

Abstract
Interleukin (IL)-1β is a proinflammatory cytokine implicated in various pathophysiological conditions of the CNS involving NMDA receptor activation. Circumstantial evidence suggests that IL-1β and NMDA receptors can functionally interact. Using primary cultures of rat hippocampal neurons, we investigated whether IL-1β affects NMDA receptor function(s) by studying (1) NMDA receptor-induced [Ca2+]i increase and (2) NMDA-mediated neurotoxicity. IL1β (0.01-0.1 ng/ml) dose-dependently enhances NMDA-induced [Ca2+]i increases with a maximal effect of ∼45%. This effect occurred only when neurons were pretreated with IL-1β, whereas it was absent if IL-1β and NMDA were applied simultaneously, and it was abolished by IL-1 receptor antagonist (50 ng/ml). Facilitation of NMDA-induced [Ca2+]i increase by IL-1β was prevented by both lavendustin (LAV) A (500 nm) and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (1 μm), suggesting an involvement of tyrosine kinases. Increased tyrosine phosphorylation of NMDA receptor subunits 2A and 2B and coimmunoprecipitation of activated Src tyrosine kinase with these subunits was observed after exposure of hippocampal neurons to 0.05 ng/ml IL-1β. Finally, 0.05 ng/ml IL-1β increased by ∼30% neuronal cell death induced by NMDA, and this effect was blocked by both lavendustin A and PP2.
These data suggest that IL-1β increases NMDA receptor function through activation of tyrosine kinases and subsequent NR2A/B subunit phosphorylation. These effects may contribute to glutamate-mediated neurodegeneration.
Keywords: cytokines, tyrosine kinase, excitotoxicity, IL-1 receptor type I, NR2A/B subunits, hippocampal neurons
Introduction
Interleukin (IL)-1β, originally described as an immune cell mediator in the periphery, has been involved in the modulation of several neurological functions and dysfunctions (Rothwell and Hopkins, 1995; Viviani et al., 2001). Thus, administration of the naturally occurring antagonist of IL-1, namely IL-1 receptor antagonist (IL-1ra), impairs long-term potentiation (Schneider et al., 1998), attenuates normal spontaneous sleep (Takahashi et al., 1996), inhibits motor and electroencephalographic seizures (Vezzani et al., 2000), and reduces neuronal damage caused by ischemic, excitotoxic, and traumatic brain injury (Yamasaki et al., 1995; Loddick and Rothwell, 1996; Allan et al., 2000). In addition, these physiological and pathological conditions are associated with an increased expression of IL-1β in the brain (Taishi et al., 1997; Schneider et al., 1998; Vezzani et al., 1999, 2000; Allan and Rothwell, 2001). Recent studies showed that IL-1β affects various classic neurotransmitter systems (Rothwell and Hopkins, 1995; Grazia de Simoni and Imeri, 1998). Substantial evidence suggests the existence of a reciprocal functional interaction between IL-1β and NMDA receptors (NMDARs). Thus, intracerebral injection of NMDA in perinatal rat brain stimulates the production of IL-1β (Hagan et al., 1996) and in adult rat brain IL-1β exacerbates the neuronal damage induced by NMDA application (Loddick and Rothwell, 1996). In addition, intrahippocampal injection of IL-1β induces proconvulsant actions in rats that are blocked by selective antagonism of NMDA receptors (Vezzani et al., 1999).
NMDA receptors are glutamate-gated ion channels consisting of an essential subunit, NR1, and one or more components of a second family of subunits termed NR2A-D (Hollmann and Heinemann, 1994) and NR3A/B (Chatterton et al., 2002). NMDA channel activity is modulated by phosphorylation by several kinases: protein kinase A (PKA) and protein kinase C (PKC) phosphorylate NR1, NR2A, and NR2B subunits at serine-threonine sites (Leonard and Hell, 1997); calcium/calmodulin-dependent protein kinase II (CaMKII), which is part of the NMDA receptor complex (Gardoni et al., 1998), as well as the Src family of kinases phosphorylate NR2A and NR2B (Moon et al., 1994; Lau and Huganir, 1995; Omkumar et al., 1996; Gardoni et al., 1998). Phosphorylation at tyrosine residues by the Src family of kinases leads to NMDA current increase (Yu et al., 1997), whereas dephosphorylation by tyrosine phosphatases determines a decrease in the probability of NMDA channel openings (Wang and Salter, 1994). Furthermore, recent evidence indicates that PKC action may involve the activation of protein tyrosine kinase 2 signaling cascade upstream of Src kinase (Lu et al., 1999; Grosshans and Browning, 2001). The Src family of kinases therefore plays a central role in the modulation of the NMDA function.
In this study, we investigated whether IL-1β enhances NMDA receptor function and the involvement of the Src family of tyrosine kinases in this process. For this purpose, we studied the effect of IL-1β on (1) intracellular Ca2+ increase induced by NMDA receptor activation and (2) the phosphorylation of NMDA receptor NR2A/B subunits using primary cultures of hippocampal neurons. Finally, to evaluate the functional consequences of IL-1β and NMDA receptor interactions, we studied whether IL-1β increased NMDA-mediated neuronal death in vitro.
Materials and Methods
Primary cultures of hippocampal neurons. Neuronal cultures were established from the hippocampus of 18-d-old rat fetuses. Briefly, brains were removed and freed from meninges, and the hippocampus was isolated. Cells were then dispersed by incubation for 5 min at 37°C in a 2.5% trypsin solution followed by trituration. The cell suspension was diluted in Neurobasal media supplemented with 1% B-27 (Life Technologies, Rockville, MD) and plated on polyornithine-coated coverslips at a density of 80,000 cells per coverslip. For immunocytochemistry, dissociated rat hippocampal neurons were grown using the Goslin and Banker method (1998). Both of these culture conditions allowed us to grow differentiated neuronal cultures with >96% homogeneity, as assessed by immunocytochemistry of microtubule-associated protein 2 and glial fibrillary acidic protein (Molecular Probes, Eugene, OR). Hippocampal neurons were used after a culturing period of 10-14 d.
IL-1β and drug applications. Rat recombinant IL-1β (Sigma, St. Louis, MO) was used in all of the experiments. Neurons were exposed to drugs in HBSS containing (in mm): 137 NaCl, 5.4 KCl, 1.8 CaCl2, 4.2 NaHCO3, 5 glucose, 10 HEPES. To measure the effect of IL-1β on NMDA-induced [Ca2+]i increase, IL-1β (0.01-1 ng/ml) was added together with or 6 min before 10 μm NMDA applied for 3 min. Lavendustin A (500 nm) and 1 μm 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (30 min) (Calbiochem, La Jolla, CA), 50 ng/ml human recombinant IL1ra (15 min) (a generous gift from Dr. P. Ghezzi, Mario Negri Institute, Milan, Italy), 50 μm 2-aminoethoxydiphenylborane (2-APB) plus 10 μm dantrolene (10 min), 1 μm nitrendipine, and 1 μm tetrodotoxin (2 min) (Tocris, Bristol, UK) were preincubated with neurons at the times shown in parentheses before addition of IL-1β and kept in the incubation medium during subsequent stimulation with NMDA. MK801 (10 μm) (Tocris) was added 1.5 min after NMDA stimulation. Control neuronal cultures were run in parallel in each experimental condition and incubated for the appropriate times in HBSS.
