

Abstract
Neurofibromatosis type 1 (NF1) patients are predisposed to learning disabilities, macrocephaly, and brain tumors as well as abnormalities on magnetic resonance imaging that are postulated to result from abnormal myelination. Here we show that Nf1+/- spinal cords in adult mice have more than twofold-increased numbers of NG2+ progenitor cells. Nf1-/- embryonic spinal cords have increased numbers of Olig2+ progenitors. Also, cultures from Nf1 mutant embryos with hemizygous and biallelic Nf1 mutations have dramatically increased numbers of CNS oligodendrocyte progenitor cells. In medium that allows growth of neuroepithelial cells and glial progenitors, mutant cells hyper-respond to FGF2, have increased basal and FGF-stimulated Ras-GTP, and fail to accumulate when treated with a farnesyltransferase inhibitor. Cell accumulation results in part from increased proliferation and decreased cell death. In contrast to wild-type cells, Nf1-/- progenitors express the glial differentiation marker O4 while retaining expression of the progenitor marker nestin. Nf1 mutant progenitors also abnormally coexpress the glial differentiation markers O4 and GFAP. Importantly, Nf1-/- spinal cord-derived oligodendrocyte progenitors, which are amplified 12-fold, retain the ability to form oligodendrocytes after in vivo transplantation. The data reveal a key role for neurofibromin and Ras signaling in the maintenance of CNS progenitor cell pools and also suggest a potential role for progenitor cell defects in the CNS abnormalities of NF1 patients.
Keywords: NF1, progenitor, oligodendrocyte, FGF, brain, Ras
Introduction
Neuroepithelial (NEP) cells give rise to neurons, astrocytes, and oligodendrocytes in the CNS. The lineage relationships among CNS progenitor cells and their differentiated progeny are under intense investigation (Temple and Alvarez-Buylla, 1999; Sauvageot and Stiles, 2002). Little is known about signaling cascades that regulate CNS progenitor cells in general and oligodendrocyte progenitors in specific, in vivo, or cell culture. Markers such as NG2 and Olig2 that identify oligodendrocyte progenitors in embryos can also be expressed by neuronal progenitors and in postnatal rodent NG2+ cells that are multipotent (Miller et al., 1999; Rao, 1999; Rowitch et al., 2002; Belachew et al., 2003). Adult NG2+ cells are present in uninjured CNS and multiple sclerosis lesions and can remyelinate after injury (Redwine and Armstrong, 1998; Chang et al., 2000; Watanabe et al., 2002). PDGF and FGF2 stimulate proliferation and inhibit differentiation in NG2+ cells in vivo that develop into oligodendrocytes (Armstrong et al., 2002; Frost et al., 2003; Oh et al., 2003) and drive oligodendrocyte progenitor growth in vitro (McKinnon et al., 1990; Mujtaba et al., 1999).
Neurofibromin is a tumor suppressor that is a GTPase-activating protein (GAP) for Ras (for review, see Donovan et al., 2002) and acts as a negative regulator of Ras signaling. In many cell types, loss of neurofibromin is correlated with increased GTP-bound active Ras. Neurofibromin is widely expressed in the developing brain (Daston and Ratner, 1992). In the adult brain, neurofibromin is present in some neurons and oligodendrocytes (Daston et al., 1992; Nordlund et al., 1993). After brain injury, astrocytes upregulate neurofibromin (Giordano et al., 1996). Neurofibromin is of particular interest to brain function. Humans with NF1 gene mutations are predisposed to learning disabilities, macrocephaly, and abnormalities on T2-weighted magnetic resonance (MR) imaging postulated to be focally severe patches of a widespread myelin disorder (DiPaolo et al., 1995; Ozonoff, 1999; Wang et al., 2000; Eastwood et al., 2001; North and Ratner, 2002). Macrocephaly results from enlarged white matter tracts (Steen et al., 2001). Pilocytic astrocytomas occur in ∼25% of children with NF1, and cells in these benign tumors express the oligodendroglial lineage marker PEN5 (Listernick et al., 1997; Li et al., 2001). Astrogliosis is a feature of NF1 patient brains (Nordlund et al., 1995). Thus, NF1 patients may have altered CNS glia, but glial lineages have not been evaluated in NF1 disease models. Neurofibromin may be important in progenitor cells, because hematopoietic progenitors from Nf1 mutant mice are hypersensitive to cytokines resulting from defective Ras and phosphoinositide-3 kinase signaling (Bollag et al., 1996; Largaespada et al., 1996; Zhang et al., 1998; Birnbaum et al., 2000).
Mice that completely lack Nf1 die in utero at or before embryonic day (E) 13 (Brannan et al., 1994; Jacks et al., 1994). Mice with hemizygous Nf1 mutations show profound brain dysfunction including astrogliosis (Gutmann et al., 1999; Rizvi et al., 1999; Bajenaru et al., 2001; Zhu et al., 2001) and learning defects (Silva et al., 1997). Here we show that the development of the oligodendrocyte lineage is abnormal in cells from mice with one or two mutant Nf1 alleles. We suggest that brain dysfunction in NF1 results from effects on progenitor cell pools and demonstrate a key role for Ras signaling in oligodendrocyte progenitors.
Materials and Methods
Animals and genotyping
C57BL/6 wild-type female mice (breeders) were obtained from Harlan Bioproducts for Science (Indianapolis, IN). The Nf1 gene was targeted in mice (Brannan et al., 1994). For genotyping, DNA was isolated, and the targeted allele was determined by PCR as described previously (Brannan et al., 1994). Nf1+/- mice had been back-crossed at least 10 generations onto the C57BL/6 background at the time of these experiments. Wild-type, Nf1+/-, and Nf1-/- embryos were obtained 12.5 d after mating Nf1+/- C57BL/6 males with Nf1+/- C57BL/6 females.
Immunohistochemistry for NG2
Wild-type and Nf1 animals (three per genotype) were perfused with 0.9% saline followed by 2% paraformaldehyde, 0.01 m sodium meta-periodate, and 0.1 m lysine fixative (Wu et al., 2000). The spinal cord was dissected out, postfixed in the same fixative overnight, and then cryoprotected overnight in 20% sucrose. Forty micrometer thick free-floating coronal serial sections, at the level of the cervical enlargement, were processed for immunohistochemistry using a biotinylated goat anti-rabbit secondary and the ABC-DAB method for visualization (Vector Laboratories, Burlingame, CA). Rabbit anti-NG2 (Chemicon, Temecula, CA) antibody was used at a dilution of 1:1000. NG2+ cells were counted in gray and white matter at 200× under bright-field optics.
Immunohistochemistry for Olig2
Wild-type, Nf1+/-, and Nf1-/- E12.5 embryos (three per genotype) were removed and fixed overnight at 4°C in 4% paraformaldehyde in PBS. Embryos were then cryoprotected overnight in 20% sucrose. Ten micrometer thick cryostat coronal serial sections, at the level of the cervical enlargement, were processed for immunohistochemistry using a biotinylated goat anti-rabbit secondary (Jackson ImmunoResearch, West Grove, PA) and the ABC-DAB method for visualization (Vector Laboratories). Rabbit anti-Olig2 (a kind gift from D. Rowitch and C. Stiles, Dana-Farber Cancer Institute, Boston, MA) was used at a dilution of 1:2000. Olig2+ cells were counted at 400× under bright-field optics.
Cell cultures
Glial restricted precursor cells.Twelve-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ) were double coated with fibronectin (Becton Dickinson) and laminin (Becton Dickinson). Fibronectin (250 μg/ml) was applied to the wells and immediately withdrawn, and the plates were allowed to dry for 1 hr before coating with laminin (20 μg/ml) overnight at 4°C (Rao and Mayer-Proschel, 1997). Spinal cords were dissected from E12.5 mouse embryos using sharpened number 5 forceps in Leibovitz's L-15 (Invitrogen, Rockville, MD) medium. Spinal cords, with dorsal root ganglia removed, were incubated at 37°C in 0.25% trypsin (Invitrogen) for 30 min with agitation. Cords were subsequently washed by placing them in DMEM (Invitrogen) with 10% fetal bovine serum (FBS) and penicillin-streptomycin and centrifuging at 800 rpm for 5 min at room temperature (RT). The supernatant was then aspirated and the pellet was resuspended in 0.5 ml of NEP cell media (Kalyani et al., 1997) (see below) by gently triturating with a glass Pasteur pipette. Laminin was aspirated from the wells and replaced with 1.5 ml of NEP medium just before the addition of resuspended cells. Cells were cultured at a density of one cord per five wells and maintained at 37°C in 7.5% CO2 with growth factors added or omitted as described in this study.
NEP medium. Based on the Bottenstein and Sato N2 medium, NEP medium contains DMEM-F12 (Invitrogen), N2 additives (progesterone, putresine, selenium, insulin, transferrin) plus complex B27 supplements (Invitrogen), 1% BSA, FGF2 (25 ng/ml, or at doses specified below; R & D Systems, Minneapolis, MN), and 10% chick embryo extract (Invitrogen). Growth factors and medium were replaced every 3 d. In some experiments, PDGFAA (10 ng/ml; R & D Systems) and farnesyltransferase inhibitor (1 μm; L744,832; Merck, Darmstadt, Germany) were added as specified in this study.
Colony counts. Four random fields (1.9 mm) were selected from each well and marked 1 d after plating. Colonies of progenitor-like cells were identified by morphology (small, phase-dark cells), counted, and grouped by colony size. Colony sizes were as follows: <40 cells (small), 40 -150 (medium), and >150 (large). Counts were performed daily at days 1-5 after plating.
