid receptors and fibrogenic markers in HSCs
As described above, chronic Schistosoma J. infection increased CB1 expression and led to the fibrogenic phenotype of HSCs. In another series of studies, we tested whether changes in CB1 expression and fibrogenic markers are associated with stimulation of Schistosoma J. eggs since they are widely known to stimulate liver cirrhosis during Schistosoma J. infection due to antigen-induced immunological or adaptive responses. We treated normal HSCs with 50 μg/ml SEA, a concentration selected based on previous reports [22]. By real-time RT-PCR, basal mRNA levels of CB1 and CB2 were detected in these HSCs (Fig. 3D). When HSCs were treated with SEA for 24 h, CB1 mRNA expression significantly increased compared to untreated HSCs, while CB2 mRNA levels remained unchanged. To further verify this SEA-induced increase in CB1 expression but not CB2 mRNA, Western blot analysis was performed, where the expression of CB1 in HSCs was found to be obviously augmented on SEA stimulation (Fig. 3E). We next addressed whether SEA had any direct effect on induction of the fibrogenic phenotype of HSCs. As shown in Fig. 3F, both fibrotic markers collagen I and TIMP-1 increased when HSCs were treated with SEA. It is noted that both Schistosoma J. infection and SEA had more pronounced increases in protein level of CB1 than its mRNA level, which may suggest that posttranscriptional and/or posttranslational regulations are also changed in HSCs. Taken together, these results showed that SEA was able to increase the expression of CB1 and promote the fibrogenic phenotype of HSCs.
CB1 silencing blocked fibrotic changes induced by SEA
We next investigated whether the CB1 receptor is involved in the SEA-induced fibrogenic phenotype change of HSCs. With previous reports suggesting a profibrotic role for CB1, we determined whether CB1 gene silencing could prevent SEA-induced fibrotic changes in HSCs. We transfected normal HSCs with CB1 siRNA followed by treatment with SEA for 24 h, and then analyzed HSC protein lysates by Western blot. The efficiency of CB1 gene silencing by CB1 siRNA was about 65% (Fig. 4A). We also demonstrated that CB1 siRNA inhibited typical signaling pathway such as Akt phosphorylation following CB1 receptor activation by its ligand anadamide (Supplementary Fig. S3). As shown in Fig. 4B, the protein expression of α-SMA, collagen I, and TIMP-1 increased after HSCs were treated with SEA, which was significantly attenuated when CB1 siRNA was transfected.
Fig. 4.
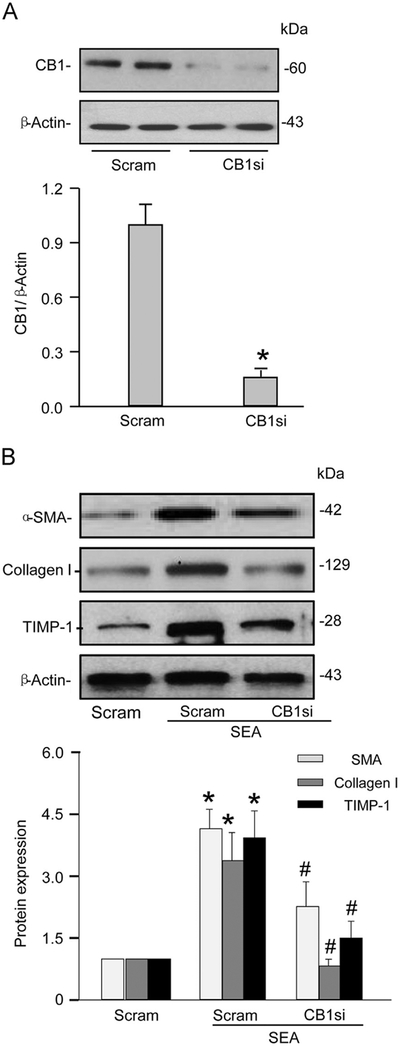
CB1 gene silencing blocked fibrotic changes in HSCs induced by SEA. (A) Western blot gels showing the efficiency of CB1 siRNAs to specifically silence corresponding genes in normal HSCs. The following bar summarized densitometric analysis from Western blot gel documents. (B) Western blot gels showing that CB1 siRNA prevented SEA-induced expression of collagen I, α-SMA, and TIMP-1. The following bar summarized densitometric analysis from Western blot gel documents of collagen I, α-SMA, and TIMP-1, respectively (n=6). *P<0.05 vs Scram siRNA, # P<0.05 vs Scram-SEA.
