

Abstract
BCL-2 family proteins are known to regulate cell death during development by influencing the permeability of mitochondrial membranes. The anti-apoptotic BCL-2 family protein BCL-xL is highly expressed in the adult brain and localizes to mitochondria in the presynaptic terminal of the adult squid stellate ganglion. Application of recombinant BCL-xL through a patch pipette to mitochondria inside the giant presynaptic terminal triggered multiconductance channel activity in mitochondrial membranes. Furthermore, injection of full-length BCL-xL protein into the presynaptic terminal enhanced postsynaptic responses and enhanced the rate of recovery from synaptic depression, whereas a recombinant pro-apoptotic cleavage product of BCL-xL attenuated postsynaptic responses. The effect of BCL-xL on synaptic responses persisted in the presence of a blocker of mitochondrial calcium uptake and was mimicked by injection of ATP into the terminal. These studies indicate that the permeability of outer mitochondrial membranes influences synaptic transmission, and they raise the possibility that modulation of mitochondrial conductance by BCL-2 family proteins affects synaptic stability.
Keywords: BCL-2, BCL-xL, squid, neurotransmitter release, mitochondria, ATP
Introduction
Regulation of the size of a releasable pool of neurotransmitter and its probability of release contribute to both short-term and long-term changes in the strength of neurotransmission. Sustained elevations in presynaptic calcium during rapid, repetitive neuronal firing have been correlated with enhancement of synaptic transmission in a number of studies (Swandulla et al., 1991; Dittman and Regehr, 1998; Wang and Kaczmarek, 1998; Augustine, 2001; Sakaba and Neher, 2001). Mitochondria are known to play a role in calcium handling in secretory cells (Budd and Nicholls, 1996; Babcock and Hille, 1998; Giovannucci et al., 1999) and in synaptic potentiation (Friel and Tsien,1994; Nguyen and Atwood, 1994; Nguyen et al., 1997; Tang and Zucker, 1997; Calupca et al., 2001; Billups and Forsythe, 2002), although the specific molecular mechanisms that define the role of this organelle in calcium and metabolite management during high-frequency presynaptic activity are not yet known.
Repetitive firing produces a change in conductance on presynaptic mitochondrial membranes (Jonas et al., 1999). The conductance change requires extracellular calcium and occurs after a delay, suggesting that calcium entry through voltage-gated channels reaches mitochondria during high-frequency firing, when calcium begins to accumulate at a distance from the active zone. The identity of the channel responsible for this mitochondrial conductance is not yet known; however, possible modulators of mitochondrial conductances are BCL-2 family proteins (Kroemer, 1997; Reed, 1997). In response to cell death or survival signals, the activation of these proteins either promotes or prevents the death of a cell by regulating outer mitochondrial membrane permeability to cytochrome c and other pro-apoptotic factors (Kluck et al., 1997; Polster et al., 2001; Joza et al., 2002).
Previous studies have found that both pro-apoptotic [e.g., BAX (BCL-2-associated X protein)] and anti-apoptotic (e.g., BCL-xL) members of the BCL-2 family can produce ion channel activity in synthetic lipid bilayers (Antonsson et al., 1997; Minn et al., 1997; Schlesinger et al., 1997; Schendel et al., 1998; Basanez et al., 1999, 2001) and in mitochondrial membrane preparations that have been combined with synthetic liposomes (Pavlov et al., 2001). These studies in artificial lipids may mimic in vivo functions, but direct evidence for intracellular ion channels or pores formed by BCL-2 family proteins is lacking thus far. The potential for BCL-xL to modulate other channels in the outer mitochondrial membrane is suggested by the finding that BCL-xL regulates the open state of the mitochondrial voltage-dependent anion channel (VDAC) in synthetic membranes (Degterev et al., 2001; Vander Heiden et al., 2001). The ion channel activities induced by BCL-2 family proteins may be related to their anti- and proapoptotic functions but may also have physiological functions that have not yet been defined.
BCL-xL, a potent inhibitor of apoptosis (Boise et al., 1993), is widely expressed in the adult brain where little physiological apoptosis is occurring (Krajewski et al., 1994; Frankowski et al., 1995; González-Garcia et al., 1995; Blömer et al., 1998) and is bound to the outer mitochondrial membrane (Krajewski et al., 1993; González-Garcia et al., 1994). BCL-xL contains four BCL-2 homology domains (BH1-BH4) and two central α helices between BH1 and BH2 that are suggested to constitute the pore of an ion channel (Muchmore et al., 1996; Kroemer, 1997; Kelekar and Thompson, 1998). In particular, the structure of BCL-xL, and all other BCL-2 family members to date, reveals a protein fold in this region similar to the B domain of diphtheria toxin, a polypeptide that is responsible for translocating the toxin A domain across membranes. The C terminus of BCL-xL serves as a transmembrane domain and as a targeting domain for the outer mitochondrial membrane (Kaufmann et al., 2003).
Here we provide evidence that manipulation of mitochondrial membrane conductance by BCL-xL in the presynaptic terminal of the squid giant synapse enhances postsynaptic responses to presynaptic stimulation, and it also enhances recovery from synaptic depression. We found that the changes in synaptic responsiveness are mimicked by injection of ATP and suggest that BCL-xL regulation of metabolite flux through the mitochondrial outer membrane may regulate the size, refilling, or release probability of the readily releasable pool of synaptic vesicles.
Materials and Methods
Intracellular membrane patch-clamp recordings. Experiments were performed on small Loligo pealii at the Marine Biological Laboratory (Woods Hole, MA). Stellate ganglia were isolated by severing the large presynaptic nerve from its cell body after it has exited the head and by freeing from the underlying mantle the ganglion containing the synapse between presynaptic axon and postsynaptic axon. The isolated ganglia were placed in a Lucite chamber and pinned to Sylgard with fine cactus needles. The bathing solution contained the following (in mm): 466 NaCl, 54 MgCl2, 11 CaCl2, 10 KCl, 3 NaHCO3, 10 HEPES, pH 7.2, and was cooled, oxygenated with 99.5% O2, 0.5% CO2, and perfused over the ganglia. Intracellular membrane pipettes (20-80 MΩ) were filled with intracellular solution that contained the following (in mm): 570 KCl, 1.2 MgCl2, 10 HEPES, 0.07 EGTA, 0.046 CaCl2, and 2 ATP, pH 7.2. The inner electrode had an outer diameter of 1.0 mm (Kwik-Fil; World Precision Instruments, Sarasota, FL) and was mounted in a standard electrode holder with a side port attached to manual suction. The outer electrode was made of 1.5 mm capillary tubing (100 μl micropipettes; Drummond Scientific, Broomall, PA), it was not polished, and only the tip was filled with intracellular solution. The inner electrode was inserted manually into the outer electrode, and microscope oil was placed between the two electrodes. The inner and outer electrodes were operated independently by separate micromanipulators. Both electrodes were moved in unison toward the cell, after which they punctured the presynaptic terminal ∼50-100 μm from its distal end during monitoring of membrane potential. The outer microelectrode was then retracted, exposing the inner tip (Jonas et al., 1999). Gigaohm seals were obtained in ∼40% of attempts and formed either spontaneously or in response to slight negative pressure. Each presynaptic terminal was used only for a single-patch experiment. The polarities of potentials reported here refer to those of the patch pipette relative to that of the ground electrode, which was placed in the external medium. The sample rate was 20 kHz and data were filtered at 500-1000 Hz.