Ras activation assay. Cells from wild-type, Nf1+/-, and Nf1-/- embryos were cultured in 60 mm culture dishes, as described above, in the presence of FGF2 (25 ng/ml) until generally confluent (7 d). FGF2 was then removed from the media for 24 hr. One-half of the dishes were then stimulated with FGF2 (25 ng/ml) for 10 min. The cells were immediately processed using the Ras activation assay kit (catalog #17-218; Upstate Biotechnology, Lake Placid, NY) according to the instructions of the manufacturer.
Immunocytochemistry
For nonexpanded marker analysis, E12.5 mouse spinal cords were dissociated and prepared as described above and plated directly onto eight-well RS-treated Labtek II chamber slides (Nalge Nunc International, Naperville, IL) at a density of 5000 cells per well, fixed with 4% paraformaldehyde in PBS at 24 hr, and then labeled as described below. For marker analysis of expanded spinal cord cultures 5 d after plating, cultures were treated with a nonenzymatic cell dissociation medium (Sigma, St. Louis, MO) for 15 min at 37°C. With a glass Pasteur pipette, the medium was gently washed over the cells to dislodge them from the dish. The cells were washed in DMEM plus 10% FBS by centrifugation at 800 rpm for 5 min. The medium was aspirated, and the pellet was resuspended in 0.5 ml of NEP media with FGF2 plus PDGFAA. Cell number was determined on a hemacytometer, and the cells were diluted to a density of 10,000 cells per milliliter. Fibronectin-laminin-coated eight-well RS-treated Labtek II chamber slides were prepared as described above. Five thousand cells were placed in each well and maintained in 7.5% CO2 at 37°C for 3 d. On day 3, the media was aspirated, and the cells were gently rinsed once with PBS and fixed in 4% paraformaldehyde for 15 min at RT. For nestin, NG2, and GFAP, cells were treated with 0.05% Triton X-100 in PBS for 15 min before incubation with the primary antibody. For lipid antigens (O4, A2B5), cells were not exposed to detergent. Cells were incubated with primary antibody for 1 hr at RT, washed three times (5 min each) in PBS, incubated in the dark with the appropriate secondary antibody for 45 min at RT, and washed three times in PBS. Cells were then stained with 5 μg/ml bis-Benzimide (Sigma) for 15 min, washed once in PBS, and coverslipped using Fluoromount G (Electron Microscopy Sciences, Ft. Washington, PA). In cases of double labeling, cell-surface antigens were stained first and then cells were refixed in 4% paraformaldehyde before permeabilization and staining internal antigens. The following antibodies and dilutions were used: Rat-401 (nestin) mouse IgG conditioned medium (CM) (Developmental Studies Hybridoma Bank, Iowa City, IA), A2B5 mouse IgM CM (Eisenbarth et al., 1979), O4 mouse IgM CM (cells from T. Gard, University of South Alabama College of Medicine, Mobile, AL) (Sommer and Schachner, 1981; Gard and Pfeiffer, 1990), GFAP rabbit IgG 1:1000 (Dako, Carpenteria, CA), 5A5 [embryonic neural cell adhesion molecule (E-NCAM)] mouse IgM CM (Developmental Studies Hybridoma Bank), PDGF receptor (PDGFR)-α rabbit IgG 1:500 (Upstate Biotechnology), and NG2 rabbit IgG 1:150 (Chemicon). Rhodamine [tetramethylrhodamine iso-thiocyanate (TRITC)]-conjugated secondary antibodies (Jackson ImmunoResearch) were used in single-labeling experiments. For double labeling, internal antigens were stained with TRITC-conjugated secondary antibodies, and cell-surface antigens with FITC-conjugated secondary antibodies (Jackson ImmunoResearch). This minimized overlap of bis-Benzimide nuclear staining with the internal antigen staining. Quantitation was performed on a Zeiss (Thornwood, NY) Axiophot fluorescent microscope by counting randomly selected fields of bis-Benzimide-stained nuclei and then determining the percentage of cells positive for a specific antigen. At least 100 cells were counted per condition.
DNA fragmentation assay. Cells from E12.5 wild-type and Nf1+/- mouse spinal cords were cultured as described above in the presence of FGF2 (25 ng/ml) until nearly confluent. The cells were then plated at a density of 20,000 cells per well onto fibronectin-laminin-coated eight-well RS-treated Labtek II chamber slides, as described previously, in the presence or absence of FGF2 (25 ng/ml) and maintained for 3 d at 37°C. The cells were fixed in 4% paraformaldehyde for 15 min at RT and processed using the fluorescein-FragEL DNA fragmentation detection kit (Oncogene Research Products, Boston, MA) according to the instructions of the manufacturer. The kit uses terminal deoxynucleotidyl transferase (TdT) to attach fluorescein-conjugated deoxynucleotides to free 3′-OH groups at the end of DNA fragments resulting from endogenous endonucleases cleaving cellular DNA as the cell undergoes apoptosis. In brief, the slides were immersed in PBS for 15 min at RT and then permeabilized with 20 ng/ml of proteinase K in 10 mm Tris, pH 8.0, for 5 min. The slides were rinsed three times in PBS and then incubated at RT in TdT equilibration buffer for 30 min. The TdT-labeling reaction mix was applied to the specimens, and slides were then incubated at 37°C in a humidified chamber for 90 min. The slides were rinsed three times for 1 min each in fresh PBS and coverslipped using the supplied fluorescein-FragEL mounting media. Cells were visualized as described above. The number of fragmented nuclei was compared with the total number of nuclei for each condition and genotype.
Bromodeoxyuridine labeling. Cells were cultured until nearly confluent, as described above, and then plated at a density of 5000 cells per well on fibronectin-laminin-coated eight-well RS-treated Labtek II chamber slides in the absence of FGF2 for 48 hr. FGF2 (10 ng/ml) was added for 24 hr, and the cells were pulsed with 10 μm bromodeoxyuridine (BrdU; Sigma) for the final 4 hr. The cells were fixed in 3.7% formaldehyde in PBS for 15 min at RT and rinsed with PBS. The cells were permeabilized in 0.3% Triton X-100 (Fisher Scientific, Pittsburgh, PA) in PBS for 15 min at RT. Rat anti-BrdU (Accurate Chemicals, Westbury, NY) was applied at 1:500 for 45 min at 37°C in the following mixture: immunofluorescence (IF) buffer (0.5% NP-40, 5 mg/ml BSA in PBS), 20 mm MgCl2, and 200 U/ml DNase 1 (Calbiochem, San Diego, CA). The slides were washed three times for 5 min in PBS and then incubated with TRITC-conjugated donkey anti-rat (Jackson ImmunoResearch) at 1:100 in IF buffer with 5 μg/ml bis-Benzimide (Sigma) for 45 min at 37°C. The slides were washed three times for 5 min in PBS, coverslipped, and visualized as described above. The number of BrdU-positive nuclei were counted and compared with the total number of nuclei in each condition and genotype. A minimum of 100 cells was counted for each sample.
Oligodendrocyte progenitor cells. A2B5-immunoreactive oligodendrocyte progenitor cells were isolated from spinal cords by immunoselection as described previously (McKinnon et al., 1990). Purified cells were plated on Falcon culture dishes (Becton Dickinson) precoated by incubating with a solution of 100 μg/ml poly-l-ornithine (Sigma) in 15 mm boric acid, pH 8.4, and then extensively washing in sterile water. The cells were cultured in DMEM containing 4.5 gm/l d-glucose (Invitrogen), penicillin (50 U/ml), streptomycin and transferrin (50 μg/ml each), sodium selenite and triiodothyronine (30 nm each), 50 ng/ml bovine insulin, and 0.5% FBS (Invitrogen). The cells were expanded as secondary cultures (McKinnon and Zazanis, 1996) by supplementing with B104 neuroblastoma-conditioned medium (B104 cm; 20% v/v) (Schubert, 1974; Louis et al., 1992). The cells were subcultured as described for the oligodendrocyte line CG-4 (Louis et al., 1992) using ATV trypsin solution (Irvine Scientific, Irvine, CA) to dislodge the cells from the culture flasks. Transplantations were done with these secondary oligodendrocyte progenitor cultures maintained in vitro for <15 passages (at a 1:3 split ratio).
Grafting of oligodendrocyte progenitor cells into postnatal day 2 mouse brain
When oligodendrocyte progenitor cells (OPCs) became confluent, they were labeled with the fluorescent dye PKH26 (Sigma), and 10,000 -20,000 pooled cells were injected into postnatal day (P) 2 shiverer mutant mouse brains lacking myelin basic protein (MBP). Cell grafts were placed into the mid-thalamus by injection through the cortex and right lateral ventricle (Osterhout et al., 1997). Shi/+ and shi/shi mouse were used as positive and negative controls for MBP staining, respectively. Pups were killed at P14 except one Shi/Shi host, which was killed at P21. Brains were removed, fixed in 4% paraformaldehyde, and cryoprotected in 20% sucrose. Free-floating coronal serial sections (40 μm) were cut on a freezing microtome and processed for immunofluorescence using a polyclonal rabbit anti-MBP (1:400) overnight (gift from Dr. D. Colman, Montreal Neurologic Institute, Montreal, Canada). Sections were then rinsed, incubated in goat anti-rabbit FITC for 90 min, mounted on gelatin-coated slides, air-dried overnight, and coverslipped with Fluoromount G. Sections were viewed under the fluorescent microscope using a TRITC filter to visualize PKH26 and a FITC filter to visualize MBP staining.
Results
We tested whether increased numbers of progenitors might reside in the Nf1+/- adult nervous system. NG2 has been used as a marker of oligodendrocyte progenitors in uninjured adult mouse spinal cord (Wu et al., 2000). Cross sections of adult wild-type and Nf1+/- cervical spinal cords were cut and labeled with anti-NG2. Figure 1, A and B, shows examples of stained cells. Labeled cells in both genotypes had characteristic small cell bodies and multiple branched processes characteristic of oligodendrocyte progenitors (Watanabe et al., 2002). Counts of NG2+ cells revealed a significant increase (p = 0.001) in NG2+ cells in both gray and white matter in Nf1+/- spinal cord (Fig. 1C).
