

Abstract
Cdk5/p35 has been implicated in cytoskeletal protein phosphorylation in normal brain and in many human neurodegenerative disorders. Yet, mouse models of cdk5/p35 hyperactivity have not yielded corresponding changes in cytoskeletal protein phosphorylation. To elucidate the relationship between p35, cdk5, and the neuronal cytoskeleton, we deleted the p35 gene in mice having a pure C57BL/6 background. We found that p35 deficiency leads to a 38% reduction of cdk5 activity in adult brain. In addition, loss of p35 causes an anterograde redistribution of cdk5 toward peripheral neuronal processes. The unusual presence of nonphosphorylated neurofilament (NF) in aberrant axon fascicles and the relocation of tau and MAP2B from cell bodies and proximal neuronal processes to more distal sites of the neuropil in p35-/- mouse brain implicate p35 in neuronal trafficking, particularly in dynein-driven retrograde transport. In many axons of normal brain, cdk5 fails to colocalize with phosphorylated cytoskeletal protein epitopes. This observation, together with an unexpected increase of NF, tau, and MAP2B phosphoepitopes accompanying the decreased cdk5 activity in p35-/- mice, supports the idea that cdk5 does not phosphorylate cytoskeletal proteins directly. Rather, in structures where cdk5 does colocalize with phosphorylated cytoskeletal protein epitopes, it may function as a negative regulator of other proline-directed kinases that directly phosphorylate the proteins. Evidence for increased glycogen synthase kinase 3β (GSK3β) activity in p35-/- mice suggests that GSK3β may be one such kinase regulated by cdk5. Our studies illustrate that p35 regulates the subcellular distribution of cdk5 and cytoskeletal proteins in neurons and that cdk5 has a hierarchical role in regulating the phosphorylation and function of cytoskeletal proteins.
Keywords: cdk5, p35, tau phosphorylation, neurofilament phosphorylation, MAP2 phosphorylation, cytoskeletal pathology, trafficking, transport
Introduction
Cyclin-dependent kinase (cdk) 5 is a member of the cdk family that, along with the mitogen-activated protein (MAP) kinase, stress-activated protein kinase, c-Jun N-terminal kinase (JNK), and glycogen synthase kinase 3 (GSK3), constitutes the proline-directed serine/threonine kinases (PDKs). Unlike most cdks that function typically in dividing cells, cdk5 is active principally in postmitotic neurons. As with all members of the cdk family, full activation of cdk5 requires association with a regulatory subunit, three of which have been identified in brain: p35, p39, and p67 (Ishiguro et al., 1994; Lew et al., 1994; Tsai et al., 1994; Shetty et al., 1995; Tang et al., 1995). The best understood role of the enzyme is in normal brain development, in which it regulates neuronal migration, axodendritic organization, and laminar architecture (Nikolic et al., 1996; Ohshima et al., 1996, 1999; Chae et al., 1997; Paglini et al., 1998; Kwon et al., 1999; Philpott et al., 1999). cdk5 is also thought to function in neuronal trafficking and transport (Ratner et al., 1998; Smith and Tsai, 2002) and phosphorylates several neuronal cytoskeletal elements, including neurofilaments (NFs) and the microtubule-associated proteins tau and MAP2, in vitro or in vivo (for review, see Grant et al., 2001).
Localization of cdk5 to neurodegenerative lesions and increases in cdk5 activity have been demonstrated in several diseases, including Alzheimer's disease (Liu et al., 1995; Yamaguchi et al., 1996; Pei et al., 1998; Lee et al., 1999; Patrick et al., 1999; Takahashi et al., 2000a), Parkinson's disease or diffuse Lewy body disease (Brion and Couck, 1995; Nakamura et al., 1997a; Takahashi et al., 2000b), progressive supranuclear palsy (Borghi et al., 2002), amyotrophic lateral sclerosis (Nakamura et al., 1997b; Bajaj et al., 1998, 1999; Nguyen et al., 2001), multiple system atrophy (Nakamura et al., 1998; Honjyo et al., 2001), and Niemann-Pick type C (Bu et al., 2002), that are characterized by increased phosphorylation of cytoskeletal proteins. It has been proposed that this hyperphosphorylation is a preliminary step in the formation of cytoskeletal lesions, which are lethal to neurons (Buee et al., 2000). Aberrant phosphorylation of the neuronal cytoskeleton could alter cellular trafficking and transport and contribute to neuronal dysfunction and death (Ratner et al., 1998; Smith and Tsai, 2002). Cleavage of p35 to a more powerful cdk5 activator, p25, may be a critical step in this pathological cascade; however, a causal relationship between cdk5 and cytoskeletal protein phosphorylation in neurodegenerative diseases has yet to be established definitively. Additionally, the role of cdk5 in normal adult brain has been difficult to ascertain because cdk5-deficient mice are not viable past the perinatal period (Ohshima et al., 1996).
To better understand the function of cdk5 in mature brain neurons, we generated and characterized p35-/--deficient mice, which are consequently incapable of producing p25. Apart from the striking axonal derangement and architectural abnormalities that have been described previously in similar mice (Chae et al., 1997; Ohshima et al., 2001), we have found alterations in the normal subcellular distribution of cdk5 in p35-/- neurons. Moreover, despite a significant decrease in brain cdk5 activity, we report an increase in phosphorylation of cytoskeletal proteins, which suggests that cdk5 may not be the kinase that normally phosphorylates these epitopes.
Materials and Methods
p35 null mutant mice. Mice lacking the p35 gene were generated using standard targeted deletion techniques (Thomas and Capecchi, 1987). Briefly, an ∼2.5 kb fragment containing the coding region for p35 was excised from an ∼16.5 kb mouse genomic clone by HindIII/NotI restriction digest and replaced with an ∼900 bp EcoRI/HindIII fragment of pMC1neo (Stratagene, La Jolla, CA) containing the neomycin resistance cassette in reverse orientation. This resulted in the deletion of ∼1.6 kb of genomic DNA. WW6 embryonic stem (ES) cells (Ioffe et al., 1995) that had undergone homologous recombination with the targeting vector were identified by Southern blot by hybridizing an ∼1 kb 3′EcoRI/XbaI fragment of the p35 genomic clone to ES cell genomic DNA digested with BamHI (Fig. 1A). Initial identification of mice with germ line transmission of the targeting vector was also performed by Southern blot. Mice carrying the targeting vector were backcrossed extensively to C57BL/6J (B6) (Jackson Laboratory, Bar Harbor, ME) mice (N10-12) to produce animals with >99% pure B6 genetic background, allowing comparisons to either wild-type (+/+) littermates or B6 mice. Experimental animals were genotyped by PCR analysis using the following primers: p35 forward (5′-ACCTCTGCAGGGACACCCAAACG-3′) plus p35 reverse (5′-GTGGGTCGGCATTGATCTGCAGC-3′), yielding a 440 bp fragment; and neo forward (5′-GATCTGGACGAAGAGCTCAGGG-3′) plus neo reverse (5′-CGTCAAGAAGGCGATAGAAGGCG-3′), yielding a 316 bp fragment (Fig. 1). Heterozygous (p35+/-) mice were mated to generate both p35-/- mice and p35+/+ littermates. The University of Washington Animal Use and Care Committee approved housing and all protocols.
Figure 1.