Immunocytochemistry and Western analysis. For immunocytochemistry, ganglia were fixed in 4% paraformaldehyde and 0.4 m sucrose in artificial seawater. Fixed ganglia were mounted in tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC), frozen rapidly at -20°C, and cut into 25 or 50 μm sections. These were permeabilized with 1% Triton X-100-PBS for 10 min and incubated for 4 hr with primary antibody (2 μg/ml rabbit α BCL-xL; Upstate Biotechnology, Lake Placid, NY) in PBS-5% nonfat dry milk. Sections were then incubated for 2 hr in secondary antibody [goat anti-rabbit IgG, fluoresceinconjugated (Pierce, Rockford, IL)] at 1:100 in PBS-5% nonfat dry milk at room temperature in the dark. Sections were mounted with Vectashield mounting media (Vector Laboratories, Burlingame, CA). Preincubation of α BCL-xL with its antigen BCL-xL eliminated the punctate pattern of staining. For staining tissue for mitochondria, live dissected ganglia were placed in 200 nm oxygenated MitoTracker Red CMXRos (Molecular Probes, Eugene, OR) stain in bathing solution for 30 min. Tissue was then rinsed once in oxygenated bathing solution and then fixed as above. Control experiments using either BCL-xL immunolocalization or Mitotracker Red alone demonstrated that no cross-contamination of these signals occurred during fluorescence microscopy.
For Western analysis, baby hamster kidney (BHK) cells infected with recombinant Sindbis viruses expressing either green fluorescent protein or human BCL-xL were harvested 24 hr after infection. Infected BHK cells and squid stellate ganglia were lysed in radioimmunoprecipitation assay buffer (150 mm NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, and 50 mm Tris, pH 7.5) and loaded onto 12% SDS-PAGE gels, followed by transfer onto Immobilon membranes and incubation with an anti-chicken BCL-xL antibody (1:1000) or an anti-human BCL-xL (PharMingen, San Diego, CA).
Isolation of mitochondria. Mitochondria were isolated from squid stellate ganglia using methods developed for subcellular fractionation of squid heart and rat brain (Mommsen and Hochachka, 1981; Rosenthal et al., 1987). Ganglia were minced in ice-cold isolation buffer (500 mm mannitol, 150 mm KCl, 2 mm sodium-EDTA, 1% fatty acid-free BSA, and 25 mm HEPES, adjusted to pH 7.4 with HCl at 25°C), homogenized with 12 strokes of a 2 ml Dounce homogenizer (Fisher Scientific, Houston, TX), and centrifuged at 2800 × g for 3 min at 4°C. The supernatant was centrifuged at 15,000 × g for 10 min at 4°C, washed, and centrifuged. Mitochondria were stored frozen at -20°C. After thawing for immunoblot analysis, 1 μl of general use protease inhibitor mixture (Sigma, St. Louis, MO) was added, and protein concentration was measured by the BCA protein assay (Pierce).
Production of BCL-xL proteins. Human (Mr 26 kDa) and ΔN76-truncated BCL-xL used in this study were produced as glutathione S-transferase (GST) fusion proteins in Escherichia coli, using a pGEX system (Amersham Biosciences, Arlington Heights, IL). Fusion proteins were induced with isopropyl-β-d-thiogalactopyranoside and purified by affinity chromatography, using glutathione-Sepharose 4B. BCL-xL was cleaved from GST by thrombin, equilibrated in PBS (150 mm NaCl and 20 mm Na2PO4, pH 7.2), and stored at -80°C.
Injection of presynaptic terminal and measurement of postsynaptic responses. Intracellular microinjection pipettes were filled with intracellular squid solution and either recombinant proteins (20 μg/ml BCL-xL or its mutant, 20 μg/ml ΔN76 BCL-xL) or 20 mm ATP and inserted into the presynaptic terminal. Pulses of positive pressure (20-40 psi, 100 msec) were given with a Picospritzer (General Valve, Fairfield, NJ). Fast green was coinjected with proteins in separate experiments to ensure filling of the terminal. Synaptic transmission was evoked by stimulating an external suction electrode attached to the presynaptic nerve. The nerve was stimulated at 0.033 Hz, 20 V, 0.01 msec to elicit single action potentials. The postsynaptic responses were recorded by an electrode containing 3 m KCl inserted into the postsynaptic nerve. Transmitter release was measured by recording the initial rate of rise of the postsynaptic response (Swandulla et al., 1991; Morgan et al., 2001). Tetani were given at 50 Hz for 2 sec, and recovery was measured by stimulating single postsynaptic responses at 10 and 30 sec after the last postsynaptic potential in the train or by stimulating continuously at 2 Hz before and after the tetanus. The initial rates of rise of the postsynaptic responses were calculated using pClamp 8.0 Clampfit software (Axon Instruments, Foster City, CA) by placing a cursor at the first onset of the synaptic response, determined by eye, and a second cursor at a time point 100-300 μsec later, before any detectable regenerative response occurred. Electrophysiological traces show individual representative examples, and all group data are given as mean ± SEM.