Figure 1.
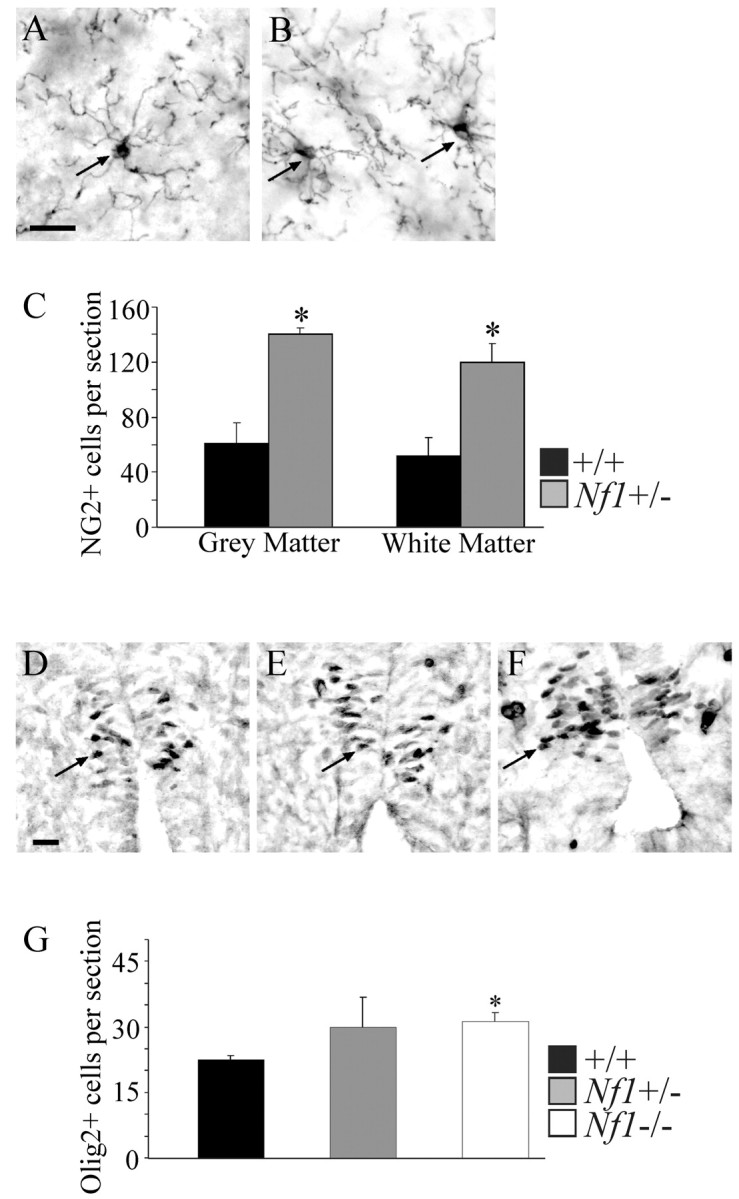
NG2 labeling of adult mouse spinal cords and Olig2 labeling of E12.5 spinal cords. A, B, Photomicrographs of transverse sections of adult wild-type (A) and Nf1+/- (B) spinal cords showing immunostained NG2+ cells (arrows). Scale bar, 20 μm. C, Significantly more NG2+ cells were found in the Nf1+/- (n = 3) versus wild-type (n = 3) spinal cords in both gray matter (*p = 0.01; paired Student's t test) and white matter (*p = 0.001; paired Student's t test). D--F, Photomicrographs of transverse sections of E12.5 wild-type (D), Nf1+/- (E), and Nf1-/- (F) spinal cords showing Olig2+ cells (arrows). Scale bar, 20 μm. G, Significantly more Olig2+ cells were found in the Nf1-/- (n = 3) versus wild-type (n = 3) spinal cords (*p = 0.01; paired Student's t test). Nf1+/- (n = 3) spinal cords were not significantly different from either wild-type or Nf1-/- spinal cords.
Increased Nf1 mutant progenitors exist in vivo and after acute dissociation of E12.5 spinal cords
We tested whether increased numbers of glial progenitors present in vivo in the adult are also present in the developing CNS in E12.5 spinal cord. The earliest known markers for oligodendrocyte progenitors are the basic helix-loop-helix transcription factors Olig1 and Olig2 (Lu et al., 2002; Zhou and Anderson, 2002). We used the spinal cord as a model system because, at E12.5, glial progenitors developed in significant numbers, whereas in brain and optic nerve, these cells arise after embryonic lethality. Spinal cords were sectioned and labeled with anti-Olig2 (kindly provided by D. Rowitch and C. Stiles). Sections were not labeled for Olig1 because the available antibody is not as reliable (J. Alberta, personal communication). Figure 1, D, E, and F, shows examples of stained cells. Counts of Olig2+ cells revealed a significant increase (p = 0.01) in Nf1-/- spinal cords compared with wild-type spinal cords (Fig. 1G).
It is now clear that Olig2 and NG2 can mark cells capable of becoming neurons as well as oligodendrocytes at certain times during development (Sun et al., 2001; Belachew et al., 2003). To define progenitors amplified in Nf1mutants, we performed marker analysis on cells from E12.5 mutant spinal cord after acute dissociation. Cells were dissociated, plated, fixed within 24 hr, and then stained for markers of CNS progenitors. As shown in Table 1, similar percentages of E-NCAM-positive putative neuronal progenitors were present across genotypes. In contrast, the percentage of cells expressing nestin, A2B5 or NG2 (progenitor and glial lineage markers in vitro, respectively), was increased threefold to fourfold in Nf1-/- spinal cords. Again, Nf1+/- spinal cord cells had intermediate levels of precursor accumulation, supporting the idea that there is an expanded pool of glial progenitors in Nf1 mutant spinal cords.
Table 1.
Marker analysis of E12.5 embryonic spinal cord cells
|
|
+/+ |
Nf1 +/− |
Nf1 −/− |
|
|---|---|---|---|---|
| Antigen | Percentage of cells expressing antigen | |||
| Nestin | 17 ± 1 | 30 ± 1 | 54 ± 2 | |
| A2B5 | 17 ± 5 | 37 ± 2 | 65 ± 1 | |
| ENCAM | 12 ± 1 | 17 ± 1 | 15 ± 1 | |
| NG2
|
8 ± 1
|
16 ± 2
|
35 ± 2
|
|
Cells of designated genotype were plated and immunostained 24 hr after dissociation. Cells from three embryos of each genotype were analyzed. Data are ± SE.
In NEP medium, Nf1 mutant cells give rise to glia but not neurons
To determine how Nf1 mutation results in increased numbers of oligodendrocyte progenitors, we turned to some well studied in vitro systems. In vitro, it is possible to grow spinal cord cells under conditions that maintain them as relatively undifferentiated neuroepithelial cells. This medium also allows the expansion of oligodendrocyte progenitors (Kalyani et al., 1997; Rao and Mayer-Proschel, 1997; Rao et al., 1998; Rao, 1999; Herrera et al., 2001; Gregori et al., 2002).
We tested whether cell populations are altered when cells derived from Nf1 mutant spinal cords are cultured in this medium, as described for mouse cells (Mujtaba et al., 1999). Small dark putative progenitors overtook other cell types in Nf1-/- cultures, forming a monolayer by day 6. In contrast, wild-type and Nf1+/- cultures failed to reach this level of growth even by day 14 (data not shown). For all three genotypes, the cells expanding under these culture conditions had an antigenic phenotype (A2B5+, NG2-, PDGFRα-) characteristic of glial lineage cells (Table 2).
Table 2.
Immunocytochemical marker analysis of expanded embryonic spinal cord cells
|
|
NEP |
GRP |
O2A |
Oligo |
Type 1 astrocyte |
Type 2 astrocyte |
+/+ |
Nf1 +/− |
Nf1 −/− |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Antigen | Percentage of cells expressing antigen | ||||||||||
| Nestin | + | + | + | − | + | + | 44 ± 3 | 78 ± 2 | 87 ± 3 | ||
| A2B5 | − | + | + | − | − | + | 56 ± 2 | 55 ± 2 | 74 ± 2 | ||
| E-NCAM | − | − | − | − | + | − | 0 | 0 | 0 | ||
| NG2 | − | −* | + | + | 7 ± 2 | 7 ± 1 | 3 ± 1 | ||||
| PDGFR-α | − | −* | + | + | N.D. | 0 | |||||
| 04 | − | − | +* | + | − | − | 49 ± 3 | 56 ± 2 | 72 ± 2 | ||
| GFAP | − | − | − | − | + | + | 33 ± 2 | 36 ± 1 | 48 ± 1 | ||
| 04/GFAP | − | − | − | − | − | − | <1 | 6 ± 1 | 17 ± 1 | ||
| Pyknotic | 17 ± 2 | 10 ± 1 | 5 ± 1 | ||||||||
| 04/GFAP plus Pyknotic
|
|
|
|
|
|
|
0
|
1 ± 1
|
20 ± 2
|
||
Eight days after dissociation from E12.5 spinal cords, cells were stained with designated markers. For comparison, antigenic phenotype of cells defined in the literature is shown. Antigenic characterization of NEP cells, GRPs, 02A, type 1 astrocytes, and type 2 astrocytes is reviewed by Rao (1999). *Do not initially express, but can gain expression maintaining multipotentiality (Gregori et al., 2002). Data is ± SE. N.D., Not determined.