Determination of the cytosolic free Ca2+concentration [Ca2+]i. Neurons were loaded with 10 μm fura-2 AM (Sigma) for 1 hr at 37°C in their culture medium supplemented with 1% bovine serum albumin, 0.1% pluronic (Sigma). [Ca2+]i measurement was performed in HEPES buffer, pH 7.4, as described previously (Viviani et al., 2001).
The fura-2 fluorescence ratio signal was measured in a Perkin-Elmer LS 50 B double-wavelength fluorimeter and calibrated in terms of [Ca2+]i.
Immunocytochemistry and confocal imaging. Coverslips containing neurons were lifted from the astrocyte monolayer, washed by immersion in PBS, and fixed in ice-cold 4% paraformaldehyde (Merck, Darmstadt, Germany) for 30 min. The coverslips were then incubated for 10 min at room temperature in PBS containing 0.25% Triton X-100 (Sigma). Non-specific sites were blocked by incubation in PBS containing 10% normal goat serum. For double immunostaining, the coverslips were incubated in 2% normal goat serum containing a 1:500 dilution of a rabbit polyclonal antibody against NR2A/B and a hamster monoclonal antibody against IL-1RI for 2 hr at 37 C. Specific binding was detected using secondary antibodies conjugated to AlexaFluor dyes (594: red, for IL-1RI; 647: blue for NR2A/B) (Molecular Probes). Images were collected on a Delta Vision Optical Sectioning Microscope consisting of an Olympus IX-70 microscope equipped with a mercury arc lamp. A photometrics CH 350 cooled CCD camera and a high-precision motorized XYZ stage were used to acquire multiple consecutive optical sections at a 0.2 μm interval for each of the fluorescent probes. A UPAPLO 60× objective was used to collect the images.
Metabolic labeling and immunoprecipitation. Hippocampal cultures were preincubated for 2 hr in phosphate-free minimal essential medium (Sigma) and then labeled with [32P]orthophosphate (0.5 mCi/ml; Amersham Biosciences, Buckinghamshire, UK) in the same medium for 2 hr at 37°C. The cultures were pretreated for 30 min with 500 nm lavendustin A or 1 μm PP2 or HBSS (control) and then incubated for 6 min with 0.05 ng/ml IL-1β. After two quick washes with cold HBSS, the cells were harvested into 0.5 ml of ice-cold solubilization buffer containing 50 mm HEPES, pH 7.4, 0.5 M NaCl, 0.5% Triton X-100, 0.1% deoxycholate, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 10 mm NaF, in the presence of a complete set of proteases inhibitors (Complete, Boehringer Mannheim, Mannheim, Germany). Each solubilized sample was incubated overnight with NR2A/B polyclonal antibody and then for 2 hr at 4°C with protein A-Sepharose beads. After this incubation, the supernatants were removed, and the beads were washed five times with solubilization buffer. After the final wash, the beads were resuspended in SDS sample buffer and centrifuged briefly; the supernatants were loaded on 7% SDS-PAGE gels.
To quantify 32P incorporation in NR2A/B, the optical density of the band corresponding to the 170 kDa NR2A/B-receptor subunits was measured on autoradiograms, and the values were corrected for the amount of 32P incorporation in total proteins. To this aim, aliquots of neuronal homogenates were spotted on P81 paper and analyzed for total 32P protein. The results were then expressed in each experiment as a percentage of control values. Western blot analysis was performed by computer-assisted imaging (Quantity-One System; Bio-Rad, Richmond, CA).
Surface expression experiments. Cultures were biotinylated by means of the reversible membrane impermeable derivate of biotin sulfo-NHS-S-S-biotin (Pierce, Rockford, IL) (1 mg/ml) for 15 min at 37°C; unreacted biotinylation reagents was quenched by three successive 5 min washes in ice-cold TBS (50 mm Tris-HCl, pH 7.5, 200 mm NaCl). Cells were harvested in TBS containing 2 mm EDTA, 2 mm EGTA, and pelleted at 14,000 × g for 30 min. Cell pellet was lysed and sonicated in 50 mm HEPES, pH 7.5, 0.1 mm EDTA, 1% Triton X-100 in the presence of a complete set of protease inhibitors (Complete, Boehringer Mannheim) and phosphatase inhibitors. Cell lysates were cleared by centrifugation at 14,000 × g for 15 min. UltraLink immobilized NeutrAvidin beads (Pierce) was added to the supernatant, and the samples were rotated at 4°C for 2 hr. Next the NeutrAvidin beads were washed five times with lysis buffer, and bound proteins were eluted into SDS sample buffer by boiling for 15 min. Biotinylated proteins were resolved by SDS-PAGE and subjected to immunoblot analysis with IL1-R1 antibody.
Reverse transcriptase-PCR. Total RNA was isolated using a commercially available single-step isolation kit (Trizol, Invitrogen, Paisley, UK) and extracted following supplier's instructions. Reverse transcriptase (RT)-PCR reactions were performed as described previously (Viviani et al., 2001). PCR primers for IL-6 and glyceraldehyde-6-phosphate dehydrogenase (GPDH) were synthesized by Primm (Milan, Italy) and contained the following sequences: IL-6 sense, 5′-ATGAAGTTCCTCTCTGCAAGAGACT-3′; IL-6 antisense, 5′-CACTAGGTTTGCCGAGTAGATCTC-3′; GPDH sense, 5′-TGAAGGTCGGAGTCAACGGATTT-3′; GPDH antisense, 5′-CATGTGGGCCATGAGGTCCACCAC-3′.
The amplified PCR products are 638 bp for IL-6 and 983 bp for GPDH. In preliminary experiments, RNA concentrations and PCR cycles were titrated to establish standard curves to document linearity and permit semiquantitative analysis of signal strength (80 and 200 ng for GPDH and IL-6, respectively). The PCR was conducted using a Perkin Elmer thermocycler, with the following conditions: 35 cycles of denaturation at 95°C for 30 sec, annealing at 65°C for 30 sec, extension at 72°C for 30 sec, followed by a final 7 min extension at 72°C. The PCR products were visualized by UV illumination after electrophoresis through 1.5% agarose at 80 V and staining in 1× TBE buffer containing 0.5 μg/ml ethidium bromide. Gels were photographed with type 667 film (Polaroid, Cambridge, MA). The image of the PCR products was acquired with a Nikon CCD video camera module (Nikon, Melville, NY). The optical density of the bands was calculated, and peak area of a given band was analyzed by means of the Image 1.61 program for digital image processing (Wayne Rasband, Research Service Branch, National Institutes of Health, Bethesda, MD).