Results
Localization of BCL-xL in squid neurons
We adopted the squid giant synapse as a convenient model system for examining a potential role for BCL-xL in the modulation of synaptic transmission. Antibodies that detect BCL-xL in a wide range of species were used to verify that BCL-xL is present in adult squid neurons and localized to mitochondria. Low-power images of sections of squid stellate ganglia stained for BCL-xL showed the presence of immunoreactivity in neuronal tissue. The most intense staining was found in the large presynaptic terminal fingers of the axo-axonal synapse onto the giant postsynaptic axon (Fig. 1A). As is typical for mitochondrial staining in large neurons and axons (Nguyen and Atwood, 1994; Nguyen et al., 1997), staining was found throughout the axoplasm, but the density of immunoreactivity was greatest close to the plasma membrane, particularly that apposed to the postsynaptic axon. At higher power, striations in BCL-xL staining were apparent (Fig. 1B). These striations are likely to correspond with the location of the structures that represent contact points between the presynaptic terminal and the postsynaptic axon (Martin and Miledi, 1975; Llinás, 1999). Observation at still higher power revealed a punctate cytoplasmic pattern that is characteristic of mitochondria and that colocalized with the mitochondrial dye MitoTracker (red), shown in Figure 1C, for the terminal itself and for neuronal somata within the stellate ganglion. Immunofluorescent staining with antibody preadsorbed with BCL-xL failed to specifically stain these structures (data not shown).
Figure 1.
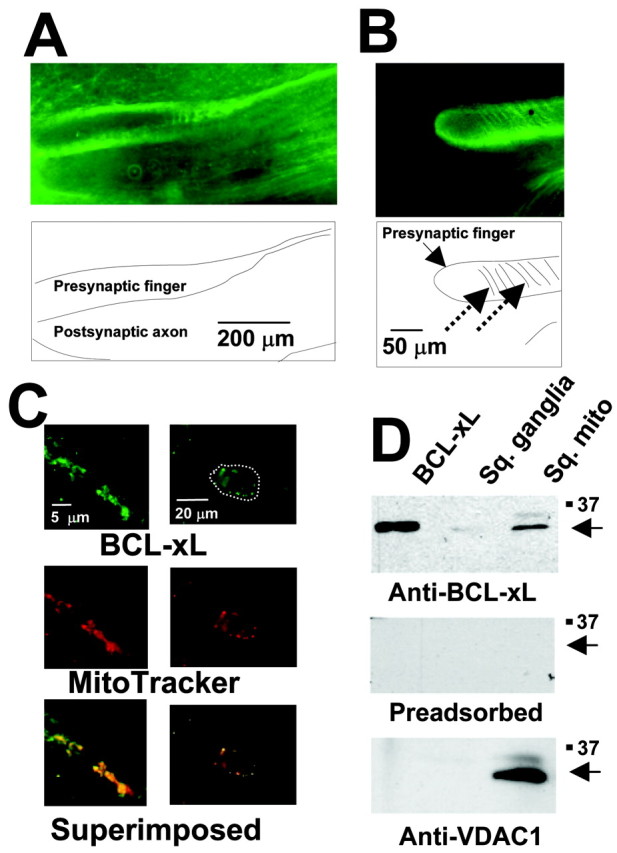
Presence of BCL-xL immunoreactivity in neuronal mitochondria. A, BCL-xL immunoreactivity in a section through the giant presynaptic terminal. B, At higher magnification, staining appears more punctate, and striations in staining (dashed arrows) are apparent. C, Colocalization of BCL-xL immunoreactivity with mitochondria at even higher magnification in the presynaptic terminal (left panels) and in neuronal somata (right panels) of the stellate ganglion. Mitochondria were stained using MitoTracker Red (middle), and the bottom panels show superposition of BCL-xL and MitoTracker staining. The outline of a single soma is marked with the white dotted line in the top panel. D, Immunoblot analysis of recombinant human BCL-xL protein, squid stellate ganglion (Sq. ganglia), and mitochondria (Sq. mito; purified from squid stellate ganglia) was performed using anti-BCL-xL antibody (top). The middle panel shows an immunoblot of the same samples using preadsorbed antibody. The bottom panel shows immunoblot analysis using an anti-VDAC1 antibody.
Additional evidence for the mitochondrial localization of BCL-xL in squid was obtained by preparation of a purified mitochondrial fraction from the stellate ganglion. Immunoblot analysis of this fraction showed the presence of squid BCL-xL that comigrated with recombinant human BCL-xL (Fig. 1D). Also detected in these fractions was the mitochondria outer membrane protein VDAC1. BCL-xL immunoreactivity was eliminated by preabsorption with recombinant human BCL-xL, demonstrating specificity of the immunoreactivity (Fig. 1D). The level of detection of both BCL-xL and VDAC1 by immunoblot analysis was greatly enhanced in the purified mitochondria, compared with similar amounts of tissue homogenates of stellate ganglia (Fig. 1D), verifying highly specific localization to these intracellular organelles.
BCL-xL induces channel activity in mitochondria
Ion channel activity of mitochondrial membranes can be recorded within the intact presynaptic terminal of squid giant stellate ganglia by entering the terminal using a patch-clamp electrode contained inside an outer ensheathing microelectrode and then making a high-resistance seal with the patch pipette on intracellular membranes (Jonas et al., 1997, 1999). Mitochondria and synaptic vesicles are the dominant intracellular organelles in the terminal, and only a very small proportion (less than ∼3%) of the intracellular membrane cannot be identified as belonging to one of these organelles. Scanning electron microscopy of the tips of the patch pipettes has shown that their tip diameter is ∼180-190 nm, which is compatible with seal formation on the mitochondria (∼300-1200 nm in diameter) but too large for the synaptic vesicles (∼40 nm) (Jonas et al., 1999). The most prevalent activity in control recordings of mitochondrial membranes, followed for up to 1 hr in ganglia perfused with oxygenated seawater, had a conductance of <30 pS (Fig. 2A). In these control patches, the overall probability of opening of larger conductances (>180 pS) was very small (0.018 ± 0.013; n = 16). Because no steps were taken to disrupt the outer mitochondrial membrane (Sorgato et al., 1987), it is likely that these recordings represent channel activity in the outer membrane.
Figure 2.

Induction of multiconductance channel activity on mitochondrial membranes by BCL-xL. A, Typical recordings of intracellular membrane activity in a squid presynaptic terminal using patch pipettes containing control intracellular solution (left) or intracellular solution containing 8.0 μg/ml BCL-xL (middle and right). Part of the control trace (left) is also shown on an expanded time and current scale to illustrate typical small conductance openings detected in control recordings. The center panel shows BCL-xL-induced activity that is maintained at a single conductance level, whereas the right panel shows typical transitions between conductance levels. Recordings were made at patch potentials of 100 (left), 40 (middle), and 100 mV (right). B, Amplitude histogram of multiconductance activity induced by BCL-xL (+20 mV) in a different patch from those shown in A. The closed state is marked C, and two prominent peaks in the histogram are labeled P1 and P2. C, Current-voltage relationship of unitary currents for the two conductance states shown in B. Amplitudes were determined from peaks in the amplitude histograms at each potential. In this patch, the mean slope conductances over the voltage range 0-60 mV were 240 and 462 pS for P1 and P2, respectively.