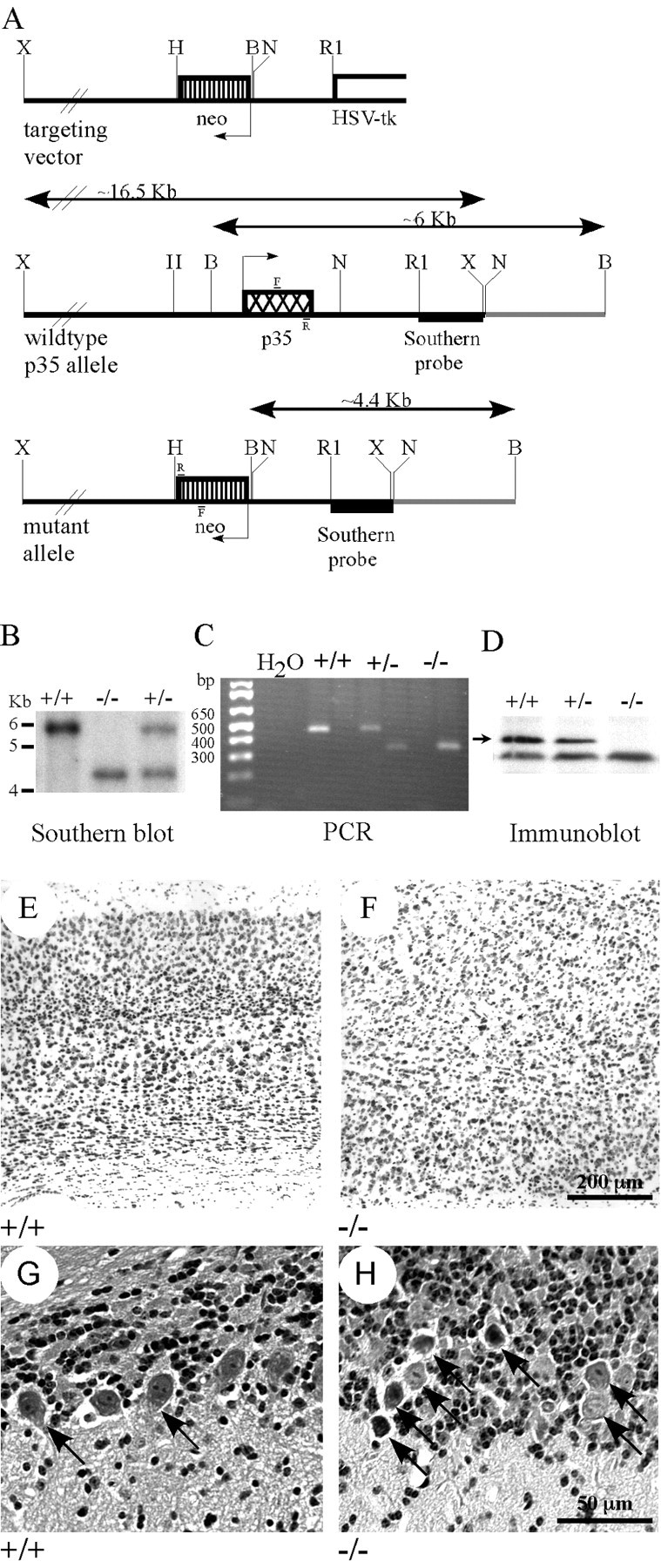
p35-/- mice. A, Diagrams of the targeting vector used to generate p35-/- mice and wild-type and mutant p35 alleles, indicating the locations of probes and primers (F and R) used for identification of null mutants. B, Identification of p35+/+ (∼6 kb band), p35-/- (∼4.4 kb band), and p35+/- (both bands) mice by Southern blot. C, Identification of p35+/+ (∼450 bp band), p35-/- (∼300 bp band), and p35+/- (both bands) mice by PCR amplification of genomic DNA. D, Western blot demonstrating normal levels of p35 in p35+/+ homogenized mouse brain supernatants, ∼50% of normal in p35+/- brains, and no p35 protein in p35-/- mouse brains. E-H, Images of hematoxylin and eosin-stained sagittal sections of p35+/+ (E, G) and p35-/- (F, H) mouse brains. E, In p35+/+ brains, the cortex displays normal layered structure. F, The normal laminar character of the cortex is disrupted in p35-/- brains. G, In p35+/+ cerebellum, a single row of Purkinje cells (arrows) is distributed at the border between the granule cell layer (top) and the molecular layer (bottom). H, In p35-/- cerebellum, Purkinje cells (arrows) are abnormally distributed within the granule cell layer, and rather than forming a single row, the Purkinje cells are often stacked one on top of another (stacks of 2 and 4 cells are shown).
Antibodies and other reagents. Antibodies to p35, cdk5, syntaxin, tau, NF, MAP2, GSK3β, and neuronal nuclei used in immunoblotting, kinase activity, and immunohistochemical studies are listed in Table 1. Subtype-specific secondary antibodies conjugated to HRP or biotin and streptavidin-HRP were from Southern Biotechnology (Birmingham, AL). Fluorescent labels (Alexa 594-conjugated goat anti-mouse IgG and streptavidin-Alexa 488) were from Molecular Probes (Eugene, OR). 4′,6-Diamidino-2-phenylindole (DAPI), hematoxylin, DAB, sodium ATP, protease inhibitor mixture, protein kinase A inhibitor (PKI), and ECL chemicals were from Sigma (St. Louis, MO). Histone H1 was purchased from Calbiochem (San Diego, CA), and recombinant human tau was from Invitrogen (Carlsbad, CA). γ-32P-ATP was obtained from DuPont NEN (Boston, MA). Bradford protein assay reagents were from Bio-Rad (Hercules, CA). Protein A-Sepharose was from Amersham Pharmacia Biotech (Piscataway, NJ). Protran nitrocellulose membrane for immunoblotting was purchased from Intermountain Scientific (Kaysville, UT).
Table 1.
Antibodies
|
Group |
Antibody |
Isotype |
Epitope |
Western dilution |
IHC dilution |
Source |
|---|---|---|---|---|---|---|
| cdk5 | cdk5 (C-8) | Rabbit polyclonal | cdk5 C terminus | 1:100 | 1:100 | Santa Cruz Biotechnology |
| p35 | p35 (C-19) | Rabbit polyclonal | p35 C terminus | 1:100 | nd | Santa Cruz Biotechnology |
| p67 | p67 | Rabbit polyclonal | p67 N terminus | 1:5000 | nd | H. Pant |
| AP18 | Mouse IgG1 | MAP2B phosphorserine 136 | 1:1000 | 1:1000 | L. Binder | |
| MAP2 | AP20 | Mouse IgG1 | Total MAP2A, 2B and 2C | 1:1000 | 1:500 | Sigma |
| R39 | Rabbit polyclonal | Total NF-H, NF-M and NF-L | 1:2000 | nd | H. Pant | |
| SMI31 | Mouse IgG1 | Phospho-NF-H and NF-M | 1:1000 | 1:500 | Sternberger Monoclonals | |
| SMI34 | Mouse IgG1 | Phospho-NF-H | 1:1000 | nd | Sternberger Monoclonals | |
| NFs | SMI32 | Mouse IgG1 | Nonphosphorylated NF-H | 1:500 | 1:500 | Sternberger Monoclonals |
| TG5 | Mouse IgG1 | Total tau | 1:10 | nd | I. Vincent | |
| CP22 | Mouse IgM | Tau phosphothreonine 175 | 1:10 | nd | P. Davies | |
| CP13 | Mouse IgG1 | Tau phosphoserine 202 | 1:10 | 1:10 | P. Davies | |
| MC6 | Mouse IgG1 | Tau phosphoserine 235 | 1:10 | nd | P. Davies | |
| MC15 | Mouse IgM | Tau phosphoserine 235 | 1:10 | nd | P. Davies | |
| Tau | PHF1 | Mouse IgG1 | Tau phosphoserine 396/404 | 1:20 | 1:10 | P. Davies |
| Neuronal antigen | NeuN | Mouse IgG1 | Neuronal nuclei | 1:250 | nd | Chemicon |
| Syntaxin | SP6 | Mouse IgG1 | Syntaxin | 1:20 | 1:10 | W. Honer |
| GSK3β | 9332 | Rabbit polyclonal | Total GSK3β | 1:500 | nd | Cell Signaling Technology |
| β-Catenin
|
9562
|
Rabbit polyclonal
|
Total β-catenin
|
1:500
|
nd
|
Cell Signaling Technology
|
nd, Not determined.
Immunoblotting. Two-month-old (p35+/+, n = 4; p35-/-, n = 4) or 6-month-old (B6, n = 3; p35-/-, n = 3) old mice were euthanized using CO2, followed by cervical dislocation; brains were removed quickly and frozen at -80°C. Frozen brains were homogenized, aliquoted, and frozen, then supernatants were prepared as described previously (Bu et al., 2002). Samples (5-50 μg of total protein as indicated in figure legends) were electrophoresed on 6-8% (NF and MAP2 antibodies) or 10% (cdk5, p67, tau, GSK3β, and β-catenin antibodies) SDS-PAGE gels, transferred to nitrocellulose, and blocked in 5% milk in TBS (10 mm Tris, pH 7.5, and 150 mm NaCl) for 30 min. Two to three separate blots using different aliquots from the same set of mouse brains were performed for each antibody used. Blots were incubated overnight at 4°C with antibodies diluted as shown in Table 1 in 5% milk in TBS. After three washes in TBS, blots were incubated with subtype-specific secondary antibodies conjugated to HRP at room temperature for 1-2 hr, washed again, incubated with ECL reagent, and apposed to film. The NeuN antibody raised against neuronal antigens (Mullen et al., 1992) was used as a loading control for all immunoblotting experiments, and band intensity for each epitope examined was normalized to NeuN band intensity. Films were scanned on a UMAX Astra 2400S scanner (Dallas, TX), band density was analyzed using ImageQuant software (Molecular Dynamics, Sunnyvale, CA), statistical analysis (Student's t test) was performed using Excel software (Microsoft, Redmond, WA), and images assembled in Photoshop (Adobe, San Jose, CA).
Kinase activity assays. Immunoprecipitation (IP) kinase activity assays were performed as described previously for cdk5 (Bu et al., 2002). Briefly, cdk5 was immunoprecipitated from 100 μg of total protein from whole-brain supernatants (p35+/+, n = 4; p35-/-, n = 4) using the polyclonal cdk5 antibody at 4°C for 2 hr with rocking. One hundred microliters of protein A-Sepharose were added to each sample, and rocking continued for 1 hr. Samples were washed twice with 1 ml of cold TBS and once with 1 ml of cold kinase buffer. A reaction mix containing HEPES kinase buffer, 10 μm ATP, 10 μg of histone H1, and 0.5 μCi of γ-32P-ATP was added to the samples and incubated at room temperature for 30 min. Reactions were halted by the addition of loading buffer, and samples were then electrophoresed on 12% SDS-PAGE gels. Histone bands were visualized by Coomassie blue staining, and gels were dried, then apposed to a phosphorimager screen and scanned on a Storm 840 PhosphorImager (Molecular Dynamics); band density was analyzed using ImageQuant (Molecular Dynamics), and statistical analysis (Student's t test) was performed using Excel (Microsoft).