Antibodies. Antibody against NR2A/B was purchased from Chemicon International (Temecula, CA); p1472-NR2B antibody was purchased from Calbiochem; p416-active-Src monoclonal antibody was purchased from Alexis (Lausen, Switzerland); IL-1RI (clone JAMA-147) was purchased from PharMingen (San Diego, CA); IL-1RI and NR2A/B secondary antibodies conjugated to AlexaFluor dyes were from Molecular Probes.
Viability assay. For viability assays, neurons were pretreated for 30 min with 500 nm lavendustin A or 1 μm PP2 or HBSS and then exposed to 10 μm NMDA only or to 0.05 ng/ml IL-1β for 6 min and subsequently to 10 μm NMDA for 3 hr. Finally, all drugs were removed and neurons were returned to their culture medium for 24 hr. Cell viability was measured by the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Denizot and Lang, 1986). Briefly, MTT tetrazolium salt was dissolved in serum-free medium to a final concentration of 0.75 mg/ml and added to the cells before the end of the experiment for 3 hr at 37°C. The medium was then removed and the formazan was extracted with 1N HCl/isopropanol (1:24). Absorbance at 560 nm was read on a Multiscan reader.
Statistical analysis. The statistical significance of differences was determined by one-way or two-way ANOVA followed by a multiple comparison test (Tukey's test). A significance level of 95% (p < 0.05) was accepted.
Results
IL-1β effect on NMDA-induced intracellular Ca2+ ([Ca2+]i) increase in primary cultures of hippocampal neurons
[Ca2+]i increase induced by NMDA was used as a well established index of functional activation of this receptor. To evaluate whether IL-1β modulates the NMDA receptor function, primary cultures of hippocampal neurons were pre-exposed to increasing concentrations of IL-1β (0.01-1 ng/ml) and subsequently stimulated with 10 μm NMDA (Fig. 1). Pre-exposure of neurons to IL-1β at the above concentrations did not affect basal [Ca2+]i levels, which were between 67 ± 9.7 and 75 ± 3.4 nm in the presence of IL-1β, and 73 ± 5.3 nm in the absence of IL-1β.
Figure 1.
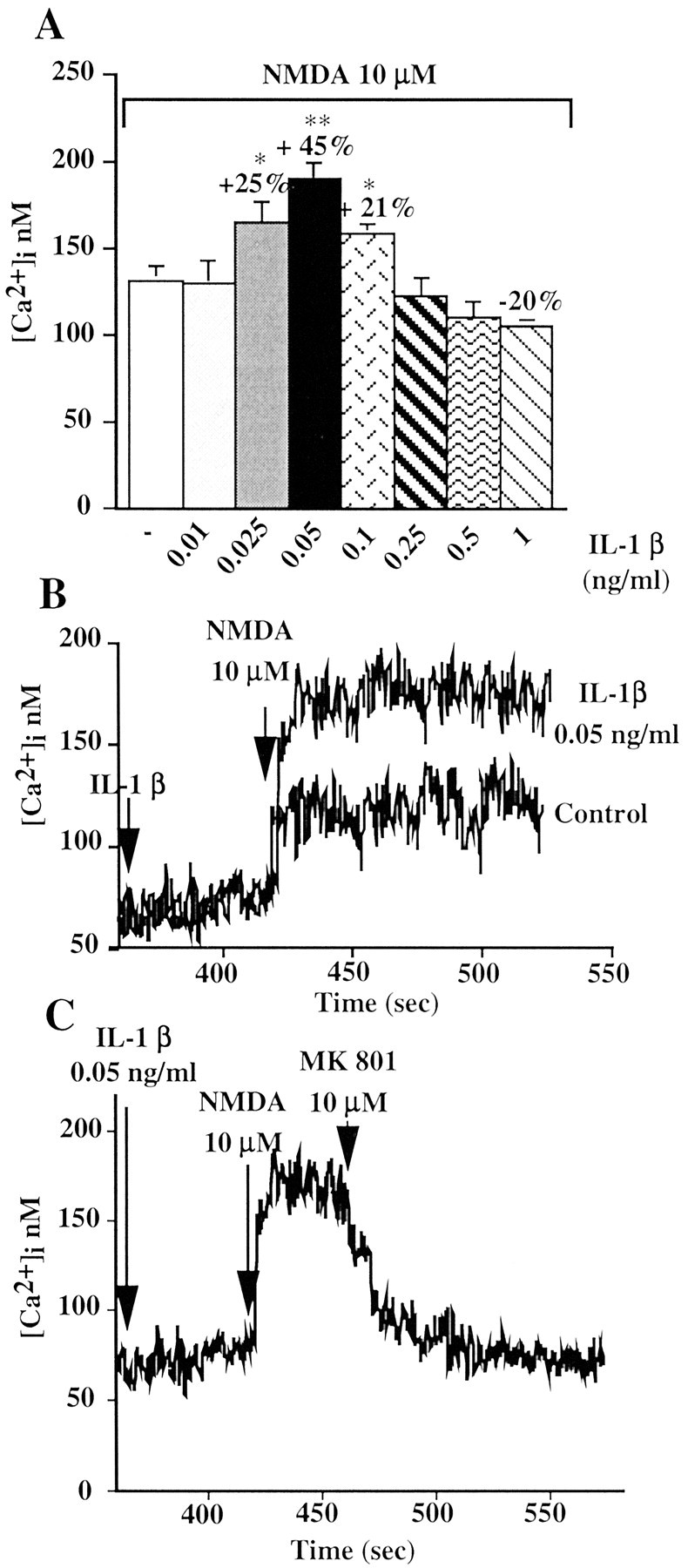
IL-1β effect on NMDA-induced [Ca2+]i increase in primary cultures of hippocampal neurons. A, Bar graph represents the average peak Ca2+ response to the application of 10 μm NMDA alone or in the presence of increasing concentration of IL-1β. Values are the mean ± SE of 7-13 independent samples. Percentage values represent the IL-1β effect expressed as percentage increase (+) or decrease (-) of the NMDA response measured in the absence of IL-1β. Hippocampal neurons were exposed to IL-1β (0.01-1 ng/ml) for 6 min and subsequently stimulated with 10 μm NMDA. [Ca2+]i increase was determined by loading hippocampal neurons with fura-2 AM and measuring fura-2 fluorescence ratio signal in a double-wavelength fluorimeter. **p < 0.01; *p < 0.05 versus NMDA alone; ANOVA followed by Tukey's test. B, Representative Ca2+ traces showing the response of hippocampal neurons exposed to 10 μm NMDA alone or with 6 min pretreatment with 0.05 ng/ml IL-1β. C, Representative Ca2+ trace showing the rapid decay in the response of hippocampal neurons when 10 μm MK801 was applied 1.5 min after NMDA stimulation. Neurons were exposed for 6 min to 0.05 ng/ml IL-1β and then stimulated with 10 μm NMDA.
