

Abstract
Opiate abuse causes adaptive changes in several processes of synaptic transmission in which the glutamatergic system appears a critical element involved in opiate tolerance and dependence, but the underlying mechanisms remain unclear. In the present study, we found that glutamate uptake in hippocampal synaptosomes was significantly increased (by 70% in chronic morphine-treated rats) during the morphine withdrawal period, likely attributable to an increase in the number of functional glutamate transporters. Immunoblot analysis showed that expression of GLT1 (glutamate transporter subtype 1) was identified to be upregulated in synaptosomes but not in total tissues, suggesting a redistribution of glutamate transporter expression. Moreover, the increase in glutamate uptake was reproduced in cultured neurons during morphine withdrawal, and the increase of uptake in neurons could be blocked by dihydrokainate, a specific inhibitor of GLT1. Cell surface biotinylation and immunoblot analysis showed that morphine withdrawal produced an increase in GLT1 expression rather than EAAC1 (excitatory amino acids carrier 1), a neuronal subtype, at the cultured neuronal cell surface, whereas no significant change was observed in that of cultured astrocytes. Electron microscopy also revealed that GLT1 expression was markedly increased in the nerve terminals of hippocampus and associated with the plasma membrane in vivo. These results suggest that GLT1 in hippocampal neurons can be induced to translocate to the nerve terminals and express on the cell surface during morphine withdrawal. The translocation of GLT1 at synapses during morphine withdrawal provides a neuronal mechanism for modulation of excitatory neurotransmission during opiate abuse.
Keywords: morphine, rat, hippocampus, glutamate transporter, GLT1, opiate withdrawal
Introduction
Opiate abuse causes long-lasting neural changes in the brain that underpin the behavioral abnormalities associated with cognitive deficits, tolerance, and dependence (Nestler and Aghajanian, 1997; Robbins and Everitt, 1999; Williams et al., 2001). Recently, a modification in neuronal plasticity at glutamatergic synapses has been suggested to account for the neural changes in drug abuse (Nestler, 2001). Importantly, increasing evidence demonstrates that opiates significantly alter glutamatergic synaptic transmission and neuronal plasticity in hippocampus (Haas and Ryall, 1980; Mansouri et al., 1997, 1999; Pu et al., 2002), a memory center possessing a high level of glutamatergic synaptic transmission. Additional studies also show that hippocampal glutamatergic transmission is functionally involved in withdrawal-like behavior mediated by μ-opioid receptors (Isaacson and Lanthorn, 1981; Hong et al., 1987, 1988) and in cocaine-seeking behavior (Vorel et al., 2001), suggesting that it may be one of the key systems involved in drug abuse. However, the molecular and cellular mechanism underlying the alteration of glutamatergic transmission by abused drugs is still unclear.
Several findings show that glutamate uptake may be involved in memory formation from vertebrate to invertebrate (Ng et al., 1997; Levenson et al., 2000; Maleszka et al., 2000) and is associated with induction of long-term potentiation (Levenson et al., 2002), suggesting that regulation of the glutamate transporter system may be a general component of plasticity at glutamate mediated synapses. Five subtypes of transporters EAAT1–EAAT5 (excitatory amino acids transporters 1–5) have been identified (Danbolt, 2001), three of which, GLAST [(glutamate/aspartate transporter) (EAAT1)] GLT1 [glutamate transporter subtype 1) (EAAT2)], and EAAC1 (EAAT3), are highly expressed in forebrain (Rothstein et al., 1994; Lehre et al., 1995) with differing cellular distributions. Although GLT1 and GLAST have been shown to be expressed in glial cells normally and EAAC1 in neurons, a neuron-expressed variant form of GLT1 has been found (Chen et al., 2002; Schmitt et al., 2002). Recent studies have shown that glutamate transporters play a critical role in the development of morphine tolerance, abnormal pain sensitivity, and withdrawal syndrome (Nakagawa et al., 2001; Mao et al., 2002), suggesting that glutamate transporters may contribute to the neural mechanisms of opiate abuse, but how the activity of glutamate transporters in brain is regulated during opiate abuse remains to be further investigated.
In the present study, we showed that glutamate uptake capacity in hippocampus synaptosomes was significantly increased during morphine withdrawal, and this was because of an upregulation of the number of glutamate transporters in the membrane. Furthermore, the increase in capacity was at least in part correlated to the translocation of neuron-expressed glutamate transporter GLT1 to nerve terminals and the induced surface expression of GLT1 at synapses. These results suggest that the glutamate transport mediated by neuronal glutamate transporter GLT1 may be inducible and significant in the altered excitatory neurotransmission during opiate abuse.
Materials and Methods
Preparations of rat brain tissue. Male Sprague Dawley rats (200–220 gm) were obtained from the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). Rats were housed in groups and maintained on a 12 hr light/dark cycle with food and water available ad libitum. All treatments were strictly in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were treated with morphine (10 mg/kg, s.c., twice per day at 12 hr intervals) or saline for 10 d (Trujillo and Akil, 1991; Fan et al., 1999; Pu et al., 2002). Then the rats were decapitated 12 hr after the last injection of morphine or 1 hr after morphine treatment (10 mg/kg, s.c.) plus naloxone (1 mg/kg, i.p.), and the brains were removed. The hippocampus, prefrontal cortex, cerebellum, and brainstem were rapidly dissected, and the subcellular fractions were prepared, respectively, according to the standard methods described previously (Ortiz et al., 1995). Brain samples were homogenized briefly in ice-cold 0.32 m sucrose and the following (in mm): 4 Tris, pH 7.4, 1 EDTA, and 10 glucose in a glass–Teflon homogenizer. Homogenates were centrifuged (900 × g for 10 min; 4°C), and the supernatants were spun at 9000 × g for 10 min in a microcentrifuge at 4°C. The pellets constituted the crude synaptosomal fraction. The crude fractions were resuspended in 1 ml of Krebs'–Ringer's–HEPES (KRH) medium containing the following (in mm): 120 NaCl, 4.7 KCl, 2.2 CaCl2, 25 HEPES, 1.2 MgSO4, 1.2 KH2PO4, and 10 glucose, pH 7.4, to give a protein concentration of ∼0.5 mg/ml that was determined by Bradford methods (Bradford, 1976). This suspension was used in the uptake assay and Western blot analysis described below.
Primary cell culture. Primary hippocampal cultures were prepared from 1 d postnatal Sprague Dawley rats using methods similar to those described previously (Mennerick et al., 1998; Wang et al., 1998a). Single cell suspensions were obtained by trituration in DMEM containing 10% horse serum, 10% fetal bovine serum (FBS), 17 mm d-glucose, 400 μm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells (150,000–200,000/ml) were plated onto poly-l-lysine-coated 24-well tissue culture plates. To obtain pure neuronal cultures, the culture medium was replaced at 4–6 hr later with Neurobasal-A medium for hippocampal neuronal culture containing B27 serum-free supplement (Invitrogen, Grand Island, NY). Twenty-four hours later, the cultures were treated with 5 μm cytosine arabinoside in vitro for 72 hr. The cultures were maintained in a humidified, 5% CO2 incubator at 37°C for 7 d and then were treated with morphine (0.01–10 nm) or saline for 7 d. The cultures for astrocytes were prepared as described previously (Swanson et al., 1997; Duan et al., 1999). In brief, the dissociated cells were plated in 24-well tissue culture plates in Eagle's MEM containing 10% FBS and 2 mm glutamine. At confluence (days 12–15), the cultures were treated for 48 hr with 10 μm cytosine arabinoside to prevent proliferation of other cell types, and the medium was replaced with MEM containing 2 mm glutamine, 3% FBS, and 0.15 mm dibutyryl cAMP to induce differentiation. The cultures were maintained in the medium containing dibutyryl cAMP and then treated with morphine at the 30th day. After 7 d of morphine treatment, the culture was used for glutamate uptake and biotinylation.
Glutamate uptake assay. Glutamate uptake of synaptosomes was initiated by adding 3H-glutamate (10 nm, 45.0 Ci/mmol; Amersham Biosciences, Buckinghamshire, UK) and 30 μm unlabeled glutamate in a final volume of 500 μl of KRH medium (Ullensvang et al., 1997). After incubation at 37°C for 5 min, the uptake was terminated by filtration on glass-fiber filters (Whatman, Maidstone, UK) using a tissue harvester under vacuum, and the filter was washed five times with 10 ml of cold nonradioactive KRH medium. Glutamate uptake of cultured neurons was determined according to the previous method (Duan et al., 1999). After 7 d morphine treatment, the morphine was washed out, the cells were preincubated with KRH medium for 60 min, and then the uptake assays were initiated in a way similar to that mentioned above. After 5 min incubation, the uptake was terminated by two ice-cold washes with 500 μl of KRH medium, followed by immediate lysis in ice-cold 0.1 N NaOH. Finally, filters containing synaptosomal particles or neuronal lysates were processed for scintillation counting (Beckman Instruments, Fullerton, CA). Nonspecific uptake was determined with sodium-free media that was prepared by replacing NaCl with choline chloride.