Activity assays for GSK3β were performed as described previously (Watcharasit et al., 2002). GSK3β was immunoprecipitated from whole-brain supernatants (2-month-old mice: p35+/+, n = 3; p35-/-, n = 4; 6-month-old mice: B6, n = 3; p35-/-, n = 3), similar to the cdk5 activity assays but using the polyclonal GSK3β antibody, 75 μg of total protein from whole-brain supernatants, and 40 μl of protein A-Sepharose. The reaction mix contained Tris kinase buffer, 250 μm ATP, 0.1 μg/μl tau, 1.4 μCi γ-32P-ATP, 1 mM EGTA, and 4 μm of PKI. Reactions were electrophoresed on 10% SDS-PAGE gels and transferred to nitrocellulose, then blots were apposed to film. Analyses were performed as above.
Immunohistochemistry. Two-month-old mice (p35+/+, n = 3; p35-/-, n = 4) were euthanized using CO2 and transcardially perfused with 4% paraformaldehyde in PBS; then, brains were removed, halved sagittally, and immersed in 4% paraformaldehyde for 1 d at 4°C. Brain halves were embedded in paraffin and cut in 10 μm sagittal sections. Immunohistochemistry was performed as reported previously (Bu et al., 2002), except sections were pretreated with 50 mm ammonium chloride for 30 min (Kerjaschki et al., 1984) before blocking, and BSA/goat serum (5% BSA plus 3% goat serum in TBS) was used for blocking and antibody dilutions. Briefly, sections were incubated with primary antibodies (Table 1) for 18-42 hr at 4°C, followed by biotin-conjugated subtype-specific secondary antibodies for 1-2 hr at room temperature, then with streptavidin-HRP for 1-2 hr at room temperature. DAB was then added to visualize antibody binding, followed by hematoxylin counterstaining. For double immunofluorescent staining, sections were incubated with both primary antibodies (polyclonal anti-cdk5 plus one mouse monoclonal IgG1) simultaneously, followed by incubation with biotin-conjugated goat anti-rabbit secondary antibodies for cdk5, then streptavidin-Alexa 488 and Alexa 594-conjugated goat anti-mouse IgG secondary antibodies for the other primary antibody used. Sections were counterstained with DAPI to label nuclei. Images were collected on an Eclipse TE200 microscope (Nikon, Melville, NY) equipped with a Spot RT CCD camera (Diagnostic Instruments, Sterling Heights, MI), and figures were assembled using Photoshop (Adobe).
Results
p35-/- gross phenotype
Wild-type (+/+), heterozygous (+/-), and p35 homozygous null (-/-) mice were identified by Southern blotting or PCR analysis (Fig. 1B,C). Western blotting detected about half of the normal amount of p35 protein in brains of p35+/- mice but no p35 protein in the brains of p35-/- mice (Fig. 1D). Histological staining and analysis revealed that the p35-/- mice lack the normal cortical lamination found in p35+/+ mice (Fig. 1E,F). Purkinje neurons of the cerebellum, which normally form a single layer at the boundary between the granule cell layer and the molecular layer (Fig. 1G), are localized ectopically within the granule cell layer (Fig. 1H) and, in some instances, aggregate with a multilayered appearance. In addition, numerous granule neurons were mislocalized within the molecular layer (see Figs. 5L, 6J).
Figure 5.

Distribution of MAP2 in p35+/+ and p35-/- brains. Images of 10 μm sagittal sections of p35+/+ (A, C, E, G, I, K) and p35-/- (B, D, F, H, J, L) brains incubated with the AP18 antibody to phospho-Ser136 in MAP2B (A, B, E, F, I, J) or the AP20 nonphospho-MAP2 antibody (C, D, G, H, K, L). A, E, I, Phospho-MAP2B (AP18) stains neuronal somata (arrows) and apical dendrites (arrowheads) in p35+/+ cortex (A), hippocampus (E), and pons (I). B, F, J, In p35-/- brains, AP18 staining is diminished greatly or not detected in neuronal somata (arrows) and dendrites (arrowheads) in the cortex (B), hippocampus (F), and pons (J). Dendritic organization is disrupted (B, F), and staining is sometimes shifted to the neuropil (F, asterisk). The granule cell layer of the CA2/CA3 region of the hippocampus is disorganized in p35-/- brains (F). C, G, K, The AP20 nonphospho-MAP2 antibody stains neuronal somata (arrows) and apical dendrites (arrowheads) in p35+/+ cortex (C), hippocampus (G), and cerebellum (K). D, H, L, In p35-/- brains, AP20 staining is shifted from neuronal somata and apical dendrites to more distal processes (asterisks), and dendritic organization is disrupted in cortex (D) and hippocampus (H). Disorganization of the granule cell layer is apparent (H). In the cerebellum (L), AP20 staining in Purkinje cell somata is decreased (arrow), whereas staining in more distal dendritic arborizations is robust.
Figure 6.

Distribution of phosphotau epitopes in p35+/+ and p35-/- brains. Images of 10 μm sagittal sections from p35+/+ (A, C, E, G, I) and p35-/- (B, D, F, H, J) brains incubated with antibodies to the CP13 or PHF-1 phosphotau epitopes. A, C, The CP13 antibody to phospho-Ser202 in tau stains primarily neuronal somata (arrows) in p35+/+ pons (A) and cerebellum (C) and is also seen in some dendritic processes and in the neuropil in the pons (A, arrowhead). B, D, In p35-/- brains, CP13 staining is increased in neuronal somata in the pons (B) and cerebellum (D), and dendritic and neuropil staining is diminished (B). E, G, I, The PHF-1 antibody to tau phospho-Ser396/404 stains scattered neuronal (arrows) and glial (<) somata and nuclei, and axons (arrowhead) in p35+/+ thalamus (E), cortex (G), and cerebellum (I). F, H, J, In p35-/- brains, PHF-1 staining is diminished in neuronal somata in the thalamus (F, arrow) and cerebellum (J, arrow), and staining is instead shifted to the neuropil (F, J, arrowhead and asterisks). In the cortex, PHF-1 stains the atypical axon fascicles (H, arrow).
cdk5 activity and localization are altered in p35-/- mice
The total levels of cdk5 protein in whole brain were similar in both p35+/+ and p35-/- mice (Fig. 2A), indicating that there is no compensatory increase in cdk5 expression in p35-/- mice. The levels of p39 and p67 were also unchanged (data are shown for p67) (Fig. 2A). IP of cdk5, followed by kinase activity assays using histone H1 as substrate, revealed that cdk5 activity was decreased by 38% in the brains of p35-/- animals compared with p35+/+ brains (Fig. 2B; p = 0.03).
Figure 2.
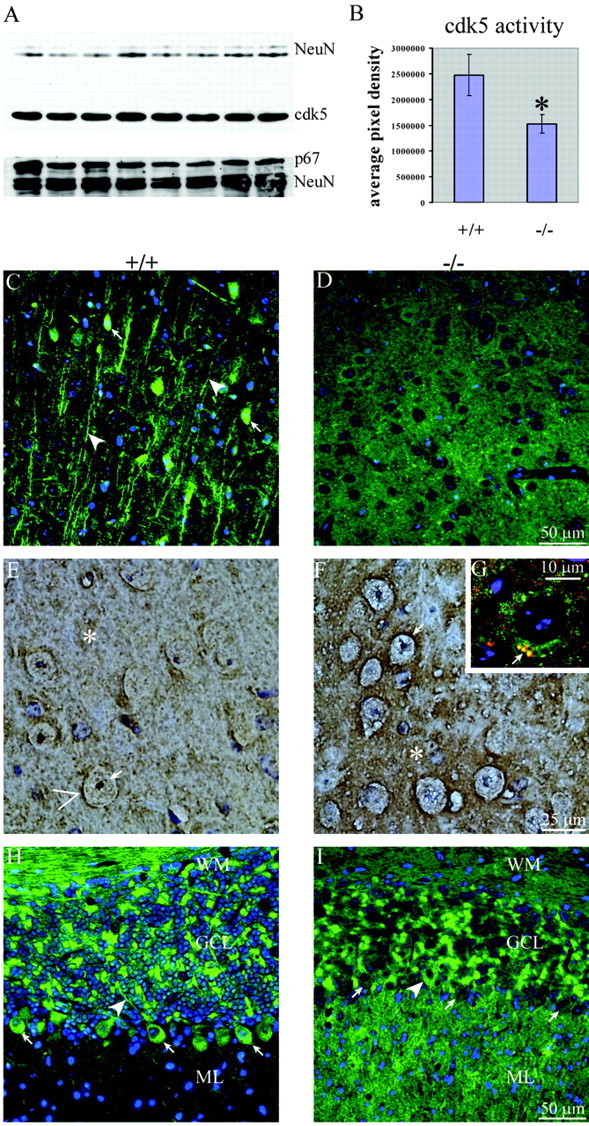
cdk5 levels, activity, and distribution in p35-/- mouse brains. A, Western blot showing similar levels of cdk5 (50 μg of total brain protein) and p67 (25 μg of total brain protein) in p35+/+ and p35-/- mice. An antibody to neuronal antigens, NeuN, was used as a loading control. B, Graph generated from IP kinase activity assays demonstrating an ∼38% decrease in cdk5 activity in p35-/- brains compared with p35+/+ brains. C-I, Images of sagittal sections of p35+/+ (C, E, H) and p35-/- (D, F, G, I) brains incubated with polyclonal antibodies to cdk5 (C, D, G-I, green). In G, the section was coincubated with antibodies to cdk5 and to the synaptic protein syntaxin (red). C, In p35+/+ cortex, cdk5 stains neuronal somata (arrows) and proximal axons (arrowheads). D, Neither somatic nor proximal axonal cdk5 staining is detected in p35-/- cortex. E, In p35+/+ thalamus, cdk5 faintly stains neuronal somata (arrows) and the neuropil (asterisk), with accumulations adjacent to neuronal somata (>). F, Somatic staining is diminished in p35-/- thalamus, whereas generalized neuropil staining (asterisk) and staining surrounding neuronal somata (arrows) are increased. G, In p35-/- cortex, cdk5 (green) and syntaxin (red) are both distributed in the neuropil and often colocalize at sites adjacent to neuronal somata (arrow). H, In p35+/+ cerebellum, cdk5 stains Purkinje cell somata (arrows) and axons (arrowhead), axon bundles in the white matter (WM), and neurons in the granule cell layer (GCL). I, Staining for cdk5 is diminished in cerebellar WM and is rarely detected in Purkinje cell somata (arrows indicate Purkinje cells lacking cdk5 staining, and the arrowhead indicates a single Purkinje cell positive for cdk5) or axons in p35-/- cerebellum; however, staining is robust in the neuropil of the molecular layer (ML).