It is known that BCL-xL translocates from cytosol to the outer mitochondrial membrane, where it is normally localized, and that when added to isolated mitochondria in vitro, it also enters this membrane (Hsu et al., 1997; Clem et al., 1998; Vander Heiden et al, 2001; Shirane and Nakayama, 2003). To determine the action of BCL-xL on mitochondrial membrane conductance, we tested the effect of including BCL-xL in the patch-pipette solution. Characteristic activity with multiple conductances was readily detected in mitochondrial patches within 1 min after forming seals with recombinant purified human BCL-xL protein inside the patch pipette. Unitary openings corresponded to conductances between 100 and 760 pS (Fig. 2A,B). Activity typically switched between different conductance levels every few seconds, but a single conductance level could also be maintained occasionally for several minutes. A series of rapid-voltage steps to potentials between -100 and 100 mV during BCL-xL-induced channel activity at each conductance level revealed that current-voltage relations were linear or very slightly outwardly rectifying, with reversal potentials between -40 and 0 mV (Fig. 2C). In recordings made with BCL-xL in the patch pipette, the probability of channel openings with conductances >180 pS was 0.32 ± 0.08 (n = 10), which is significantly greater than in control recordings (p < 0.0002).
BCL-xL enhances synaptic transmission
Because BCL-xL produces a change in mitochondrial membrane conductance, we tested its effects on the strength of synaptic transmission. BCL-xL (20 μg/ml) was injected into presynaptic termini, while suprathreshold stimuli were given to the presynaptic nerve once every 30 sec (0.033 Hz). The strength of synaptic transmission was assayed by measuring the rate of rise of the postsynaptic response (Swandulla et al., 1991; Morgan et al., 2001). Control postsynaptic responses were followed over 10 min (n = 6). These cells showed either no change or a slight rundown of the postsynaptic response (Fig. 3E). In contrast, injection of BCL-xL into the presynaptic terminal enhanced the rate of rise of the postsynaptic response (Fig. 3D) (n = 10), resulting in an earlier latency for evoked action potentials (Fig. 3A). BCL-xL was effective in both healthy synapses and in those in which transmission had run down to the point that the postsynaptic potential no longer triggered postsynaptic action potentials. Under these conditions, injection of BCL-xL into the terminal could enhance the amplitude of the postsynaptic potential, restoring suprathreshold responses, and again producing a progressive decrease in their latency (Fig. 3B).
Figure 3.

BCL-xL injection enhances synaptic transmission. A, Evoked postsynaptic responses before and 20 min after presynaptic injection of BCL-xL (20 μg/ml in pipette solution). B, Effects of BCL-xL injection into the presynaptic terminal in a synapse in which the postsynaptic response had failed to reach threshold for action potential firing. The left panel shows three superimposed postsynaptic responses before BCL-xL injection. The right panel shows responses at increasing times (1, 2, and 3) after injection up to 17 min. C, Typical time courses of effect of BCL-xL injection on the slope of the postsynaptic response evoked by stimulation at 0.033 Hz (left) or 2.0 Hz (right). D, Bar graphs of all experiments showing the peak postsynaptic response before and after BCL-xL injection (n = 10; **p < 0.005). E, Percentage change in the slope of the postsynaptic response over 10 min in untreated synapses (n = 6) compared with the percentage change in response of BCL-xL-injected terminals (20 min after injection; *p < 0.015).
A typical time course for the action of BCL-xL is shown in Figure 3C (left). Enhancement of transmission began immediately after injection in some synapses and in some cells, lasted as long as 45 min. The peak response occurred on average at 20 ± 6 min after injection, after which responses began to decline. In addition to experiments in which stimulation was applied every 30 sec, we injected BCL-xL into presynaptic terminals that were stimulated at 2 Hz, a higher frequency that produces a significant degree of synaptic depression (Swandulla et al., 1991). At this stimulus frequency, BCL-xL also enhanced transmission (Fig. 3C, right). Injection of intracellular buffer alone did not enhance the postsynaptic response (see Fig. 7A). Moreover, despite the fact that BCL-xL was injected into the presynaptic terminal, it did not affect the kinetics or amplitude of the presynaptic action potential (n = 6; data not shown), suggesting that its effects on synaptic transmission are unlikely to be caused by alterations of plasma membrane currents that could alter calcium influx.
Figure 7.

ATP injection mimics BCL-xL injection. A, Postsynaptic responses shown before and 20 min after injection of squid intracellular solution without ATP. Bottom traces display the rate of rise of the postsynaptic responses on an expanded scale and show that there is no change in the postsynaptic response. B, Postsynaptic responses shown before and 20 min after injection of 20 mm ATP in squid intracellular solution. Bottom traces display postsynaptic responses on an expanded scale, and the superimposed dashed lines are the fits to the initial slope of the postsynaptic responses. C, Time course of response to ATP injection. D, BCL-xL fails to enhance transmission in the presence of injected ATP. The left panel shows three superimposed postsynaptic responses before ATP injection in a synapse with a postsynaptic response that had failed to reach threshold for action potential firing. The middle panel shows responses at increasing times (5, 8, and 20 min corresponding to traces 1, 2, and 3, respectively) after injection. The third panel shows the response immediately before BCL-xL injection [20 min after ATP injection (200 mm)], together with the traces recorded 5 and 10 min after introduction of BCL-xL. No enhancement of synaptic potentials in response to BCL-xL injection was detected.
Cleavage of BCL-xL near the N terminus by caspase-3 occurs in apoptotic cells and can convert anti-apoptotic BCL-xL into a pro-death protein (Clem et al., 1998). Expression of an engineered deletion mutant of BCL-xL that lacks the N-terminal BH4 domain (ΔN76 BCL-xL), mimicking the cleavage fragment, potently induces cell death and cytochrome c release from mitochondria. We therefore also injected recombinant human ΔN76 BCL-xL into the presynaptic terminal and determined its actions on postsynaptic responses. In contrast to BCL-xL, ΔN76 BCL-xL produced a decrease in the rate of rise of synaptic responses, resulting in rundown of the postsynaptic response (Fig. 4A,B). The time course of this action was similar to that of BCL-xL (Fig. 4C).
