

Abstract
A member of the neurotrophin family, brain-derived neurotrophic factor (BDNF) regulates neuronal survival and differentiation during development. Within the adult brain, BDNF is also important in neuronal adaptive processes, such as the activity-dependent plasticity that underlies learning and memory. These long-term changes in synaptic strength are mediated through alterations in gene expression. However, many of the mechanisms by which BDNF is linked to transcriptional and translational regulation remain unknown. Recently, the transcription factor NFATc4 (nuclear factor of activated T-cells isoform 4) was discovered in neurons, where it is believed to play an important role in long-term changes in neuronal function. Interestingly, NFATc4 is particularly sensitive to the second messenger systems activated by BDNF. Thus, we hypothesized that NFAT-dependent transcription may be an important mediator of BDNF-induced plasticity. In cultured rat CA3-CA1 hippocampal neurons, BDNF activated NFAT-dependent transcription via TrkB receptors. Inhibition of calcineurin blocked BDNF-induced nuclear translocation of NFATc4, thus preventing transcription. Further, phospholipase C was a critical signaling intermediate between BDNF activation of TrkB and the initiation of NFAT-dependent transcription. Both inositol 1,4,5-triphosphate (IP3)-mediated release of calcium from intracellular stores and activation of protein kinase C were required for BDNF-induced NFAT-dependent transcription. Finally, increased expression of IP3 receptor 1 and BDNF after neuronal exposure to BDNF was linked to NFAT-dependent transcription. These results suggest that NFATc4 plays a crucial role in neurotrophin-mediated synaptic plasticity.
Keywords: BDNF, TrkB, NFATc4, IP3R1, hippocampus, CREB, neurotrophin, NGF, NT-3, NT-4/5
Introduction
A critical feature of the nervous system is its ability to modulate synaptic transmission in response to particular spatial and temporal patterns of neuronal firing. In fact, activity-dependent synaptic plasticity is thought to underlie such diverse processes as the refinement of neuronal connections during development to cell survival, chronic pain, drug addiction, and learning and memory. One mediator of these neuronal adaptive processes is brain-derived neurotrophic factor (BDNF), whose expression and release are regulated by neuronal activity (Zafra et al., 1990, 1991; Ghosh et al., 1994; Kruttgen et al., 1998). Although known primarily for its critical role in shaping neuronal connections during development (Huang and Reichardt, 2001), BDNF also triggers long-lasting modulation of synapses within the adult brain (Lewin and Barde, 1996; Schuman, 1999; Schinder and Poo, 2000; Xu et al., 2000; Guillin et al., 2001; Poo, 2001; Pezet et al., 2002).
The highest levels of BDNF expression in the mammalian brain are found within the hippocampus (Ayer-LeLievre et al., 1988; Ernfors et al., 1990; Hofer et al., 1990; Murer et al., 2001), an area associated with spatial learning and memory. As such, BDNF has been implicated in inducing long-term modifications in both hippocampal synaptic structure and function. BDNF increases the number of dendritic spines, translating to an increased number of hippocampal excitatory synapses (Tyler and Pozzo-Miller, 2001; Tolwani et al., 2002). BDNF exposure also generates long-lasting enhancement in synaptic neurotransmission (Kang and Schuman, 1995, 1996; Levine et al., 1995; Kang et al., 1997). Moreover, the induction of long-term potentiation is impaired in BDNF knock-out mice (Korte et al., 1995; Patterson et al., 1996; Pozzo-Miller et al., 1999).
To establish many lasting changes in synaptic efficacy, alterations in gene expression must first occur (Frey et al., 1988; Bailey et al., 1996; Dubnau and Tully, 1998). Despite the considerable evidence demonstrating that BDNF exerts long-term changes in neuronal function, very little is known regarding the mechanisms by which this neurotrophin induces gene expression. Thus far, only the transcription factor cAMP response element-binding protein (CREB) has been linked to BDNF-induced changes in neuronal plasticity (Finkbeiner et al., 1997; Minichiello et al., 2002; Ying et al., 2002). Nonetheless, it is becoming increasingly clear that diverse forms of synaptic plasticity require the coordinated activation of multiple transcription factors (West et al., 2002; Deisseroth et al., 2003; Dolmetsch, 2003).
The transcription factor NFATc4 (nuclear factor of activated T-cells isoform 4) may be another mediator of BDNF-induced plasticity. Recently identified within the hippocampus, NFATc4 initiates gene expression after periods of heightened synaptic activity and is hypothesized to play a crucial role in shaping long-term changes in cell excitability (Graef et al., 1999). After increases in intracellular calcium, NFATc4 is dephosphorylated by calcineurin (CaN), thereby promoting translocation of the transcription factor from the cytosol into the nucleus (Beals et al., 1997; Rao et al., 1997). Once in the nucleus, NFATc requires the cooperative binding of a phosphorylated nuclear partner to initiate transcription. Because multiple proteins may serve this role, they are generically termed NFATn. In hippocampal neurons, NFATn is activated by protein kinase C (PKC) (Graef et al., 1999).
Intriguingly, BDNF signaling appears ideally suited to activate NFAT-dependent transcription. On binding to TrkB, BDNF activates several signaling cascades, resulting in both elevations in intracellular calcium and activation of PKC. Consequently, the aims of this study were threefold: (1) to determine whether BDNF induces NFAT-dependent transcription; (2) to delineate the major signaling pathways by which BDNF activates the NFATc4-NFATn transcriptional complex; and (3) to identify potential genes regulated by BDNF through activation of NFAT-dependent transcription.
Materials and Methods
Cell culture. Hippocampal pyramidal neurons were cultured from 1- to 2-d-old rat pups as previously described (Mermelstein et al., 2000), using a protocol approved by the Animal Care and Use Committee at the University of Minnesota. Chemicals were obtained from Sigma (St. Louis, MO) unless otherwise stated. After decapitation, the CA3-CA1 region of the hippocampus was isolated in ice-cold modified HBSS containing 20% fetal bovine serum (FBS; Hyclone, Logan, UT) and (in mm): 4.2 NaHCO3 and 1 HEPES, pH 7.35, 300 mOsm. The tissue was washed and digested for 5 min in a trypsin solution (type XI, 10 mg/ml) containing (in mm): 137 NaCl, 5 KCl, 7 Na2HPO4, and 25 HEPES, with 1500 U of DNase, pH 7.2, 300 mOsm. After additional washing, the tissue was dissociated using Pasteur pipettes of decreasing diameters. The cell suspension was pelleted twice to remove contaminants, plated on 10 mm coverslips (treated previously with Matrigel to promote adherence; BD Biosciences, San Jose, CA), and incubated for 15 min at room temperature. Two milliliters of minimum essential media (MEM; Invitrogen, Grand Island, NY) containing (in mm): 28 glucose, 2.4 NaHCO3, 0.0013 transferrin (Calbiochem, La Jolla, CA), 2 glutamine, and 0.0042 insulin, with 1% B-27 supplement (Invitrogen) and 10% FBS, pH 7.35, 300 mOsm, were added to each coverslip. Twenty-four hours later, 1 ml of media was replaced with a similar solution containing 4 μm cytosine 1-β-d-arabinofuranoside and 5% FBS. Seventy-two hours later, 1 ml of media was again replaced with the modified MEM solution containing 5% FBS. Media solutions contained 2 μg/ml gentamicin (Invitrogen) to prevent bacterial growth.