Biotinylation. Biotinylation of cell surface proteins was performed as described previously (Davis et al., 1998; Duan et al., 1999) and with some modifications. After morphine washout and 1 hr preincubation, the cultured cells on 10 cm tissue culture plates were rinsed twice with PBS–Ca 2+–Mg 2+ containing the following (in mm): 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, and 0.1 CaCl2, pH 7.3. The cells were then incubated in sulfo–N-hydroxysulfosuccinimide–biotin solution (1 mg/ml in PBS–Ca 2+/Mg 2+; Pierce, Rockford, IL) for 20 min at 4°C. Biotinylation was terminated by washing twice in a quenching solution of PBS–Ca 2+–Mg 2+, in which there was an equimolar substitution of 100 mm glycine for NaCl. This was followed by an additional 45 min of incubation in the quenching solution at 4°C. Quenching solution was removed, and the cells were lysed with 1 ml of radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors (100 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm iodoacetamide, and 250 mm phenylmethylsulfonyl fluoride plus 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mg/ml leupeptin, and 5 mg/ml aprotinin) for 1 hr at 4°C with vigorous shaking. The lysates were centrifuged at 14,000 gm for 15 min at 4°C. One hundred fifty microliters of the supernatant were taken for Western blot analysis as the whole-cell fraction. The rest of the supernatant (850 μl) was incubated with 150 μl of avidin bead suspension (Pierce) at 4°C overnight with gentle shaking. The avidin–lysate solution was then centrifuged for 15 min at 14,000 × g, and the supernatant was taken for Western blot analysis as the intracellular fraction. The pellet was washed four times with 1 ml of RIPA buffer and resuspended in 150 μl of loading buffer (62.4 mm Tris-HCl, pH 6.8, 2% SDS, 20% glycerol, and 5% 2-mercaptethanol) for 1 hr with gentle shaking at room temperature. After centrifugation for 15 min at 14,000 × g, the supernatant was taken for Western blot analysis as the biotinylated (plasma membrane) fraction.
Western blot analysis. The samples (0.5 mg/ml, 20 μl) from brain tissue or cultured cells of control and chronic morphine-treated groups were loaded, then subjected to 10% SDS-PAGE, and electroblotted onto nitrocellulose membrane using a minigel and mini transblot apparatus (Bio-Rad, Hercules, CA). The membranes were blocked in blocking buffer (20 mm Tris-HCl, pH 7.5, 137 mm NaCl, 0.1% Tween 20, and 15% nonfat milk) at room temperature for 1 hr. Then the membranes were incubated with antibodies of goat anti-GLAST (1.3 μg/ml; C-19 amino acids; Santa Cruz Biotechnology, Santa Cruz, CA) or goat anti-GLT1 (0.4 μg/ml; N-19 amino acids; Santa Cruz Biotechnology), or rabbit anti-EAAC1 (1:1000; provided by Dr. Jian Fei, Institute of Biochemistry and Cell Biology, Shanghai, China) or actin (1:10,000; Santa Cruz Biotechnology), respectively, overnight at 4°C. The blots were washed and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-goat secondary antibody for 1 hr at room temperature (Sigma, St. Louis, MO). Finally, the blots were visualized with enhanced chemiluminescence (Amersham Biosciences). For the quantification of the Western blot data, the developed films were scanned, the immunoreactive bands were digitized, and the densitometry was performed using Scion Image for Windows (Scion, Frederick, MD). The signal for each lane was calculated by summing the area × intensity of immunoreactivity (gray level of immunoreactive band–background level) of the discrete monomer and multimer bands produced by GLT1 and EAAC1 and normalized with internal control bands (actin).
In situ hybridization. The oligonucleotide probes for GLT1, 5′-CAG CCC GCC ACA TAC TGC TCC CAG GAT GAC ACC AAA CA-3′, were synthesized by Invitrogen on the basis of the rat GLT1 (GenBank accession number U15098). In situ hybridization was performed according to a previous method (Zhang et al., 1994b). The oligonucleotide was labeled at the 3′ end with [α- 35S]dATP (NEN, Boston, MA) using terminal transferase enzyme (Amersham Biosciences). Six rats per group were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and killed. The brains were removed, and the sections (14 μm) of hippocampus were cut in a cryostat. Slide-mounted sections were hybridized overnight at 42°C in a probe concentration of 40 ng/μl in hybridization buffer containing 40% deionized formamide, 3× SSC, 10% dextran sulfate, 5× Denhardt's solution, and 75–100 μg/ml salmon sperm DNA. Then the sections were washed in 1× SSC at 55°C for 30 min. After the washes, the sections were dehydrated and placed in a phosphate screen for 4–5 d, dipped in Hypercoat LM-1 emulsions (Amersham Biosciences), and exposed for periods of 3–5 weeks. After development in Kodak D19, the sections were counterstained with 1% toluidine blue, dehydrated and coverslipped, and photographed under bright-field illumination.
For quantitative analysis of the data, at least three rats were used in each group. GLT1 mRNA-positive neurons were identified as the cells (20–30 μm in diameter) that contained the number of silver grains threefold greater than the background in the pyramidal cell layer of CA1 and CA3 or granule cell layer of dentate gyrus (DG) region in bright-field micrographs. The GLT1 mRNA expression levels in neurons were determined by both the hybridization signals in the neurons and the percentage of positive neurons. A total of ∼300 neurons per hippocampal region were measured in six sections in each group.
Immunohistochemistry. The brains of rats were prepared for electron microscopy as described previously (Zhang et al., 1995, 1998). Twelve hours after termination of morphine or saline treatment, three rats per group were deeply anesthetized with sodium pentobarbital. The rats were perfused intra-aortically with 50 ml of warm saline (preincubated at 37°C), followed by 50 ml of warm mixture of 4% freshly depolymerized paraformaldehyde, 0.1% glutaraldehyde, 0.2% picric acid in 0.1 m phosphate buffer, pH 7.4, and 200 ml of the same ice-cold fixative for 5 min. The brains were removed and postfixed (1.5 hr, 4°C). The sections of hippocampus were cut at 50 μm with a vibrating slicer, and the sections in the same position were used. The sections were immersed in 20% sucrose solution for 2 hr and subjected to freeze–thaw treatment three times. Then the sections were incubated in antibodies against GLT1 (0.4 μg/ml) for 36 hr at 4°C, followed by biotinylated rabbit anti-goat IgG (1:200; Vector Laboratories, Burlingame, CA) and then stained with avidin–biotin–peroxidase complex (1:100; Vector ABC kit; Vector Laboratories). For the immunolabeling with gold–silver particles, rabbit antigoat IgG conjugated with 1.4 nm gold particles (1:100; Nanoprobes, Stony Brook, NY) were used to incubate for 2 hr and then enhanced with silver for 10 min (Nanoprobes). The sections for electron microscopy were postfixed in 1% osmium tetroxide at 4°C for 30 min. Dehydration was performed in increasing concentrations of ethanol. After passing through propylene oxide and incubation in Epon 812, the sections were flat embedded in Epon 812 between two sheets of plastic film. After polymerization, the sections were examined under light microscope to ensure that the identical area (1 × 1mm 2) of CA1, CA3, and DG in each group was selected for capsule embedding and ultrathin sectioning. Ultrathin sections were cut on an LKB (Bromma, Sweden) Nova ultratome and counterstained with uranyl acetate and lead citrate. These sections were examined in a Hitachi (Tokyo, Japan) H-600 electron microscope.
To determine the percentage of the GLT1-positive nerve terminals in the regions, the data from sections with the identical area of CA1, CA3, and DG in each group were selected, and immunolabeled nerve terminals that form synapses were counted in three sections from different rats in each group. For gold–silver particle quantification, the numbers of gold–silver particles in the nerve terminals and the membrane-associated gold–silver particles per synapse were calculated, respectively. Gold–silver particle associated with membrane was defined as actual contact with the plasma membrane; gold–silver particle in close proximity to the plasma membrane was considered to be intracellular. Synapses without gold–silver grains were eliminated.