Figure 4.

Injection of ΔN 76 BCL-xL into the presynaptic terminal depresses synaptic transmission. A, Evoked postsynaptic responses before and 10 min after presynaptic injection of ΔN 76 BCL-xL (20 μg/ml in pipette solution) in a neuron with a strong synaptic response. Dashed lines are fitted to the initial slope of the postsynaptic responses. B, Bar graphs of all experiments showing the rates of rise of postsynaptic responses before and after ΔN 76 BCL-xL injection (n = 6; *p < 0.03). C, Typical time course of effect of ΔN 76 BCL-xL injection on the slope of the postsynaptic response.
BCL-xL enhances recovery from synaptic depression
Because mitochondria are abundant in the synaptic terminals of neurons that are capable of firing at high frequencies (Tolbert and Morest, 1982; Rowland et al., 2000) and are known to be involved in presynaptic responses to high-frequency simulation (Friel and Tsien, 1994; Tang and Zucker, 1997), we tested the actions of BCL-xL on synaptic responses during and after tetanic stimulation. The presynaptic terminal was stimulated at a frequency of 50 Hz for a period of 2 sec. We first measured the amount of synaptic depression that occurred during the tetanus by comparing the rate of rise of the postsynaptic potential before the tetanus with that of the postsynaptic potential evoked by the last stimulus of the tetanus. There was no difference in degree of depression during such high-frequency stimulation between control neurons and those injected with BCL-xL (Fig. 5A,B,E). Moreover, the rate of depression during the tetani, as judged by single exponential fits to responses during the train, was not significantly altered by BCL-xL [τ = 165 ± 26 msec (control); 201 ± 34 msec (BCL-xL); n = 7].
Figure 5.

Effects of BCL-xL injection on recovery from a tetanus. A, Time course of synaptic depression and recovery from depression in response to a tetanus (50 Hz, 2 sec) given during basal stimulation at 2 Hz, before and 9 min after injection of BCL-xL. B, Time course of recovery from depression in response to a tetanus given during basal stimulation at 0.033 Hz, before and after injection of BCL-xL. Postsynaptic responses were evoked at 10, 30, or 60 sec after the tetanus. C, Postsynaptic responses before (1), during (2), and 30 sec after (3) a tetanus during basal stimulation at 0.033 Hz. Data were obtained before the effect of BCL-xL. The inset shows rates of rise of postsynaptic responses on an expanded scale. D, Postsynaptic responses for the same cell recorded 50 min after injection of BCL-xL. E, Bar graphs comparing the pretetanus rate of rise of postsynaptic responses with that of the last response in the train for controls and synapses injected with BCL-xL. F, Bar graphs showing percentage recovery of postsynaptic responses 30 sec after a tetanus. BCL-xL responses were significantly increased over control (*p < 0.03). For controls, n = 10; for BCL-xL, n = 6; for ΔN 76 BCL-xL, n = 4.
We also examined the effects of BCL-xL on the rate of recovery from synaptic depression using two different paradigms that measure different components of recovery. In the first, stimulation was performed at 2 Hz before and after the tetanus (Fig. 5A). As reported previously (Swandulla et al., 1991), this basal rate of stimulation produces synaptic depression, and recovery from the tetanus often occurs over a declining baseline. Recovery of synaptic responses occurs relatively rapidly after the tetanus and was not significantly affected by injection of BCL-xL (Fig. 5A). Exponential fits to the rates of recovery provided time constants of 1.19 ± 0.5 sec (control) and 1.12 ± 0.37 sec (BCL-xL) (n = 6).
In the second paradigm, we measured the recovery from tetanic stimulation when basal stimulation was applied only once every 30 sec. Under these conditions, full recovery of synaptic responses after the tetanus is slower (Fig. 5B). The amount of recovery measured within 30 sec of the end of the tetanus, however, was significantly enhanced by BCL-xL injection when compared with controls (Fig. 5B-D,F). Thus, although synaptic depression is unaffected by BCL-xL, a slow component of the time course of recovery of the readily releasable pool of transmitter appears to be sensitive to the actions of BCL-xL.
Calcium release from mitochondria is known to play a role in synaptic plasticity; specifically, re-release of calcium from mitochondria is responsible for the long tail of residual calcium that causes post-tetanic potentiation at many synapses (Friel and Tsien, 1994; Tang and Zucker, 1995; Jonas et al., 1999). As stated above, however, at the squid giant synapse in physiological solutions, the calcium that enters the terminal during repeated action potentials produces strong synaptic depression, thought to result from depletion of synaptic vesicles (Swandulla et al., 1991). Thus, it is unlikely that the enhancement of transmission by BCL-xL, particularly at higher-stimulus frequencies (e.g., 2Hz), results from additional elevation of calcium levels in the presynaptic terminal. To examine the potential role of mitochondrial calcium flux in enhancement of synaptic transmission during BCL-xL injection, we treated neurons with ruthenium red, an agent that is taken up by neurons and inhibits uptake of calcium into mitochondria (Tapia and Velasco, 1997; Sanchez et al., 2001). In some cases, ruthenium red also inhibits calcium channels, an effect that would also decrease subsequent calcium uptake into mitochondria (Tapia and Velasco, 1997). As a positive control, we first tested the actions of ruthenium red (3 μm) on post-tetanic potentiation, which can be recorded at the squid synapse in external media containing low calcium (1.75 mm) (Swandulla et al., 1991; Jonas et al., 1999). We found that ruthenium red induced a slight increase in rundown of transmission and that it eliminated posttetanic potentiation without blocking augmentation of synaptic responses during the tetanus (n = 2; data not shown). Nevertheless, in six of seven cells in normal calcium-containing medium, BCL-xL potentiated transmitter release even in the presence of ruthenium red [either bath-applied (3 μm; n = 5) (Fig. 6) or injected directly into the terminal (1 mm at the pipette tip; n = 2)]. The rate of rise of the postsynaptic response in these experiments increased from 16.2 ± 4.3 mV/msec before injection to 24.1 ± 5.2 mV/msec after injection of BCL-xL (p < 0.005). In addition, the BCL-xL-induced percentage change in slope of the postsynaptic response in the presence of ruthenium red (93 ± 43%) did not differ from that in control medium (Fig. 3E). These results suggest that the actions of BCL-xL do not require calcium uptake by mitochondria.
Figure 6.