Drugs. The following drugs were used: d(-)AP-5 (AP-5, 25 μm; Tocris, Ellisville, MO); recombinant human BDNF (1-500 ng/ml; Alomone Labs, Jerusalem, Israel); bisindolylmaleimide I (500 nm; Calbiochem); cyclosporin A (CsA, 1 μg/ml); FK506 (200 ng/ml; Calbiochem); K252a (100 nm; Alomone Labs); recombinant human neurotrophin 3 and neurotrophin-4/5 (NT-3 and NT-4/5, 100 ng/ml; Alomone Labs); nifedipine (5 μm); tetrodotoxin (TTX, 1-2 μm); thapsigargin (1 μm; Calbiochem); and U73122 (1 μm).
Luciferase-based gene reporter assays. Cultured neurons were transfected ∼7 d in vitro (d.i.v.) with a luciferase-based reporter (1 μg of DNA/coverslip) of NFAT-dependent transcription (Shaw et al., 1988; Graef et al., 1999) using a calcium-phosphate method previously described that results in >95% of the transfected cells being neurons (Deisseroth et al., 1998; Weick et al., 2003). Although response magnitudes varied across experiments, BDNF consistently increased NFAT-dependent transcription (p < 0.001). To control for nonspecific effects on transcription, a constitutively active pGL3 luciferase vector was transfected (0.5 μg of DNA/coverslip) into neurons (Promega, Madison, WI). In another study, neurons were transfected (1 μg of DNA/coverslip) with a reporter construct in which luciferase expression was dependent on ∼500 bp of the murine inositol 1,4,5-triphosphate receptor 1 (IP3R1) promoter (a gift from A. Genazzani, University of Cambridge, Cambridge, UK). After transfection, cells were placed in serum-free DMEM (Invitrogen) containing an insulin-transferrin-selenium-A (ITS) supplement (Invitrogen) and 1% B-27 supplement, pH 7.35, 300 mOsm. The DMEM, ITS, and B-27 also contained 2 μg/ml gentamicin to prevent bacterial growth and 2 μm TTX to minimize NFAT-dependent transcription triggered by synaptic activity. Twenty-four hours later, unstimulated neurons were readministered TTX; all other groups were treated with TTX in addition to the specified drug(s) outlined in Results. Notably, treatment groups receiving intracellular signaling inhibitors were pretreated with the inhibitor for 30 min before neurotrophin stimulation. Approximately 16 hr after stimulation, cells were lysed and assayed for luciferase expression using a standard luminometer (Monolight 3010; PharMingen, San Diego, CA). Each treatment group within a single experiment comprised 7-10 coverslips. Furthermore, all experiments were replicated to verify the consistency of the results. Statistical differences between groups were comparable across each experiment.
Immunocytochemistry. Immunocytochemistry procedures closely followed those previously described (Mermelstein et al., 2001). Before a 15 min exposure to BDNF (100 ng/ml), cultured neurons (7-9 d.i.v.) were preincubated for 3 hr at room temperature in a Tyrode solution containing 1 μm TTX. Where stated, FK506 and CsA were present 15 min before and during BDNF treatment. After stimulation, cells were fixed for 20 min with ice-cold 4% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in Dulbecco's PBS containing 4 mm EGTA. Cells were washed three times with PBS and then permeabilized for 5 min with 0.1% Triton X-100 (VWR Scientific, West Chester, PA). After another PBS wash, cells were incubated for 30 min at 37°C in a PBS-based blocking solution containing 1% BSA and 2% goat serum (Jackson ImmunoResearch, West Grove, PA). Afterward, cells were left overnight at 4°C in a block solution containing a polyclonal antibody directed against NFATc4 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) and a monoclonal antibody directed against α-tubulin (Molecular Probes, Eugene, OR). The next day, cells were washed with PBS and incubated for 1 hr at 37°C with FITC- and CY3-conjugated secondary antibodies (diluted in block solution) recognizing NFATc4 and α-tubulin, respectively (Jackson ImmunoResearch). After the final PBS wash, coverslips were mounted on cover slides using the antiquenching reagent Citifluor (Ted Pella, Redding, CA). Acquisition of the fluorescent intensities (n = 15-21 cells per group) was performed using a Bio-Rad (Hercules, CA) MRC 1024 confocal workstation. Data were quantified using Metamorph software version 4.6 (Universal Imaging, Downingtown, PA). Briefly, using the α-tubulin staining as a guide, a confocal section through the approximate midline (z-axis) of the neuron was obtained. Two regions of interest (ROIs) were drawn: one of the nucleus and the other of the surrounding soma, avoiding the interface between the two. The ROIs were then transferred to the NFATc4-stained section. The ratio of the average fluorescence within each ROI was then determined.
Semiquantitative single-cell reverse transcription-PCR. Single-cell reverse transcription (RT)-PCR was performed essentially as described previously (Song et al., 1998; Mermelstein et al., 1999; Song et al., 2000; Tkatch et al., 2000; Yan et al., 2001) with a few modifications. To remove RNase contamination, electrode glass was heated to 200°C for at least 4 hr and allowed to return to room temperature before being pulled with a micropipette puller (P-97; Sutter Instruments, Novato, CA). Where indicated, neurons were transfected with an enhanced green fluorescent protein (EGFP)-tagged, constitutively nuclear NFATc4 construct (EGFP-NFATc4 S → A; provided by I. Graef, K. Stankunas, and G. Crabtree, Howard Hughes Medical Institute, Chevy Chase, MD, and Stanford University, Stanford, CA). Control neurons were exposed to the transfection conditions in the absence of plasmid DNA. Twenty-four hours later, mock-transfected or individual pyramidal neurons exhibiting EGFP-NFATc4 S → A fluorescence were aspirated into electrodes containing ∼5.0 μl of diethylpyrocarbonate (DEPC)-treated water. The contents of each electrode were then ejected into a thin-walled PCR tube (MJ Research, Waterton, MA) placed on ice containing (in μl): 5.0 of DEPC-treated water, 0.5 of RNaseOut (40 U/μl), 0.5 of dithiothreitol (DTT, 0.1 m), and 1.0 of oligo-dT (0.5 μg/ml). The tube was heated to 70°C for 10 min to linearize the mRNA and then returned to ice for at least 1 min. Linearized mRNA was reverse-transcribed into single-stranded cDNA with the addition of (in μl): 1.0 of SuperScript II reverse transcriptase (200 U/μl), 2.0 of 10× PCR buffer (200 mm Tris-HCl and 500 mm KCl), 2.0 of MgCl2 (50 mm), 1.0 of dNTPs (10 μm), 0.5 of RNaseOut, and 1.5 of DTT, followed by heating of the reaction mixture to 42°C for 50 min. Incubation at 70°C for 15 min terminated the RT reaction. To eliminate RNA, 1.0 μl of RNase H (2 U/μl) was added to each tube before incubation at 37°C for 20 min. All RT reagents were obtained from Invitrogen (Carlsbad, CA).