Early oligodendrocyte progenitors lack expression of the PDGF receptor and require FGF2 for growth (Rao and Mayer-Proschel, 1997; Rao, 1999). We first determined whether FGF2 was necessary for the appearance of mutant progenitors. Cultures were generated in NEP medium either without growth factors, with FGF2 (5 ng/ml) alone, with PDGFAA (10 ng/ml) alone, or with FGF2 and PDGFAA in combination, and colony number and size were counted daily for 5 d (Fig. 2). Colonies did not appear in large numbers when cells were maintained in the absence of growth factors or with PDGFAA alone. In contrast, large colonies appeared when FGF2 was present, and the combination of FGF2 and PDGFAA was not significantly different from FGF2 alone.
Figure 2.
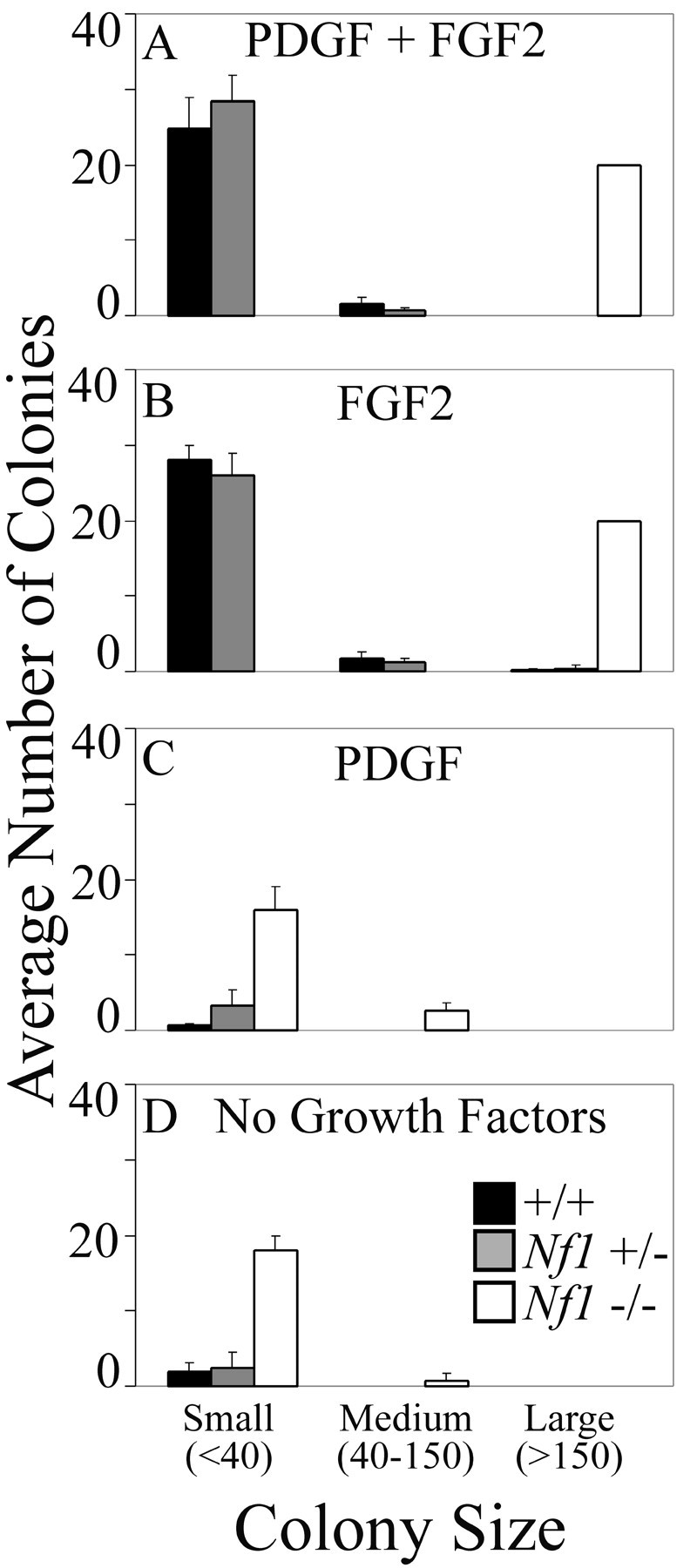
Growth factor requirements of Nf1 mutant glial progenitors. A--D, Colony number and size from wild-type (n = 9), Nf1+/- (n = 6), and Nf1-/- (n = 2) E12.5 spinal cord cultures at day 5 in the presence of FGF2 plus PDGF (A), FGF2 (B), PDGF (C), and no growth factors (D).
Because some Nf1 mutant cell types are hypersensitive to certain growth factors, we varied the dose of FGF2 and assayed colony number. If Nf1-deficient cells are hypersensitive to FGF2, the low dose used in the above study might have masked an Nf1+/- phenotype. We therefore varied the concentration of FGF2 between 1 and 25 ng/ml, and colony counts were again performed daily for 5 d (Fig. 3A). At the lowest doses tested (1-2.5 ng/ml), there was little or no colony formation with cells from any genotype. At 5 ng/ml, in keeping with the above study, abundant cell accumulation was observed in Nf1-/- cultures, and significantly less growth was observed in Nf1+/- and wild-type cultures. At 10 ng/ml of FGF2, Nf1+/- cultures developed significantly more colonies than did wild-type cells (Fig. 3B). At 25 ng/ml, colonies formed in cells of all three genotypes, although they appeared earlier in Nf1 mutant cultures; Nf1-/- cultures became confluent at day 3, Nf1+/- cells at day 4, and wild-type cultures by day 5.
Figure 3.

FGF2 dose-response curves. A, Number of large (<150 cells) colonies observed at day 5 of wild-type (n = 3), Nf1+/- (n = 3), and Nf1-/- (n = 3) E12.5 spinal cord cultures grown in the presence of 1, 2.5, 5, 10, and 25 ng/ml FGF2. B, Day 5 photographs of wild-type, Nf1+/-, and Nf1-/- E12.5 spinal cord cultures grown in the presence of 10 ng/ml of FGF2.
The hypersensitivity of Nf1+/- and Nf1-/- cells to FGF2 suggested a negative role for Nf1 in regulation of FGF signaling. The best-defined neurofibromin-signaling pathway is control of Ras-GTP levels. NF1 returns active Ras-GTP to its inactive (GDP) state, and in some cell types (likely when Ha-Ras is important; see Discussion), Ras activation can be blocked by farnesylprotein transferase inhibitors (FTI) (Yan et al., 1995; Kim et al., 1997; Costa et al., 2002). To begin to examine whether Ras-GTP was required for the accumulation of progenitors, cells were maintained in the presence of 5 ng/ml FGF2 (and PDGFAA), either with or without 1 μm of the FTI, L744,832 (a gift from Merck Research Labs) (Kohl et al., 1995). The presence of FTI completely inhibited the aberrant accumulation of cells in the Nf1-/- cultures (Fig. 4A,B). In contrast, the drug did not affect the appearance of the wild-type cultures (Fig. 4A,B).
Figure 4.
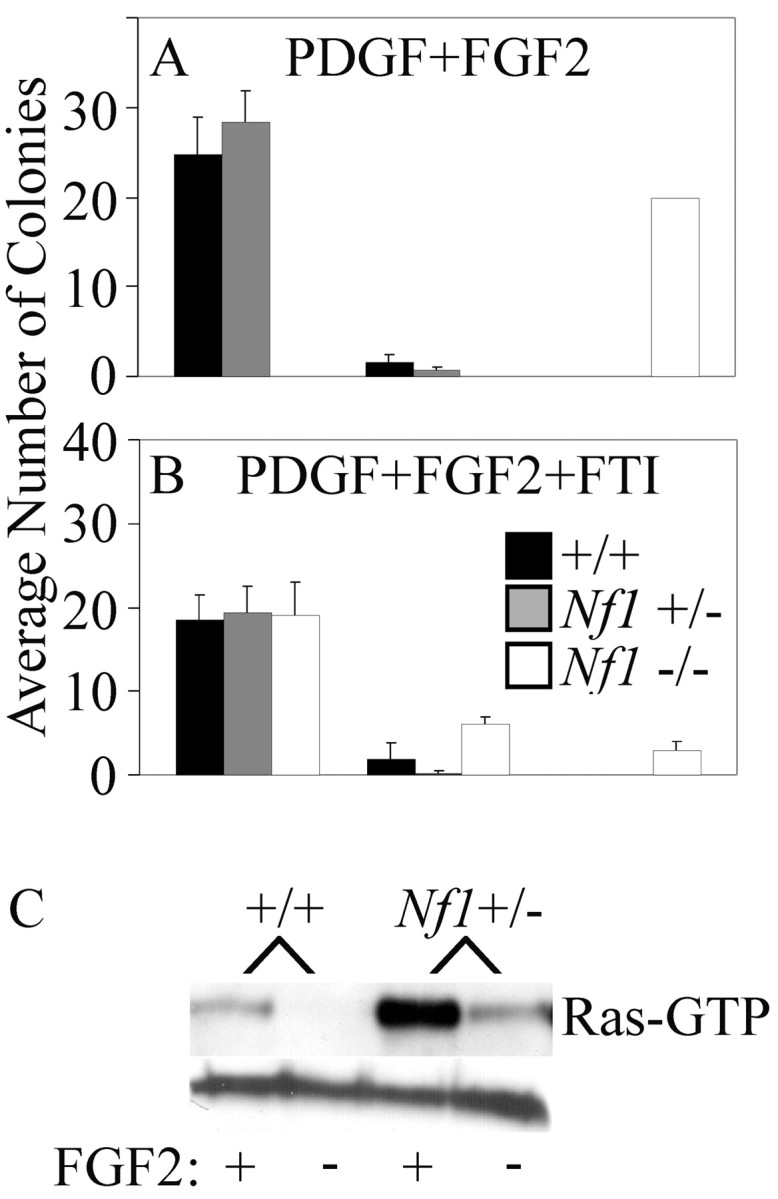
Inhibition of growth by FTI and Ras activation assay. A, B, Colony number and size from wild-type (n = 9), Nf1+/- (n = 6), and Nf1-/- (n = 2) E12.5 spinal cord cultures at day 5 in the presence of FGF2 plus PDGF (A) and FGF2, PDGF, and FTI (B). C, Immunoblot of a Ras-GTP pull-down assay showing increased basal and FGF2-stimulated Ras-GTP in Nf1+/- cells compared with wild-type cells from E12.5 mouse spinal cord cultures.