Ruthenium red does not block the enhancement of the postsynaptic response by injection of BCL-xL. The time course of the rate of rise of postsynaptic responses is shown in a synapse with strong basal transmission. The preparation was treated with ruthenium red (3 μm). Period of injection with BCL-xL (20 μm in pipette solution) is shown by a bar.
Work with non-neuronal cells has suggested that BCL-xL regulates the flux of metabolites across the outer mitochondrial membrane to facilitate transport of ATP into the cytosol after a death stimulus (Vander Heiden et al., 2000, 2001). To test whether the mechanism of action of BCL-xL on synaptic activity could involve an enhancement of the release of ATP from mitochondria, we measured the effect of direct microinjection of ATP on synaptic transmission. Although control injections of intracellular solution produced no enhancement of the rate of rise of postsynaptic potentials (n = 3) (Fig. 7A), pressure injection of ATP (20-200 mm in the pipette) into the presynaptic terminal effectively enhanced the postsynaptic responses (n = 7) (Fig. 7B). As with injections of BCL-xL, the latency of evoked action potentials was shortened by the enhanced postsynaptic potentials, and injection of ATP could restore transmission in synapses in which the postsynaptic potential no longer triggered action potentials (Fig. 7D). The effects of ATP injection were evident within 3-4 min of injection, and like the effects of BCL-xL, persisted for tens of minutes (Fig. 7C).
To test whether the actions of injected ATP could occlude those of BCL-xL, we injected BCL-xL into terminals that had previously been injected with ATP. No enhancement in the rate of rise of synaptic responses over those obtained with ATP alone could be detected (Fig. 7D) (n = 4). In two of four experiments, however, a small response to BCL-xL injection could be detected after the effects of ATP on the postsynaptic potential had abated. Thus, the ability of ATP to occlude the effects of BCL-xL is consistent with the possibility that BCL-xL may enhance synaptic activity by triggering release of ATP from mitochondria.
Discussion
BCL-xL is required for embryonic development of the mammalian nervous system, but unlike BCL-2, BCL-xL protein is abundantly expressed even in mature neurons, especially in the cortex and hippocampus (Frankowski et al., 1995). The role of BCL-xL in mature neurons is less obvious than in the developing nervous system, where apoptosis is more prevalent. In the adult nervous system, anti-apoptotic proteins may prevent neuronal death during insults such as ischemia. Nevertheless, the high levels of expression of these anti-apoptotic molecules in adult neural tissue suggest that they have other functions. Our findings that BCL-xL enhances synaptic responses and increases the rate of recovery from high-frequency neuronal activity provide a potentially important function for this molecule in neuronal plasticity and suggest that some BCL-2 family proteins may have actions distinct from their known cell death regulatory functions (Chen et al., 1997). Alternatively, the effects of BCL-2 family proteins on synaptic activity could also underlie or at least contribute to their effects on neuron survival. A recent study of BAK-deficient mice revealed that endogenous BAK protects mice from seizure-induced neuronal apoptosis and protects cultured neurons from excitotoxicity (Fannjiang et al., 2003). Consistent with the protective effect of BAK against seizure and excitotoxic agents, BAK-deficient neurons exhibited altered spontaneous and evoked neurotransmitter release.
Our results in the squid presynaptic terminal are consistent with the hypothesis that BCL-xL influences synaptic transmission through its actions on mitochondrial membrane conductance, although we cannot eliminate the possibility that it also acts on other, as yet uncharacterized, targets. The permeability of the outer mitochondrial membrane is regulated by at least two types of proteins, the BCL-2 family proteins and VDAC. VDAC is known to regulate the release of ions and metabolites, such as ATP, from mitochondria (Colombini et al., 1996; Rostovtseva and Colombini, 1996), and recent evidence suggests that BCL-xL may also control the flux of ATP and phosphocreatine across the outer membrane (Vander Heiden et al., 2000, 2001). Indeed, the permeability of the outer membrane to these substances may involve the concerted action of both classes of proteins. It has been shown that mitochondria isolated from cells undergoing apoptosis no longer have the ability to conduct ATP, ADP, or phosphocreatine through the outer membrane (Vander Heiden et al., 2000). Addition of recombinant BCL-xL to these mitochondria results in efficient incorporation of this molecule into the outer membrane and facilitates release of the trapped metabolites from the intermembrane space (Vander Heiden, 2001). Our finding that the actions of BCL-xL could be mimicked and occluded by direct injection of ATP into the presynaptic terminal suggests that a similar induction of ATP release across the outer membrane may account for the observed enhancement of synaptic transmission.
Our findings also indicate that a truncated pro-apoptotic fragment of BCL-xL produced some attenuation of synaptic transmission. The mechanism of action of this fragment is unknown but could be related to the finding that pro-apoptotic proteins are able to release large molecules (70 kDa) from lipid vesicles and produce increased permeability of the outer mitochondrial membrane to large molecules, such as cytochrome c, leading to a disruption of mitochondrial function (Basanez et al., 2001; Hardwick and Polster, 2002; Kuwana et al., 2002).
Brief tetanic stimulation of the presynaptic terminal causes synaptic depression (Swandulla et al., 1991), a phenomenon that can be attributed to the depletion of neurotransmitter-containing vesicles (Kusano and Landau, 1975; Heuser et al., 1979; Zucker, 1989). We found that BCL-xL increased the rate of recovery of synaptic responses from tetanic stimulation, but only if the tetanus was applied during low-frequency basal stimulation (0.033 Hz). At a higher basal rate of stimulation (2 Hz), during which substantial synaptic depression has already occurred (Kusano and Landau, 1975; Swandulla et al., 1991), recovery from a tetanus was rapid and unaffected by BCL-xL injection. In contrast, at the lower rate of basal stimulation, which itself produces very little or no synaptic depression, there appear to be two components of recovery: a rapid component and a slower component that recovers only over tens of seconds. Under these conditions, BCL-xL enhanced recovery. This difference could be explained if release at the high versus low basal frequencies is mediated by different subsets of vesicles with rates of recovery regulated by distinct biochemical mechanisms, as has been described previously for the calyx of Held synapse (Sakaba and Neher, 2001).