Single-stranded cDNA was amplified by PCR through the addition of 1.0, 2.0, 3.0, 4.0, or 8.0 μl of RT template to a thin-walled PCR tube containing (in μl): 5.0 of 10× PCR buffer, 2.5 of MgCl2 (25 μm), 1.0 of dNTPs (25 mm), 2.5 of upper primer for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or BDNF (20 μm), 2.5 of lower primer (20 μm), 28.0-35.0 of autoclaved water, and 0.5 of Taq polymerase (5 U/μl). All PCR reagents were obtained from Invitrogen. PCR amplification was performed using a peltier thermal cycler (PTC-200; MJ Research). The thermal cycling program for BDNF amplification was 94°C for 0.75 min, 50°C for 0.75 min, and 72°C for 1.5 min for 45 cycles. The program for GAPDH amplification was 94°C for 0.75 min, 55°C for 0.75 min, and 72°C for 1.5 min for 40 cycles. PCR products were separated by electrophoresis in 1.5% agarose gels and visualized with ethidium bromide. Gels were digitally imaged and processed with an Eastman Kodak Co. (Rochester, NY) 1D system, version 3.5.3, to determine whether amplicons of the expected size were present. Amplicons from single neurons were also sequenced to verify that the PCR reactions were specific for BDNF and GAPDH.
The upper and lower primer sequences for BDNF (GenBank accession number D10938) were 5′-GAG AAA GTC CCG GTA TCA AA-3′ (nucleotides 2631-2650) and 5′-CCA GCC AAT TCT CTT TTT-3′ (nucleotides 2796-2813), yielding a predicted product of 182 bp. The primers for GAPDH (GenBank accession number AF106860) were 5′-ACC ACA GTC CAT GCC ATC AC-3′ (nucleotides 566-588) and 5′-TCC ACC ACC CTG TTG CTG TA-3′ (nucleotides 998-1017), yielding a predicted size of 451 bp. The primers were synthesized by Invitrogen.
To ensure that extraneous DNA did not contaminate the single-cell RT-PCR experiments, water was used as a PCR template. To confirm that genomic DNA was not being amplified, reverse transcriptase was omitted from one RT reaction. Furthermore, as a positive control, cDNA generated from whole hippocampal tissue (see below) was used as a PCR template. Each of these controls reliably yielded the expected results.
Extraction of total hippocampal RNA. To generate cDNA from whole tissue, hippocampi from 2-d-old rats were homogenized in an RNase-free tube containing Trizol reagent (1 ml/50-100 mg of tissue; Invitrogen). After incubation at room temperature for 5 min, 200 μl of chloroform/ml of Trizol was added to the homogenate, and the tube was shaken vigorously. After an additional incubation at 4°C for 5 min, the solution was centrifuged (12,000 rpm) at 4°C for 15 min. The aqueous phase was transferred to another Eppendorf tube containing 500 μl of isopropyl alcohol/ml of Trizol, shaken, and incubated on ice for 15 min. The solution was centrifuged (12,000 rpm) at 4°C for 15 min. The supernatant was discarded, and the pellet was washed with 80% ethanol. After an additional centrifugation (12,000 rpm) at 4°C for 8 min, the ethanol was removed, and the pellet was allowed to air dry. The RNA was then dissolved in 200 μl of DEPC-treated water, and ∼2 μg of RNA was added to a tube containing 2 μlof10× DNase I reaction buffer, 2 μl of DNase I (1 U/μl; Invitrogen), and DEPC-treated water for a total volume of 20 μl. The reaction mixture was incubated at room temperature for 15 min, and then 2 μl of 25 mm EDTA was added. After a 10 min incubation at 65°C, hippocampal mRNA was reverse-transcribed using methods described above.
Western blot. Cultured hippocampal neurons (∼8 d.i.v.) were placed in DMEM, ITS, B-27, and, where indicated, BDNF (100 ng/ml) for 48 hr and then lysed in radioimmunoprecipitation assay buffer containing a protease inhibitor mixture (Complete Mini EDTA-free; Roche Molecular Biochemicals, Indianapolis, IN). Lithium dodecyl sulfate loading buffer and β-mercaptoethanol were added to the lysate (25 and 10% of the total lysate volume, respectively), and the mixture was heated to 95°C for 5 min. Cellular protein was separated on a 4-12% Bis-Tris gel (Mini-Sub electrophoresis cell; Bio-Rad) and transferred to a nitrocellulose membrane. The membrane was then blocked at room temperature for 1 hr in a Tris-buffered saline (TBS) solution containing 10% milk and 1% BSA. Overnight incubation with an antibody against BDNF (1:1000; Chemicon, Temecula, CA), extracellular signal-regulated kinase (ERK, 1:15,000; Upstate Biotechnology, Lake Placid, NY), or IP3R1 (1:1000; a gift from I. Bezprozvanny, University of Texas Southwestern Medical Center, Dallas, TX) diluted in TBS containing (in %): 1 milk, 1 BSA, and 0.1 Tween 20 was performed at 4°C. The following day, the membrane was washed with 0.1% Tween 20 and TBS and incubated with a secondary antibody conjugated to horseradish peroxidase (1:25,000; Pierce, Rockford, IL). After incubation in a chemiluminescent substrate (Super Signal; Pierce), the membrane was exposed to X-Omat XB-1 film (Kodak). Each experiment was repeated two times to verify results. ERK was used as a loading control because its expression is not altered with BDNF signaling (Patterson et al., 2001; Sato et al., 2001; Minichiello et al., 2002; Ying et al., 2002).
Statistics. Luciferase assays and immunocytochemistry experiments were analyzed using ANOVAs (F values) and Fisher's protected least significant difference post hoc (p values) tests. The single-cell RT-PCR data were analyzed using a Kruskal-Wallis H test. p < 0.05 were considered a priori as significant.
Results
BDNF induces NFAT-dependent transcription
To determine whether BDNF signaling activates NFAT-dependent transcription, cultured hippocampal CA3-CA1 pyramidal neurons were transfected with a luciferase-based gene expression reporter construct (Shaw et al., 1988; Graef et al., 1999). Stimulation with BDNF (1, 10, 100, and 500 ng/ml) significantly increased NFAT-mediated luciferase expression in a concentration-dependent manner (p < 0.001, except for 1 ng/ml, p < 0.05; Fig. 1A). The EC50 for activation of NFAT-dependent transcription was 3.2 ng/ml (120 pM). Because 100 ng/ml (3.7 nm) BDNF was the lowest concentration found to achieve maximal NFAT responses, this amount was used in the remainder of the experiments. To verify that the effect of BDNF on the luciferase expression was dependent on NFAT, a separate group of neurons were transfected with a constitutively active reporter construct. Under these conditions, BDNF application failed to result in a change in luciferase expression [control, 131,285 ± 8893 reflective light units (RLU); BDNF, 144,242 ± 6895 RLU; p > 0.05], demonstrating that BDNF influences on NFAT-dependent transcription were not attributable to differences in cell survival, general transcription, or a nonspecific effect on luciferase translation or stability.
Figure 1.

BDNF activates NFAT-dependent transcription. A, BDNF significantly increased NFAT-mediated luciferase expression in a concentration-dependent manner (F = 14.41; p < 0.05 for 1 ng/ml BDNF; p < 0.001 at all other concentrations). B, BDNF induction of NFAT-dependent transcription relies on the activation of calcineurin because pretreatment with FK506 (200 ng/ml) and CsA (1 μg/ml) significantly (F = 37.90; **p < 0.001) attenuated luciferase expression. Data represent mean ± SEM.