Contribution of Nox1 or Nox4 to SEA-induced superoxide production in normal HSCs
To explore the mechanism by which SEA or Schistosoma J. infection increased CB1 expression, we tested the role of given the implication of a redox signaling pathway in CB1-mediated actions [23]. By ESR measurement, the production of was significantly increased in HSCs from chronically infected mice and SEA-treated HSCs compared to normal HSCs without treatment of SEA (Fig. 5A). In the presence of a pharmacological NADPH oxidase inhibitor, DPI, SEA-induced production was substantially attenuated (Fig. 5B). To determine whether SEA-induced increase in production is mediated by Nox1 or Nox4, two major isoforms of NADPH oxidase in HSCs [16,24], we transfected HSCs and efficiently inhibited the expression of Nox1 and Nox4 with the corresponding siRNA (Fig. 5C and D). Interestingly, knockdown of Nox1 decreased Nox4 expression, while knockdown of Nox4 had no effect on Nox1 expression (Fig. 5C and D). In contrast, CB1 siRNA transfection did not affect the expression of either Nox1 or Nox4. (Fig. 5E). After transfection, HSCs were treated with SEA for 24 h and then analyzed for production. As shown in Fig. 5F, both Nox1 and Nox4-mediated production on SEA stimulations was markedly attenuated in the siRNA-transfected HSCs. However, CB1 siRNA had no significant effect on the increase in production induced by SEA in HSCs.
Fig. 5.

Superoxide production in Schistosoma J.-infected or SEA-stimulated HSCs. (A) Representative ESR traces of superoxide trapped by CMH using NADPH as a substrate. The bar summarized ESR data showing that SEA stimulated greater production in HSCs during chronic and acute infection (n=6). *P<0.05 vs Ctrl. (B) Summarized ESR data showing that NADPH oxidase pharmacologic inhibitor DPI (20 μM) blocked its activity. *P<0.05 vs Ctrl; # <0.05 vs SEA. (C–E) Western blot gels and summarized data showing the effects of Nox1 siRNA, Nox4 siRNA, or CB1 siRNA on protein expression of Nox1 or Nox4 in normal HSCs (n=4–6). *P<0.05 vs Scram. (F) Summarized data showing that Nox1 and Nox4 siRNA, but not CB1 siRNA, blocked SEA-induced production (n=5–6). *P<0.05 vs Scram Ctrl; # P<0.05 vs Scram-SEA. DPI, diphenyleneiodonium; Nox, NADPH oxidase; Scram: Scramble siRNA.
Upregulation of CB1 expression associated with Nox1 and Nox4
We next tested whether NADPH oxidase-mediated ROS is responsible for the upregulation of CB1 expression stimulated by SEA, thereby mediating the action of SEA-induced hepatic fibrogenic phenotypes of HSCs. The siRNAs specific to both NADPH oxidase isoforms and the pharmacologic inhibitor DPI were used to reduce the expression and activity of NADPH oxidase, followed by SEA stimulation of HSCs. By Western blot analysis it was found that SEA-induced increases in the expression of CB1 in HSCs were blocked by DPI (Fig. 6A), Nox1 siRNA, and Nox4 siRNA (Fig. 6B). However, none of these treatments had effects on the expression of CB2 receptors (Fig. 6A and B). In contrast, Nox2 gene silencing did not affect SEA-induced CB1 upregulation in HSCs (Supplementary Fig. S4).
Fig. 6.

Effects of NADPH oxidase inhibition and gene silencing on SEA-induced CB1 and CB2 expression in HSCs. (A) Western blot gels demonstrating that DPI (20 μM) blocked SEA-induced increased expression of CB1 in normal HSCs. The following bars separately summarized densitometric analysis from Western blot gel documents of CB1 and CB2 protein expression (n=4). *P<0.05 vs Vehl Ctrl; # P<0.05 vs SEA alone. (B) Representative Western blot gels showing that both Nox1 and Nox4 gene silencing by siRNA blocked SEA-induced increased expression of CB1 in normal HSCs. The below bars summarized densitometric analysis from Western blot gel documents of CB1 and CB2 (n=4). *P<0.05 vs Scram-Vehl; # P<0.05 vs Scram-SEA.