Together, results from these experiments suggest that the accumulation of cells in both Nf1-/- and Nf1+/- cultures depends on FGF2 and may depend on activation of the Ras-signaling pathway. To test directly whether there is increased Ras activation in the Nf1+/- cells, a Ras-activation assay was performed. We chose to use Nf1+/- cells rather than Nf1-/- cells as a stringent test of possible alterations in Ras-GTP levels. We compared Nf1+/+ with Nf1+/- cells. Wild-type and Nf1+/- cells were FGF2-starved for 24 hr and then stimulated with FGF2 for 10 min. Cell extracts were incubated with a bead-conjugated fragment of Raf1 with high affinity for Ras-GTP but not Ras-GDP, and bound protein eluted from beads and levels of GTP-bound Ras proteins were determined by immune blotting with a Ras10 antibody. Wild-type cells had low levels of Ras-GTP in uninduced conditions and readily detectable levels after FGF stimulation (Fig. 4C). In contrast, Nf1+/- cells had elevated levels of Ras-GTP in uninduced conditions and showed super maximal stimulation during FGF stimulation (Fig. 4C). These results are consistent with NF1 acting as a Ras-GAP in the FGF2 signaling pathway and with the interpretation that a decrease in neurofibromin function leads to a hypersensitivity to FGF2.
Marker analysis of expanded embryonic spinal cord cultures
Cells from Nf1+/-, Nf1-/-, and wild-type cultures were stained with markers that distinguish among CNS progenitor types defined in vitro (Rao, 1999) (Table 2). In general, the Nf1 mutant cells were similar to wild-type cells cultured under the same conditions. Neither cell type expressed E-NCAM, a marker of neuronal precursors (Mujtaba et al., 1999), PDGFRα, or NG2 chondroitin sulfate, which is expressed in concert with PDGFR. Absence of these markers is characteristic of early oligodendrocyte precursors (Rao, 1999) and consistent with the lack of response of mutant cells to PDGFAA (see above). Both Nf1 mutant and wild-type cells also expressed antigenic determinants of early oligodendrocyte lineage phenotype, including nestin and A2B5 immunoreactivity, and the ability to generate O4-immunoreactive cells (Table 2).
Several important differences were observed between cells in Nf1 mutant and wild-type cultures. Almost all Nf1-/- cultures were positive for the progenitor marker nestin (87%) and A2B5 (74%), whereas only approximately one-half the cells expressed these markers in wild-type cultures. Wild-type cells generally lose nestin expression before acquiring O4 immunoreactivity, a marker for the oligodendrocyte lineage (Rao, 1999). In contrast, 72% of Nf1-/- cells were positive for O4, most of which (87%) retained nestin expression (Table 2). Nf1+/- cells were intermediate between wild-type and Nf1-/- cells in marker expression.
In Nf1 mutant cultures, 16% of the cells coexpressed O4 and GFAP immunoreactivity (Fig. 5A; Table 2). Wild-type cultures did not contain cells with this phenotype; Nf1+/- cells were intermediate. This mixed glial phenotype was observed under culture conditions (FGF2) predicted to prevent cells from acquiring astrocytic characteristics (Rao, 1999). Similar phenotypes have been reported previously in pathologic white matter (Dyer et al., 2000). Thus, immunolabeling studies indicate that abnormal progenitor cells arise in large numbers in Nf1 mutant cultures.
Figure 5.
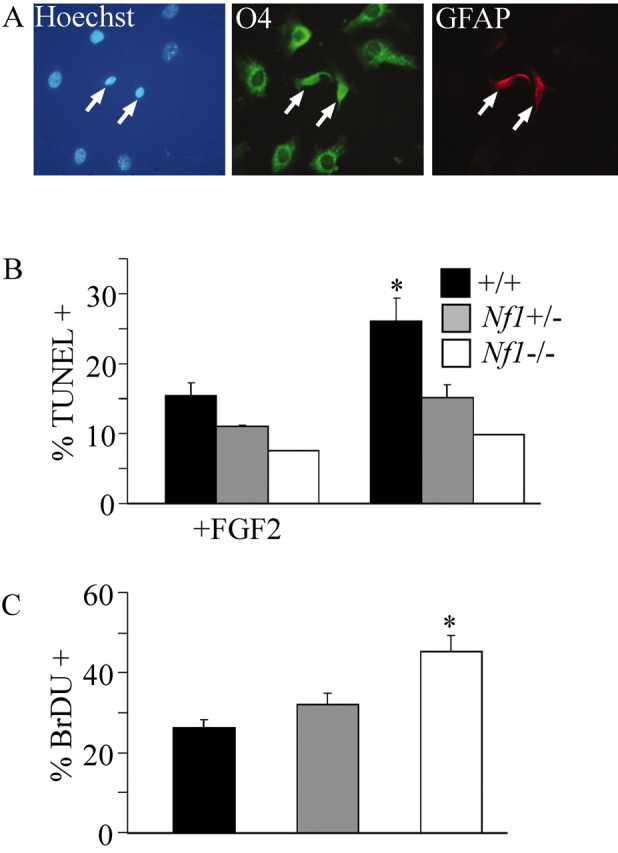
Nf1-/- mixed glial cells, DNA fragmentation, and cell proliferation assays. A, Triple labeling of Nf1-/- spinal cord cultures showing O4+/GFAP+ cells (arrows). Expanded cultures of E12.5 mouse spinal cord cells were assayed for DNA fragmentation and BrdU incorporation. B, Significantly fewer (*p = 0.03; paired Student's t test) Nf1+/- (n = 3) cells were undergoing cell death in the unstimulated condition versus wild-type cells (n = 3). A significant difference was not found when stimulated with 25 ng/ml FGF2. Fewer Nf1-/- (n = 1) cells were dying in each condition. C, A 4 hr pulse revealed a significant increase (*p = 0.02; paired Student's ttest) in BrdU incorporation in the Nf1-/- (n = 3) cultures versus both Nf1+/- (n = 3) and wild-type (n = 3) cultures after 24 hr of FGF2 stimulation (10 ng/ml). There was a slight increase in BrdU incorporation in the Nf1+/- cultures versus wild-type cultures, but it failed to reach significance.
Cell proliferation and death
The accumulation of large numbers of cells in Nf1 mutant cultures must result from an increase in cell proliferation or a decrease in cell death. To begin to test whether numbers of dying cells were different in wild-type and mutant cells, Hoechst-labeled nuclei were visualized. Overall, there were three times fewer pyknotic nuclei in Nf1-/- cultures versus wild-type cultures (Table 2). Strikingly, O4 -GFAP double-labeled Nf1-/- cells exhibited a high level of pyknotic nuclei (20%).
To confirm these results, we performed a fluorescent DNA fragmentation assay on cultures of wild-type, Nf1+/-, and Nf1-/- cells. Primary cells grown to 90% confluence were re-plated and allowed to grow for 3 d in the presence or absence of FGF2 and then examined for cell death (Fig. 5B). In the unstimulated condition, there were significantly fewer dying Nf1+/- mutant cells compared with wild-type cells (p = 0.03), suggesting that activated Ras-GTP promoted increased survival. In FGF2-stimulated conditions, both wild-type and Nf1+/- cultures had fewer TUNEL-labeled cells, although the results were not significantly different from the untreated Nf1 mutant cultures. Nf1-/- cells showed an additional decrease in TUNEL-labeled cells. Thus, Nf1 mutation is sufficient to promote increased survival.
To test whether proliferation might also contribute to the accumulation of Nf1 mutant cells, we performed a BrdU incorporation assay on wild-type, Nf1+/-, and Nf1-/- cells. Primary cells grown to 90% confluence were subcultured for 3 d in the absence of FGF2 and then pulsed with FGF2 (10 ng/ml) for 24 hr with 10 nm BrdU present for the final 4 hr. The number of nuclei that incorporated BrdU was then counted (Fig. 5C). FGF stimulation resulted in increased numbers of BrdU-positive nuclei in Nf1-/- cells (p = 0.02; one-tailed Student's t test). Therefore, both increased proliferation and increased cell survival likely contribute to increases in oligodendrocyte progenitors in Nf1 mutant cells.
OPC colony formation
To confirm that Nf1 mutant oligodendrocyte lineage cell expansion was not dependent on NEP medium, we tested a second well studied OPC culture system, in which OPCs proliferate and do not differentiate in medium containing FGF2 (McKinnon et al., 1990; Miller et al., 1999) or PDGF and FGF2 (Bogler et al., 1990). In media containing uncharacterized mitogens from neuroblastoma B104-conditioned media, OPCs expand as focal colonies (Louis et al., 1992). Equal numbers of cells from E12.5 wild-type and Nf1 mutant spinal cords were plated in the presence of B104 mitogens, and colonies were observed in all cultures after several weeks of continuous culture (Fig. 6A). The frequency of colony formation varied from 2.5 × 10-3 (Nf1+/+) to 14.1 × 10-3 (Nf1+/-) and 28.5 × 10-3 (Nf1-/-). Consistent with the cells being primarily oligodendroglial lineage, colonies contained either A2B5+ OPCs or mixtures of OPCs and A2B5+, GFAP+ cells (type 2 astrocyte; a cell type only found in culture); only a few GFAP+/A2B5- type 1 astrocytes were found (Fig. 6B).