A key regulatory step in the signaling pathway that leads to NFAT-dependent transcription is the activation of CaN. CaN dephosphorylates NFATc4, causing its rapid translocation from the cytosol into the nucleus, whereupon it can bind DNA and stimulate transcription via cooperation with its nuclear partner, NFATn. Therefore, by preventing NFATc4 dephosphorylation, BDNF-induced NFAT-dependent transcription should be eliminated. Consistent with this hypothesis, pretreatment with the CaN inhibitors FK506 and CsA abolished the BDNF-induced increase in NFAT-dependent transcription (p < 0.001; Fig. 1B).
To examine the activity of endogenous NFATc4 further, we directly visualized the subcellular distribution of the transcription factor by pairing immunocytochemistry with confocal microscopy. Neurons were double-labeled with α-tubulin to clearly identify the nuclear-cytoplasmic boundary. In unstimulated pyramidal neurons, the distribution of immunolabeled NFATc4 was predominantly cytoplasmic (Fig. 2A). Interestingly, in neurons stimulated with BDNF, the distribution of immunolabeled NFATc4 was profoundly altered, localizing primarily in the nucleus (Fig. 2B). As predicted, pretreatment with FK506 and CsA blocked BDNF-induced nuclear translocation of NFATc4 (Fig. 2C). Quantification of the confocal images revealed an increase (p < 0.001) in the nuclear-to-cytoplasm fluorescence ratio of immunolabeled NFATc4 in neurons treated with BDNF; the effect was abolished by FK506 and CsA (Fig. 2D). Collectively, these results establish that BDNF induces NFAT-dependent transcription within hippocampal pyramidal neurons.
Figure 2.

BDNF triggers the rapid nuclear translocation of NFATc4. A, Confocal images of immunolabeled NFATc4 (top) and α-tubulin (bottom) in cultured hippocampal neurons. In unstimulated cells (No Stim), the majority of NFATc4 is localized within the cytoplasm. B, Fifteen minutes after BDNF exposure, NFATc4 is observed primarily within the nucleus. C, Pretreatment with FK506 and CsA blocks the nuclear translocation of NFATc4 after BDNF stimulation. D, Quantification of the confocal images demonstrates that BDNF significantly increases the nuclear-to-cytoplasm ratio of immunolabeled NFATc4 (F = 45.00; **p < 0.001).
TrkB mediates BDNF-induced activation of NFAT-dependent transcription
BDNF signals through TrkB, a high-affinity receptor-tyrosine kinase, and p75, a low-affinity pan-neurotrophin receptor (Meakin and Shooter, 1992; Bothwell, 1995). Based on the dose-response data (see Fig. 1), BDNF stimulation of NFAT-dependent transcription is consistent with a TrkB-mediated event. To determine whether the actions of BDNF on NFAT-dependent transcription were in fact mediated through TrkB, neurons were treated with the tyrosine kinase inhibitor K252a. Application of K252a (p < 0.001) attenuated the BDNF-generated increase in NFAT-mediated gene expression (Fig. 3A). However, because of the nonselective nature of K252a, we confirmed the necessity of TrkB for induction of NFAT-dependent gene transcription by stimulating neurons with two other TrkB ligands: NT-3 and NT-4/5. Although TrkB binds BDNF and NT-4/5 with equal affinity, it has a lower affinity for NT-3 (Klein et al., 1991), the cognate neurotrophin for TrkC (Lamballe et al., 1991). BDNF and NT-4/5 were equally effective in stimulating NFAT-dependent transcription (Fig. 3B). NT-3 also significantly increased NFAT-dependent transcription (p < 0.05), although to a lesser extent (p < 0.001) than the other two neurotrophins (Fig. 3B). These data strongly suggest that induction of NFAT-dependent transcription by BDNF is mediated via TrkB signaling.
Figure 3.

BDNF activates NFAT-dependent transcription via TrkB receptors. A, Pretreatment with the tyrosine kinase inhibitor K252a (100 nm) abolished the BDNF-induced increase in NFAT-dependent transcription (F = 14.61; **p < 0.001). B, Consistent with a TrkB mediated event, BDNF and NT-4/5 (100 ng/ml) were equally efficacious in stimulating NFAT-mediated luciferase expression; NT-3 (100 ng/ml) was less effective (F = 30.94; **p < 0.001).
L-type calcium channels and NMDA receptors are not required for BDNF-induced NFAT-dependent transcription
Previously, we have shown that calcium entry specifically through L-type calcium channels initiates NFAT-dependent transcription (Graef et al., 1999). However, during many forms of synaptic neurotransmission, NMDA receptors are responsible for generating the membrane depolarizations necessary for activation of these voltage-gated calcium channels and therefore are critical for triggering gene expression themselves (Graef et al., 1999; Nakazawa and Murphy, 1999; Rajadhyaksha et al., 1999). Previous work has demonstrated that BDNF signaling can result in the enhancement of NMDA receptor activity (Levine et al., 1998). Thus, to determine whether BDNF-induced NFAT-dependent transcription is mediated by facilitation of NMDA receptors, leading to increased L-type calcium channel openings, BDNF-stimulated hippocampal neurons were pretreated with either the NMDA receptor antagonist AP-5 or the L-type calcium channel blocker nifedipine. Interestingly, BDNF induction of NFAT-dependent transcription was unaffected by either drug (Fig. 4), implicating a second mechanism by which activation of NFAT-dependent transcription can occur within neurons.
Figure 4.

NMDA receptors and L-type calcium channels are not required for BDNF activation of NFAT-dependent transcription. Pretreatment with the NMDA receptor antagonist AP-5 (25 μm; A) or the L-type calcium channel blocker nifedipine (5 μm; B) had no effect on BDNF-induced NFAT-dependent transcription.
Activation of phospholipase C is critical for BDNF-induced NFAT-dependent transcription
On BDNF binding, the dimerized TrkB receptor activates several signaling pathways, including the phospholipase C (PLC) pathway (Kaplan and Miller, 2000). Activation of PLC cleaves phosphotidylinositol-4,5-biphosphate to generate IP3 and diacylglycerol (DAG). IP3 binds to intracellular receptors in the endoplasmic reticulum, leading to increases in intracellular calcium, whereas DAG activates PKC. Initiation of NFAT-dependent transcription requires both increases in intracellular calcium, to activate NFATc4 via CaN, and PKC activation, which stimulates NFATn (Graef et al., 1999). Therefore, we hypothesized that PLC signaling underlies BDNF-mediated activation of NFAT-dependent transcription. Consistent with this hypothesis, pretreatment with the PLC inhibitor U73122 reduced (p < 0.001) NFAT-dependent transcription mediated by BDNF (Fig. 5A).
Figure 5.