Polyclonal antibodies against cdk5 were used on sagittal brain sections to compare the subcellular location of cdk5 in p35+/+ and p35-/- mice. Our results on the localization of cdk5 in p35+/+ mice were similar to those reported previously (Tsai et al., 1993; Matsushita et al., 1996; Pigino et al., 1997; Veeranna et al., 1997; Terada et al., 1998), showing strongest cdk5 staining in proximal axons (arrowheads) and neuronal somata (arrows) (Fig. 2C,E,H). Some neuropil staining was also detected in p35+/+ brains (Fig. 2E, asterisk and >). This pattern was altered dramatically in p35-/- mice, with little or no cdk5 detected in proximal axons and neuronal somata (Fig. 2D,F,I). Instead, p35-/- mice exhibited a strong increase in neuropil staining (Fig. 2F, asterisk) with intense labeling around neuronal somata (Fig. 2F, arrow). Double labeling with anti-cdk5 and an antibody specific for the presynaptic protein syntaxin showed colocalization of both proteins, presumably at presynaptic sites of p35-/- mice (Fig. 2G, arrow). cdk5 staining of large myelinated axon tracts of the corpus callosum (data not shown) and the white matter of the cerebellum of p35-/- mice appeared diminished (Fig. 2I). In the cerebellum, a marked increase in intensity was observed in the molecular layer (Fig. 2I), which is relatively unstained in p35+/+ mice (Fig. 2H). These data indicate that the subcellular distribution of cdk5 is altered in p35-/- mice.
Altered cytoskeletal protein phosphorylation in p35-/- mice
The complex polar morphology of neurons is determined by the neuronal cytoskeleton, which comprises a number of structural elements including microtubules, microfilaments, and intermediate filaments as well as associated proteins such as molecular motors (kinesins and dyneins) and microtubule-associated proteins like MAP2 and tau, which help regulate microtubule assembly. One of the major mechanisms for regulating cytoskeletal function is via phosphorylation of cytoskeletal components such as NFs, MAP2, and tau (for review, see Billingsley and Kincaid, 1997; Sanchez et al., 2000; Miller et al., 2002). We examined the extent of phosphorylation of these proteins using antibodies specific for sites that have been demonstrated to be produced by the cdk5 kinase. In all cases shown below, the levels of protein were normalized to the levels of the neuronal antigen NeuN.
NF phosphorylation
NFs are composed of three protein subunits of different molecular masses: NF-H (heavy) (∼200 kDa), NF-M (medium) (∼140-160 kDa), and NF-L (light) (∼70 kDa). The C-terminal domains of NF-H and NF-M contain repetitive Lys-Ser-Pro cdk5 consensus motifs that are phosphorylated by cdk5 in vitro (for review, see Grant et al., 2001), and there are at least two distinct phosphoepitopes within this repeat region as recognized by the SMI31 and SMI34 antibodies (Shea and Beermann, 1993; Bajaj et al., 1999). NF-H C-terminal domains also contain nonconsensus phosphorylation sites outside the repeat motifs that are phosphorylated by cdk5, and the SMI32 antibody recognizes these sites only when they are not phosphorylated (Bajaj et al., 1999). Immunoblotting of whole-brain supernatants found similar levels of total NF in p35-/- mice compared with p35+/+ mice (Fig. 3A, R39) but detected an increase in SMI32 immunoreactivity with NF-H, consistent with a decrease in phosphorylation of this site (Fig. 3A, SMI32; 143.9 ± 6.5%; p = 0.0001). No change in SMI31 immunoreactivity with NF-H was observed, but SMI31 immunoreactivity with NF-M was increased (Fig. 3A, SMI31; 29.2 ± 8.2%; p = 0.02). The SMI34 antibody displayed increased immunoreactivity with NF-H, indicating increased phosphorylation of this site (Fig. 3A, SMI34; 276.8 ± 28.75%; p = 0.04). Although both NF-H and NF-M contain similar motifs in their C-terminal domains, these data indicate that these two subunits can be phosphorylated differentially in vivo.
Figure 3.

Western blots demonstrating phosphorylation status of cytoskeletal proteins in p35+/+ and p35-/- mouse brains. The amount of total protein in supernatants from whole-brain homogenates is shown in parentheses for each antibody used. Blots incubated with an antibody to neuronal antigens, NeuN, are shown as loading controls. A, NFs. Total levels of NF-H and NF-M are similar in p35+/+ and p35-/- brains (R39; 10 μg). Phosphorylation of NF-H at sites in the C terminus but outside the Lys-Ser-Pro repeat region is lower in p35-/- brains, as shown by an increase in bands detected by an antibody to only nonphosphorylated NF-H (SMI32; 50 μg). Phosphorylation of NF-H at repetitive Lys-Ser-Pro motifs is similar in p35+/+ and p35-/- brains, but phosphorylation of these repetitive motifs is increased in NF-M in p35-/- brains (SMI31; 5 μg). Phosphorylation of NF-H at other sites in the C-terminal repeat region is increased in p35-/- brains (SMI34; 5 μg). B, MAP2. Similar levels of total MAP2A and 2B are detected in p35+/+ and p35-/- brains (AP20; 10 μg), but there is a significant increase in phospho-MAP2B levels in p35-/- brains (AP18; 25 μg). C, Tau. Total tau levels are increased in p35-/- brains compared with p35+/+ brains (TG5; 25 μg). Phosphorylation of tau increases at Ser202 (CP13; 25 μg), Ser235 (MC15; 25 μg), and Ser 396/404 (PHF-1; 25 μg) in p35-/- brains. D, Antibodies to phosphotau epitopes recognize an epitope in a protein that migrates at the same rate as NF-H, and levels of these phosphoepitopes increase in p35-/- brains.
MAP2B phosphorylation
The microtubule-stabilizing and -nucleating activities of MAP2 are altered by phosphorylation of Ser136 and serine/threonine residues in the tubulin-binding domain (for review, see Sanchez et al., 2000), and the Ser136 residue has been identified as a substrate for PDKs, including the cdks, both in vivo and in vitro (Berling et al., 1994). We found similar levels of total MAP2 in p35+/+ and p35-/- brains (Fig. 3B, AP20), but phospho-Ser136 MAP2B levels were significantly higher in p35-/- brains (Fig. 3B, AP18; 661.1 ± 34.6%; p = 0.02).
Tau phosphorylation
Tau is a microtubule-stabilizing protein that is hyperphosphorylated in Alzheimer's disease. Of the ∼30 serine/threonine phosphorylation sites identified in tau, most are proline directed (Buee et al., 2000). At least four of these proline-directed sites in tau are thought to be produced by cdk5: Ser202, Thr205, Ser235, and Ser396/404 (Michel et al., 1998). We have examined tau phosphorylation at three of these sites (Ser202, Ser235, and Ser396/404).
The levels of total tau as detected with TG5 were increased in the brains of p35-/- mice (Fig. 3C, TG5; 89.4 ± 16.2% increase; p = 0.006). Phosphorylation of tau was also significantly increased at residues Ser202 (Fig. 3C, CP13; 188.1 ± 26.6%; p = 0.01), Ser235 (Fig. 3C, MC15; 371.7 ± 39.8%; p = 0.04), and Ser396/404 (Fig. 3C, PHF-1; 82 ± 24.1%; p = 0.04). Tau hyperphosphorylation in p35-/- mice was also evident with TG5 by way of intensification of higher molecular weight species relative to lower-weight isoforms in p35+/+ brains (Fig. 3C). The increase in tau levels may be secondary to hyperphosphorylation, because phosphorylated tau is more resistant to proteolysis (Litersky and Johnson, 1992; Vincent et al., 1994).