Previous work has clearly established a role for calcium uptake and re-release by mitochondria during high-frequency neuronal firing. For example, at many synapses, the rate of repletion of the readily releasable pool can be enhanced by an elevation of intracellular calcium levels (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998; Sakaba and Neher, 2001). Furthermore, posttetanic potentiation in many types of synapses has been attributed to elevated residual calcium levels produced by calcium uptake and prolonged release from mitochondria after the tetanus (Friel and Tsien, 1994; Tang and Zucker, 1997; Jonas et al., 1999). The uptake of calcium into mitochondria is tightly coupled to the production of ATP, and electrical stimulation of synaptosomes produces an increase in the rates of both glycolysis and respiration (De Belleroche and Bradford, 1972). Mechanistically, calcium uptake into the mitochondrial matrix that occurs during tetanic stimulation or steady plasma membrane depolarization (Friel and Tsien, 1994; Babcock and Hille, 1998; Kaftan et al., 2000) may increase the activity of mitochondrial enzymes, such as pyruvate dehydrogenase, TCA cycle enzymes, and electron transport proteins, resulting in an increase in the rate of production of ATP (McCormack et al., 1990; Sparagna et al., 1995; Zazueta et al., 1999).
An increase in the conductance of outer mitochondrial membranes alone could regulate the release of ATP or other metabolites by mitochondria during both low- and high-frequency firing in the absence of net mitochondrial calcium uptake. There are multiple ATP-dependent steps both in the preparation of vesicles for release and in the subsequent recycling of vesicular membranes (Faundez and Kelly, 2000; Heidelberger, 2001; Morgan et al., 2001; Tobaben et al., 2001). Although mitochondrial calcium uptake and release clearly represent potential mechanisms for modifying presynaptic transmitter release, our finding that the actions of BCL-xL on synaptic transmission could be detected in either the presence or absence of ruthenium red suggests that the uptake of calcium into the mitochondrial matrix is not required for this effect of BCL-xL.
The activity of BCL-2 family proteins is regulated by diverse signaling pathways and can involve phosphorylation, insertion into mitochondrial membranes, conformational changes, and proteolytic cleavage (Desagher et al., 1999; Griffiths et al., 1999; Martinou and Green, 2001; Wang, 2001). The factors that regulate the activity of BCL-xL at synaptic terminals are not yet known, but regulation of the activity or concentration of BCL-xL in the outer mitochondrial membrane may regulate the release of ions or metabolites that, in turn, influence the strength of neurotransmission. BCL-xL also protects neurons from apoptosis, an event that occurs at neuronal somata. Because most of the neuronal mitochondria are located in synaptic endings, at a distance from the soma, our findings also raise the possibility that BCL-xL and other BCL-2 family proteins may regulate the stability or retraction of a presynaptic ending, without influencing the survival of the entire neuron.
Footnotes
This research was supported by an American Heart Established Investigator Award (E.A.J.) and National Institutes of Health Grants NS 34702 (J.M.H.) and NS 18492 (L.K.K).
Correspondence should be addressed to Leonard K. Kaczmarek, Department of Pharmacology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: leonard.kaczmarek@yale.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238423-09$15.00/0
References
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC ( 1997) Inhibition of Bax channel-forming activity by Bcl-2. Science 277 : 370-372. [DOI] [PubMed] [Google Scholar]
- Augustine GJ ( 2001) How does calcium trigger neurotransmitter release? Curr Opin Neurobiol 11 : 320-326. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Hille B ( 1998) Mitochondrial oversight of cellular Ca2+ signaling. Curr Opin Neurobiol 8 : 398-404. [DOI] [PubMed] [Google Scholar]
- Basanez G, Nechushtan A, Drozhinin O, Chanturiya A, Choe E, Tutt S, Wood K, Hsu Y, Zimmerberg J, Youle RJ ( 1999) Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci USA 96 : 5492-5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, Zimmerberg J ( 2001) Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem 276 : 31083-31091. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID ( 2002) Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22 : 5840-5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blömer U, Kafri T, Randolph-Moore L, Verma IM, Gage FH ( 1998) Bcl-xL protects adult septal cholinergic neurons from axotomized cell death. Proc Natl Acad Sci USA 95 : 2603-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, González-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nûñez G, Thompson CB ( 1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74 : 597-608. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG ( 1996) A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem 66 : 403-411. [DOI] [PubMed] [Google Scholar]
- Calupca MA, Prior C, Merriam LA, Hendricks GM, Parsons RL ( 2001) Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. J Physiol (Lond) 532 : 217-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DF, Schneider GE, Martinou JC, Tonegawa S ( 1997) Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385 : 434-439. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EHY, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM ( 1998) Modulation of cell death by Bcl-xL through caspase interaction. Proc Natl Acad Sci USA 95 : 554-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M, Blachly-Dyson E, Forte M ( 1996) VDAC, a channel in the outer mitochondrial membrane. Ion Channels 4 : 169-202. [DOI] [PubMed] [Google Scholar]
- De Belleroche JS, Bradford HF ( 1972) Metabolism of beds of mammalian cortical synaptosomes: response to depolarizing influences. J Neurochem 19 : 585-602. [DOI] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J ( 2001) The channel of death. J Cell Biol 155 : 695-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC ( 1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 144 : 891-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG ( 1998) Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to the Purkinje cell synapse. J Neurosci 18 : 6147-6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Sappington AL, Dyke S, Kerr DA, Huganir RL, Jonas EA, Kaczmarek LK, Hardwick JM ( 2003) BAK affects neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell 4 : 575-585. [DOI] [PubMed] [Google Scholar]
- Faundez VV, Kelly RB ( 2000) The AP-3 complex required for endosomal synaptic vesicle biogenesis is associated with a casein kinase Iα-like isoform. Mol Biol Cell 10 : 2591-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowski H, Missotten M, Fernandez PA, Martinou I, Michel P, Sadoul R, Martinou JC ( 1995) Function and expression of the Bcl-x gene in the developing and adult nervous system. NeuroReport 6 : 1917-1921. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW ( 1994) An FCCP-sensitive store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]I J Neurosci 14 : 4007-4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci DR, Hlubek MD, Stuenkel EL ( 1999) Mitochondria regulate the Ca2+-exocytosis relationship of bovine adrenal chromaffin cells. J Neurosci 19 : 9261-9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Garcia M, Perez-Ballestro RP, Ling L, Duan L, Boise LH, Thompson CB, Nûñez G ( 1994) Bcl-xl is the major mRNA form expressed during murine development and its product localizes to mitochondria. Development 120 : 3033-3042. [DOI] [PubMed] [Google Scholar]
- González-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nunez G ( 1995) bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA 92 : 4304-4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA ( 1999) Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 144 : 903-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Polster BM ( 2002) Bax, along with lipid conspirators, allows cytochrome c to escape mitochondria. Mol Cell 10 : 963-965. [DOI] [PubMed] [Google Scholar]
- Heidelberger R ( 2001) ATP is required at an early step in compensatory endocytosis in synaptic terminals. J Neurosci 21 : 6467-6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L ( 1979) Synaptic vesicle endocytosis captured by quick freezing and correlated with quantal transmitter release. J Cell Biol 81 : 275-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ ( 1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 94 : 3668-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Knox RJ, Kaczmarek LK ( 1997) Gigaohm seals on intracellular membranes: a technique for studying intracellular ion channels in intact cells. Neuron 19 : 7-13. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Buchanan J, Kaczmarek LK ( 1999) Prolonged activation of mitochondrial conductances during synaptic transmission. Science 286 : 1347-1350. [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM ( 2002) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410 : 549-554. [DOI] [PubMed] [Google Scholar]
- Kaftan EJ, Xu T, Abercrombie RF, Hille B ( 2000) Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J Biol Chem 275 : 25465-25470. [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C ( 2003) Characterization of the signal that directs Bcl-xL, but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 160 : 53-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB ( 1998) Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 8 : 324-330. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD ( 1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275 : 1132-1136. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC ( 1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53 : 4701-4714. [PubMed] [Google Scholar]
- Krajewski S, Krajewska M, Shabaik A, Wang H-G, Irie S, Fong L, Reed JC ( 1994) Immunohistochemical analysis of in vivo patterns of bcl-x expression. Cancer Res 54 : 5501-5507. [PubMed] [Google Scholar]
- Kroemer G ( 1997) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3 : 614-620. [DOI] [PubMed] [Google Scholar]
- Kusano K, Landau EM ( 1975) Depression and recovery of transmission at the squid giant synapse. J Physiol (Lond) 245 : 13-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD ( 2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111 : 331-342. [DOI] [PubMed] [Google Scholar]
- Llinás RR ( 1999) The squid giant synapse: a model for chemical transmission. New York: Oxford UP.