[Ca2+]i increase induced by 10 μm NMDA was enhanced by 6 min preincubation with IL-1β in a dose-dependent manner (Fig. 1A). Thus, 0.025-0.1 ng/ml IL-1β enhanced NMDA-induced response, reaching a maximal effect (+45 ± 2.36%; n = 13) at 0.05 ng/ml (Fig. 1A,B). Higher concentrations of IL-1β (0.25-0.5 ng/ml) were ineffective, whereas 1 ng/ml was slightly inhibitory (Fig. 1A).
We then focused our attention on the facilitating action of IL-1β on NMDA-induced response, because this effect could explain several of the in vivo actions of this cytokine.
The potentiating effect of IL-1β on NMDA-induced increase in [Ca2+]i was caused predominantly by Ca2+ influx through the NMDA receptor channel. This is supported by the following evidence: (1) 10 μm MK801, an open channel blocker that binds only to activated NMDA receptors, when added 1.5 min after NMDA application in IL-1 β (0.05 ng/ml) pretreated neurons, sharply reduced [Ca2+]i to resting levels (Fig. 1C); (2) pretreatment of neurons with 1 μm TTX, which blocks sodium channels and neurotransmission, or nitrendipine, an L-type voltage-gated Ca2+ channel blocker, did not modify IL-1β potentiation of NMDA-induced [Ca2+]i increase (Table 1); (3) pretreatment with 10 μm dantrolene, an antagonist of ryanodine receptor, together with 50 μm 2-APB, an IP3 receptor antagonist, did not alter IL-1β enhancing effect (Table 1), thus suggesting that both IP3-sensitive and -insensitive Ca2+ stores are not involved in IL-1β effect; and (4) finally, a contribution of Ca2+-permeable AMPA receptors attributable to a secondary release of glutamate was excluded, because AMPA response was not potentiated by IL-1β (AMPA 100 μm: 147 ± 8.7 nm; IL-1β plus AMPA 100 μm: 141 ± 12 nm; n = 5). The classic AMPA receptor inhibitors 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide and CNQX could not be used to exclude AMPA receptor-mediated effects, because of their interference with the fura-2 fluorescent signal.
Table 1.
IL-1β effect on NMDA-induced [Ca2+]i increase in hippocampal neurons pretreated with TTX, nitrendipine, or 2-APB and dantrolene
|
|
[Ca2+]i (nm) |
||
|---|---|---|---|
|
|
10 μm NMDA
|
0.05 ng/ml IL-1β + 10 μm NMDA
|
|
| None | 143 ± 3.7 | 193 ± 6.6 | |
| 1 μm TTX | 142 ± 18 | 199 ± 25.2 | |
| 1 μm Nitrendipine | 133 ± 23.5 | 182 ± 10.4 | |
| 50 μm 2-APB + 10 μm dantrolene
|
141 ± 7.5
|
200 ± 11.7
|
|
Values are means ± SE of four independent samples and represent the average peak Ca2+ response. TTX (1 μm, 2 min), nitrendipine (1 μm, 2 min) and APB (50 μm), plus dantrolene (10 μm, 10 min) were preincubated with neurons at the times shown in parentheses before the addition of 0.05 ng/ml IL-1β and kept in the medium during subsequent stimulation with 10 μm NMDA.
To study whether IL-1β modulation of the NMDA response was reversible after removal of this cytokine, hippocampal cells, with or without 6 min pretreatment with IL-1β, were washed in fresh HBSS after 3 min NMDA application (Fig. 2A). Removal of NMDA in control neurons or of IL-1β plus NMDA in IL-1β-pretreated neurons resulted in a rapid drop of [Ca2+]i to basal levels (Fig. 2A). The same neurons were then exposed to 10 μm NMDA alone for three consecutive times. [Ca2+]i was enhanced to a lesser extent compared with the immediately preceding NMDA application (Fig. 2A,B); however, NMDA-induced [Ca2+]i was significantly higher in neurons pre-exposed once to IL-1β (Fig. 2, trace 1) than in control neurons (Fig. 2, trace 2). This indicates that the potentiating effect of IL-1β lasts for at least 24 min even in the absence of this cytokine (Fig. 2B).
Figure 2.

Duration of the IL-1β effect on NMDA-induced [Ca2+]i increase. A, Trace 1 (gray) depicts a representative Ca2+ trace showing the response of hippocampal neurons to four consecutive applications of 10 μm NMDA, the first in the presence (6 min preincubation) and the following in the absence of 0.05 ng/ml IL-1β. Trace 2 (black) depicts a representative Ca2+ trace showing the response of hippocampal neurons to four consecutive applications of 10 μm NMDA done in the absence of IL-1β. Intracellular Ca2+ increase was determined by loading hippocampal neurons with fura-2 AM and measuring fura-2 fluorescence ratio signal in a double-wavelength fluorimeter. B, Average peak Ca2+ response to four consecutive applications of NMDA as reported in A. Values are the mean ± SE of 13 independent samples. **p < 0.01 and *p < 0.05 versus control; ANOVA followed by Tukey's test.
Effect of IL-1ra on IL-1β potentiation of NMDA receptor function
IL-1β exerts its actions by binding to IL-1 receptor type I (IL-1RI) (Dinarello, 1996). To assess whether the effect of IL-1β on NMDA receptor was receptor mediated, hippocampal neurons were incubated for 5 min with 50 ng/ml IL-1ra before treatment with 0.05 ng/ml IL-1β and the subsequent stimulation with NMDA. IL-1ra per se did not affect NMDA-induced [Ca2+]i (10 μm NMDA: 133 ± 7.5 nm; IL-1ra plus NMDA: 136 ± 17 nm; n = 5) but prevented IL-1β-induced potentiation of the NMDA response (0.05 ng/ml IL-1β plus NMDA: 190 ± 10 nm; IL-1ra plus IL-1β + NMDA: 126 ± 11.4 nm; n = 5). Double labeling of hippocampal neurons with specific antibodies against NMDA receptor 2A/B subunits and IL-1RI (Fig. 3, IL-1 RI, red, and NR 2A/B, green) showed that these neurons express both receptors with a similar distribution pattern, thus further supporting the possibility that IL-1β actions on NMDA receptors occur through IL-1RI.