Interestingly, the phosphotau antibodies CP22 (Thr175) and MC6 (Ser235) did not react well with tau but detected increased phosphorylation of a protein that migrates at the same size as NF-H. The phosphotau antibody MC15 (Ser235) also reacted strongly with this protein (shown for CP22 and MC15) (Fig. 3C). Although the identity of this protein is unknown, NF-H does contain similar motifs as those phosphorylated in tau. Regardless of the identity of this antigen, it is notable that increased phosphorylation correlates with decreased cdk5 activity in p35-/- mice.
Altered cytoskeletal phosphoprotein distribution in p35-/- mice
The cytoskeletal phosphoproteins examined in these studies occupy distinct subcellular compartments within normal neurons. Because cdk5 distribution was altered in p35-/- mice, we examined the distribution of cytoskeletal phosphoproteins in sagittal brain sections from these mice. We also used double immunofluorescence staining of sagittal brain sections to compare the distributions of cytoskeletal phosphoepitopes relative to cdk5.
NF distribution
The phosphorylated NF-H/M epitopes (SMI31) are detected primarily in axons, whereas nonphosphorylated NF-H epitopes (e.g., SMI32) are normally found in scattered somatodendritic compartments and a few thick axons (Goldstein et al., 1983). Our results of p35+/+ mouse brains were consistent with this pattern (Fig. 4A,C,E). In p35-/- mice, antibodies against the phosphorylated NF epitopes highlighted the abnormal axonal organization (SMI31) (Fig. 4B,D) detected previously in p35-/- mice with silver stain (Chae et al., 1997). Aberrant axon fascicles appeared more intensely labeled with SMI31; however, this may be because of the abnormal aggregation of axons. SMI32 immunoreactivity had a similar pattern in p35+/+ and p35-/- brains but was more intense in both somata and processes in p35-/- brains (shown for the pons) (Fig. 4E,F).
Figure 4.
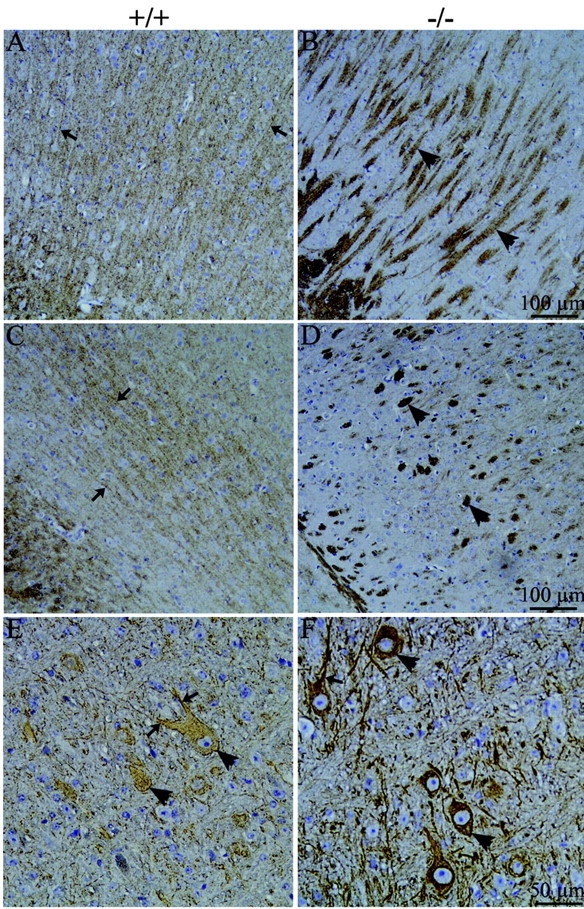
Distribution of NF in p35+/+ and p35-/- brains. Images of 10 μm sagittal sections of p35+/+ (A, C, E) and p35-/- (B, D, F) brains incubated with antibodies to NF phosphoepitopes and nonphosphoepitopes. A, C, Phosphorylated NF-H/M (SMI31) stains axons (arrows) in the frontal (A) and medial (C) cortex of p35+/+ brains. B, D, In p35-/- cortex, robust SMI31 staining is seen in the atypical axon fascicles (arrowheads). E, Nonphosphorylated NF-H is found in neuronal somata (arrowheads) and dendrites (arrow) in the pons in p35+/+ brains. F, In p35-/- pons, SMI32 staining increases in both neuronal somata (arrowheads) and dendrites (arrow).
MAP2B distribution
The phospho-Ser136 epitope recognized by AP18 in MAP2B and the nonphosphorylated MAP2 epitope recognized by AP20 are localized to somata and dendrites of normal mice (Binder et al., 1986). Our results with p35+/+ mice agree with this localization and show that AP18 predominates in apical dendrites (Fig. 5A,E,I), whereas AP20 seems to predominate in somata (Fig. 5C,G,K). In contrast, little or no phospho-MAP2B was detected in somata of p35-/- mice (Fig. 5B,J), and a marked disorganization of dendritic structure and polarity was observed (Fig. 5B,F). Abnormal fascicles had no AP18 immunoreactivity (data not shown), confirming that these structures are composed of axons. In p35-/- mice, AP20 immunoreactivity diminished in apical dendrites but intensified in distal processes (Fig. 5D,H). In the cerebellum, AP20 staining of Purkinje cell somata and proximal dendrites was diminished, but more distal dendritic arborizations became prominent (Fig. 5L). Collectively, these data demonstrate a redistribution of MAP2 from somata to more distal neuronal processes in the brains of p35-/- mice.
Tau distribution
The subcellular locations of tau epitopes in normal mouse brains has been difficult to analyze, possibly because of sensitivity of tau to paraformaldehyde fixation. Using ammonium chloride pretreatment (see Materials and Methods), CP13 immunoreactivity was detected primarily in neuronal somata (Fig. 6A,C) but not in axons. In p35-/- brains, the intensity of somatic staining for the CP13 epitope was increased, whereas neuropil staining was diminished (Fig. 6B,D). Staining with this antibody again highlighted the disrupted cerebellar morphology in p35-/- brains (Fig. 6D). In contrast to CP13, the PHF-1 phosphoepitope was detected primarily in axons and in scattered neuronal and glial somata and nuclei of p35+/+ mice (Fig. 6E,G,I). In p35-/- brains, less PHF-1 phosphotau was detected in neuronal somata, most notably in Purkinje cells (Fig. 6J), whereas neuropil staining intensified mainly around cell bodies (Fig. 6F). In general, axonal staining was less, although the abnormal fascicles appeared more intense (Fig. 6H).
cdk5 and cytoskeletal protein phosphorylation
Of the various epitopes studied here, the only epitope that exhibited a decrease in phosphorylation correlating directly with a decrease in cdk5 activity was the SMI32 nonphosphorylated NF-H epitope (increased levels indicate decreased phosphorylation). Consistent with this relationship, we detected increased SMI32 immunoreactivity (Fig. 4F), corresponding with a loss of cdk5 immunoreactivity in neuronal cell bodies (Fig. 2D) in p35-/- brains. In contrast, SMI32 immunoreactivity was abnormally present in axon fascicles in p35-/- cortices in which cdk5 was enriched (Fig. 7A). These structures also had abundant SMI31 (Fig. 7B) and PHF-1 immunoreactivity (Fig. 7C). Yet, when one considers the overall reduction of brain cdk5 activity and the concomitant invariance of SMI31 NF-H immunoreactivity and increased PHF-1 immunoreactivity in immunoblots, it is unlikely that cdk5 acts directly on these epitopes. This conclusion is supported by our observations of normal brain in which cdk5, SMI31, and PHF-1 all localized to axons but little, if any, overlap was observed between cdk5 and SMI31 (Fig. 7D) or cdk5 and PHF-1 (Fig. 7E). A more striking discrepancy in localization of cdk5 with these phosphorylated cytoskeletal epitopes was in the large myelinated fiber tracts of the basal ganglia, in which cdk5 immunoreactivity was undetectable (Fig. 7F) but SMI31 (Fig. 7G) and PHF-1 (Fig. 7H) immunoreactivities were robust. The CP13 phosphotau epitope contains a classic cdk5 consensus sequence but was, surprisingly, lacking in axons (Fig. 6A), in which cdk5 immunoreactivity was robust in p35+/+ brains (Fig. 2C). Additionally, CP13 immunoreactivity had an inverse relationship with cdk5: CP13 immunoreactivity increased when cdk5 activity decreased in immunoblots, and CP13 was intensified in neuronal somata (Fig. 6B,D) of p35-/- mice, where cdk5 was reduced (Fig. 2D,I). The MC15 antibody recognizing the Ser235 phosphoepitope in tau also displayed an increase in p35-/- mice, but immunohistochemical analysis with this antibody was complicated by its cross-reactivity with a protein migrating at the same apparent size as NF-H. Colocalization of AP18 phospho-MAP2B immunoreactivity with cdk5 was noted in neuronal somata of normal mice, but in neuronal processes, phospho-MAP2B segregated to dendrites, and cdk5 to axons (Fig. 7I). Additionally, decreased cdk5 activity correlated with an increase in AP18 immunoreactivity in p35-/- brains. Collectively, our data indicate that with the possible exception of the SMI32 epitope, cdk5/p35 does not directly phosphorylate the majority of cytoskeletal protein epitopes but affects their phosphorylation indirectly by regulating their interaction with other PDKs.