Activation of Nrf2 mediates SEA-induced CB1 upregulation
We further examined whether activation of nuclear factor E2-related factor 2 (Nrf2), a ROS-sensitive transcription factor, by Nox1/Nox4 contributes to SEA-induced CB1 upregulation in HSCs. Stimulation of HSCs with SEA resulted in a nuclear translocation of Nrf2 (Fig. 7A) in HSCs, a characteristic event of Nrf2 activation, which was blocked by either Nox1 siRNA or Nox4 siRNA transfection. Nrf2 activity was further assayed using a Nrf2 transcription factor assay kit (Cayman Chemical) following the manufacturer’s instruction. Consistently, SEA-induced Nrf2 activation in HSCs was inhibited by Nox1 or Nox4 gene silencing (Fig. 7B). Moreover, SEA-induced CB1 upregulation was abolished in HSCs by Nrf2 siRNA (Fig. 7C).
Fig. 7.

NADPH oxidase-mediated CB1 upregulation requires Nrf2 activity in HSCs on SEA stimulation. (A) Immunofluorescence images showing the effect of SEA stimulation on the nuclear translocation of Nrf2 in normal HSCs with scram or Nox1/4 siRNAs (n=6). (B) Nrf2 activity in scram, or Nox1/4 siRNAs-transfected HSCs with or without SEA stimulation (n=6). (C) Western blot gels and summarized data showing the effects of Nrf2 siRNA on the protein expression of CB1 in normal HSCs with or without SEA stimulation (n=4–6). *P<0.05 vs Scram Vehl; # P<0.05 vs Scram SEA.
Effects of DPI or Nox1 and Nox4 silencing on SEA-induced fibrotic phenotype changes in HSCs
Thus far, we have demonstrated that CB1 mediates SEA-induced fibrotic phenotype changes in HSCs and that NADPH oxidase-derived can regulate the expression of CB1, but not CB2. We next investigated the role of NADPH oxidase activity in SEA-induced fibrotic changes in HSCs. As shown in Fig. 8A, the fibrotic markers α-SMA, collagen I, and TIMP-1 increased in HSCs on SEA stimulation, which was blocked by pretreatment of HSCs with DPI. Similarly, these SEA-induced increases in the expression of fibrotic markers were blocked in HSCs transfected with either Nox1 siRNA or Nox4 siRNA (Fig. 8B).
Fig. 8.

Blockade of NADPH oxidase by DPI and Nox1 or Nox4 gene silencing inhibited SEA-induced fibrotic changes in HSCs. (A) Western blot gels and summarized data showing that DPI blocked the effect of SEA-induced increased α-SMA, collagen I, and TIMP-1 protein expression (n=6). *P<0.05 vs Vehl Ctrl; # P<0.05 vs Ctrl SEA. (B) Western blot gels showing that Nox4 and Nox1 siRNA blocked the effect of SEA-induced increases in fibrotic markers α-SMA, collagen I, and TIMP-1 (n=4). *P<0.05 vs Scram Vehl; # P<0.05 vs Scram SEA.
Discussion
There is accumulating evidence that the endocannabinoid system is activated in response to a number of fibrogenic stimuli in the liver and thereby participates in the development of liver fibrosis or cirrhosis [12,25–28]. This action of the endocannabinoid system in liver cirrhosis has been reported to be associated with the upregulation of CB1 receptors, which may affect ECM metabolism, thereby leading to fibrosis [26,29–32]. Abnormal ECM metabolism depends on both excessive synthesis and deregulated degradation of ECM components [33–36]. However, the cell and molecular mechanisms mediating this CB receptor-associated abnormal ECM metabolism remain unknown. The present study used isolated HSCs from the liver of mice with Schistosoma J. infection to explore the possible mechanisms mediating a CB1 receptor-mediated derangement of ECM metabolism. We demonstrated that Schistosoma J. infection or Schistosoma J. egg antigen extracts activate NADPH oxidase to produce , leading to redox activation of CB1 receptor upregulation, which causes the abnormal metabolism of ECM in HSCs. These HSCs with phenotype changes may constitute an important fibrogenic mechanism in the liver after Schistosoma J. infection.