Figure 3.

Immunofluorescence of IL-1RI and NR2A/B subunits of the NMDA receptor in primary cultures of hippocampal neurons. Detection of IL-1RI and NR2A/B subunits was obtained by double immunostaining with specific antibodies (see Materials and Methods) and AlexaFluor-conjugated secondary antibodies. Images were collected on a Delta Vision Optical Sectioning Microscope. IL-1RI and NR2A/B subunits are localized on the same neurons and showed a similar distribution pattern.
IL-1β and tyrosine kinase-dependent phosphorylation of NMDA receptors
Potentiation of the NMDA response by IL-1β required that hippocampal neurons were exposed to 0.05 ng/ml IL-1β before NMDA addition; thus their simultaneous application did not enhance NMDA effect (Fig. 4A). This suggests that activation of intracellular cascade(s) downstream to IL-1β and IL-1RI interaction is mandatory to mediate this effect. In accordance with the involvement of Tyr-phosphorylation processes, we found that pretreatment of hippocampal neurons with lavendustin A (500 nm), a tyrosine kinase inhibitor, or the Src family tyrosine kinase inhibitor PP2 (1 μm) prevented IL-1β enhancement of NMDA-mediated [Ca2+]i rise, without affecting the NMDA response per se (Fig. 4B). To investigate whether NR2A/B subunits were phosphorylated in the presence of IL-1β, hippocampal neurons were labeled with [32P]orthophosphate and exposed to 0.05 ng/ml IL-1β for 6 min. 32P incorporation was quantified on neuronal lysate by immunoprecipitation with a polyclonal antibody raised against NR2A/B. Our data show that IL-1β treatment of hippocampal neurons significantly increased the phosphorylation of NMDA receptor subunits NR2A/B, and this effect was blocked by either lavendustin A or PP2 (Fig. 5A,B).
Figure 4.
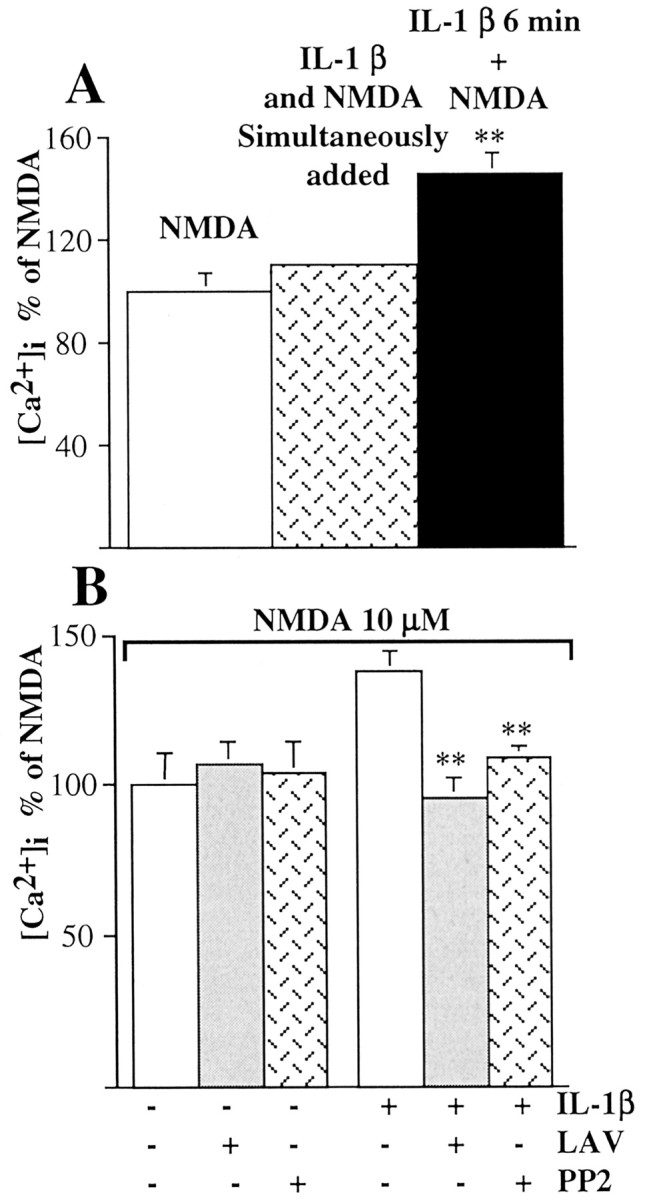
Involvement of tyrosine kinases in IL-1β facilitation of the NMDA response. A, Hippocampal neurons were exposed simultaneously to 0.05 ng/ml IL-1β and 10 μm NMDA (dotted column) or pretreated for 6 min with 0.05 ng/ml IL-1β and then stimulated with 10 μm NMDA (black column). Values are means ± SE of five independent samples and represent the average peak Ca2+ response. **p < 0.01 versus control; ANOVA followed by Tukey's test. B, Average peak Ca2+ increase as percentage of the NMDA response in control neurons ± lavendustin A or PP2 and in IL-1β-pretreated neurons ± lavendustin A or PP2. Hippocampal neurons were pretreated with 500 nm lavendustin A or 1 μm PP2 for 30 min and then exposed to NMDA alone or 0.05 ng/ml IL-1 β for 6 min and subsequently stimulated with 10 μm NMDA. Values are means ± SE of five independent samples. **p < 0.01 versus IL-1 plus NMDA; ANOVA followed by Tukey's test.
Figure 5.

IL-1β-induced phosphorylation of NR2A/B subunits through the Src family of tyrosine kinases. A, Metabolic labeling and immunoprecipitation of NR2A/B subunits. Hippocampal neurons were incubated for 30 min in HBSS plus 500 nm lavendustin A (LAV) or 1 μm PP2 and then exposed to 0.05 ng/ml IL-1β for 6 min. Controls (C) were hippocampal neurons incubated in HBSS for 36 min. B, Histogram shows the 32P incorporation in NR2A/B subunits in the various experimental conditions, expressed as percentage of control. Values were normalized as reported in Materials and Methods. Lavendustin and PP2 alone were not different from the control. Values are mean ± SE (n = 6). **p < 0.01 IL-1 versus control; §§p < 0.01 IL-1 + LAV and IL-1 + PP2 versus IL-1 by ANOVA followed by Tukey's test. C, Representative autoradiography showing the presence of a phosphorylated protein band of apparent molecular weight 65 kDa in anti-NR2A/B immunoprecipitates (arrowhead). Molecular weight standards are reported to the left. D, Histograms shows 32P incorporation in a 65 kDa phospho-band in the various experimental conditions, expressed as percentage of control. Values were normalized as reported in Materials and Methods. Lavendustin and PP2 alone were not different from the control. Values are mean ± SE (n = 6). **p < 0.01 IL-1 versus control; §§p < 0.01 IL-1 + LAV and IL-1 + PP2 versus IL-1 by ANOVA followed by Tukey's test.