Figure 7.

Distribution of cytoskeletal protein phosphoepitopes relative to cdk5. Images of 10 μm sagittal sections from p35-/- (A-C) and p35+/+ (D-I) brains incubated with antibodies to cdk5 and antibodies to cytoskeletal protein phosphoepitopes. A-C, In p35-/- cortex, cdk5 (green) colocalizes with non-P-NF-H (A, SMI32, red), P-NF-H/M (B, SMI31, red), and phosphotau (C, PHF1, red) in the abnormal fascicles (arrows) characteristic of these mice. D, E, In p35+/+ cortex, axons display immunoreactivity for cdk5 (D, E, green arrows), phospho-NF-H/M (D, SMI31, red arrowheads), and phosphotau (E, PHF-1, red arrowheads), but there is little, if any, overlap in the distribution of cdk5 with either phosphoepitope. F-H, In p35+/+ mice, large myelinated fiber tracts in the basal ganglia (arrows) are negative for cdk5 staining (F) but heavily stained for P-NF-H/M (G-SMI31) and phosphotau (H-PHF-1). I, In p35+/+ cortex, cdk5 (green) and phospho-MAP2B (AP18, red) both stain neuronal somata, but AP18 segregates to apical dendrites (arrow) and cdk5 to proximal axons (arrowhead).
Elevated GSK3β activity in p35-/- mice
GSK3β is another PDK that has been implicated in the production of the PHF-1 phosphotau epitope (for review, see Billingsley and Kincaid, 1997). Immunoblotting experiments demonstrated decreased levels of total GSK3β protein in p35-/- brains compared with p35+/+ brains (Fig. 8A, GSK3β; 39.1 ± 13.4%). Yet, we found GSK3β activity was increased in p35-/- brains. One known substrate of GSK3β is β-catenin, and GSK3β-mediated phosphorylation of this protein targets it for degradation via the ubiquitin-proteasome pathway (for review, see Cohen and Frame, 2001). Immunoblotting experiments demonstrated decreased levels of β-catenin in p35-/- brains compared with p35+/+ brains (Fig. 8A, β-catenin; 36.5 ± 13%; p = 0.004), indicating increased GSK3β activity. Additionally, IP kinase activity assays using recombinant human tau as a substrate revealed increased GSK3β activity in p35-/- brains compared with p35+/+ brains (Fig. 8B; 68.5 ± 13.7%; p = 0.01). This increased GSK3β activity in p35-/- mice supports the idea that cdk5 may negatively regulate GSK3β, either directly or indirectly.
Figure 8.
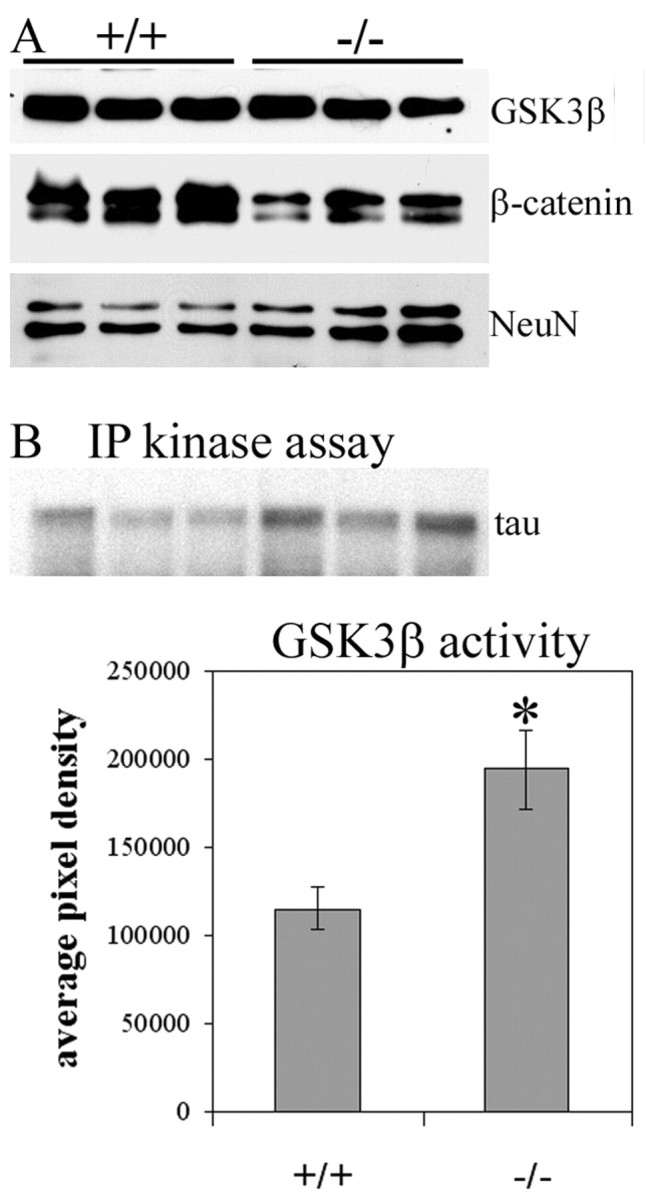
GSK3β levels and activity. A, Western blots showing slightly decreased levels of total GSK3β protein but decreased levels of β-catenin in p35+/+ and p35-/- whole-brain supernatants (25-50 μg of total protein). B, Autoradiograph of GSK3β IP kinase assay using tau as a substrate, and graph generated from IP kinase activity assays demonstrating a 68.5% increase in GSK3β activity in p35-/- brains compared with p35+/+ brains.
Discussion
Our studies of p35 deficiency in mice having a B6 genetic background substantiate the crucial role of cdk5/p35 in neuronal axodendritic polarity and the cytoarchitecture of the cerebral cortex, hippocampus, and cerebellum (Chae et al., 1997; Ohshima et al., 2001; present study). In addition, we report two novel findings: (1) p35 deficiency causes redistribution of cdk5 from its normal somatic and proximal axonal locations to distal neuronal processes; and (2) a decrease in adult brain cdk5 activity is accompanied, paradoxically, by a generalized increase in phosphorylation of neuronal cytoskeletal proteins.
The reduced cdk5 activity and brain architectural defects in all three p35-/- lines confirm that a significant fraction of brain cdk5 activity is dependent on p35 and cannot be compensated for by p39 or p67. Surprisingly, Ko et al. (2001) observed p39 upregulation without loss of cdk5-associated activity in p35-/- neonates, but Chae et al. (1997) were unable to detect histone H1 phosphorylation in cdk5 IPs from adult brains of the very same knock-out line. Ohshima et al. (2001) observed 80-90% reductions of kinase activity in cdk5 IPs from their adult p35-/- mice using a KSPXK repeat polypeptide from NF-H as substrate. These previous results would suggest that p35 is the dominant cdk5 activator in adult brain, an idea consistent with the lack of overt phenotype in p39-/- mice (Ko et al., 2001). In contrast, it seems unexpected that mice having no or 10-20% residual cdk5 activity could survive relatively normally (over 20 months), with only 15% dying from sporadic seizure activity. Indeed, mice lacking cdk5 do not survive postnatally (Ohshima et al., 2001). Our result of a 38% reduction of total brain cdk5 activity, in the absence of upregulation of p39 or p67 implies that more than one-third of total brain cdk5 activity is p35 dependent. This leaves a substantial balance to be modulated by other activators, or other mechanisms such as phosphorylation (Sharma et al., 1999; Zukerberg et al., 2000).
Despite barely detectable cdk5 activity in p35-/- (Chae et al., 1997), p35-/-;p39-/- (Ko et al., 2001), or p25 transgenic; p35-/- compound mice (Patzke et al., 2003), tau phosphorylation was found previously unaltered compared with wild-type mice. In contrast, we report significant elevations of cytoskeletal protein phosphorylation in our p35-/- mice. The effects of p35 deficiency on NF-H phosphorylation are remarkable in that immunoreactivity with SMI32 increased (reduced phosphorylation), SMI31 immunoreactivity was unchanged, and SMI34 immunoreactivity increased. In contrast, phosphorylation of the SMI31 site in NF-M; the CP13, MC15, and PHF-1 epitopes in tau; the AP18 epitope in MAP2; and the CP22, MC6, and MC15 epitopes in unknown proteins, was elevated in p35-/- mice. Others have reported hyperphosphorylation of NF proteins in cdk5-/- (Ohshima et al., 1996) and p35-/- (Ohshima et al., 2001; Sharma et al., 2002) mice. The most straightforward interpretation of such hyperphosphorylation accompanying reduced cdk5 activity is that cdk5 does not produce these phosphoepitopes. This idea is supported by the lack of colocalization of cdk5 and the phosphoepitopes in many regions of normal and p35-/- brain. The atypical axon fascicles of p35-/- brains could be a site of increased phosphorylation of these epitopes, because they colocalize with cdk5 in these structures. However, given the decrease of cdk5 activity in p35-/- mice, it is unlikely that cdk5 produces the hyperphosphorylation, unless p39, p67, or some other activator were elevated in the same location. This possibility needs to be explored once more specific antibodies for p39 and p67 antibodies become available.