Figure 6.
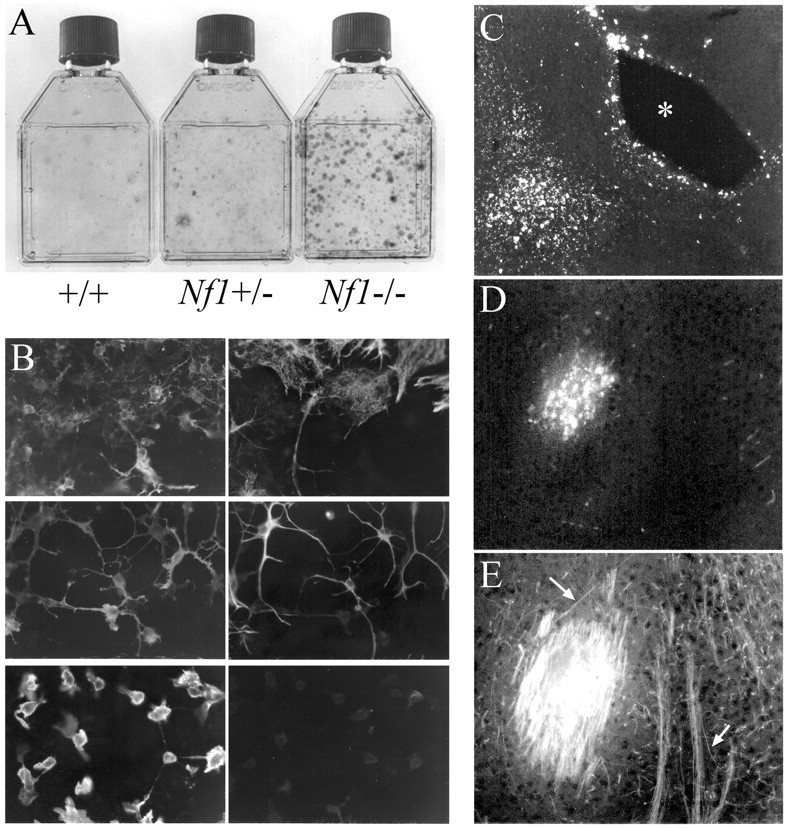
E12.5 spinal cord cultures and grafting of dye-labeled Nf1-/- OPCs (O2A cells) into the forebrain of MBP-/- mice.A, OPC cultures from wild-type, Nf1 +/-, and Nf1-/-E12.5 spinal cords stained with cresyl violet. B, Cells expanded from Nf1-/- cultures were identified by double-label immunocytochemistry using A2B5 (left) and anti-GFAP antibodies (right). Left and right images represent the same field of cells in each pair. Top, Colony containing both type 1 astrocytes (A2B5-/GFAP+) and oligodendrocyte progenitor cells (A2B5+/GFAP-). Center, Colony with type 2 (A2B5+/GFAP+) astrocytes. Bottom, Colony with only A2B5+/GFAP- oligodendrocyte progenitor cells. C, A low-magnification micrograph shows the relationship to the third ventricle (*) of dye-labeled grafted OPCs in MBP- mouse brain. A group of dye-labeled cells is shown at higher magnification in D with associated MBP+ myelin sheaths in E. Arrows point to a small group of longitudinally cut myelin sheaths.
To determine whether cells isolated from Nf1 mutant mice were capable of differentiating into oligodendrocytes, Nf1-/- OPCs were dye-labeled and then implanted into P2 myelin basic protein (MBP-/-) (shiverer) mouse brains. If MBP-expressing cells are detected, they must arise from the grafted cells. Numerous grafted cells became MBP positive and showed myelin sheath formation 12 d after grafting (Figs. 6C,D,E). Thus, cells expanded in Nf1-/- cultures can give rise to oligodendrocytes, and the absence of neurofibromin in these cells did not affect their ability to form myelin in vivo.
Discussion
The findings presented here demonstrate that loss of neurofibromin results in an abnormal accumulation of CNS progenitor cells. Oligodendrocyte progenitors are increased in number in the adult and embryonic Nf1mutant spinal cord. Marker analysis suggests that glial progenitors are enriched in vivo and immediately after dissociation from embryonic spinal cords and expand in culture. Amplified cells express markers of the oligodendrocyte lineage, including NG2, Olig2, and O4, but in vitro lack expression of the neuronal precursor marker E-NCAM. Although the full potential of the Nf1 mutant cells studied here remains to be determined, the abnormal progenitor cell population is dependent on FGF2 for expansion and forms oligodendrocytes on transplantation in vivo. Together, the data support a role for Nf1 in oligodendrocyte development.
The antigenic profile of the Nf1 mutant cells characterized shows some similarities to a cell type identified in vitro as a GRP (Rao and Mayer-Proschel, 1997; Mujtaba et al., 1999). GRPs can give rise to astrocytes and OPC cells, but not neurons, in vitro and to astrocytes and oligodendrocytes during transplantation in vivo (Herrera et al., 2001). We did not address the ability of Nf1 mutant cells grown in NEP medium to differentiate into neurons or astrocytes. Indeed, cell fate studies suggest that astrocytes and oligodendrocytes may be derived from separate progenitor cell pools in vivo (Lu et al., 2002; Zhou and Anderson, 2002), and thus the full range of phenotypic choices available to progenitors may not be fully exercised under normal developmental conditions. When grown under conditions that promote the expansion of PDGF-responsive OPCs (Louis et al., 1992; McKinnon and Zazanis, 1996), Nf1 mutant cultures generated A2B5+ progenitors and A2B5+/GFAP+ type 2 astrocytes. Because in vivo analyses have not detected type 2 astrocytes, we focused on oligodendrocyte differentiation of the mutant cells. Strikingly, during transplantation into MBP-deficient mice, Nf1 mutant progenitors differentiated into myelin-forming oligodendrocytes.
A significant fraction of cells in Nf1 mutant cultures simultaneously expressed precursor markers (nestin, A2B5) and later emerging differentiation markers (e.g., O4). In contrast, wild-type oligodendrocytes stop expressing nestin as they begin to express differentiation markers (Rao, 1999). Defects in CNS progenitors may also arise in nestin+ NEPs earlier in development, accounting for high nestin expression. Many Nf1 mutant cells also concomitantly expressed GFAP and O4, exhibiting an aberrant mixed glial phenotype (Godfraind et al., 1989; Dyer et al., 2000). Thus, Nf1 mutation uncouples the normal cessation of proliferation that occurs under in vitro differentiation conditions. However, the ability of Nf1 mutant cultures to generate myelin-forming oligodendrocytes during transplantation into MBP-deficient mice indicates that such alterations do not uniformly interfere with normal differentiation.
Cell defects driven by increased response to growth factors may be a common feature of loss of Nf1. Nf1 null embryonic peripheral neurons show that Ras-mediated hyper-responsiveness to neurotrophic factors (Vogel et al., 1995, 2000; Zhu et al., 2001) and astrocytoma cells from Nf1 patients, as well as Nf1 mutant fibroblasts, are hyper-responsive to epidermal growth factor (Atit et al., 1999; Gutmann et al., 1999). Hematopoietic progenitors from Nf1-/- murine fetal liver form abnormally high numbers of progenitor colonies in response to low levels of cytokines in vitro (Largaespada et al., 1996; Zhang et al., 1998). Our observation that FGF2-stimulated Ras-GTP is elevated in Nf1 mutant glial progenitors is the first direct demonstration of abnormal Ras activation by FGF2 in an Nf1-deficient background. Previous evidence implicated neurofibromin in modulation of response to FGF2 signaling (Kitano et al., 1992; Griesser et al., 1997; Kaufmann et al., 1999). Ras-GTP was elevated in CNS progenitors under basal conditions, as in Nf1 mutant Schwann cells (Kim et al., 1995; Sherman et al., 2000). In contrast, Nf1 mutant hematopoietic progenitors show increased Ras-GTP only after cytokine stimulation (Zhang et al., 1998). Thus, the effects of loss of Nf1 on Ras regulation among progenitor populations appear to be cell-type specific.
Although neurofibromin has poorly defined non-Ras functions (Johnson et al., 1993, 1994; Guo et al., 1997, 2000; Kim et al., 1997; Tong et al., 2002), these appear not to be relevant to phenotypes defined here, because abnormal accumulation of Nf1 mutant glial progenitor cells was abolished by the addition of FTIs. This finding suggests the involvement of aberrant Ras signaling in the mutant phenotype. Blocking farnesylation inhibits Ras processing that is required for Ras membrane localization and activation. The data are consistent with the ability of FTIs to rescue Nf1-dependent phenotypes in Schwann cells (Yan et al., 1995; Kim et al., 1997) and learning deficits in Nf1 heterozygous mice (Costa et al., 2002). We cannot presently exclude effects of FTIs on non-Ras farnesylated proteins, such as RhoB (Prendergast, 2000), in this system. However, consistent with effects of FTIs on progenitor cells being mediated through Ras, Ras-GTP was elevated in Nf1 mutant CNS progenitor cells. Together, these data support a key role for farnesylation-sensitive form(s) of Ras, such as H-Ras (Sebti and Hamilton, 1997), upstream of neurofibromin in glial progenitors.