In our experiments, HSCs isolated from Schistosoma J.-infected mice were found to express significantly higher levels of α-SMA and desmin compared to those from uninfected mice, indicating that Schistosoma J. infection activates HSCs in the liver. In these activated HSCs, the expression of CB1, but not CB2 receptors, was markedly increased at both mRNA and protein levels, which was accompanied by a significant abundance of collagen 1 and TIMP-1. The upregulation of ECM components in HSCs indicates the fibrogenic phenotype in infected HSCs, which could be substantially blocked by CB1 gene silencing using its specific siRNA. All these changes in the expression of CB1 receptors and ECM components in HSCs during Schistosoma J. infection were mimicked by stimulation of normal HSCs with SEA. To our knowledge, these findings for the first time provide evidence that Schistosoma J. infection or SEA upregulates CB1 receptors, which is responsible for the phenotype changes of HSCs to become more fibrogenic. In previous studies using immunohistochemical (IHC) staining, we indeed demonstrated that the expression of CB1 receptors was obviously enhanced in hepatocytes and hepatic sinusoids of Schistosoma J.-infected mice [15]. Although CB2 was also found to be increased in hepatocytes and hepatic sinusoids of Schistosoma J.-infected mice that previous study did not further localize such enhanced expression in HSCs. In other types of liver cirrhosis, such as alcohol-induced liver fibrosis, CB1 receptors were also demonstrated to be upregulated, and deletion of CB1 receptors protected against alcoholic-induced fibrosis in vivo in mice and in vitro in HSCs [13]. It should be noted that the present study did not find any significant changes in CB2 receptor expression in HSCs from Schistosoma J.-infected mice or HSCs exposed to SEA, which is different from some studies using other models of liver cirrhosis. In those studies, CB2 receptors were found upregulated as a compensatory antifibrogenic mechanism to counteract the action of HSCs as one of the fibrogenic determinants during liver fibrosis [37]. The reason for the failure of our studies to observe changes in CB2 receptor expression is unknown. It is possible that our experimental model with Schistosoma J. infection is at a relatively early stage of liver injury. In particular, our results from a short time stimulation of HSCs by SEA may indicate that the upregulation of CB1 receptors during Schistosoma J. egg stimulation is an early event in HSCs during liver fibrosis. In addition, recent studies have also indicated that CB2 receptors may not be necessarily required for the antifibrogenic action of cannabinoids given that there is evidence indicating that the apoptosis in activated HSCs during some fibrogenic stimuli occurs through a cannabinoid receptor-independent mechanism [38].
One of the important findings of the present study is that NADPH oxidase-derived ROS are involved in the regulation of CB1 receptors and associated fibrogenic phenotype formation in HSCs in SSLF. Chronic liver diseases are characterized by both increased ROS production and decreased activity of antioxidant systems, which may result in local oxidative stress and acute and chronic injury including fibrotic pathology [39]. It has been well known that NADPH oxidase is a multiprotein complex that generates or ROS in response to a large body of stimuli [40]. Three isoforms of NADPH oxidase (Nox1 and Nox2 and Nox4) have been involved in HSC activation and liver fibrosis [17,24,41]. Indeed, cellular oxidative stress contributes to the upregulation of CB1 receptors in human retinal pigment epithelial cells and to the activation of human and mouse HSCs [13,23]. The present study demonstrated that that blockade of NADPH oxidase activity by DPI inhibited Schistosoma J. infection-induced liver fibrosis and CB1 upregulation in mice (Fig. 1). Schistosoma J.-infected HSCs produced much more than normal HSCs and that SEA stimulation also significantly increased production. This indicates that production in HSCs occurs even when they are exposed early to Schistosoma J. eggs and therefore the redox signaling–mediated actions may represent a very early event for HSC activation and consequent conversion of these cells into a fibrogenic phenotype. Our results further showed that gene silencing of either Nox1 or Nox4 substantially blocked SEA-induced increases in production and CB1 upregulation, and fibrogenic phenotype formation. In contrast to Nox1 and Nox4, silencing of Nox2, another NADPH oxidase isoform expressed in HSCs, did not affect SEA-induced CB1 upregulation (Supplementary Fig. 