An alternative explanation for the inverse relationship between cdk5 activity and phosphorylation is that cdk5 functions as a negative regulator of other PDKs, such as MAPK, GSK3β, or JNK, which act directly on cytoskeletal protein substrates. This hypothesis is supported by the increased GSK3β activity we detected in p35-/- brains and by the demonstration of cdk5-mediated phosphorylation and inhibition of MAP kinase kinase-1 (MEK1) in vivo, resulting in downregulation of extracellular signal-regulated kinase (ERK) 1/2 (Sharma et al., 2002), a kinase that phosphorylates NF (Veeranna et al., 1998), tau (for review, see Zhu et al., 2002), and MAP2 (Berling et al., 1994). In cdk5-/- and p35-/- mice, MEK1 and ERK1/2 are hyperactivated, and SMI31 immunoreactivity for NF-M is increased (Sharma et al., 2002). Similarly, cdk5 phosphorylates and inhibits JNK-3 (Li et al., 2002), another kinase shown to phosphorylate NF (Giasson and Mushynski, 1997; Brownlees et al., 2000), tau (Anderton et al., 2001; Sato et al., 2002), and MAP2 (Chang et al., 2003). cdk5 has also been shown to phosphorylate and activate another brain kinase with homology to cdks, pctaire-1 (Cheng et al., 2002); a relationship between pctaire-1 and cytoskeletal protein phosphorylation has not been described, but a role in the regulation of neurite outgrowth has been reported (Graeser et al., 2002). Furthermore, a recent study using calpain-mediated production of p25 in primary rat neurons found elevated cdk5 activity and decreased tau phosphorylation (Kerokoski et al., 2002), which is consistent with an inverse relationship between cdk5 activity and cytoskeletal protein phosphorylation. Alternatively, changes in activity of GSK3β or other PDKs may be a developmental compensatory response to reduced cdk5 activity, and it is possible such developmental compensation may activate kinases that, under normal physiological circumstances (i.e., intact cdk5 activity), do not phosphorylate these substrates. Thus, the paradoxical results do not rule out a role for cdk5 in those modifications nor do they prove that any upregulated kinases produce those modifications.
The expectation from in vitro studies and human neurodegenerative diseases having concomitant elevations of cdk5 activity and phosphorylation is that elevated cdk5 activity causes protein hyperphosphorylation. Surprisingly, hyperphosphorylation has not been observed in most transgenic mouse models of increased cdk5 activity. Overexpression of cdk5, p35, and human tau caused elevated cdk5 activity but no tau hyperphosphorylation (Van den Haute et al., 2001). But, when p25 was overexpressed together with mutant tau, hyperphosphorylation was seen (Noble et al., 2003), perhaps as a consequence of ectopic accumulation of mutant tau in neurons. In two independent p25-overexpressing strains, no net increase in tau (or NF) phosphorylation was detected, even with 2-5 fold elevations of cdk5 activity (Ahlijanian et al., 2000; Takashima et al., 2001). Another mouse with neuronally targeted p25 overexpression displayed axonopathy and dyskinesia, but no change in protein phosphorylation (Bian et al., 2002). Thus, elevated cdk5 activity may cause alterations in subcellular distribution or protein turnover, which might be key to the development of cytoskeletal pathology. That decreased phosphorylation of cytoskeletal proteins, as would be predicted by our suggestion of a negative influence of cdk5 on other PDKs, is not observed in these mouse models implies that these PDKs are maximally inhibited in normal brain. Thus, increasing the activity of cdk5 beyond basal levels might have no significant effect on protein phosphorylation, whereas inhibiting basal cdk5 activity results in hyperphosphorylation.
The transport of NFs into neuronal processes occurs via a kinesin-dependent mechanism along microtubules (for review, see Hirokawa, 1998; Shea and Flanagan, 2001), whereas microtubules and MAPs are transported by a dynein-powered mechanism (Baas, 1999). In p35-/- mice, NFs retain their normal distribution, except for the presence of nonphosphorylated isoforms in axon fascicles. However, cdk5, PHF-1-positive tau, and MAP2 shift from neuronal somata to distal sites of the neuropil. This pattern implies an impairment of dynein-based retrograde transport in the p35-/- mice. Previous studies have shown that cdk5 phosphorylates Nudel, a dynein-interacting protein, and alters its subcellular distribution (Niethammer et al., 2000). Whether the relocations in p35-/- mouse brain are mediated by dynein and Nudel requires additional studies. In cdk5-/- mice, phosphorylated NF accumulated in neuronal soma, indicating defective NF transport (Sharma et al., 2002). We speculate that different levels of cdk5 inhibition might affect selective transport mechanisms, with dynein-driven transport being more sensitive to lower levels of cdk5 inhibition than kinesin-driven transport. Regardless, axonal transport is critical for the establishment of axodendritic polarity (Takada et al., 1988), and the aberrant axodendritic polarity in p35-/- mice reinforces the role of cdk5/p35 in this process.
The redistribution of cytoskeletal proteins could itself promote errant interactions with other PDKs. Even without hyperactivation of these PDKs, differences in the rate of turnover of phosphoproteins in their new locations could result in accumulation and “apparent” hyperphosphorylation of the proteins. Additional studies will be required to determine the relative contributions of subcellular distribution of cytoskeletal proteins and other PDKs in their phosphorylation and determine how cdk5 regulates these PDKs. In Alzheimer's disease, appearance of the CP13 epitope in the somatodendritic compartment is one of the earliest pathological signs (Braak et al., 1994; Su et al., 1994), followed by PHF-1 immunoreactivity in neurites (Su et al., 1994). We observed accumulation of CP13 and PHF-1 immunoreactivity at similar locations in p35-/- mice, suggesting that the mice will be useful for identifying the PDKs that produce these phosphoepitopes.
In summary, our analyses of p35-/- adult mice suggest that p35 is an essential regulator of the activity, distribution, and interaction of cdk5 with appropriate substrates in a given neuronal microenvironment. The function of p35, therefore, bears resemblance to its cyclin counterparts regulating cdks during cell division. In some brain regions, cdk5 may influence the activities of other neuronal kinases in a hierarchical manner to affect downstream cytoskeletal protein targets. As demonstrated by our evidence for GSK3β activation in p35-/- mice, this model is of value in identifying these kinases.
Footnotes
This work was supported by National Institute on Aging Grants AG12721 (I.V.) and AG00057 (J.L.H.) and Alzheimer's Association Grant 11RG-99-18 (I.V.). We thank Drs. Peter Davies, Harish Pant, Lester Binder, and William Honer for the generous gifts of antibodies, Drs. Jerry Wang and John Lew for p35 plasmid, and Dr. H. Jurgen Wenzel for the images of hematoxylin and eosin-stained wild-type and p35 null cortices.