Nf1 mutant cells in our experiments accumulated, at least in part, through increased cell survival. There were significantly fewer dying (pyknotic and TUNEL+) cells in Nf1+/- mutant cultures compared with wild-type cells, and a trend toward increased survival in the Nf1 mutants was stimulated with FGF2. Differences were also detected in cell proliferation rates. We conclude that survival and proliferation are both affected. Previous studies identified FGF2 as a survival factor for early glial progenitors (Yasuda et al., 1995). Increased survival of Nf1 mutant progenitors in low doses of FGF implicates Ras signaling downstream of FGF-mediated survival. In later OPCs, activation of phosphotidyl inositol 3-kinase is essential for survival (Vemuri and McMorris, 1996), and this survival pathway is activated by PDGF but not FGF (Ebner et al., 2000). The data suggest that signaling pathways required for glial progenitor survival differ as the cells undergo maturation.
It is especially relevant to note that we observed abnormalities in Nf1 hemizygous glial lineage cells, in both embryos and adult mice. Mutant human NF1+/- cells could account for some non-focal effects in the NF1 patient nervous system. Magnetic resonance spectroscopy of NF1 patients shows an increase in choline, suggestive of focal edema and vacuolization of myelin (Wang et al., 2000). Eastwood et al. (2001) showed increases in water diffusibility by MR imaging and proposed that this also reflected widespread myelin disorder in the NF1 brain. On pathologic examination, T2-hyperintense lesions in NF1 patients appeared to contain vacuolated myelin (DiPaolo et al., 1995); these lesions have even higher water diffusibility than the surrounding NF1 brain (Eastwood et al., 2001). Although nests of demyelinated cells (T2 hyperintensities) in NF1 patients may represent either heterozygous or null NF1 mutations, the global changes in brain metabolism likely result from NF1 haplo-insufficiency. It is also plausible that with increased numbers of NF1 glial progenitors, some lose the second NF1 allele, causing astrocyte tumor formation; loss of heterozygosity at NF1 is frequent in NF1 patient pilocytic astrocytomas (Gutmann et al., 2000; Kluwe et al., 2001; for review, see Cichowski and Jacks, 2001). In this context, it is of interest that astrocytes arise from cells that would normally make oligodendrocytes or motor neurons when signaling is perturbed (Zhou et al., 2002).
A striking finding in this study is the marked increase in vivo of NG2+ cells in the adult Nf1+/- mouse spinal cord. Previous studies identified proliferating NG2+ cells in the adult human and rodent CNS that are distinct from astrocytes, oligodendrocytes, and microglia (Chang et al., 2000; Wu et al., 2000; McTigue et al., 2001). Cells with these characteristics can give rise to mature oligodendrocytes in vitro (Wolswijk and Noble, 1989) and in vivo (Watanabe et al., 2002). Abnormalities in the embryonic glial progenitor pool that we defined in vitro are likely to exist in vivo and persist into the adult, accounting for the increase in adult NG2+ cells. Indeed, FGF2 maintains NG2+ progenitors in a proliferative state in adult CNS (Armstrong et al., 2002).
Our data suggest the hypothesis that abnormalities in the glial lineage contribute to brain dysfunction in NF1 patients. Small increases in brain progenitor pools result in a magnified increase in brain volume (Rakic, 1995). For example, increased progenitor numbers could contribute to the enlarged white matter tracts that underlie macrocephaly in NF1 patients.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS28840 (N.R.) and National Multiple Sclerosis Society (NMSS) Grant RG3229-A-4 (N.R.). R.M. was supported by NIH and NMSS. T.R. was supported in part by Department of Defense Grant on Neurofibromatosis (NF000023), and M.B. was supported by National Institute of Neurological Disorders and Stroke Grant 5 T32 N707453. R.M. is a member of The Cancer Institute of New Jersey. We thank Robert Miller (Case Western) for initial observations of NF1 spinal cord cells. The farnesyltransferase inhibitor used was a kind gift from Jackson Gibbs and Nancy Kohl (Merck Research Laboratories). We thank Yuan Huang for help with timed dissections, Annie Stammen and Jason Bowersock for genotyping embryos, and Johanna Hadley, Brenda Meinhardt, and Sylvie Ebner for maintenance of NF1 (J.H. and B.M.) and MBP [shiverer] (S.E.) mouse colonies. The rat 401 (nestin) and 5A5 (E-NCAM) antibodies developed by S. Hockfield (rat 401), T. M. Jessel, and J. Dodd (5A5) were obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development, and maintained by The University of Iowa Department of Biological Sciences.
Correspondence should be addressed to Nancy Ratner, Department of Cell Biology, Neurobiology, and Anatomy, University of Cincinnati College of Medicine, 3125 Eden Avenue, Cincinnati, OH 45267-0521. E-mail: nancy.ratner@uc.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237207-11$15.00/0
References
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC ( 2002) Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci 22: 8574-8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atit RP, Crowe M, Greenhalgh D, Wenstrup R, Ratner N ( 1999) The Nf1 tumor suppressor is required for mouse skin wound healing, fibroblast proliferation and collagen deposited by fibroblasts. J Invest Dermatol 112: 835-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Donahoe J, Corral T, Reilly KM, Brophy S, Pellicer A, Gutmann DH ( 2001) Neurofibromatosis 1 (NF1) heterozygosity results in a cell-autonomous growth advantage for astrocytes. Glia 33: 314-323. [DOI] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V ( 2003) Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol 161: 169-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum RA, O'Marcaigh A, Wardak Z, Zhang YY, Dranoff G, Jacks T, Clapp DW, Shannon KM ( 2000) Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol Cell 5: 189-195. [DOI] [PubMed] [Google Scholar]
- Bogler O, Wren D, Barnett SC, Land H, Noble M ( 1990) Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA 87: 6368-6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K ( 1996) Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12: 144-148. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG ( 1994) Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 8: 1019-1029. [DOI] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp D ( 2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20: 6404-6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Jacks T ( 2001) NF1 tumor suppressor gene function: narrowing the GAP. Cell 104: 593-604. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ ( 2002) Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415: 526-530. [DOI] [PubMed] [Google Scholar]
- Daston MM, Ratner N ( 1992) Neurofibromin, a predominantly neuronal GTPase activating protein in the adult, is ubiquitously expressed during development. Dev Dyn 195: 216-226. [DOI] [PubMed] [Google Scholar]
- Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N ( 1992) The protein product of the neurofibromatosis type I gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 8: 415-428. [DOI] [PubMed] [Google Scholar]
- DiPaolo DP, Zimmerman RA, Rorke LB, Zackai EH, Bilaniuk LT, Yachnis AT ( 1995) Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain. Radiology 195: 721-724. [DOI] [PubMed] [Google Scholar]
- Donovan S, Shannon KM, Bollag G ( 2002) GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 1602: 23-45. [DOI] [PubMed] [Google Scholar]
- Dyer CA, Kendler A, Jean-Guillaume D, Awatramani R, Lee A, Mason LM, Kamholz J ( 2000) GFAP-positive and myelin marker-positive glia in normal and pathologic environments. J Neurosci Res 60: 412-426. [DOI] [PubMed] [Google Scholar]
- Eastwood JD, Fiorella DJ, MacFall JF, Delong DM, Provenzale JM, Greenwood RS ( 2001) Increased brain apparent diffusion coefficient in children with neurofibromatosis type 1. Radiology 219: 354-358. [DOI] [PubMed] [Google Scholar]
- Ebner S, Dunbar M, McKinnon RD ( 2000) Distinct roles for PI3K in proliferation and survival of oligodendrocyte progenitor cells. J Neurosci Res 62: 336-345. [DOI] [PubMed] [Google Scholar]
- Eisenbarth GS, Walsh FS, Nirenberg M ( 1979) Monoclonal antibody to a plasma membrane antigen of neurons. Proc Natl Acad Sci USA 76: 4913-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost EE, Nielsen JA, Le TQ, Armstrong RC ( 2003) PDGF and FGF2 regulate oligodendrocyte progenitor responses to demyelination. J Neurobiol 54: 457-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE ( 1990) Two proliferative stages of the oligodendrocyte lineage (A2B5+04- and 04+GalC-) under different mitogenic control. Neuron 5: 615-625. [DOI] [PubMed] [Google Scholar]
- Giordano MJ, Mahadeo DK, He YY, Geist RT, Hsu C, Gutmann DH ( 1996) Increased expression of the neurofibromatosis 1 (NF1) gene product, neurofibromin, in astrocytes in response to cerebral ischemia. J Neurosci Res 43: 246-253. [DOI] [PubMed] [Google Scholar]
- Godfraind C, Friedrich VL, Holmes KV, Dubois-Dalcq M ( 1989) In vivo analysis of glial cell phenotypes during a viral demyelinating disease in mice. J Cell Biol 109: 2405-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori N, Proschel C, Noble M, Mayer-Proschel M ( 2002) The tripotential glial-restricted precursor (GRP) cell and glial development in the spinal cord: generation of bipotential oligodendrocyte-type-2 astrocyte progenitor cells and dorsal-ventral differences in GRP cell function. J Neurosci 22: 248-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesser J, Kaufmann D, Maier B, Mailhammer R, Kuehl P, Krone W ( 1997) Post-transcriptional regulation of neurofibromin level in cultured human melanocytes in response to growth factors. J Invest Dermatol 108: 275-280. [DOI] [PubMed] [Google Scholar]
- Guo HF, The I, Hannan F, Bernards A, Zhong Y ( 1997) Requirement for Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276: 795-798. [DOI] [PubMed] [Google Scholar]
- Guo HF, Tong J, Hannan F, Luo L, Zhong Y ( 2000) A neurofibromatosis- 1-regulated pathway is required for learning in Drosophila Nature 403: 895-898. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Loehr A, Zhang Y, Kim J, Henkemeyer M, Cashen A ( 1999) Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene 18: 4450-4459. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Donahoe J, Brown T, James CD, Perry A ( 2000) Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol 26: 361-367. [DOI] [PubMed] [Google Scholar]