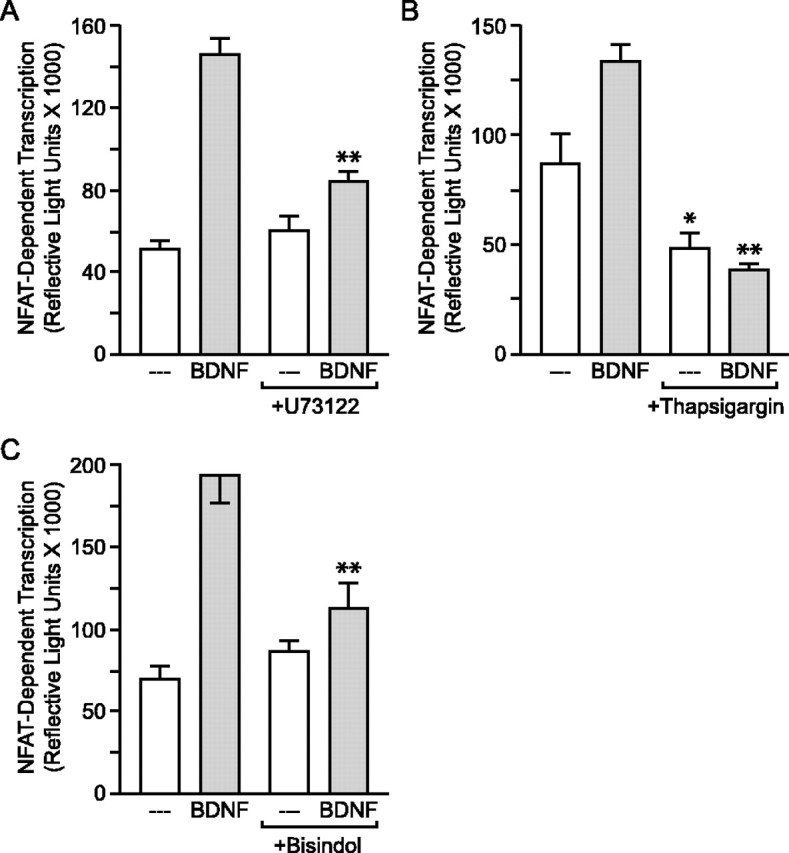
BDNF activation of NFAT-dependent transcription is dependent on PLC signaling. A, Pretreatment with the PLC inhibitor U73122 (1 μm) significantly (F = 53.21; **p < 0.001) attenuated BDNF-induced NFAT-mediated luciferase expression. B, Depletion of calcium from intracellular stores with thapsigargin (1 μm) significantly reduced both basal (F = 28.35; *p < 0.05) and BDNF-stimulated (**p < 0.001) NFAT-dependent transcription. C, BDNF-mediated NFAT-dependent transcription was also significantly attenuated (F = 17.53; **p < 0.001) by the PKC inhibitor bisindolylmaleimide (500 nm).
In the next set of experiments, we separately examined the two distinct signaling pathways downstream of PLC activation with regard to their role in triggering NFAT-dependent transcription. Depleting intracellular stores of calcium via application of thapsigargin abolished BDNF-induced NFAT-dependent transcription (p < 0.001; Fig. 5B). Furthermore, pretreatment with the PKC inhibitor bisindolylmaleimide also (p < 0.001) attenuated BDNF-induced NFAT-dependent transcription (Fig. 5C). Thus, the activation of both branches of the PLC signaling pathway appears important for BDNF-mediated NFAT-dependent transcription.
BDNF regulation of IP3R1
The genes regulated by the NFATc class of transcription factors have primarily been determined outside the nervous system. As such, there is a paucity of information concerning the neuronal genes regulated by NFATc4. Previous experiments strongly suggested that the gene encoding the neuronal-specific IP3R1 is regulated by NFATc4 (Carafoli et al., 1999; Genazzani et al., 1999; Graef et al., 1999). Thus, to determine whether BDNF triggers IP3R1 expression, neurons were transfected with a luciferase-based reporter construct driven by the IP3R1 promoter (a gift from A. Genazzani). After BDNF stimulation, a significant (p < 0.001) increase in luciferase expression occurred (Fig. 6A). Consistent with an NFAT-regulated event, pretreatment with FK506 and CsA (p < 0.001) attenuated the BDNF-induced increase in gene expression. Furthermore, to assess whether these changes in gene expression reflected an increase in protein, Western blot studies were performed using an antibody directed against IP3R1 (provided by I. Bezprozvanny). Cell lysates from neurons treated with BDNF exhibited increased expression of IP3R1 (Fig. 6B). The effect was reduced by FK506 and CsA, suggesting the BDNF-generated increase in IP3R1 expression is in part mediated through NFAT-dependent transcription.
Figure 6.

BDNF and CaN regulation of IP3R1 expression: a role for NFAT-dependent transcription. A, BDNF increased (F = 15.53; p < 0.001) luciferase expression driven by the IP3R1 promoter. The effect was significantly (**p < 0.001) attenuated by pretreatment with FK506 and CsA. B, Similar to mRNA expression, BDNF-mediated increases in IP3R1 protein are partially dependent on CaN. No Stim, Unstimulated cells.
NFAT-dependent transcription leads to BDNF expression
A positive feedback mechanism in which BDNF administration leads to the upregulation of BDNF expression has been described (Saarelainen et al., 2001), and although regulation of the BDNF gene has not yet been fully characterized because of its multiple promoter regions (and thus the use of luciferase reporter assays impractical), it does contain several putative NFAT binding sites (Timmusk et al., 1993; Nakayama et al., 1994). Thus, to determine whether BDNF mRNA is upregulated through NFATc4, as an alternative approach, we performed a semiquantitative method of single-cell RT-PCR (scRT-PCR). Detection of BDNF cDNA was compared between mock-transfected neurons and those transfected with an EGFP-tagged NFATc4 construct, rendered constitutively nuclear by several serine-to-alanine substitutions (EGFP-NFATc4 S → A). Previous experiments determined that expression of EGFP-NFATc4 S → A potentiates NFAT-dependent transcription (I. A. Graef and P. G. Mermelstein, unpublished observations). Thus, if NFATc4 regulates transcription of the gene encoding BDNF, an increase in BDNF mRNA should be detected in transfected cells.
To determine whether the mRNA extracted from individual neurons was successfully reverse-transcribed, 15% of the total cellular cDNA obtained from each cell was used as a PCR template using primers specific to GAPDH (predicted size, 451 bp). In neurons in which the PCR product for GAPDH was observed (29 of 30), the threshold for detecting BDNF cDNA was determined by serially diluting the remaining template for use in PCR reactions containing primers specific to BDNF (predicted size, 182 bp). Figure 7A displays the results from a mock-transfected control neuron, as well as a cell transfected with EGFP-NFATc4 S → A. In the control cell, the PCR product for BDNF was only observed when using 40% of the total cDNA template. In contrast, the BDNF amplicon was observed in the EGFP-NFATc4 S → A-transfected cell when only 5% of the cDNA template was used. In approximately one-third of the neurons, regardless of experimental condition, PCR products for BDNF were not detected (10 of 29). However, in the remaining cells, expression of EGFP-NFATc4 S → A significantly decreased the threshold for detection of BDNF (p < 0.001; Fig. 7B).
Figure 7.

BDNF expression is regulated through NFAT-dependent transcription. A, Data taken from scRT-PCR experiments. In the control neuron, 40% of the total cellular cDNA template was required to detect BDNF expression. In comparison, in the example neuron expressing a constitutively nuclear NFATc4 construct, amplicons for BDNF were observed when using 40, 20, 10, and 5% of the cDNA template. GAPDH was used as a positive control for RT. B, In neurons where BDNF cDNA was detected (n = 19 of 29), there was a shift (H = 11.27; p < 0.001) in the threshold for detection in cells expressing the constitutively nuclear NFATc4 construct. ND, Not detected. C, BDNF-induced increases in BDNF protein are dependent on CaN, suggestive of an NFAT-mediated event. No Stim, Unstimulated cells.