Statistical analysis. The results were presented as mean ± SEM. Statistics differences were determined by Student's t test for two-group comparisons or ANOVA followed by Duncan's multiple range test for multiple comparisons among more than two groups.
Results
Increase in glutamate uptake in hippocampus during morphine withdrawal
To test the potential changes in the glutamate uptake machinery during opiate abuse, rats were injected with morphine (10 mg/kg, s.c.) twice per day for 10 d, a procedure known to produce significant morphine tolerance and dependence (Trujillo and Akil, 1991; Fan et al., 1999). The crude synaptosomal fractions of rat brain were prepared for glutamate uptake analysis after cessation of morphine treatment. We found that the glutamate uptake in the synaptosomal fraction of hippocampus was markedly increased rather than some other regions, such as prefrontal cortex, cerebellum, and brainstem, 12 hr after cessation of 10 d of morphine treatment (Fig. 1A,B). The increase in glutamate uptake required chronic exposure of morphine and was not observed in acute morphine-treated rats 12 hr after morphine injection (Fig. 1C). After the termination of the 10 d morphine treatment, glutamate uptake was increased to the highest level (170% of the control level) 12 hr later and then recovered gradually (Fig. 1D). Reexposure of the animals to morphine (10 mg/kg) at 12 hr time point restored the increased glutamate uptake to the normal level 1 hr later, which was compared with the animals treated with saline (Fig. 1E). Moreover, when the morphine-treated rats were treated with morphine plus naloxone (1 mg/kg, i.p.), a nonspecific opioid receptor antagonist documented to lead to morphine withdrawal (Trujillo and Akil, 1991; Fan et al., 1999), an increase in glutamate uptake was observed 1 hr later (Fig. 1E). This suggested that the change of glutamate uptake in chronic morphinetreated rats correlated to morphine withdrawal after termination of 10 d of morphine treatment.
Figure 1.

Increase of glutamate uptake in hippocampal synaptosomes during withdrawal in chronicmorphine-treatedrats. The results were presented as mean±SEM of four to six rats per group, each tested by triplicate reconstitutions. *p < 0.05 and **p < 0.01 compared with the saline group. A, B, Effect of chronic morphine treatment (Mor) and saline treatment (Sal) on different brain regions. After subcutaneous injection of morphine (10 mg/kg) twice per day at 12 hr intervals for 10 d, the glutamate uptake in the synaptosomal particles of hippocampus (Hip) was increased significantly, whereas that of other regions such as prefrontal cortex (PC), cerebellum (Cer), and brainstem (BS) were decreased. C, Glutamate uptake in synaptosomal particles of hippocampus was increased time dependently after 1,5,10 dmorphine exposure.D, The glutamate uptake was increased to the highest level 12 hr after the termination of morphine treatment and then decreased gradually 24, 36, and 48 hr later. E, Reexposure of the animals to morphine (10 mg/kg) at the 12 hr time point restored the increased glutamate uptake to the normal level 1 hr later, and the increase was initiated again by additional treatment of naloxone (Nal) (1 mg/kg).
To distinguish between increases in affinity and the number of functional transporters, Km and Vmax were determined. Morphine withdrawal increased the Vmax from 332 ± 17.8 to 586 ± 62.3 pmol · mg-1 · min-1 but did not induce any significant changes in Km values, which were 20.8 ± 3.21 and 22.4 ± 6.36 μm for the control and morphine-treated groups, respectively (Fig. 2A). This indicated that the change in glutamate uptake was likely attributable to the increased number of functional transporters.
Figure 2.
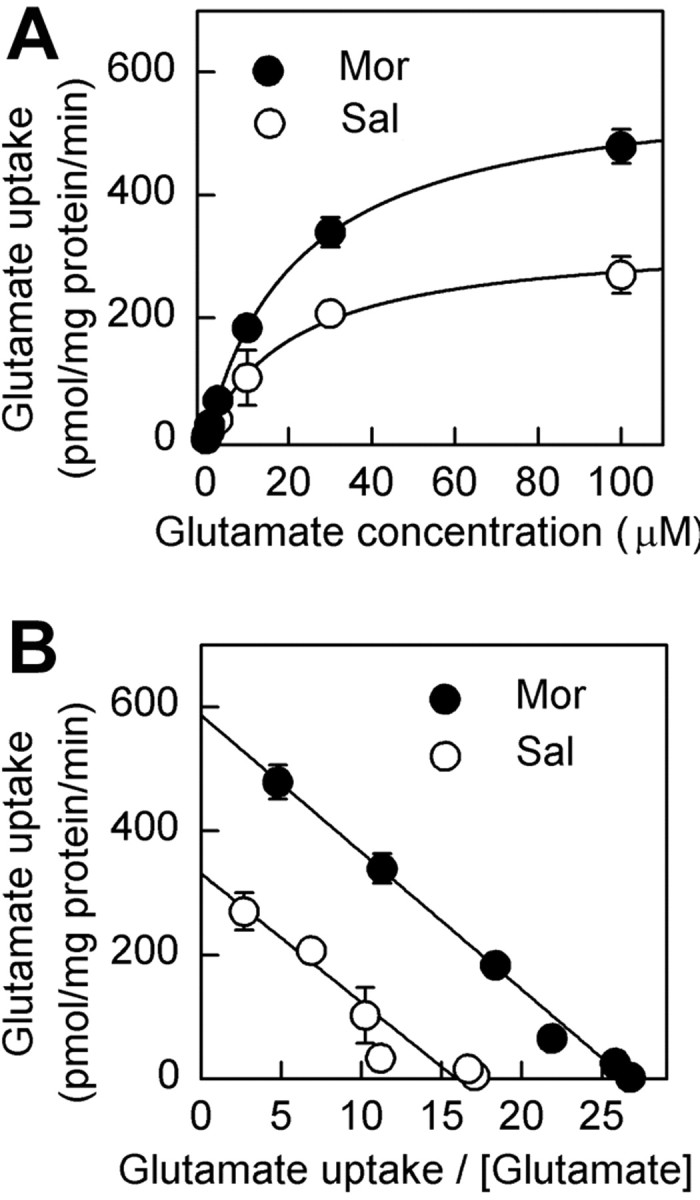
Saturation analysis for glutamate uptake during morphine withdrawal by incubating the synaptosomal fractions in different glutamate concentrations. A, Glutamate uptake capacity was increased in synaptosomes during morphine withdrawal. The experiment was replicated for three times, and data were presented as mean±SEM of replicate samples in total (n = 6). B, Data as shown in Eadie-Hofstee transformations indicate an increase in Vmax of glutamate uptake. Sal, Saline treatment; Mor, morphine treatment.
Differential regulation of the expression of glutamate transporter subtypes in hippocampus
To further pinpoint the subtype of glutamate transporters, antibodies of anti-GLAST, anti-GLT1 (N terminal), and anti-EAAC1 were used to detect the amount of protein expressed in hippocampus. As shown in Figure 3A, the antibodies of anti-GLAST and anti-GLT1 each produced bands at ∼66 and ∼220 kDa in hippocampal samples. It is likely that the higher-mass bands represent a multimeric form of the proteins (Duan et al., 1999). The specificity of the two antibodies was tested by preabsorption of antibody with the peptide antigen (Santa Cruz Biotechnology), respectively, and, in each case, the peptide completely blocked the appearance of the bands at 66 and 220 kDa (Fig. 3A). The protein samples from hippocampus and HEK 293 cells expressing EAAC1 protein were also immunoblotted with anti-EAAC1 and anti-GLT1 antibodies, respectively, and the anti-GLT1 antibody could not recognize EAAC1 protein expressed in HEK 293 cells (Fig. 3B).
Figure 3.