- Herrera J, Yang H, Zhang SC, Proschel C, Tresco P, Duncan ID, Luskin M, Mayer-Proschel M ( 2001) Embryonic-derived glial-restricted precursor cells (GRP cells) can differentiate into astrocytes and oligodendrocytes in vivo Exp Neurol 171: 11-21. [DOI] [PubMed] [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA ( 1994) Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 7: 353-361. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Look AT, DeClue JE, Valentine MB, Lowy DR ( 1993) Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP-Ras. Proc Natl Acad Sci USA 90: 5539-5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MR, DeClue JE, Felzmann S, Vass WC, Xu G, White R, Lowy DR ( 1994) Neurofibromin can inhibit ras-dependent growth by a mechanism independent of its GTPase-accelerating function. Mol Cell Biol 14: 641-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyani A, Hobson K, Rao MS ( 1997) Neuroepithelial stem cells from the embryonic spinal cord: isolation, characterization, and clonal analysis. Dev Biol 186: 202-223. [DOI] [PubMed] [Google Scholar]
- Kaufmann D, Junge I, Bartelt B, Lattke H, Muller R ( 1999) On the lysosomal degradation of neurofibromin and its phosphorylation in cultured melanocytes. Biol Chem 380: 1071-1078. [DOI] [PubMed] [Google Scholar]
- Kim H, Rosenbaum T, Marchionni M, Ratner N, DeClue J ( 1995) Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of Schwann cell proliferation and morphologic changes. Oncogene 11: 325-335. [PubMed] [Google Scholar]
- Kim H, Ling B, Ratner N ( 1997) Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol 17: 862-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano Y, Okamoto E, Saito K, Okano Y ( 1992) Effects of several growth factors on cultured neurofibroma cells. J Dermatol Sci 3: 137-144. [DOI] [PubMed] [Google Scholar]
- Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, von Deimling A, Mautner VF ( 2001) Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol 60: 917-920. [DOI] [PubMed] [Google Scholar]
- Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton K, Handt LK, Hartmen GD, Koblan KS, Kral AM, Miller PJ, Mosser SD, O'Neill TJ, Rands E, Schaber MD, Gibbs JB, et al. ( 1995) Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1: 792-797. [DOI] [PubMed] [Google Scholar]
- Largaespada DA, Brannan CI, Jenkins NA, Copeland NG ( 1996) Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukemia. Nat Genet 12: 137-143. [DOI] [PubMed] [Google Scholar]
- Li J, Perry A, James CD, Gutmann DH ( 2001) Cancer-related gene expression profiles in NF1-associated pilocytic astrocytomas. Neurology 56: 885-890. [DOI] [PubMed] [Google Scholar]
- Listernick R, Louis DN, Packer RJ, Gutmann DH ( 1997) Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol 41: 143-149. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S ( 1992) CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res 31: 193-204. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH ( 2002) Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75-86. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Zazanis GA ( 1996) Transplantation of genetically engineered primary cells for the analysis of gene function in CNS development. Methods 10: 332-342. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA ( 1990) FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 5: 603-614. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT ( 2001) Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci 21: 3392-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH, Hayes JE, Dyer KL, Sussman CR ( 1999) Mechanisms of oligodendrocyte commitment in the vertebrate CNS. Int J Dev Neurosci 17: 753-763. [DOI] [PubMed] [Google Scholar]
- Mujtaba T, Piper DR, Kalyani A, Groves AK, Lucero MT, Rao MS ( 1999) Lineage-restricted neural precursors can be isolated from both the mouse neural tube and cultured ES cells. Dev Biol 214: 113-127. [DOI] [PubMed] [Google Scholar]
- Nordlund M, Gu X, Shipley MT, Ratner N ( 1993) Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci 13: 1588-1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund ML, Rizvi TA, Brannan CI, Ratner N ( 1995) Neurofibromin expression and astrogliosis in neurofibromatosis (Type 1) brains. J Neuropathol Exp Neurol 54: 588-600. [DOI] [PubMed] [Google Scholar]
- North K, Ratner N ( 2003) The central nervous system in neurofibromatosis type 1. In: Genetics and genomics of neurobehavioral disorders (Fisch GS, ed), pp 97-131. Totowa, NJ: Humana.
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R ( 2003) Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci 23: 883-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterhout DJ, Ebner S, Xu J, Ornitz DM, Zazanis GA, McKinnon RD ( 1997) Transplanted oligodendrocyte progenitor cells expressing a dominant-negative FGF receptor transgene fail to migrate in vivo J Neurosci 17: 9122-9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S ( 1999) Cognitive impairment in neurofibromatosis type 1. Am J Med Genet 89: 45-52. [PubMed] [Google Scholar]
- Prendergast GC ( 2000) Farnesyltransferase inhibitors: antineoplastic mechanism and clinical prospects. Curr Opin Cell Biol 12: 166-173. [DOI] [PubMed] [Google Scholar]
- Rakic P ( 1995) A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 18: 383-388. [DOI] [PubMed] [Google Scholar]
- Rao MS ( 1999) Multipotent and restricted precursors in the central nervous system. Anat Rec 257: 137-148. [DOI] [PubMed] [Google Scholar]
- Rao MS, Mayer-Proschel M ( 1997) Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Dev Biol 188: 48-63. [DOI] [PubMed] [Google Scholar]
- Rao MS, Noble M, Mayer-Proschel M ( 1998) A tripotential glial precursor is present in the developing spinal cord. Proc Natl Acad Sci USA 95: 3996-4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redwine JM, Armstrong RC ( 1998) In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J Neurobiol 37: 413-428. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Akunuru S, de Courten-Myers G, Switzer III RC, Nordlund ML, Ratner N ( 1999) Region-specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res 816: 111-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Lu QR, Kessaris N, Richardson WD ( 2002) An “oligarchy” rules neural development. Trends Neurosci 25: 417-422. [DOI] [PubMed] [Google Scholar]
- Sauvageot CM, Stiles CD ( 2002) Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol 12: 244-249. [DOI] [PubMed] [Google Scholar]
- Schubert D ( 1974) Induced differentiation of clonal rat nerve and glial cells. Neurobiology 4: 376-387. [PubMed] [Google Scholar]
- Sebti S, Hamilton AD ( 1997) Inhibitors of prenyl transferases. Curr Opin Oncol 9: 557-561. [DOI] [PubMed] [Google Scholar]
- Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N ( 2000) Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem 275: 30740-30745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Frankland PW, Marowitz Z, Friedman E, Lazlo G, Cioffi D, Jacks T, Bourtchuladze R ( 1997) A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet 15: 281-284. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M ( 1981) Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83: 311-327. [DOI] [PubMed] [Google Scholar]
- Steen RG, Taylor JS, Langston JW, Glass JO, Brewer VR, Reddick WE, Mages R, Pivnick EK ( 2001) Prospective evaluation of the brain in asymptomatic children with neurofibromatosis type 1: relationship of macrocephaly to T1 relaxation changes and structural brain abnormalities. AJNR Am J Neuroradiol 22: 810-817. [PMC free article] [PubMed] [Google Scholar]
- Sun T, Echelard Y, Lu R, Yuk DI, Kaing S, Stiles CD, Rowitch DH ( 2001) Olig bHLH proteins interact with homeodomain proteins to regulate cell fate acquisition in progenitors of the ventral neural tube. Curr Biol 11: 1413-1420. [DOI] [PubMed] [Google Scholar]
- Temple S, Alvarez-Buylla A ( 1999) Stem cells in the adult mammalian central nervous system. Curr Opin Neurobiol 9: 135-141. [DOI] [PubMed] [Google Scholar]
- Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y ( 2002) Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci 5: 95-96. [DOI] [PubMed] [Google Scholar]
- Vemuri GS, McMorris FA ( 1996) Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development 122: 2529-2537. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF ( 1995) Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell 82: 733-742. [DOI] [PubMed] [Google Scholar]
- Vogel KS, El-Afandi M, Parada LF ( 2000) Neurofibromin negatively regulates neurotrophin signaling through p21ras in embryonic sensory neurons. Mol Cell Neurosci 15: 398-407. [DOI] [PubMed] [Google Scholar]
- Wang PY, Kaufmann WE, Koth CW, Denckla MB, Barker PB ( 2000) Thalamic involvement in neurofibromatosis type 1: evaluation with proton magnetic resonance spectroscopic imaging. Ann Neurol 47: 477-484. [PubMed] [Google Scholar]
- Watanabe M, Toyama Y, Nishiyama A ( 2002) Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res 69: 826-836. [DOI] [PubMed] [Google Scholar]
- Wolswijk G, Noble M ( 1989) Identification of an adult-specific glial progenitor cell. Development 105: 387-400. [DOI] [PubMed] [Google Scholar]
- Wu Q, Miller RH, Ransohoff RM, Robinson S, Bu J, Nishiyama A ( 2000) Elevatated levels of the chemoking GRO-1 correlate with elevated oligodendrocyte progenitor proliferation in the Jimpy mutant. J Neurosci 20: 2609-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Ricca C, Fletcher J, Glover T, Seizinger BR, Manne V ( 1995) Farnesyltransferase inhibitors block the neurofibromatosis type I (NF1) malignant phenotype. Cancer Res 55: 3569-3575. [PubMed] [Google Scholar]
- Yasuda T, Grinspan J, Stern J, Franceschini B, Bannerman P, Pleasure D ( 1995) Apoptosis occurs in the oligodendroglial lineage, and is prevented by basic fibroblast growth factor. J Neurosci Res 40: 306-317. [DOI] [PubMed] [Google Scholar]
- ZhangYY, Vik TA, Ryder JW, Srour EF, Jacks T, Shannon K, Clapp DW ( 1998) Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med 187: 1893-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ ( 2002) The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109: 61-73. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF ( 2001) Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15: 859-876. [DOI] [PMC free article] [PubMed] [Google Scholar]