As a final experiment, we determined whether BDNF stimulation would result in an increase in BDNF protein. Sixteen hours after BDNF application, cell lysates from hippocampal cultures exhibited an increase in BDNF expression. The effect was not attributed to detection of the exogenous BDNF administration because the increase in BDNF protein was abolished by FK506 and CsA (Fig. 7C). The results from the scRT-PCR and Western blot experiments implicate that BDNF, acting through NFATc4, increases BDNF mRNA and protein expression.
Discussion
Through alterations in gene expression, BDNF regulates a variety of activity-dependent neuroadaptive processes, ranging from cell survival and differentiation during development to modification of synaptic connections in the adult. Here we delineate a novel pathway whereby BDNF signaling, through activation of the transcription factor NFATc4, triggers the induction of gene expression. Specifically, BDNF was found to activate NFAT-dependent transcription in a concentration-dependent, calcineurin-sensitive manner. The primary mechanism underlying BDNF activation of the NFAT transcriptional complex involved TrkB-mediated PLC signaling to IP3 and PKC. Finally, expression of IP3R1 and BDNF were regulated by BDNF through activation of NFAT-dependent transcription. Collectively, these results support a model wherein activation of NFAT-dependent transcription underlies a significant component of BDNF-induced gene expression and long-term changes in neuroplasticity.
In a previous study examining the regulation of NFAT-dependent transcription in brain, Graef et al. (1999) found that the L-type voltage-gated calcium channel activates both partners of the NFAT transcriptional complex. Indeed, after brief periods of heightened synaptic activity, calcium entry through L-type calcium channels not only triggered CaN-mediated dephosphorylation of NFATc4 but also activated NFATn through stimulation of PKC. In addition, activation of L-type calcium channels led to the inactivation of glycogen synthase kinase 3β (GSK-3β), a kinase responsible for the phosphorylation and nuclear export of activated NFATc4.
BDNF-mediated activation of NFAT-dependent transcription occurred independent of L-type calcium channels, instead relying on PLC signaling. Interestingly, this second pathway revealed to stimulate NFAT-dependent transcription in neurons more closely matches the intracellular signaling cascades originally characterized as critical mediators of NFAT activation in the immune system (Rao et al., 1997; Zhu and McKeon, 2000; Crabtree and Olson, 2002). In T-lymphocytes, where NFAT-dependent transcription was first described, activation of antigen receptors leads to PLC signaling. This in turn leads to the rapid nuclear import of NFATc after IP3-mediated calcium release from intracellular stores, with concurrent activation of NFATn via DAG-activated PKC.
One potential difference between NFATc signaling in neurons and nonexcitable cells is that for gene expression to occur in non-neuronal tissue, sustained increases in intracellular calcium concentrations are required (Dolmetsch et al., 1997, 1998). These prolonged calcium elevations are achieved via calcium release-activated calcium (CRAC) channels (Rao et al., 1997; Crabtree, 1999). Because NFAT-dependent transcription in neurons does not require sustained calcium elevations (Graef et al., 1999), it is unknown whether the neuronal equivalents to CRAC channels play a role in NFAT-mediated gene expression.
In addition to PLC, TrkB couples to two other major signaling pathways via activation of phosphatidylinositol-3 kinase (PI3K) and the adaptor Src homology 2 and collagen protein (SHC) (Segal and Greenberg, 1996; Gottschalk et al., 1999; Huang and Reichardt, 2001). Interestingly, both signaling pathways may play a secondary role in modulating NFAT-dependent transcription. First, after dephosphorylation, NFATc4 remains active until it is phosphorylated and exported from the nucleus. As mentioned, one protein kinase shown to promote the export of NFATc4 is GSK-3β. Intriguingly, the effector of PI3K signaling is protein kinase B (or Akt), which is known to inhibit the activity of GSK-3β (Cross et al., 1995; Pap and Cooper, 1998). As such, activation of PI3K should prolong the nuclear localization of NFATc4 and thus potentiate NFAT-dependent transcription. Second, phosphorylated SHC is coupled to a series of protein kinases in the Ras-MAPK (mitogen-activated protein kinase) pathway. There is well documented cross-talk between Ras-MAPK and PKC signaling, as well as Ras-MAPK activation of NFATn (Ueda et al., 1996; Schreiber and Crabtree, 1997; Isakov and Altman, 2002). Thus, activation of SHC may also enhance NFAT-dependent transcription. Future experiments will test whether these two important signaling pathways have secondary effects on NFAT-dependent transcription.
In the search for potential genes regulated by NFAT-dependent transcription, several pieces of information have pointed to the neuron-specific IP3R1 gene as a likely candidate. IP3R1 protein expression is regulated by L-type calcium channels and CaN (Carafoli et al., 1999; Genazzani et al., 1999; Graef et al., 1999), stimuli known to activate NFAT-dependent transcription. There are also several putative NFATc binding sites within the IP3R1 promoter (Furutama et al., 1996), and NFAT transcription factors will cooperatively bind to these DNA sites (Graef et al., 1999). Moreover, mice lacking NFATc4 exhibit attenuated IP3R1 expression (I. A. Graef, personal communication). Here we not only extend the implication that IP3R1 expression is regulated by NFAT-dependent transcription but also provide evidence that BDNF stimulates this signaling pathway. Specifically, BDNF increased IP3R1 promoter-driven gene expression, as well as IP3R1 protein expression in a calcineurin-sensitive manner. The implication is that BDNF-induced NFAT-dependent transcription plays a role in synaptic plasticity because expression of IP3R1 has been shown to influence enduring changes in cell excitability, such as long-term depression (Kasono and Hirano, 1995; Reyes and Stanton, 1996; Inoue et al., 1998). Notably, inhibition of NFAT signaling only partially reduced BDNF-induced expression of IP3R1. This is likely attributable to neurotrophin activation of multiple transcription factors, several which may bind and activate the IP3R1 promoter independent of NFAT.
BDNF is known to regulate its own expression (Saarelainen et al., 2001). Within hippocampal neurons, overexpression of constitutively nuclear NFATc4 increased the relative abundance of BDNF mRNA, determined using a semiquantitative scRT-PCR approach. Furthermore, expression of BDNF was strongly dependent on CaN. These data are consistent with BDNF regulation of BDNF expression through NFATc4. Previous studies in cortical neurons have demonstrated that BDNF expression is also regulated by the transcription factors CREB and calcium regulatory factor (Timmusk et al., 1993; Shieh et al., 1998; Tao et al., 1998, 2002). This is not surprising because the rat BDNF gene contains four distinct promoter regions (Timmusk et al., 1993; Nakayama et al., 1994). The presence of multiple promoters in the BDNF gene allows for differential mechanisms of activation, as well as tissue-specific expression within the CNS (Falkenberg et al., 1992; Metsis et al., 1993; Kokaia et al., 1994; Timmusk et al., 1995). Consequently, differing patterns of neuronal activity might stimulate BDNF expression through use of distinct transcription factors. Because of its central role in a diverse array of activity-dependent processes, it is not surprising to find a variety of mechanisms in place to regulate the expression of BDNF. Of significance, putative NFAT binding sites are located within each of the four BDNF promoters. (To examine the global net effect of NFATc4 activation on BDNF expression, the PCR primers were specifically designed not to distinguish among the different BDNF transcripts.) In contrast to IP3R1 expression, inhibition of NFAT signaling led to a complete ablation of BDNF expression, suggesting that although multiple transcription factors may regulate expression of the neurotrophin, concurrent NFAT activation may be necessary for gene expression.