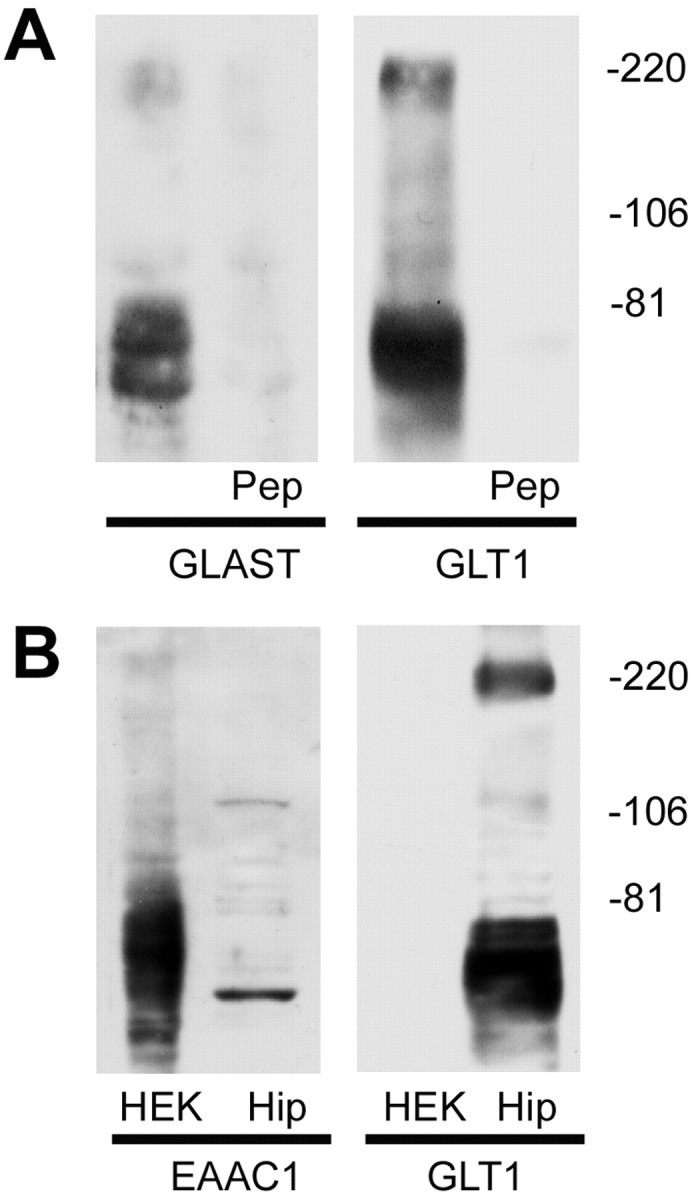
Immunoblot characterization of glutamate transporters antibodies. A, Protein samples from the hippocampus of adult rat were analyzed by immunoblot analysis using anti-GLAST (left) and anti-GLT1 (right), respectively. Both of the antibodies recognized bands at ∼66 and ∼220 kDa. Immunoreactivity was completely abolished when each antibody was preabsorbed with 90 μm of the peptide antigen (Pep), respectively. Control experiments were done with heterologous peptides (anti-GLT1 antibody preabsorbed with GLAST peptide and anti-GLAST antibody preabsorbed with GLT1 peptide), which had no effect on the immunoreactivity in hippocampal sample immunoblots. B, Immunoblot analysis showed that the anti-GLT1 antibody could not recognize EAAC1 protein. Protein samples from HEK 293 cells expressing EAAC1 protein (HEK) and hippocampus (Hip) were subjected to SDS-PAGE and immunoblotted with the antibodies of EAAC1 and GLT1, respectively. Antibody to EAAC1 recognized bands in EAAC1 cDNA-transfected HEK 293 cells as well as in rat hippocampal tissue (left), whereas GLT1 antibody only recognized the bands in rat hippocampal tissue (right). The immunoblots were representative of three independent experiments.
Then we examined the changes of the protein level of glutamate transporter subtypes in the hippocampal synaptosomes 12 hr after cessation of morphine. Among three subtypes, the amount of expressed GLT1 was significantly increased in synaptosomal particles in the morphine group compared with that of the saline group, whereas no significant change in GLAST or EAAC1 was observed under the same conditions (Fig. 4). Moreover, no change of GLT1 expression was seen in total lysates of tissue sample during morphine withdrawal, suggesting that the increase of GLT1 expression in synaptosomes might not be attributable to an upregulation of GLT1 in total content.
Figure 4.
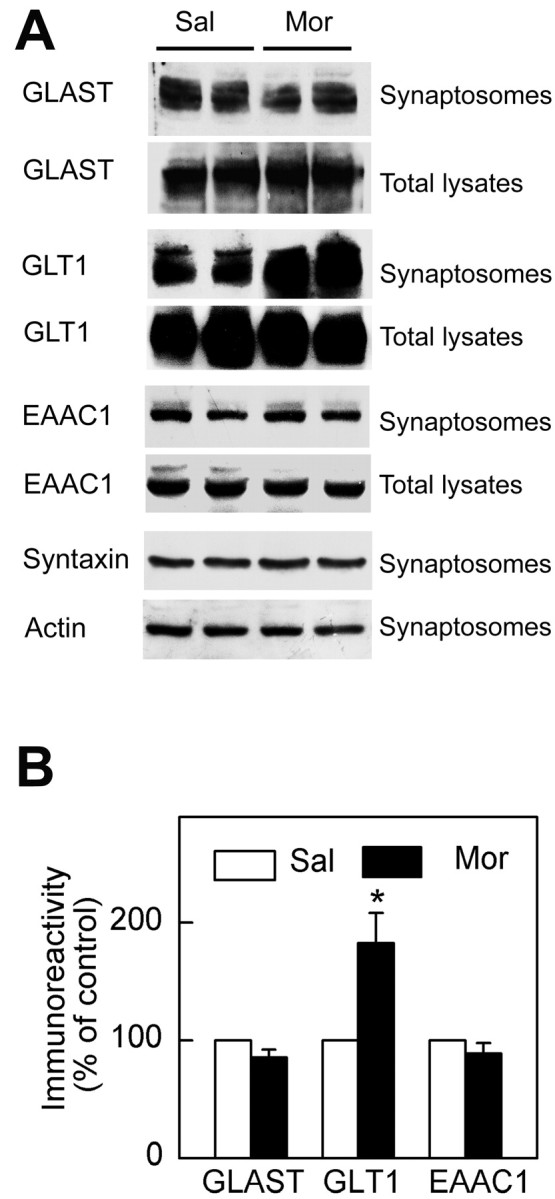
Differential regulation of glutamate transporter subtypes in hippocampus after morphine withdrawal. A, During morphine withdrawal, the synaptosomal protein levels of GLT1 (n = 6) were significantly increased in hippocampus compared with the saline control group rather than that of GLAST (n = 5) and EAAC1 (n = 5). The monomer bands at ∼66 kDa were presented for each subtype. No significant changes were observed in the expression of either syntaxin or actin in synaptosomal fractions. B, The density of glutamate transporter immunoreactivity of synaptosomal fractions of morphine-treated rats was expressed as a percentage of that of control group (mean±SEM). *p < 0.05 compared with the saline group. Sal, Saline treatment; Mor, morphine treatment.
Upregulation of glutamate uptake and surface expression of GLT1 in cultured neurons
Two isoforms of GLT1, GLT1a and GLT1b, varying in the C terminus are identified in brain so far (Chen et al., 2002; Reye et al., 2002; Schmitt et al., 2002), and both isoforms may be detected by the anti-GLT1 antibody used in our study. GLT1a is primarily expressed in astrocytes (Rothstein et al., 1994; Lehre et al., 1995), whereas GLT1b is shown to express in both neurons and astrocytes recently (Chen et al., 2002; Schmitt et al., 2002). Thus, it was essential to determine whether the increase in glutamate uptake was attributable to the contamination of astrocytes in the synaptosomal preparation (Danbolt, 2001) or really occurred in nerve terminals. Next, pure hippocampal neurons and differentiated astrocytes by dibutyryl cAMP, respectively, were cultured according to the previous studies (Mennerick et al., 1998; Wang et al., 1998a), and both types of cells were reported to express GLT1 protein and to reuptake glutamate in the presence of Na+ (Swanson et al., 1997; Chen et al., 2002). Then the cell cultures were subjected to chronic morphine treatment (0.01–10 nm).
As shown in Figure 5, an increase of glutamate uptake was observed in the neurons 1 hr after morphine withdrawal, and this increase was dose dependent and time dependent on chronic morphine treatment (Fig. 5A,B). We also tested the changes in glutamate uptake in cultured neurons 0–2 hr after morphine (10 nm) washout, and the glutamate uptake in the 1 hr time point was the highest (data not shown). In the astrocytes, however, no significant change could be observed in the morphine-treated group (Fig. 5A,B). The glutamate uptake in the astrocytes could be blocked by l-trans-pyrrolidine-2, 4-dicarboxyic acid (PDC) (Tocris Cookson, Bristol, UK), a nonselective glutamate transporter blocker, but not by dihydrokainate (DHK) (Tocris Cookson), a specific inhibitor of GLT1 (Fig. 5C). This is consistent with previous studies that glutamate uptake in astrocytes is primarily mediated by GLAST subtype (Swanson et al., 1997). In contrast, the glutamate uptake observed in the neurons of both groups could be blocked by treatment of either PDC or DHK (Fig. 5D), suggesting possible involvement of neuronal GLT1 in the glutamate uptake.