Among tyrosine kinases, the nonreceptor Src family tyrosine kinases have been implicated in NR2A and NR2B subunit phosphorylation (Yu et al., 1997). Under our experimental conditions, immunoprecipitation of NR2A/B induces coprecipitation of a phosphorylated band at ∼65 kDa (Fig. 5C,D), corresponding to the phosphorylated members of the Src family as reported previously (Yu et al., 1997; Gardoni et al., 2002). 32P incorporation into this band was induced by IL-1β and completely blocked by pretreatment with either lavendustin A or PP2. Src family members Sre activated after Tyr416 phosphorylation in the autocatalytic site (Cooper and MacAuley, 1988). Six minutes of exposure to 0.05 ng/ml IL-1β significantly increased the amount of phosphorylated Tyr416-Src coimmunoprecipitated with the NR2A/B subunits (Fig. 6A,B) as revealed by an anti-phospho-Tyr416 Src antibody. As expected, both lavendustin A and PP2 were able to block Tyr416 phosphorylation. No difference in NR2A/B immunoreactivity was observed among the various treatments (Fig. 6A).
Figure 6.

IL-1β-induced phosphorylation of Src-Tyr-416 and NR2B-Tyr-1472. A, Proteins from hippocampal cell lysates were immunoprecipitated (IP) with a polyclonal antibody raised against NR2A/B. Western blot was done in the immunoprecipitated material using the NR2A/B antibody (top) or the phospho-specific p416Src antibody (bottom). Histograms in B show the quantification of the Tyr416-phosphorylated Src coimmunoprecipitated with the NR2A/B subunits. Data are normalized for NR2A/B immunoreactivity. Values are mean ± SE of two independent experiments (**p < 0.01 IL-1 vs control; §p < 0.05 IL-1 + LAV and IL-1 + PP2 vs IL-1; ANOVA followed by Tukey's test). C, Representative Western blot depicting increased phosphorylation of NR2B-Tyr-1472 after exposure to IL-1 β (bottom). D, Quantification of Tyr-1472 phosphorylation in the various experimental conditions. Data are normalized for NR2A/B immunoreactivity (top) in the respective samples, which did not differ among treatments. Values are mean ± SE (n = 6) obtained from two experiments, each including three independent samples (**p < 0.01 IL-1 vs control; §p < 0.05 IL-1 + LAV and IL-1 + PP2 vs IL-1; ANOVA followed by Tukey's test).
Several tyrosine residues in the C-terminal cytoplasmic region of NR2B have been shown to be phosphorylated by Src family tyrosine kinases in vitro (Nakazawa et al., 2001). Among these, Tyr-1472 is the major phosphorylation site. Using a well characterized antibody to phospho-Tyr-1472 (Nakazawa et al., 2001), we next investigated whether this particular tyrosine phosphosite was modulated by IL-1β treatment. As shown in Figure 6C, IL-1β induced NR2B Tyr-1472 phosphorylation (Fig. 6D), and this effect was blocked by either lavendustin A or PP2. The total amount of NR2B protein was not modified by the various treatments (Fig. 6C).
By using membrane-impermeant biotinylation techniques, we found that the IL1-R1 concentration on the cell surface was not affected by either lavendustin (89.2 ± 14.1%; LAV vs controls) and PP2 (92.4 ± 20.5%; PP2 vs controls) (Fig. 7), therefore excluding a direct role of tyrosine kinase inhibitors on IL1-R1 receptor availability.
Figure 7.

IL-1RI surface expression after lavendustin and PP2 exposure. Hippocampal neurons were preincubated for 30 min with HBSS plus 500 nm lavendustin A or 1 μm PP2 or to HBSS for 30 min (control). IL-1RI surface expression was measured by membrane-impermeant biotinylation techniques. A, Representative Western blot showing IL-1RI cell surface expression in the various experimental conditions. Histograms in B show the quantification of IL-1RI concentrations on cell surface in the various experimental conditions. Data are normalized for IL-1RI concentrations in the total cell lysates. Values are mean ± SE of three independent samples.
To exclude the possibility that the Src family of tyrosine kinases could directly facilitate IL-1RI-mediated signal transduction, we studied the effect of the selective Src family kinases inhibitor PP2 on IL-1β-induced IL6 expression. Hippocampal neurons were pretreated for 30 min with 1 μm PP2 and then exposed for 6 min to 0.05 ng/ml IL-1β. IL-6 mRNA expression was measured 3 hr later by RT-PCR. As shown in Figure 8, IL-1β strongly increased IL-6 mRNA expression in hippocampal neurons and PP2 did not alter this effect.
Figure 8.

Lack of effect of PP2 on IL-1β-induced IL-6 mRNA expression. Hippocampal neurons were preincubated for 30 min with HBSS ± 1 μm PP2 and then exposed to 0.05 ng/ml IL-1β for 6 min. Control neurons were exposed to HBSS for the duration of the experiment. IL-6 mRNA expression was measured 3 hr later by RT-PCR.
IL-1β effect on NMDA-induced neuronal death
We studied whether IL-1β potentiation of NMDA-induced [Ca2+]i increase results in an enhancement of NMDA-mediated death of hippocampal neurons. Treatment of hippocampal neurons with 10 μm NMDA for 3 hr resulted in ∼20% of neuronal death over the next 24 hr (Fig. 9). Pre-exposure of neurons for 6 min to 0.05 ng/ml IL-1β had no toxic effects per se but significantly increased NMDA-induced neuronal death, and this effect was prevented by pretreatment with 500 nm lavendustin A or 1 μm PP2 (Fig. 9). Lavendustin A and PP2 reduced NMDA-triggered neuronal cell death in IL-1β-pretreated neurons below control values, but this effect was not significant.
Figure 9.

IL-1β effect on NMDA-induced neurotoxicity. Hippocampal neurons were preincubated for 30 min with HBSS ± 500 nm lavendustin A or 1 μm PP2 and then exposed to NMDA only or to 0.05 ng/ml IL-1β for 6 min and subsequently to 10 μm NMDA for 3 hr. Control neurons were exposed to HBSS for the duration of the experiment. At the end of this period, neurons were returned to conditioned culture medium for 24 hr. MTT test was performed to assay cell viability. The enhancing effect of IL-1β on NMDA-induced neuronal cell death was prevented by both lavendustin A and PP2. Both lavendustin A and PP2 did not affect cell viability per se (% of control; controls: 100 ± 7.8; lavendustin A: 104 + 9.2; PP2: 101 ± 13.5). Values are mean ± SE of six independent experiments: *p < 0.05 IL-1 versus control; §p < 0.05 IL-1 plus LAV or plus PP2 versus IL-1; ANOVA followed by Tukey's test.