The regulation of BDNF and IP3R1 by NFAT-dependent transcription is intriguing because both proteins participate in a signaling pathway that ultimately results in their expression. This positive feedback loop provides a means by which synaptic neurotransmission may be enhanced. There may also exist an endogenous negative feedback loop, whereby NFAT proteins regulate the expression of one isoform of the calcineurin inhibitor, modulatory calcineurin interacting protein (MCIP1.4), which in humans is located within the Down syndrome critical region (Fuentes et al., 1997). Previous work has demonstrated that calcineurin activity increases MCIP1.4 expression (Yang et al., 2000), presumed via activation of NFAT-dependent transcription, because several NFAT sites have been identified within the MCIP1.4 promoter (Rothermel et al., 2003). Multiple influences on cell function may shift the balance between these positive and negative feedback loops initiated by NFAT-mediated gene expression, resulting in either increased or decreased cell excitability or even pathological states. One example may be Alzheimer's disease, in which MCIP1 expression is increased severalfold (Ermak et al., 2001), and BDNF expression levels are diminished (Phillips et al., 1991; Holsinger et al., 2000). In the future, it will also be critically important to understand how this balance of NFAT-mediated feedback loops is adjusted within “normal” ranges of gene expression and the impact on brain function.
Another area that will require further attention is the extent to which NFATc4 mediates other forms of neurotrophin-mediated synaptic plasticity. In addition to BDNF, the neurotrophin family includes nerve growth factor (NGF), NT-3, and NT-4/5. Each of these neurotrophins signals through a high-affinity receptor-tyrosine kinase that activates signaling cascades similar to TrkB. As shown, both NT-4/5 and NT-3 activate NFAT-dependent transcription; it remains to be seen whether NGF can also signal to NFATc4. Thus, it is plausible that NFATc4 mediates, at least in part, the gene expression initiated by these other neurotrophins in diverse areas of the nervous system to affect such processes as development, regeneration, drug addiction, and chronic pain. Consistent with this hypothesis are recent data demonstrating that neurotrophin- and netrin-induced axonal outgrowth is compromised in NFATc knock-out mice (Graef et al., 2003).
The NFATc class of transcription factors was originally characterized in lymphocytes, playing an essential role in the adaptation of the immune system in response to antigens (Yoshida et al., 1998). Recently, the NFATc family of transcription factors has been found in a variety of other cell types, regulating such diverse activities as musculoskeletal development (Chin et al., 1998), cardiac valve formation (de la Pompa et al., 1998; Ranger et al., 1998), chondrogenesis (Ranger et al., 2000; Tomita et al., 2002), adipogenesis (Ho et al., 1998), and patterning of the vasculature (Graef et al., 2001) and implicated in cardiac hypertrophy (Molkentin et al., 1998; van Rooij et al., 2002). Add to this impressive list recent data suggesting that NFATc4 is involved in synaptic plasticity within the brain. Here we provide evidence that NFAT-dependent transcription underlies some of the effects of neurotrophins within the nervous system.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS41302 and a Whitehall Foundation grant (P.G.M.). R.D.G. is supported by NIH Training Grant DA07234. We thank Dr. Virginia Seybold, Dr. Stan Thayer, and Dr. Mark Thomas for scientific input, Dr. Armando Genazzani for the IP3R1 luciferase reporter, Dr. Ilya Bezprozvanny for the IP3R1 antibody, and Bryan Becklund, Marissa Iden, Ann Isaksen, Kathryn Klammer, Sidney Kuo, Leigh Ann Mrotek, and Jason Weick for technical support.
Correspondence should be addressed to Paul G. Mermelstein, Department of Neuroscience, University of Minnesota, 6-145 Jackson Hall, 321 Church Street, Southeast, Minneapolis, MN 55455. E-mail: pmerm@umn.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238125-10$15.00/0
References
- Ayer-LeLievre C, Olson L, Ebendal T, Seiger A, Persson H ( 1988) Expression of the beta-nerve growth factor gene in hippocampal neurons. Science 240: 1339-1341. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER ( 1996) Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA 93: 13445-13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals CR, Clipstone NA, Ho SN, Crabtree GR ( 1997) Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev 11: 824-834. [DOI] [PubMed] [Google Scholar]
- Bothwell M ( 1995) Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci 18: 223-253. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Genazzani A, Guerini D ( 1999) Calcium controls the transcription of its own transporters and channels in developing neurons. Biochem Biophys Res Commun 266: 624-632. [DOI] [PubMed] [Google Scholar]
- Chin ER, Olson EN, Richardson JA, Yang Q, Humphries C, Shelton JM, Wu H, Zhu W, Bassel-Duby R, Williams RS ( 1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev 12: 2499-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR ( 1999) Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell 96: 611-614. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN ( 2002) NFAT signaling: choreographing the social lives of cells. Cell [Suppl] 109: S67-S79. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA ( 1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW ( 1998) Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 392: 182-186. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW ( 1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198-202. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW ( 2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13: 354-365. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R ( 2003) Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE 2003: PE4. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI ( 1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386: 855-858. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS ( 1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933-936. [DOI] [PubMed] [Google Scholar]
- Dubnau J, Tully T ( 1998) Gene discovery in Drosophila: new insights for learning and memory. Annu Rev Neurosci 21: 407-444. [DOI] [PubMed] [Google Scholar]
- Ermak G, Morgan TE, Davies KJA ( 2001) Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem 276: 38787-38794. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Wetmore C, Olson L, Persson H ( 1990) Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 5: 511-526. [DOI] [PubMed] [Google Scholar]
- Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N ( 1992) Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett 138: 153-156. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME ( 1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031-1047. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H ( 1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57-65. [DOI] [PubMed] [Google Scholar]
- Fuentes JJ, Pritchard MA, Estivill X ( 1997) Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics 44: 358-361. [DOI] [PubMed] [Google Scholar]
- Furutama D, Shimoda K, Yoshikawa S, Miyawaki A, Furuichi T, Mikoshiba K ( 1996) Functional expression of the type 1 inositol 1,4,5-trisphosphate receptor promoter-lacZ fusion genes in transgenic mice. J Neurochem 66: 1793-1801. [DOI] [PubMed] [Google Scholar]
- Genazzani AA, Carafoli E, Guerini D ( 1999) Calcineurin controls inositol 1,4,5-trisphosphate type 1 receptor expression in neurons. Proc Natl Acad Sci USA 96: 5797-5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME ( 1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618-1623. [DOI] [PubMed] [Google Scholar]
- Gottschalk WA, Jiang H, Tartaglia N, Feng L, Figurov A, Lu B ( 1999) Signaling mechanisms mediating BDNF modulation of synaptic plasticity in the hippocampus. Learn Mem 6: 243-256. [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR ( 1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401: 703-708. [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR ( 2001) Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863-875. [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR ( 2003) Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113: 657-670. [DOI] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P ( 2001) BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 411: 86-89. [DOI] [PubMed] [Google Scholar]