Figure 5.

Effect of morphine withdrawal on glutamate uptake in chronic morphine-treated hippocampal neurons and astrocytes. A, B, After morphine withdrawal, the increase in glutamate uptake in cultured neurons was morphine dose (A) and time (B) dependent, whereas no significant change was seen in cultured astrocytes. The glutamate uptake in the morphinetreated group was presented as percentage of that of control group. C, Glutamate uptake in cultured astrocytes could be blocked by PDC but not by DHK treatment and was not significantly altered during morphine withdrawal after 7 d morphine (10 nm) treatment. D, Glutamate uptake in cultured neurons was increased after morphine withdrawal and could be blocked by either PDC or DHK treatment. Glutamate, 10 μm. The control rates in the cultured astrocytes and neurons were 1070 ± 93 and 748 ± 48 pmol · mg -1 · min -1 protein, respectively. The data were presented as mean ± SEM of three independent experiments performed in duplicate. Sal, Saline treatment; Mor, morphine treatment.
The question asked then was whether the increase in glutamate uptake in cultured neurons was related to an expression of GLT1 on the cell surface. As an essential control, the possible changes of EAAC1 expression during morphine withdrawal should also be examined considering its expression in the neurons. On the other hand, it is proved that glutamate uptake in astrocytes is mediated by GLAST but not GLT1 (Swanson et al., 1997), so transport studies in astrocytes do not reflect changes in GLT1. Therefore, immunoblot analysis of membrane fraction from cultured astrocytes is also required to test translocation of GLT1.
The effects of morphine withdrawal are apparent on the Western blots after surface biotinylation shown in Figures 6 and 7. The blots were probed with anti-actin antibody and with antibodies to transporter. Morphine withdrawal produced no significant change in biotinylated (cell surface) or total cell GLT1 expression in cultured astrocytes (Fig. 6). We also tested the effect of morphine on undifferentiated astrocytes without dibutyryl cAMP treatment, in which GLT1 protein is shown to be undetectable (Swanson et al., 1997). The GLT1 protein was not seen in either the saline-treated or morphine-treated group (data not shown), which suggested that chronic morphine could not increase the GLT1 expression in the astrocytes. In contrast to astrocytes, however, in cultured neurons during morphine withdrawal, an increase in biotinylated GLT1 protein and a decrease in nonbiotinylated (intracellular) GLT1 were observed. There was no significant change in total GLT1 protein (Fig. 7). In addition, no change in EAAC1 was observed in cultured neurons under the same conditions.
Figure 6.
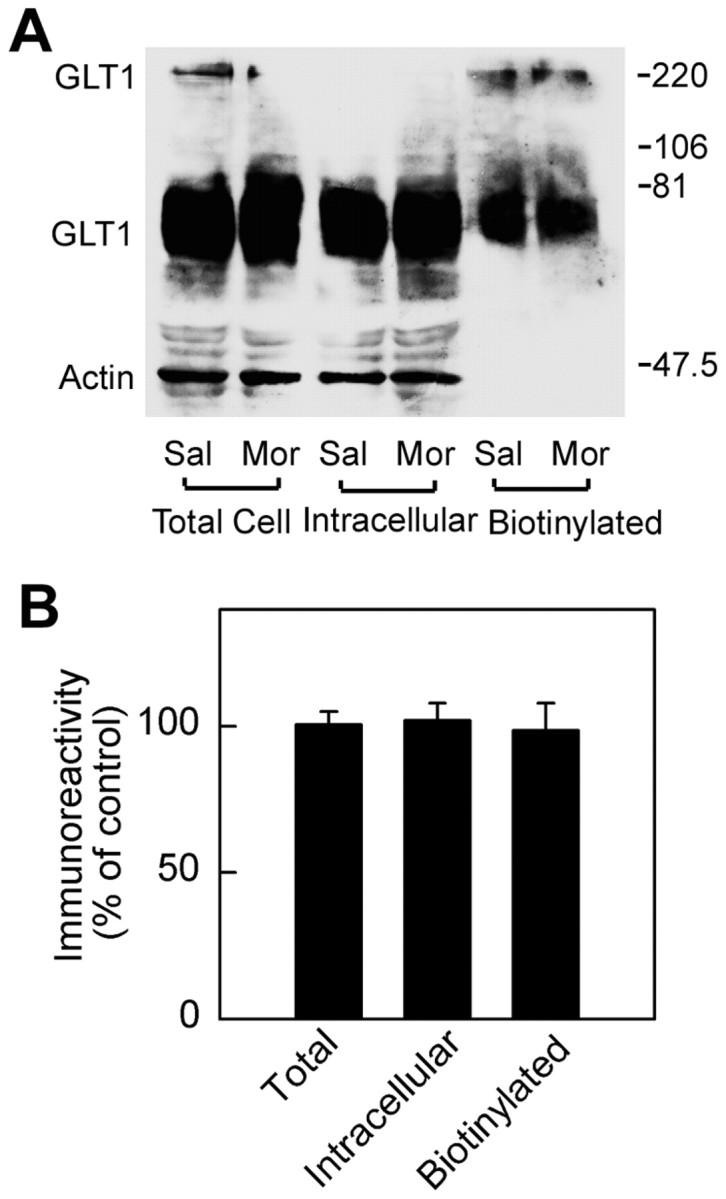
Analysis of GLT1 expression in cultured astrocytes during morphine withdrawal. A, After 7 d morphine treatment, the culture medium containing morphine was withdrawn. The astrocytes were preincubated with 0.01m PBS for 1 hr and were then biotinylated. Westernblots were probed with antibodies to GLT1 and actin. The actin bands provided an index of intracellular proteins. B, Quantitation of immunoblot of GLT1 bands was pooled from three independent experiments. GLT1 immunoreactivity values were normalized to actin in the lysate fraction. The data were expressed as a percentage of the saline-treated group for each fraction (mean ± SEM). There was no significant change in total cell or cell surface GLT1 expression. Sal, Saline treatment; Mor, morphine treatment.
Figure 7.

Analysis of GLT1 and EAAC1 expression in cultured neurons during morphine withdrawal. After 7 d morphine treatment, the culture medium containing morphine was withdrawn. The cultured neurons were preincubated with 0.01 m PBS for 1 hr and were then biotinylated. A, Western blots were probed with antibodies to GLT1 and actin. B, The membranes were then striped and reprobed to EAAC1 and actin. The actin bands provided an index of intracellular proteins. The immunoblot of GLT1 and EAAC1 bands was pooled from three independent experiments. Sal, Saline treatment; Mor, morphine treatment.
Translocation and surface expression of GLT1 at nerve terminals in vivo during morphine withdrawal
A previous study with Northern blot has shown that the total mRNA of GLT1 is upregulated in several brain regions such as striatum and thalamus but not in hippocampus during morphine withdrawal (Ozawa et al., 2001). However, whether distribution of GLT1 mRNA is also unchanged at the cellular level in hippocampus neurons needs to be further clarified. Therefore, we used in situ hybridization to further examine the possible change of GLT1 mRNA levels in hippocampal neurons during morphine withdrawal. In the slice counterstained with toluidine blue, the neuronal cell bodies were large in size (∼20–30 μm in diameter) and distributed mainly in the pyramidal cell layer in the CA1 and CA3 regions or in the granule cell layer in the DG region. As shown in Figure 8, these neurons contained GLT1 mRNA, and the number of GLT1 mRNA-positive neurons in CA3 regions was more than that in the CA1 or DG regions (Fig. 8A,C), which was consistent with the previous studies (Schmitt et al., 1996; Sutherland et al., 1996). However, no obvious change in the intensity of in situ hybridization signal was detected in GLT1 mRNA-positive neurons (Fig. 8 A,B,D), nor did the change of the percentage of the GLT1 mRNA-positive neurons in the regions of the morphine-treated group (Fig. 8C) compared with that in the control group.
Figure 8.