4), excluding a role of Nox2 in CB1-mediated fibrogenic changes in SSLF. Thus, these results suggest that stimulation of HSCs by SEA may stimulate Nox1 or Nox4 to thereby produce which then upregulates CB1 receptors to lead to HSCs change to a more fibrogenic phenotype. It should be noted that Nox1 regulated Nox4 expression in HSCs since Nox1 gene silencing decreased Nox4 expression (Fig. 5C), while knockdown of Nox4 had no effect on Nox1 expression (Fig. 5D), suggesting that Nox1-mediated action on production and consequent fibrogenic phenotype changes may be through regulation of Nox4. Previous studies have suggested that activation of CB receptors can be an upstream signaling mechanism regulating Nox2 and Nox4-derived oxidative stress in human leukemia cells or mouse kidney [42,43]. However, in the present study, silencing of CB1 receptor did not alter either the expression of Nox1/Nox4 or the SEA-induced production (Fig. 5E and F). Thus, our data support the view that Nox1/Nox4-derived production is upstream of CB1 upregulation in SSLF. The mechanism by which Schistosoma J. activates NADPH oxidase in HSCs is currently unknown. In this regard, toll-like receptors (TLRs) including TLR2 and TLR9 were involved in the recognition of Schistosome antigens, especially SEA, in regulatory T cells and dendritic cells [44–46], or in the host-pathogen interaction in mouse liver [47]. Given the expression of these TLRs in HSCs and their coupling with NADPH oxidase activity in many other mammalian cells [48,49], it is possible that TLR2/TLR9 are involved in activation of NADPH oxidase in SSLF. Taken together, the NADPH oxidase-CB1 redox signaling axis may be an important early mechanisms triggering fibrogenesis in HSCs during Schistosoma J. infection.
Our data further suggest that activation of transcription factor Nrf2 by Nox1/Nox4-mediated ROS production is crucial for CB1 upregulation in SSLF. Nrf2 is a lucine zipper-type transcription factor that plays a key role in maintaining cellular redox homeostasis via its interaction with redox-sensitive protein Kelch-like ECH-associated protein 1 (Keap1) [50]. Under resting conditions, Nrf2 interacts with Keap1 to form a protein complex, which is degraded by proteasomes. Increased oxidative stress may cause conformational changes of Keap1 or phosphorylation of Nrf2, resulting in dissociation of Nrf2 from the Nrf2–Keap1 complex, translocation of Nrf2 to the nucleus, and induction of expression of a variety of genes including antioxidant proteins and anti-inflammatory factors [50]. Here, we demonstrated that SEA-induced nuclear translocation of Nrf2 and increase in its activity in HSCs were both blocked by Nox1 or Nox4 gene silencing. These data suggest that Nrf2 activation depends on Nox1/Nox4-derived ROS. More importantly, when Nrf2 gene was silenced, CB1 upregulation by SEA was markedly inhibited in HSCs. Thus, the present study, for the first time, links Nrf2 activation with CB1 upregulation implicating a detrimental role of Nrf2 in the development of SSLF. Interestingly, several recent studies have indicated that the Nrf2-signaling pathway may play a protective role in none-SSLF liver fibrosis models [51,52]. Nrf2 deficiency caused defective repair of the liver injury, enhanced inflammatory and profibrotic response, and aggravated liver fibrosis in hepatotoxin carbon tetrachloride-treated mice [51]. Further, pharmacological activation of Nrf2 by sulforaphane was reported to inhibit the development of early stage hepatic fibrosis in bile duct-ligated mice, accompanied by reduced expression of profibrogenic genes or HSC activation markers [53]. Taken together, these findings from previous studies and our study suggest that Nrf2 may play both detrimental and protective roles in the pathophysiology of liver fibrosis depending on the animal models used (SSLF vs none-SSLF models). Therefore, the potential application of Nrf2 as a therapeutic target to prevent and treat liver diseases should be cautious.
Previous studies have demonstrated that genetic or pharmacological inactivation of CB1 receptors decreased fibrogenesis by lowering hepatic transforming growth factor (TGF)-β1 and reducing accumulation of fibrogenic cells in the liver of mice [25]. However, we did not observe an increase TGFβ1 release or cellular TGFβ1 level in HSCs on SEA stimulation with or without CB1 silencing (Supplementary Fig. S5). Our data suggest that the role of CB1 upregulation in SSLF is independent of TGFβ1 production. Further, our data also exclude the possibility of this cytokine in the regulation of NADPH oxidase activity and phenotypic properties in the HSCs by SEA stimulation.