Discussion
The major finding of this study is that IL-1β induces tyrosine phosphorylation of NR2A/B subunits of the NMDA receptor in hippocampal neurons. In accordance with data showing that the activation of this pathway enhances NMDA receptor-mediated Ca2+ fluxes (Wang and Salter, 1994), we observed that (1) concentrations of IL-1β that trigger NMDA receptor phosphorylation also facilitate NMDA-induced [Ca2+]i increase and (2) both lavendustin A, an inhibitor of the tyrosine kinase pathway, and PP2, a more specific inhibitor for the Src family of tyrosine kinase, prevents this effect.
The potentiation of NMDA response occurs via increased Ca2+ entry through the receptor-associated ion channel because MK-801, a blocker that binds only to activated NMDA receptors, inhibited this effect. In addition, AMPA receptors, voltage-dependent Ca2+ channels, intracellular Ca2+ stores, or neurotransmitters released by neuronal depolarization do not appear to play any significant role in mediating the effect of IL-1β, which remained unchanged in the presence of specific blockers of these pathways.
IL-1β-mediated potentiation of the NMDA receptor function persists for several minutes after removal of this cytokine. This evidence therefore suggests that even a transient exposure of neurons to this cytokine may favor long-lasting activation of the NMDAR function. This may have functional relevance for either neuronal excitability or excitotoxicity. Thus, our results suggest that hippocampal neurons exposed to IL-1β are more susceptible to glutamatergic excitation through the NMDA receptor component. This may give insights into the molecular mechanisms underlying several of the in vivo effects exerted by this cytokine in the hippocampus. In particular, the recent observation that IL-1β prolongs seizure activity in vivo and this effect is blocked by a selective NMDA receptor antagonist (Vezzani et al., 1999) is compatible with the present findings. Our results also support previous in vivo evidence suggesting that this cytokine is a pathogenic mediator of ischemic, traumatic, or excitotoxic brain damage in rats, which are pathological conditions that depend primarily on NMDA receptor function (Yamasaki et al., 1995; Loddick and Rothwell, 1996). Thus, we show that concentrations of IL-1β that cause NMDA receptor phosphorylation and potentiate NMDA-induced [Ca2+]i increase also exacerbate NMDA-induced neuronal death.
With regard to the specific molecular pathway that mediates IL-1β effect on NMDA receptor function, we provide evidence for the involvement of the Src family of kinases. Src and Fyn are lead members of a family of protein tyrosine kinases expressed in the CNS that is involved in NR2A/B subunit phosphorylation and the consequent functional upregulation of NMDA receptors (Moon et al., 1994; Kohr and Seeburg, 1996; Lu et al., 1999). Accordingly, lavendustin A and PP2 prevented both IL-1β-induced 32P incorporation into NR2A/B subunits and phosphorylation of the Src family specific NR2B-Tyr-1472 site, and they blocked the enhanced NMDA-dependent Ca2+ influx.
It is unlikely that the Src family of tyrosine kinases may directly facilitate IL-1RI signal transduction thereby modulating IL-1β effect. Thus, Hazeki et al. (2003) reported that PP2 does not affect IL-1 receptor-associated kinase (a serine-threonine kinase) activity after MyD88 recruitment by Toll-like receptors. In addition, we found that PP2 did not affect the expression of IL6 mRNA induced by IL-1β in hippocampal neurons. Thus, the Src family of kinases appears to be selectively involved in the downstream cascade of events resulting in IL-1β potentiation of NMDAR functions.
Activation of tyrosine kinases appears to be a crucial pathway for the occurrence of neuronal cell death in the presence of IL-1β plus NMDA, because lavendustin A and PP2 prevented IL-1β-mediated potentiation of neuronal damage. Accordingly, tyrosine phosphorylation of the NMDA receptor has been previously implicated in exacerbation of neuronal cell loss induced by the human immunodeficiency virus-1 protein Tat (Haughey et al., 2001) or by zinc (Manzerra et al., 2001), and receptor phosphorylation is increased after ischemic brain damage (Takagi et al., 1997).
It should be noted that this pathway is also involved in physiological functions mediated by NMDA receptors, such as the induction of LTP (Salter, 1998) and the regulation of gene transcription (Takasu et al., 2002). Thus, tyrosine phosphorylation of the NMDA receptor appears to be relevant for regulating both physiological and pathological processes involving glutamatergic neurotransmission, and IL-1β may modulate both of these pathways.
Recently it has been shown that the different subcellular localization of NMDA receptors can be relevant in governing the ultimate neuronal fate. Thus, NMDA synaptic complexes promote gene expression and cell survival, whereas NMDA extrasynaptic complexes are involved in cell death (Hardingham et al., 2002). We observed that (1) the effects of IL-1 β on the NMDA receptors are mediated via IL-1RI and (2) IL-1RI is expressed with NR2A/B receptor subunits in hippocampal neurons, suggesting a direct functional cross-talk between IL1β, IL-1RI, and the NMDA receptor. Although we did not address the subcellular localization of IL-1RI and NR2A/B in this paper, it will be worth studying whether IL-1β modulates a pathological rather than a physiological event by selectively enhancing the function of extrasynaptic or synaptic NMDA receptors.
Our finding that IL-1β enhances NMDA-receptor function in vitro is only in apparent contradiction with previous data reporting an inhibitory effect of this cytokine on NMDA receptor-mediated EPSP (Coogan and O'Connor, 1997) and on excitotoxic neuronal cell death (Strijbos and Rothwell, 1995; Pringle et al., 2001). Thus, the effects of IL-1β on neurons depend on several factors, including the functional state and the type of neurons, the concentration of IL-1β, and the duration of time that the neuron is exposed to this cytokine. We indeed found a dual effect of IL-1β on NMDA receptor function depending on its concentration. Thus, relatively low concentrations of IL-1β (0.01-0.1 ng/ml) increased NMDA-receptor function, whereas 1 ng/ml had an inhibitory effect on NMDA-mediated increases in [Ca2+]i. These findings are compatible with previous evidence showing that dose-response effects of IL-1β often display a bell-shaped curve (Rothwell and Hopkins, 1995; Allan and Rothwell, 2001).
There is a major difference in the length of time that the neurons were pre-exposed to IL-1β in our experimental conditions showing exacerbation of neuronal death (i.e., 6 min) and previous evidence reporting inhibitory effects of IL-1β on neuronal damage (i.e., 24 hr). Thus, exposure of neurons to IL-1β for 24 hr before NMDA application or induction of injury may have induced the synthesis and release from neurons of various factors (i.e., anti-inflammatory cytokines, neuropeptides, growth factors) that in turn are responsible for changing neuronal susceptibility to damage. In accordance, Strijbos and Rothwell (1995) suggest that IL-1β reduction of excitatory amino acid-induced neurodegeneration in vitro may be in part mediated via nerve growth factor.