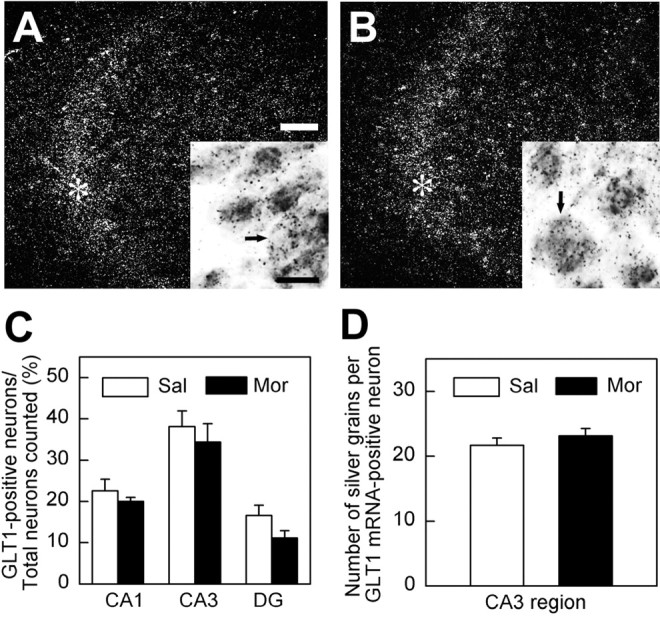
Analysis of GLT1 mRNA in hippocampal neurons during morphine withdrawal, as revealed by in situ hybridization. Dark-field micrographs showed GLT1 mRNA distribution in hippocampal CA3 regions in the saline control group (A) and in the morphine group (B). Brightfield micrographs in the insets of A and B presented the positive pyramidal neurons containing GLT1 mRNA (arrows). Asterisks in the dark field of saline group (A) and morphine group (B) indicated the region of these neurons (arrows) located. Scale bars: A, B, 100 μm; insets in A, B, 20 μm.C, The percentage of GLT1-positive neurons in the pyramidal cell layer of CA1 and CA3 or granule cell layer of DG region was quantified, respectively, and no significant change was observed. The results were pooled from the sections (n = 4–6) of three animals in each group. D, The number of silver grains contained in GLT1-positive neurons in CA3 regions was not significantly changed during morphine withdrawal (n = 124 in control group; n = 118 in morphine-treated group). Sal, Saline treatment; Mor, morphine treatment.
The present immunoblot studies demonstrated that GLT1 protein expression was upregulated in the synaptosomal particles and on the cell surface of cultured neurons during morphine withdrawal. However, the possibility still existed that the isolated synaptosomes may be contaminated by astrocytes, and the cell surface expression of GLT1 in cultured neurons was possibly caused by some aspects of the culture procedure in vitro. Then we used both preembedding immunoperoxidase and immunonanogoldsilver labeling to localize GLT1 in vivo and examined the labeling by using electron microscopy. In CA1 and CA3 regions of hippocampus of controlrat, the intensive immunolabeling of GLT1 was primarily seen in astrocytes and their processes (Fig. 9B,C). GLT1 labeling was also seen in the cytoplasm of a few nerve terminals but only occasionally associated with the cell surface of the nerve terminals (Fig. 9C,G,H). Twelve hours after morphine treatment, the intensity of GLT1 labeling in the nerve terminals was increased markedly (Fig. 9D–F). It is striking that gold–silver labeling of GLT1 was often seen on the cell surface of nerve terminals and could associate with the presynaptic membrane (Fig. 9F,H). The quantification of the GLT1 immunolabeling showed that the number of GLT1-positive nerve terminals was increased from ∼1 to ∼10% in both CA1 and CA3 regions (Fig. 9G). The number of gold–silver particles in axonal terminals or dendrites and the number of cell surface-associated gold–silver particles per GLT1-positive synapse were also significantly increased in the morphine-treated group compared with the saline group (Fig. 9H). These results demonstrate that there is indeed an apparent increase in GLT1 protein level and surface expression in nerve terminals after morphine withdrawal.
Figure 9.
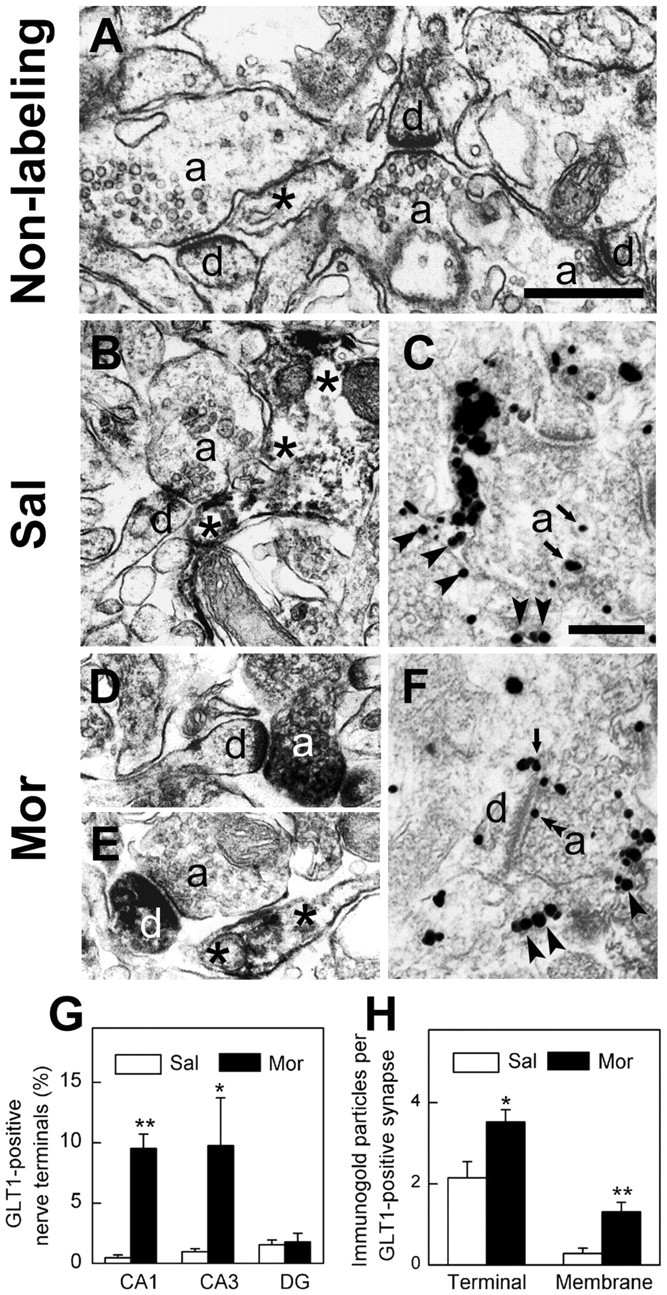
Translocation and surface expression of GLT1 at hippocampal nerve terminals after morphine withdrawal as revealed by electron microscopy. The morphological results of the CA3 region of the hippocampus were presented as representatives in A–F. A, The ultrastructure without immunolabeling of hippocampal CA3 regions, in which synapses were formed by axonal terminals (a) and dendrites (d). A process of astrocyte was indicated with an asterisk. B, A GLT1-positive astrocyte (asterisks) was present to be juxtaposed to a synapse in the CA3 region of the saline group.C, Anaxonal terminal (a) containing gold–silver particles for GLT1 labeling was seen adjacent to a GLT1-positive astrocyte process (arrowheads) in the saline group. The gold–silver particles (arrows) in the axonal terminal were in the cytoplasm.D, AGLT1-positive axonal terminal(a) made synaptic contact with an unlabeled dendrite(d) in the morphine-treated group.E, Near a GLT1-positive as trocyte(asterisks), an unlabeled axonal terminal (a) formed a synapse with a GLT1-positive dendrite (d) in the morphinetreated group. F, A hippocampal axonal terminal (a) containing GLT1 labeling was shown close to a GLT1-positive astrocyte (arrowheads). Arrow points to gold–silver particle located in the plasma membrane of a dendrite. Double arrowheads point to the labeling in the presynaptic membrane. Scalebars:(inA) A,B,D,E, 1 μm; (in C) C,F, 0.5 μm. G, Quantitative analysis showed that the number of GLT1-positive nerve terminals was increased in both CA1 and CA3 region but not in DG region. The data were pooled from three sections of different rats in each group. The total numbers of terminals counted in CA1, CA3, and DG regions: 496, 519, and 381 in the saline group; 428, 494, and 326 in the morphine group. H, The number of gold–silver particles in nerve terminals that form synapses (Terminal) as well as associated with the plasma membrane per GLT1-positive synapse(Membrane) were quantified in the saline (n=28) and morphine-treated (n=110) groups, respectively. Both particle numbers were significantly increased in the morphine-treated group compared with the control group. *p < 0.05 and **p < 0.01 compared with the saline group. Sal, Saline treatment; Mor, morphine treatment.