In summary, the present study demonstrated that Schistosoma J. infection and SEA stimulation significantly upregulated CB1 receptors in mouse HSCs, which was dependent on production by activation of NADPH oxidase. CB1 upregulation by ROS contributes to the action and phenotype changes in HSCs during Schistosoma J. infection or on SEA stimulation, implicating a role of CB1 upregulation in the development of SSLF. NADPH oxidase-mediated redox signaling in HSCs may be a novel therapeutic target for prevention or treatment of liver cirrhosis during schistosomiasis because blockade of redox upregulation of CB1 receptors is able to prevent an early event in this pathological process.
Supplementary Material
Acknowledgments
This work was supported by the Natural Science Foundation of China, No. 81071381 and grants from the National Institute of Health (HL057244, HL075316, and HL091464).
Abbreviations:
- CB1
cannabinoid receptor 1
- CMH
1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- DPI
diphenyleneiodonium
- ECM
extracellular matrix
- HSCs
hepatic stellate cells
- PBS
phosphate-buffered saline
- RT-PCR
real-time reverse transcription–polymerase chain reaction
- ROS
reactive oxygen species
- SSLF
schistosomiasis-associated liver fibrosis
- SEA
soluble egg antigen
- TGF
transforming growth factor
Footnotes
No competing financial interests exist.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2014.03.015.
References
- [1].Savioli L; Renganathan E; Montresor A; Davis A; Behbehani K Control of schistosomiasis—a global picture. Parasitol. Today 13:444–448; 1997. [DOI] [PubMed] [Google Scholar]
- [2].The control of schistosomiasis. Second report of the WHO Expert Committee. World Health Organ. Tech. Rep. Ser 830:1–86; 1993. [PubMed] [Google Scholar]
- [3].King CH; Dangerfield-Cha M The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 4:65–79; 2008. [DOI] [PubMed] [Google Scholar]
- [4].McManus DP; Gray DJ; Li Y; Feng Z; Williams GM; Stewart D; Rey-Ladino J; Ross AG Schistosomiasis in the People’s Republic of China: the era of the Three Gorges Dam. Clin. Microbiol. Rev 23:442–466; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boros DL Immunopathology of Schistosoma mansoni infection. Clin. Microbiol. Rev 2:250–269; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cioli D; Pica-Mattoccia L Praziquantel. Parasitol. Res 90(Suppl. 1):S3–S9; 2003. [DOI] [PubMed] [Google Scholar]
- [7].Southgate VR; Rollinson D; Tchuem Tchuente LA; Hagan P Towards control of schistosomiasis in sub-Saharan Africa. J. Helminthol 79:181–185; 2005. [DOI] [PubMed] [Google Scholar]
- [8].Gryseels B; Polman K; Clerinx J; Kestens L Human schistosomiasis. Lancet 368:1106–1118; 2006. [DOI] [PubMed] [Google Scholar]
- [9].Zhang JP; Zhang M; Jin C; Zhou B; Xie WF; Guo C; Zhang C; Qian DH Matrine inhibits production and actions of fibrogenic cytokines released by mouse peritoneal macrophages. Acta Pharmacol. Sin 22:765–768; 2001. [PubMed] [Google Scholar]
- [10].Du WD; Zhang YE; Zhai WR; Zhou XM Dynamic changes of type I, III and IV collagen synthesis and distribution of collagen-producing cells in carbon tetrachloride-induced rat liver fibrosis. World J. Gastroenterol 5:397–403; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kresina TF; He Q; Degli Esposti S; Zern MA Gene expression of transforming growth factor beta 1 and extracellular matrix proteins in murine Schistosoma mansoni infection. Gastroenterology 107:773–780; 1994. [DOI] [PubMed] [Google Scholar]
- [12].Trebicka J; Racz I; Siegmund SV; Cara E; Granzow M; Schierwagen R; Klein S; Wojtalla A; Hennenberg M; Huss S; Fischer HP; Heller J; Zimmer A; Sauerbruch T Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int. 31:860–870; 2011. [DOI] [PubMed] [Google Scholar]
- [13].Patsenker E; Stoll M; Millonig G; Agaimy A; Wissniowski T; Schneider V; Mueller S; Brenneisen R; Seitz HK; Ocker M; Stickel F Cannabinoid receptor type I modulates alcohol-induced liver fibrosis. Mol. Med 17:1285–1294; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen SW; Wu BY; Xu SP; Fan KX; Yan L; Gong Y; Wen JB; Wu DH Suppression of CB1 cannabinoid receptor by lentivirus mediated small interfering RNA ameliorates hepatic fibrosis in rats. PLoS One 7:e50850; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gao X; Guo Y; Liu HY; Tang WX [The expression of cannabinoid receptor 1 in fibrotic liver tissues of mice infested with Schistosoma mansoni]. Zhonghua Gan Zang Bing Za Zhi 15:389–390; 2007. [PubMed] [Google Scholar]