In summary, our findings show a novel molecular mechanism by which IL-1β, a proinflammatory cytokine produced by both glia and neurons in various CNS pathological conditions, may significantly enhance NMDA receptor function. Pharmacological intervention aimed at interfering with this cytokine pathway may thus represent an innovative approach to control excessive [Ca2+]i increase in neurons and the consequent cascade of events leading to neuronal hyperexcitability and irreversible cell injury.
Footnotes
This work was supported by the First-University of Milan, Fondo Integrato Ricerca di Base-Ministero Istruzione Universitá e Ricerca 2002, Telethon Onlus Foundation Grant GP0285/01, and Fondazione Mariani, Citizens United for Research in Epilepsy.
Correspondence should be addressed to Dr. Barbara Viviani, Department of Pharmacological Sciences, University of Milan, Via Balzaretti 9, 20133 Milan, Italy. E-mail: barbara.viviani@unimi.it.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238692-09$15.00/0
References
- Allan SM, Rothwell NJ ( 2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2: 734-744. [DOI] [PubMed] [Google Scholar]
- Allan SM, Parker LC, Collins B, Davies R, Luheshi GN, Rothwell NJ ( 2000) Cortical cell death induced by IL-1 is mediated via actions in the hypothalamus of the rat. Proc Natl Acad Sci USA 97: 5580-5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cul J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D ( 2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415: 793-798. [DOI] [PubMed] [Google Scholar]
- Coogan A, O'Connor JJ ( 1997) Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. NeuroReport 8: 2107-2110. [DOI] [PubMed] [Google Scholar]
- Cooper JA, MacAuley A ( 1998) Potential positive and negative autoregulation of p60-src by intermolecular autophosphorylation. Proc Natl Acad Sci USA 85: 4232-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denizot F, Lang R ( 1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89: 271-277. [DOI] [PubMed] [Google Scholar]
- Dinarello CA ( 1996) Biologic basis for interleukin-1 in disease. Blood 87: 2095-2147. [PubMed] [Google Scholar]
- Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M ( 1998) Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem 71: 1733-1741. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Kamal A, Bellone C, Biessels GJ, Ramakers GM, Cattabeni F, Gispent WH, Di Luca M ( 2002) Effects of streptozotocin-diabetes on the hippocampal NMDA receptor complex in rats. J Neurochem 80: 438-447. [DOI] [PubMed] [Google Scholar]
- Grazia de Simoni M, Imeri L ( 1998) Cytokine-neurotransmitter interactions in the brain. Biol Signals Receptors 7: 33-44. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Browning MD ( 2001) Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. J Neurochem 76: 737-744. [DOI] [PubMed] [Google Scholar]
- Hagan P, Poole S, Bristow AF, Tilders F, Silverstein FS ( 1996) Intracerebral NMDA injection stimulates production of interleukin-1beta in perinatal rat brain. J Neurochem 67: 2215-2218. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H ( 2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405-414. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD ( 2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78: 457-467. [DOI] [PubMed] [Google Scholar]
- Hazeki K, Masuda N, Funami K, Sukenobu N, Matsumoto M, Akira S, Takeda K, Seya T, Hazeki O ( 2003) Toll-like receptor-mediated tyrosine phosphorylation of paxillin via MyD88-dependent and -independent pathways. Eur J Immunol 33: 740-747. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S ( 1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31-108. [DOI] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH ( 1996) Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol (Lond) 492: 445-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Huganir RL ( 1995) Differential tyrosine phosphorylation of N-methyl-d-aspartate receptor subunits. J Biol Chem 270: 20036-20041. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Hell JW ( 1997) Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem 272: 12107-12115. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ ( 1996) Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab 16: 932-940. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF ( 1999) G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci 2: 331-338. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Behrens MM, Canzoniero LM, Wang XQ, Heidinger V, Ichinose T, Yu SP, Choi DW ( 2001) Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci USA 98: 11055-11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon IS, Apperson ML, Kennedy MB ( 1994) The major tyrosinephosphorylated protein in the postsynaptic density fraction is N-methyl-d-aspartate receptor subunit 2B. Proc Natl Acad Sci USA 91: 3954-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T ( 2001) Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J Biol Chem 276: 693-699. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB ( 1996) Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 271: 31670-31678. [DOI] [PubMed] [Google Scholar]
- Pringle AK, Niyadurupola N, Johns P, Anthony DC, Iannotti F ( 2001) Interleukin-1beta exacerbates hypoxia-induced neuronal damage, but attenuates toxicity produced by simulated ischaemia and excitotoxicity in rat organotypic hippocampal slice cultures. Neurosci Lett 305: 29-32. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Hopkins SJ ( 1995) Cytokines and the nervous system II: actions and mechanisms of action. Trends Neurosci 18: 130-136. [DOI] [PubMed] [Google Scholar]
- Salter MW ( 1998) Src, N-methyl-d-aspartate (NMDA) receptors, and synaptic plasticity. Biochem Pharmacol 56: 789-798. [DOI] [PubMed] [Google Scholar]
- Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO ( 1998) A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA 95: 7778-7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijbos PJ, Rothwell NJ ( 1995) Interleukin-1 beta attenuates excitatory amino acid-induced neurodegeneration in vitro: involvement of nerve growth factor. J Neurosci 15: 3468-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taishi P, Bredow S, Guha-Thakurta N, Obal Jr F, Krueger JM ( 1997) Diurnal variations of interleukin-1 beta mRNA and beta-actin mRNA in rat brain. J Neuroimmunol 75: 69-74. [DOI] [PubMed] [Google Scholar]
- Takagi N, Shinno K, Teves L, Bissoon N, Wallace MC, Gurd JW ( 1997) Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J Neurochem 69: 1060-1065. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kapas L, Fang J, Seyer JM, Wang Y, Krueger JM ( 1996) An interleukin-1 receptor fragment inhibits spontaneous sleep and muramyl dipeptide-induced sleep in rabbits. Am J Physiol 271: R101-108. [DOI] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME ( 2002) Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 295: 491-495. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG ( 1999) Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci 19: 5054-5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, De Simoni MG, Sperk G, Andell-Jonsson S, Lundkvist J, Iverfeldt K, Bartfai T ( 2000) Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci USA 97: 11534-11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Corsini E, Binaglia M, Galli CL, Marinovich M ( 2001) Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience 107: 51-58. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW ( 1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369: 233-235. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K ( 1995) Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 26: 676-680. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil II GJ, Salter MW ( 1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275: 674-678. [DOI] [PubMed] [Google Scholar]