Discussion
It is known that opiate abuse exerts extensive adaptive changes in brain functions, including many aspects of neurotransmission, such as transmitter release during morphine withdrawal (Manzoni and Williams, 1999), postsynaptic receptor activation, and subsequent excitatory synaptic transmission after chronic morphine treatment (Martin et al., 1999a,b). The present study also supported the previous studies and revealed that glutamate uptake and the expression of GLT1, a glutamate transporter subtype in hippocampal synaptosomes, were significantly upregulated during morphine withdrawal. The longer time course in synaptosomes after morphine termination may be attributable to the long-lasting time for the absorption and metabolism of morphine in vivo and the long-term response of morphine or its metabolites in brain (Stain et al., 1995). The increase in glutamate uptake was further reproduced in cultured neurons treated with morphine, and the time course was in agreement with the increase observed 1 hr after naloxone-initiated morphine withdrawal in synaptosomes, suggesting that the alteration in glutamate uptake may be primarily attributable to a direct effect of morphine on hippocampal neurons. Considering a great number of active transporters at plasma membrane of the astrocytes that is adjacent to synapses, it is still difficult to completely exclude the possibility that astrocytes may also contribute to the increased glutamate uptake. However, on the basis of the finding that GLT1 expression was markedly increased in the plasma membrane viewed by both cell surface biotinylation in cultured neurons and electron microscopy in vivo, it is reasonable to propose that there is a greater number of GLT1 active in the plasma membranes during morphine withdrawal, and at least some of this increase is neuronal.
Among the subtypes of glutamate transporters identified, GLT1 is by far the major transporter of the forebrain because, in GLT1 knock-out mice, <6% glutamate transporter activity remains in synaptosomes compared with wild-type animals (Tanaka et al., 1997). Lack of GLT1 causes selective neuronal degeneration (Tanaka et al., 1997; Rao et al., 2001a,b) and impairment of the long-term potentiation induction (Katagiri et al., 2001) in hippocampus. A recent study has demonstrated that chronic morphine induces downregulation of spinal glutamate transporters GLAST and EAAC1 (Mao et al., 2002), but GLT1 is not examined in the study. Our present data demonstrated that GLT1 could be regulated in brain, whereas the other subtype remain unchanged, suggesting that there may exist different pathways in regulation of these transporters in brain. In two GLT1 isoforms identified, GLT1a is originally found and has been shown to express in astrocytes (Rothstein et al., 1994; Lehre et al., 1995). GLT1b is demonstrated to express in both astrocytes and neurons recently (Chen et al., 2002; Schmitt et al., 2002). Both variant forms might be recognized by the anti-GLT1 antibody used in the present study, but a previous study (Lehre et al., 1995) failed to detect GLT1 immunolabeling in neurons with electron microscopy by using several different antibodies against different regions of GLT1 shared by two forms. There are three considerations for the difference between the present finding and previous data. First, antibodies against different regions of a molecule may produce very different patterns of labeling or sensitivity, which was mentioned in the previous study (Chen et al., 2002). Second, the freeze–thaw procedure (Zhang et al., 1995, 1998) may lead to a greater penetration of antibody, which would enhance the sensitivity of the immunoelectron microscopic methods. Third, immunonanogold labeling and the silver enhancement method may provide higher labeling density, better sensitivity, and greater penetration into tissues (Hainfeld and Powell, 2000). According to our results, the upregulation of GLT1 during morphine withdrawal is unlikely mediated by GLT1a because GLT1a protein has not been found in neurons in the normal adult brain (Schmitt et al., 2002), and the mRNA of GLT1 in hippocampal neurons is also unchanged in the morphine group.
It has been shown that GLT1 in neurons is not typically associated with the plasma membrane (Chen et al., 2002). We found that the amount of GLT1 labeling in the plasma membrane was significantly increased in the nerve terminals of the morphine group, whereas in the control group, the gold–silver particles for GLT1 labeling were constantly distributed in cytoplasm, which was consistent with the previous study. This is in good agreement with our result of cell surface biotinylation in cultured neurons, showing a significant increase in the cell surface expression of GLT1 protein during morphine withdrawal. The interpretation of this finding is that most of GLT1 in neurons may be normally stored in the cytoplasm until mobilized to the cell surface during morphine withdrawal. Furthermore, both in situ hybridization in the present study and Northern blot analysis in the published study (Ozawa et al., 2001) indicate that this is not attributable to the increase of the mRNA level of GLT1 in hippocampus. This is also consistent with the unchanged protein level of GLT1 in either hippocampal tissues or the cultured neurons during morphine withdrawal. Thus, these results suggest that translocation of glutamate transporters to the surface of nerve terminals may be critical for GLT1 to increase the neuronal glutamate transport during morphine withdrawal.
The cellular mechanisms of morphine withdrawal-induced surface expression of GLT1 in neurons remain to be investigated. It is possible that GLT1 could be regulated via protein kinasemediated phosphorylation because GLT1 possesses quite a few putative PKC and PKA phosphorylation sites in its intracellular loops (Kanner, 1993; Gegelashvili and Schousboe, 1997), and GLT1 phosphorylation mediated by PKC activation induces a significant enhancement of glutamate uptake (Casado et al., 1993). The upregulation of PKC and PKA activity is also shown during opiate withdrawal in previous studies (Ventayol et al., 1997; Escriba and Garcia-Sevilla, 1999; Pu et al., 2002), which might promote the phosphorylation of GLT1 and, thus, induce its translocation to the synaptic surface. Another possibility is that GLT1 expression could be regulated by extracellular glutamate directly, because glutamate-induced upregulation of transport activity mediated by membrane translocation of glutamate transporters has been reported previously (Duan et al., 1999). This possibility is supported by our unpublished observation that upregulation of glutamate release probability occurred in hippocampus during morphine withdrawal. Although the solid evidence to support the possibilities is not adequate so far, these hypotheses merit future investigation.
In summary, the present study revealed an increase of glutamate uptake in hippocampal synaptosomes and provided in vivo evidence showing upregulation and surface expression of glutamate transporters at nerve terminals during morphine withdrawal. The observed accumulation of glutamate transporters at the nerve terminals leads to several functional considerations. It is known that opiate withdrawal causes a significant increase in extracellular glutamate concentration in many brain regions (Aghajanian et al., 1994; Zhang et al., 1994a; Sepulveda et al., 1998), which may affect neurotransmission. The translocation of GLT1 to the nerve terminals and the induced surface expression of GLT1 might efficiently remove the excessive glutamate to maintain the normal synaptic functions. In fact, neuronal GLT1 is demonstrated to be required to protect cultured neurons from the excessive amount of extracellular glutamate (Wang et al., 1998a,b). Therefore, the present results provided evidence for neuronal GLT1 trafficking that possibly influenced the modulation of the excitatory transmission during opiate abuse.
Footnotes
This work was supported by Ministry of Science and Technology Grants G1999053907 and G2000077800, Chinese Academy of Sciences Grants KSCX2-2, KSCX1-SW-11, and KSCX2-SW-204, the National Natural Science Foundation of China Grants 39840160 and 30024003, Shanghai Science and Technology Committee Grant 018014015, and the K. C. Wong Education Foundation of Hong Kong. We thank Drs. Shi-Gang He and Lin-Yin Feng for their helpful suggestions, Dr.Jian Fei for providing EAAC1 antibodies, and Ya-Lan Wu, Peng Xia, Zhu Wang, Jian-Ping Mao, Wei-Qi Xu, Hai-Jiang Cai, Ji-Song Guan, and Guo-Dong Li for their technical assistance and helpful discussions.