- Martin R, Miledi R ( 1975) The presynaptic complex in the giant synapse of squid. J Neurocytol 4 : 121-129. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR ( 2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2 : 63-67. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM ( 1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70 : 391-425. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB ( 1997) Bcl-xL forms an ion channel in synthetic lipid membranes. Nature 385 : 353-357. [DOI] [PubMed] [Google Scholar]
- Mommsen TP, Hochachka PW ( 1981) Respiratory and enzymatic properties of squid heart mitochondria. Eur J Biochem 120 : 345-352. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Jin S, Augustine GJ, Lafer EM ( 2001) Uncoating of clathrin-coated vesicles in presynaptic terminals: roles for Hsc70 and auxilin. Neuron 32 : 289-300. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW ( 1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381 : 335-341. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Atwood HL ( 1994) Altered impulse activity modifies synaptic physiology and mitochondria in crayfish phasic motor neurons. J Neurophysiol 72 : 2944-2955. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Marin L, Atwood HL ( 1997) Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J Neurophysiol 78 : 281-294. [DOI] [PubMed] [Google Scholar]
- Pavlov EV, Priault M, Pietkiewicz D, Cheng EH-Y, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW ( 2001) A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol 155 : 725-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster BM, Kinnally KW, Fiskum G ( 2001) BH3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J Biol Chem 276 : 37887-37894. [DOI] [PubMed] [Google Scholar]
- Reed JC ( 1997) Double identity for proteins of the Bcl-2 family. Nature 387 : 773-776. [DOI] [PubMed] [Google Scholar]
- Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S ( 1987) Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab 7 : 752-758. [DOI] [PubMed] [Google Scholar]
- Rostovtseva T, Colombini M ( 1996) ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J Biol Chem 271 : 28006-28008. [DOI] [PubMed] [Google Scholar]
- Rowland KC, Irby NK, Spirou GA ( 2000) Specialized synapse-associated structures within the calyx of Held. J Neurosci 20 : 9135-9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E ( 2001) Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32 : 1119-1131. [DOI] [PubMed] [Google Scholar]
- Sanchez JA, Garcia MC, Sharma VK, Young KC, Matlib MA, Sheu S-S ( 2001) Mitochondria regulate inactivation of l-type Ca2+ channels in rat heart. J Physiol (Lond) 536 : 387-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendel SL, Montal M, Reed JC ( 1998) Bcl-2 family proteins as ion-channels. Cell Death Differ 5 : 372-380. [DOI] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin XM, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ ( 1997) Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic, BCL-2. Proc Natl Acad Sci USA 94 : 11357-11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI ( 2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol 5 : 28-37. [DOI] [PubMed] [Google Scholar]
- Sorgato MC, Keller BU, Stuhmer W ( 1987) Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature 330 : 498-500. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu S-S, Gunter TE ( 1995) Mitochondrial calcium uptake from physiological-type pulses of calcium. J Biol Chem 270 : 27510-27515. [DOI] [PubMed] [Google Scholar]
- Swandulla D, Hans M, Zipser K, Augustine GJ ( 1991) Role of residual calcium in synaptic depression and posttetanic potentiation: fast and slow calcium signaling in nerve terminals. Neuron 7 : 915-926. [DOI] [PubMed] [Google Scholar]
- Tang YG, Zucker RS ( 1997) Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18 : 483-491. [DOI] [PubMed] [Google Scholar]
- Tapia R, Velasco I ( 1997) Ruthenium red as a tool to study calcium channels, neuronal death and the function of neural pathways. Neurochem Int 30 : 137-147. [DOI] [PubMed] [Google Scholar]
- Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B ( 2001) A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31 : 987-999. [DOI] [PubMed] [Google Scholar]
- Tolbert LP, Morest DK ( 1982) The neuronal architecture of the anteroventral cochlear nucleus of the cat in the region of the cochlear nerve root: electron microscopy. Neuroscience 7 : 3053-3067. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB ( 2000) Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci USA 97 : 4666-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M ( 2001) Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem 276 : 19414-19419. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK ( 1998) High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394 : 384-388. [DOI] [PubMed] [Google Scholar]
- Wang X ( 2001) The expanding role of mitochondria in apoptosis. Genes Dev 15 : 2922-2933. [PubMed] [Google Scholar]
- Zazueta C, Sosa-Torres ME, Correa F, Garza-Ortiz A ( 1999) Inhibitory properties of ruthenium amine complexes on mitochondrial calcium uptake. J Bioenerg Biomembr 31 : 551-557. [DOI] [PubMed] [Google Scholar]
- Zucker RS ( 1989) Short-term synaptic plasticity. Annu Rev Neurosci 12 : 13-31. [DOI] [PubMed] [Google Scholar]