- [16].De Minicis S; Seki E; Paik YH; Osterreicher CH; Kodama Y; Kluwe J; Torozzi L; Miyai K; Benedetti A; Schwabe RF; Brenner DA Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology 52:1420–1430; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sancho P; Mainez J; Crosas-Molist E; Roncero C; Fernandez-Rodriguez CM; Pinedo F; Huber H; Eferl R; Mikulits W; Fabregat I NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One 7:e45285; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aram G; Potter JJ; Liu X; Wang L; Torbenson MS; Mezey E Deficiency of nicotinamide adenine dinucleotide phosphate, reduced form oxidase enhances hepatocellular injury but attenuates fibrosis after chronic carbon tetrachloride administration. Hepatology 49:911–919; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schafer S; Zerbe O; Gressner AM The synthesis of proteoglycans in fat-storing cells of rat liver. Hepatology 7:680–687; 1987. [DOI] [PubMed] [Google Scholar]
- [20].Huang DK; Zhang YX; Man SQ; Yu FZ; Shen JJ [Comparison of collagen fiber staining between Van-Gieson staining and Masson trichrome staining of hepatic specimens in mice with Schistosoma japonicum infection]. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 24:468–470; 2012. [PubMed] [Google Scholar]
- [21].Xu M; Li XX; Ritter JK; Abais JM; Zhang Y; Li PL Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS One 8:e71212; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu P; Wang M; Lu XD; Zhang SJ; Tang WX Schistosoma japonicum egg antigen up-regulates fibrogenesis and inhibits proliferation in primary hepatic stellate cells in a concentration-dependent manner. World J. Gastroenterol 19:1230–1238; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wei Y; Wang X; Zhao F; Zhao PQ; Kang XL Cannabinoid receptor 1 blockade protects human retinal pigment epithelial cells from oxidative injury. Mol. Vis 19:357–366; 2013. [PMC free article] [PubMed] [Google Scholar]
- [24].Paik YH; Iwaisako K; Seki E; Inokuchi S; Schnabl B; Osterreicher CH; Kisseleva T; Brenner DA The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 53:1730–1741; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Teixeira-Clerc F; Julien B; Grenard P; Tran Van Nhieu J; Deveaux V; Li L; Serriere-Lanneau V; Ledent C; Mallat A; Lotersztajn S CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat. Med 12:671–676; 2006. [DOI] [PubMed] [Google Scholar]
- [26].Tam J; Liu J; Mukhopadhyay B; Cinar R; Godlewski G; Kunos G Endocannabinoids in liver disease. Hepatology 53:346–355; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Julien B; Grenard P; Teixeira-Clerc F; Van Nhieu JT; Li L; Karsak M; Zimmer A; Mallat A; Lotersztajn S Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 128:742–755; 2005. [DOI] [PubMed] [Google Scholar]
- [28].Caraceni P; Viola A; Piscitelli F; Giannone F; Berzigotti A; Cescon M; Domenicali M; Petrosino S; Giampalma E; Riili A; Grazi G; Golfieri R; Zoli M; Bernardi M; Di Marzo V Circulating and hepatic endocannabinoids and endocannabinoid-related molecules in patients with cirrhosis. Liver Int. 30:816–825; 2010. [DOI] [PubMed] [Google Scholar]
- [29].Ruiz-Ortega M; Ruperez M; Esteban V; Rodriguez-Vita J; Sanchez-Lopez E; Carvajal G; Egido J Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol. Dial. Transplant 21:16–20; 2006. [DOI] [PubMed] [Google Scholar]
- [30].Higgins DF; Kimura K; Bernhardt WM; Shrimanker N; Akai Y; Hohenstein B; Saito Y; Johnson RS; Kretzler M; Cohen CD; Eckardt KU; Iwano M; Haase VH Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest 117:3810–3820; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wasmuth HE; Trautwein C CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Hepatology 45:543–544; 2007. [DOI] [PubMed] [Google Scholar]
- [32].Di Marzo V The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res 60:77–84; 2009. [DOI] [PubMed] [Google Scholar]