Correspondence should be addressed to either of the following: Dr. Gang Pei, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue-Yang Road, Shanghai 200031, China, E-mail: gpei@sibs.ac.cn; or Dr. Xu Zhang, Institute of Neuroscience, Shanghai Institutes for Biological Sciences, 320 Yue-Yang Road, Shanghai 200031, China, E-mail: xuzhang@ion.ac.cn.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234775-10$15.00/0
References
- Aghajanian GK, Kogan JH, Moghaddam B ( 1994) Opiate withdrawal increases glutamate and aspartate efflux in the locus coeruleus: an in vivo microdialysis study. Brain Res 636: 126–130. [DOI] [PubMed] [Google Scholar]
- Bradford MM ( 1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Casado M, Bendahan A, Zafra F, Danbolt NC, Aragon C, Gimenez C, Kanner BI ( 1993) Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem 268: 27313–27317. [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA ( 2002) Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22: 2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC ( 2001) Glutamate uptake. Prog Neurobiol 65: 1–105. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB ( 1998) Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci 18: 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA ( 1999) Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19: 10193–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriba PV, Garcia-Sevilla JA ( 1999) Parallel modulation of receptor for activated C kinase 1 and protein kinase C-alpha and beta isoforms in brains of morphine-treated rats. Br J Pharmacol 127: 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GH, Wang LZ, Qiu HC, Ma L, Pei G ( 1999) Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol 56: 39–45. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A ( 1997) High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 52: 6–15. [DOI] [PubMed] [Google Scholar]
- Haas HL, Ryall RW ( 1980) Is excitation by enkephalins of hippocampal neurones in the rat due to presynaptic facilitation or to disinhibition? J Physiol (Lond) 308: 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainfeld JF, Powell RD ( 2000) New frontiers in gold labeling. J Histochem Cytochem 48: 471–480. [DOI] [PubMed] [Google Scholar]
- Hong JS, Grimes L, Kanamatsu T, McGinty JF ( 1987) Kainic acid as a tool to study the regulation and function of opioid peptides in the hippocampus. Toxicology 46: 141–157. [DOI] [PubMed] [Google Scholar]
- Hong JS, McGinty JF, Grimes L, Kanamatsu T, Obie J, Mitchell CL ( 1988) Seizure-induced alterations in the metabolism of hippocampal opioid peptides suggest opioid modulation of seizure-related behaviors. NIDA Res Monogr 82: 48–66. [PubMed] [Google Scholar]
- Isaacson RL, Lanthorn TH ( 1981) Hippocampal involvement in the pharmacologic induction of withdrawal-like behaviors. Fed Proc 40: 1508–1512. [PubMed] [Google Scholar]
- Kanner BI ( 1993) Glutamate transporters from brain. A novel neurotransmitter transporter family. FEBS Lett 325: 95–99. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T ( 2001) Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci 14: 547–553. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC ( 1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15: 1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J, Endo S, Kategaya LS, Fernandez RI, Brabham DG, Chin J, Byrne JH, Eskin A ( 2000) Long-term regulation of neuronal high-affinity glutamate and glutamine uptake in Aplysia Proc Natl Acad Sci USA 97: 12858–12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A ( 2002) Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat Neurosci 5: 155–161. [DOI] [PubMed] [Google Scholar]
- Maleszka R, Helliwell P, Kucharski R ( 2000) Pharmacological interference with glutamate re-uptake impairs long-term memory in the honeybee, apis mellifera Behav Brain Res 115: 49–53. [DOI] [PubMed] [Google Scholar]
- Mansouri FA, Motamedi F, Fathollahi Y, Atapour N, Semnanian S ( 1997) Augmentation of LTP induced by primed-bursts tetanic stimulation in hippocampal CA1 area of morphine dependent rats. Brain Res 769: 119–124. [DOI] [PubMed] [Google Scholar]
- Mansouri FA, Motamedi F, Fathollahi Y ( 1999) Chronic in vivo morphine administration facilitates primed-bursts-induced long-term potentiation of Schaffer collateral-CA1 synapses in hippocampal slices in vitro Brain Res 815: 419–423. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT ( 1999) Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci 19: 6629–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G ( 2002) Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Przewlocki R, Siggins GR ( 1999a) Chronic morphine treatment selectively augments metabotropic glutamate receptor-induced inhibition of N-methyl-d-aspartate receptor-mediated neurotransmission in nucleus accumbens. J Pharmacol Exp Ther 288: 30–35. [PubMed] [Google Scholar]
- Martin G, Ahmed SH, Blank T, Spiess J, Koob GF, Siggins GR ( 1999b) Chronic morphine treatment alters NMDA receptor-mediated synaptic transmission in the nucleus accumbens. J Neurosci 19: 9081–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF ( 1998) Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci 18: 4490–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M ( 2001) Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol 419: 39–45. [DOI] [PubMed] [Google Scholar]
- Nestler EJ ( 2001) Neurobiology. Total recall—the memory of addiction. Science 292: 2266–2267. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK ( 1997) Molecular and cellular basis of addiction. Science 278: 58–63. [DOI] [PubMed] [Google Scholar]
- Ng KT, O'Dowd BS, Rickard NS, Robinson SR, Gibbs ME, Rainey C, Zhao WQ, Sedman GL, Hertz L ( 1997) Complex roles of glutamate in the Gibbs-Ng model of one-trial aversive learning in the new-born chick. Neurosci Biobehav Rev 21: 45–54. [DOI] [PubMed] [Google Scholar]
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW, Nestler EJ ( 1995) Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci 15: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M ( 2001) Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res 905: 254–258. [DOI] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G ( 2002) Hippocampal long-term potentiation is reduced by chronic morphine treatment and can be restored by re-exposure to opiates. J Neurosci 22: 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Bowen KK, Todd KG, Dempsey RJ ( 2001a) Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur J Neurosci 13: 119–128. [PubMed] [Google Scholar]
- Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, Rothstein JD, Dempsey RJ ( 2001b) Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci 21: 1876–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Fletcher EL, Pow DV ( 2002) Distribution of two splice variants of the glutamate transporter GLT1 in the retinas of humans, monkeys, rabbits, rats, cats, and chickens. J Comp Neurol 445: 1–12. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ ( 1999) Drug addiction: bad habits add up. Nature 398: 567–570. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW ( 1994) Localization of neuronal and glial glutamate transporters. Neuron 13: 713–725. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Puschel B, Jons T, Kugler P ( 1996) Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 71: 989–1004. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch KP, Kugler P ( 2002) A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience 109: 45–61. [DOI] [PubMed] [Google Scholar]
- Sepulveda MJ, Hernandez L, Rada P, Tucci S, Contreras E ( 1998) Effect of precipitated withdrawal on extracellular glutamate and aspartate in the nucleus accumbens of chronically morphine-treated rats: an in vivo microdialysis study. Pharmacol Biochem Behav 60: 255–262. [DOI] [PubMed] [Google Scholar]
- Stain F, Barjavel MJ, Sandouk P, Plotkine M, Scherrmann JM, Bhargava HN ( 1995) Analgesic response and plasma and brain extracellular fluid pharmacokinetics of morphine and morphine-6-beta-d-glucuronide in the rat. J Pharmacol Exp Ther 274: 852–857. [PubMed] [Google Scholar]
- Sutherland ML, Delaney TA, Noebels JL ( 1996) Glutamate transporter mRNA expression in proliferative zones of the developing and adult murine CNS. J Neurosci 16: 2191–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC ( 1997) Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci 17: 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K ( 1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H ( 1991) Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251: 85–87. [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC ( 1997) Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci 9: 1646–1655. [DOI] [PubMed] [Google Scholar]
- Ventayol P, Busquets X, Garcia-Sevilla JA ( 1997) Modulation of immunoreactive protein kinase C-alpha and beta isoforms and G proteins by acute and chronic treatments with morphine and other opiate drugs in rat brain. Naunyn Schmiedebergs Arch Pharmacol 355: 491–500. [DOI] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL ( 2001) Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292: 1175–1178. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Pratt K, Zable AC, Kavanaugh MP, Rosenberg PA ( 1998a) High affinity glutamate transport in rat cortical neurons in culture. Mol Pharmacol 53: 88–96. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Lea E, Robinson MB, Potthoff WK, Aizenman E, Rosenberg PA ( 1998b) Dihydrokainate-sensitive neuronal glutamate transport is required for protection of rat cortical neurons in culture against synaptically released glutamate. Eur J Neurosci 10: 2523–2531. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O ( 2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81: 299–343. [DOI] [PubMed] [Google Scholar]
- Zhang T, Feng Y, Rockhold RW, Ho IK ( 1994) Naloxone-precipitated morphine withdrawal increases pontine glutamate levels in the rat. Life Sci 55: PL25–PL31. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wiesenfeld-Hallin Z, Hokfelt T ( 1994) Effect of peripheral axotomy on expression of neuropeptide Y receptor mRNA in rat lumbar dorsal root ganglia. Eur J Neurosci 6: 43–57. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bean AJ, Wiesenfeld-Hallin Z, Xu XJ, Hokfelt T ( 1995) Ultrastructural studies on peptides in the dorsal horn of the rat spinal cord. III. Effects of peripheral axotomy with special reference to galanin. Neuroscience 64: 893–915. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao L, Arvidsson U, Elde R, Hokfelt T ( 1998) Localization and regulation of the delta-opioid receptor in dorsal root ganglia and spinal cord of the rat and monkey: evidence for association with the membrane of large dense-core vesicles. Neuroscience 82: 1225–1242. [DOI] [PubMed] [Google Scholar]