- Ho IC, Kim JH, Rooney JW, Spiegelman BM, Glimcher LH ( 1998) A potential role for the nuclear factor of activated T cells family of transcriptional regulatory proteins in adipogenesis. Proc Natl Acad Sci USA 95: 15537-15541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA ( 1990) Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 9: 2459-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M ( 2000) Quantification of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res 76: 347-354. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF ( 2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K ( 1998) Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18: 5366-5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov N, Altman A ( 2002) Protein kinase C(theta) in T cell activation. Annu Rev Immunol 20: 761-794. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM ( 1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658-1662. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM ( 1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: 1402-1406. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM ( 1997) Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19: 653-664. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD ( 2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Kasono K, Hirano T ( 1995) Involvement of inositol trisphosphate in cerebellar long-term depression. NeuroReport 6: 569-572. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M ( 1991) The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 66: 395-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Metsis M, Kokaia M, Bengzon J, Elmer E, Smith ML, Timmusk T, Siesjo BK, Persson H, Lindvall O ( 1994) Brain insults in rats induce increased expression of the BDNF gene through differential use of multiple promoters. Eur J Neurosci 6: 587-596. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T ( 1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruttgen A, Moller JC, Heymach Jr JV, Shooter EM ( 1998) Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci USA 95: 9614-9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamballe F, Klein R, Barbacid M ( 1991) trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 66: 967-979. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR ( 1995) Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA 92: 8074-8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR ( 1998) Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235-10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Barde YA ( 1996) Physiology of the neurotrophins. Annu Rev Neurosci 19: 289-317. [DOI] [PubMed] [Google Scholar]
- Meakin SO, Shooter EM ( 1992) The nerve growth factor family of receptors. Trends Neurosci 15: 323-331. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Foehring RC, Tkatch T, Song WJ, Baranauskas G, Surmeier DJ ( 1999) Properties of Q-type calcium channels in neostriatal and cortical neurons are correlated with β subunit expression. J Neurosci 19: 7268-7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW ( 2000) Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci 20: 266-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW ( 2001) Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci USA 98: 15342-15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsis M, Timmusk T, Arenas E, Persson H ( 1993) Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc Natl Acad Sci USA 90: 8802-8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M ( 2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36: 121-137. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN ( 1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93: 215-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R ( 2001) Brain-derived neurotrophic factor in the control human brain and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 63: 71-124. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Gahara Y, Kitamura T, Ohara O ( 1994) Distinctive four promoters collectively direct expression of brain-derived neurotrophic factor gene. Brain Res Mol Brain Res 21: 206-218. [DOI] [PubMed] [Google Scholar]
- Nakazawa H, Murphy TH ( 1999) Activation of nuclear calcium dynamics by synaptic stimulation in cultured cortical neurons. J Neurochem 73: 1075-1083. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM ( 1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273: 19929-19932. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER ( 1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137-1145. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER ( 2001) Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32: 123-140. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, McMahon SB ( 2002) BDNF: a neuromodulator in nociceptive pathways? Brain Res Rev 40: 240-249. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW ( 1991) BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron 7: 695-702. [DOI] [PubMed] [Google Scholar]
- Poo MM ( 2001) Neurotrophins as synaptic modulators. Nat Rev Neurosci 2: 24-32. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B ( 1999) Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci 19: 4972-4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadhyaksha A, Barczak A, Macías W, Leveque JC, Lewis SE, Konradi C ( 1999) L-type Ca(2+) channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci 19: 6348-6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH ( 1998) The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392: 186-190. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Gerstenfeld LC, Wang J, Kon T, Bae H, Gravallese EM, Glimcher MJ, Glimcher LH ( 2000) The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J Exp Med 191: 9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG ( 1997) Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707-747. [DOI] [PubMed] [Google Scholar]
- Reyes M, Stanton PK ( 1996) Induction of hippocampal long-term depression requires release of Ca 2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci 16: 5951-5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel BA, Vega RB, Williams RS ( 2003) The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med 13: 15-21. [DOI] [PubMed] [Google Scholar]
- Saarelainen T, Vaittinen S, Castren E ( 2001) trkB-receptor activation contributes to the kainate-induced increase in BDNF mRNA synthesis. Cell Mol Neurobiol 21: 429-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S ( 2001) NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci 21: 3797-3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinder AF, Poo M ( 2000) The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 23: 639-645. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR ( 1997) Immunophilins, ligands, and the control of signal transduction. Harvey Lectures 91: 99-114. [PubMed] [Google Scholar]
- Schuman EM ( 1999) Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol 9: 105-109. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME ( 1996) Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci 19: 463-489. [DOI] [PubMed] [Google Scholar]
- Shaw J-P, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR ( 1988) Identification of a putative regulator of early T cell activation genes. Science 241: 202-205. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A ( 1998) Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20: 727-740. [DOI] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ ( 1998) Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci 18: 3124-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Surmeier DJ ( 2000) Adenosine receptor expression and modulation of Ca(2+) channels in rat striatal cholinergic interneurons. J Neurophysiol 83: 322-332. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME ( 1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20: 709-726. [DOI] [PubMed] [Google Scholar]
- Tao X, West AE, Chen WG, Corfas G, Greenberg ME ( 2002) A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 33: 383-395. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H ( 1993) Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10: 475-489. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Lendahl U, Funakoshi H, Arenas E, Persson H, Metsis M ( 1995) Identification of brain-derived neurotrophic factor promoter regions mediating tissue-specific, axotomy-, and neuronal activity-induced expression in transgenic mice. J Cell Biol 128: 185-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatch T, Baranauskas G, Surmeier DJ ( 2000) Kv4.2 mRNA abundance and A-type K + current amplitude are linearly related in basal ganglia and basal forebrain neurons. J Neurosci 20: 579-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolwani RJ, Buckmaster PS, Varma S, Cosgaya JM, Wu Y, Suri C, Shooter EM ( 2002) BDNF overexpression increases dendrite complexity in hippocampal dentate gyrus. Neuroscience 114: 795-805. [DOI] [PubMed] [Google Scholar]
- Tomita M, Reinhold MI, Molkentin JD, Naski MC ( 2002) Calcineurin and NFAT4 induce chondrogenesis. J Biol Chem 277: 42214-42218. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD ( 2001) BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci 21: 4249-4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S ( 1996) Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem 271: 23512-23519. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Doevendans PA, de Theije CC, Babiker FA, Molkentin JD, de Windt LJ ( 2002) Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem 277: 48617-48626. [DOI] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG ( 2003) Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci 23: 3446-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME ( 2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3: 921-931. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF ( 2000) The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20: 6888-6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Flores-Hernandez J, Surmeier DJ ( 2001) Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience 103: 1017-1024. [DOI] [PubMed] [Google Scholar]
- Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS ( 2000) Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res 87: e61-e68. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR ( 2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22: 1532-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Nishina H, Takimoto H, Marengere LE, Wakeham AC, Bouchard D, Kong YY, Ohteki T, Shahinian A, Bachmann M, Ohashi PS, Penninger JM, Crabtree GR, Mak TW ( 1998) The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity 8: 115-124. [DOI] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D ( 1990) Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J 9: 3545-3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Castren E, Thoenen H, Lindholm D ( 1991) Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA 88: 10037-10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, McKeon F ( 2000) Nucleocytoplasmic shuttling and the control of NF-AT signaling. Cell Mol Life Sci 57: 411-420. [DOI] [PMC free article] [PubMed] [Google Scholar]