- [33].Iredale JP; Benyon RC; Arthur MJ; Ferris WF; Alcolado R; Winwood PJ; Clark N; Murphy G Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology 24:176–184; 1996. [DOI] [PubMed] [Google Scholar]
- [34].Iredale JP Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest 117:539–548; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Friedman SL Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 88:125–172; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Arthur MJ Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest Liver Physiol 279:G245–G249; 2000. [DOI] [PubMed] [Google Scholar]
- [37].Mallat A; Teixeira-Clerc F; Lotersztajn S Cannabinoid signaling and liver therapeutics. J. Hepatol 59:891–896; 2013. [DOI] [PubMed] [Google Scholar]
- [38].Lim MP; Devi LA; Rozenfeld R Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2:e170; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Albano E Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc 65:278–290; 2006. [DOI] [PubMed] [Google Scholar]
- [40].Babior BM NADPH oxidase: an update. Blood 93:1464–1476; 1999. [PubMed] [Google Scholar]
- [41].Jiang JX; Venugopal S; Serizawa N; Chen X; Scott F; Li Y; Adamson R; Devaraj S; Shah V; Gershwin ME; Friedman SL; Torok NJ Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 139:1375–1384; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McKallip RJ; Jia W; Schlomer J; Warren JW; Nagarkatti PS; Nagarkatti M Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol 70:897–908; 2006. [DOI] [PubMed] [Google Scholar]
- [43].Mukhopadhyay P; Pan H; Rajesh M; Batkai S; Patel V; Harvey-White J; Mukhopadhyay B; Hasko G; Gao B; Mackie K; Pacher P CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br. J. Pharmacol 160:657–668; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Layland LE; Rad R; Wagner H; da Costa CU Immunopathology in schistosomiasis is controlled by antigen-specific regulatory T cells primed in the presence of TLR2. Eur. J. Immunol 37:2174–2184; 2007. [DOI] [PubMed] [Google Scholar]
- [45].Gao Y; Chen L; Hou M; Chen Y; Ji M; Wu H; Wu G TLR2 Directing PD-L2 expression inhibit T cells response in Schistosoma japonicum infection. PLoS One 8:e82480; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ito T; Schaller M; Raymond T; Joshi AD; Coelho AL; Frantz FG; Carson W. F. t.; Hogaboam CM; Lukacs NW; Standiford TJ; Phan SH; Chensue SW; Kunkel SL Toll-like receptor 9 activation is a key mechanism for the maintenance of chronic lung inflammation. Am. J. Respir. Crit. Care Med 180:1227–1238; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Magalhaes K; Almeida PE; Atella G; Maya-Monteiro CM; Castro-Faria-Neto H; Pelajo-Machado M; Lenzi HL; Bozza MT; Bozza PT Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through Toll-like receptor-2-dependent mechanisms. J. Infect. Dis 202:1369–1379; 2010. [DOI] [PubMed] [Google Scholar]
- [48].Lee JH; Joo JH; Kim J; Lim HJ; Kim S; Curtiss L; Seong JK; Cui W; Yabe-Nishimura C; Bae YS Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res 99:483–493; 2013. [DOI] [PubMed] [Google Scholar]
- [49].Lee JG; Lee SH; Park DW; Yoon HS; Chin BR; Kim JH; Kim JR; Baek SH Toll-like receptor 9-stimulated monocyte chemoattractant protein-1 is mediated via JNK-cytosolic phospholipase A2-ROS signaling. Cell Signal. 20:105–111; 2008. [DOI] [PubMed] [Google Scholar]
- [50].Shin SM; Yang JH; Ki SH Role of the Nrf2-ARE pathway in liver diseases. Oxid. Med. Cell Longev 2013:763257; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xu W; Hellerbrand C; Kohler UA; Bugnon P; Kan YW; Werner S; Beyer TA The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab. Invest 88:1068–1078; 2008. [DOI] [PubMed] [Google Scholar]
- [52].Chen S; Zou L; Li L; Wu T The protective effect of glycyrrhetinic acid on carbon tetrachloride-induced chronic liver fibrosis in mice via upregulation of Nrf2. PLoS One 8:e53662; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Oh CJ; Kim JY; Min AK; Park KG; Harris RA; Kim HJ; Lee IK Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. Free Radic. Biol. Med 52:671–682; 2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.