Correspondence should be addressed to Dr. Inez Vincent, University of Washington, 1959 Pacific Street, Box 357705, Seattle, WA 98195. E-mail: ivincent@u.washington.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310633-12$15.00/0
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, Mc-Carthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD ( 2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 97: 2910-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderton BH, Betts J, Blackstock WP, Brion JP, Chapman S, Connell J, Dayanandan R, Gallo JM, Gibb G, Hanger DP, Hutton M, Kardalinou E, Leroy K, Lovestone S, Mack T, Reynolds CH, Van Slegtenhorst M ( 2001) Sites of phosphorylation in tau and factors affecting their regulation. Biochem Soc Symp 67: 73-80. [DOI] [PubMed] [Google Scholar]
- Baas PW ( 1999) Microtubules and neuronal polarity: lessons from mitosis. Neuron 22: 23-31. [DOI] [PubMed] [Google Scholar]
- Bajaj NP, Al-Sarraj ST, Anderson V, Kibble M, Leigh N, Miller CC ( 1998) Cyclin-dependent kinase-5 is associated with lipofuscin in motor neurones in amyotrophic lateral sclerosis. Neurosci Lett 245: 45-48. [DOI] [PubMed] [Google Scholar]
- Bajaj NP, al-Sarraj ST, Leigh PN, Anderson V, Miller CC ( 1999) Cyclin dependent kinase-5 (CDK-5) phosphorylates neurofilament heavy (NF-H) chain to generate epitopes for antibodies that label neurofilament accumulations in amyotrophic lateral sclerosis (ALS) and is present in affected motor neurones in ALS. Prog Neuropsychopharmacol Biol Psychiatry 23: 833-850. [DOI] [PubMed] [Google Scholar]
- Berling B, Wille H, Roll B, Mandelkow EM, Garner C, Mandelkow E ( 1994) Phosphorylation of microtubule-associated proteins MAP2a, b and MAP2c at Ser136 by proline-directed kinases in vivo and in vitro. Eur J Cell Biol 64: 120-130. [PubMed] [Google Scholar]
- Bian F, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KK, Walker LC ( 2002) Axonopathy, tau abnormalities, and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol 446: 257-266. [DOI] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL ( 1997) Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 323: 577-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI ( 1986) Differential localization of MAP-2 and tau in mammalian neurons in situ. Ann NY Acad Sci 466: 145-166. [DOI] [PubMed] [Google Scholar]
- Borghi R, Giliberto L, Assini A, Delacourte A, Perry G, Smith MA, Strocchi P, Zaccheo D, Tabaton M ( 2002) Increase of cdk5 is related to neurofibrillary pathology in progressive supranuclear palsy. Neurology 58: 589-592. [DOI] [PubMed] [Google Scholar]
- Braak E, Braak H, Mandelkow EM ( 1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87: 554-567. [DOI] [PubMed] [Google Scholar]
- Brion JP, Couck AM ( 1995) Cortical and brainstem-type Lewy bodies are immunoreactive for the cyclin-dependent kinase 5. Am J Pathol 147: 1465-1476. [PMC free article] [PubMed] [Google Scholar]
- Brownlees J, Yates A, Bajaj NP, Davis D, Anderton BH, Leigh PN, Shaw CE, Miller CC ( 2000) Phosphorylation of neurofilament heavy chain side-arms by stress activated protein kinase-1b/Jun N-terminal kinase-3. J Cell Sci 113: 401-407. [DOI] [PubMed] [Google Scholar]
- Bu B, Li J, Davies P, Vincent I ( 2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22: 6515-6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR ( 2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33: 95-130. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH ( 1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18: 29-42. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M ( 2003) JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of Microtubule-associated proteins. Dev Cell 4: 521-533. [DOI] [PubMed] [Google Scholar]
- Cheng K, Li Z, Fu WY, Wang JH, Fu AK, Ip NY ( 2002) Pctaire1 interacts with p35 and is a novel substrate for Cdk5/p35. J Biol Chem 277: 31988-31993. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S ( 2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769-776. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Mushynski WE ( 1997) Study of proline-directed protein kinases involved in phosphorylation of the heavy neurofilament subunit. J Neurosci 17: 9466-9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein ME, Sternberger LA, Sternberger NH ( 1983) Microheterogeneity (“neurotypy”) of neurofilament proteins. Proc Natl Acad Sci USA 80: 3101-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeser R, Gannon J, Poon RY, Dubois T, Aitken A, Hunt T ( 2002) Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite outgrowth in Neuro-2A cells. J Cell Sci 115: 3479-3490. [DOI] [PubMed] [Google Scholar]
- Grant P, Sharma P, Pant HC ( 2001) Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem 268: 1534-1546. [PubMed] [Google Scholar]
- Hirokawa N ( 1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519-526. [DOI] [PubMed] [Google Scholar]
- Honjyo Y, Kawamoto Y, Nakano S, Akiguchi L ( 2001) P39 immunoreactivity in glial cytoplasmic inclusions in brains with multiple system atrophy. Acta Neuropathol (Berl) 101: 190-194. [DOI] [PubMed] [Google Scholar]
- Ioffe E, Liu Y, Bhaumik M, Poirier F, Factor SM, Stanley P ( 1995) WW6: an embryonic stem cell line with an inert genetic marker that can be traced in chimeras. Proc Natl Acad Sci USA 92: 7357-7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K, Kobayashi S, Omori A, Takamatsu M, Yonekura S, Anzai K, Imahori K, Uchida T ( 1994) Identification of the 23 kDa subunit of tau protein kinase II as a putative activator of cdk5 in bovine brain. FEBS Lett 342: 203-208. [DOI] [PubMed] [Google Scholar]
- Kerjaschki D, Noronha-Blob L, Sacktor B, Farquhar MG ( 1984) Microdomains of distinctive glycoprotein composition in the kidney proximal tubule brush border. J Cell Biol 98: 1505-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T ( 2002) Cleavage of the cyclin-dependent kinase 5 activator p35 to p25 does not induce tau hyperphosphorylation. Biochem Biophys Res Commun 298: 693-698. [DOI] [PubMed] [Google Scholar]
- Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH ( 2001) p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci 21: 6758-6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Tsai LH, Crandall JE ( 1999) Callosal axon guidance defects in p35(-/-) mice. J Comp Neurol 415: 218-229. [DOI] [PubMed] [Google Scholar]
- Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN ( 1999) Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res 34: 21-29. [DOI] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH ( 1994) A brain-specific activator of cyclin-dependent kinase 5. Nature 371: 423-426. [DOI] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC ( 2002) Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J 21: 324-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litersky JM, Johnson GV ( 1992) Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem 267: 1563-1568. [PubMed] [Google Scholar]
- Liu WK, Williams RT, Hall FL, Dickson DW, Yen SH ( 1995) Detection of a Cdc2-related kinase associated with Alzheimer paired helical filaments. Am J Pathol 146: 228-238. [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Tomizawa K, Lu YF, Moriwaki A, Tokuda M, Itano T, Wang JH, Hatase O, Matsui H ( 1996) Distinct cellular compartment of cyclin-dependent kinase 5 (Cdk5) and neuron-specific Cdk5 activator protein (p35nck5a) in the developing rat cerebellum. Brain Res 734: 319-322. [PubMed] [Google Scholar]
- Michel G, Mercken M, Murayama M, Noguchi K, Ishiguro K, Imahori K, Takashima A ( 1998) Characterization of tau phosphorylation in glycogen synthase kinase-3beta and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim Biophys Acta 1380: 177-182. [DOI] [PubMed] [Google Scholar]
- Miller CC, Ackerley S, Brownlees J, Grierson AJ, Jacobsen NJ, Thornhill P ( 2002) Axonal transport of neurofilaments in normal and disease states. Cell Mol Life Sci 59: 323-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM ( 1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201-211. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kawamoto Y, Nakano S, Akiguchi I, Kimura J ( 1997a) p35nck5a and cyclin-dependent kinase 5 colocalize in Lewy bodies of brains with Parkinson's disease. Acta Neuropathol (Berl) 94: 153-157. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kawamoto Y, Nakano S, Ikemoto A, Akiguchi I, Kimura J ( 1997b) Cyclin-dependent kinase 5 in Lewy body-like inclusions in anterior horn cells of a patient with sporadic amyotrophic lateral sclerosis. Neurology 48: 267-270. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kawamaoto Y, Nakano S, Akiguchi I, Kimura J ( 1998) Cyclin-dependent kinase 5 and mitogen-activated protein kinase in glial cytoplasmic inclusions in multiple system atrophy. J Neuropathol Exp Neurol 57: 690-698. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP ( 2001) Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30: 135-147. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH ( 2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28: 697-711. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH ( 1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: 816-825. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K ( 2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38: 555-565. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB ( 1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93: 11173-11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB ( 1999) Migration defects of cdk5(-/-) neurons in the developing cerebellum is cell autonomous. J Neurosci 19: 6017-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Ogawa M, Veeranna, Hirasawa M, Longenecker G, Ishiguro K, Pant HC, Brady RO, Kulkarni AB, Mikoshiba K ( 2001) Synergistic contributions of cyclin-dependant kinase 5/p35 and Reelin/Dab1 to the positioning of cortical neurons in the developing mouse brain. Proc Natl Acad Sci USA 98: 2764-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Caceres A ( 1998) Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J Neurosci 18: 9858-9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH ( 1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Patzke H, Maddineni U, Ayala R, Morabito M, Volker J, Dikkes P, Ahlijanian MK, Tsai LH ( 2003) Partial rescue of the p35-/- brain phenotype by low expression of a neuronal-specific enolase p25 transgene. J Neurosci 23: 2769-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF ( 1998) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res 797: 267-277. [DOI] [PubMed] [Google Scholar]
- Philpott A, Tsai L, Kirschner MW ( 1999) Neuronal differentiation and patterning in Xenopus: the role of cdk5 and a novel activator xp35.2. Dev Biol 207: 119-132. [DOI] [PubMed] [Google Scholar]
- Pigino G, Paglini G, Ulloa L, Avila J, Caceres A ( 1997) Analysis of the expression, distribution and function of cyclin dependent kinase 5 (cdk5) in developing cerebellar macroneurons. J Cell Sci 110: 257-270. [DOI] [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST ( 1998) A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J Neurosci 18: 7717-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J ( 2000) Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61: 133-168. [DOI] [PubMed] [Google Scholar]
- Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A ( 2002) Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem 277: 42060-42065. [DOI] [PubMed] [Google Scholar]
- Sharma P, Sharma M, Amin ND, Albers RW, Pant HC ( 1999) Regulation of cyclin-dependent kinase 5 catalytic activity by phosphorylation. Proc Natl Acad Sci USA 96: 11156-11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Veeranna, Sharma M, Amin ND, Sihag RK, Grant P, Ahn N, Kulkarni AB, Pant HC ( 2002) Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J Biol Chem 277: 528-534. [DOI] [PubMed] [Google Scholar]
- Shea TB, Beermann ML ( 1993) Evidence that the monoclonal antibodies SMI-31 and SMI-34 recognize different phosphorylation-dependent epitopes of the murine high molecular mass neurofilament subunit. J Neuroimmunol 44: 117-121. [DOI] [PubMed] [Google Scholar]
- Shea TB, Flanagan LA ( 2001) Kinesin, dynein and neurofilament transport. Trends Neurosci 24: 644-648. [DOI] [PubMed] [Google Scholar]
- Shetty KT, Kaech S, Link WT, Jaffe H, Flores CM, Wray S, Pant HC, Beushausen S ( 1995) Molecular characterization of a neuronal-specific protein that stimulates the activity of Cdk5. J Neurochem 64: 1988-1995. [DOI] [PubMed] [Google Scholar]
- Smith DS, Tsai LH ( 2002) Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol 12: 28-36. [DOI] [PubMed] [Google Scholar]
- Su JH, Cummings BJ, Cotman CW ( 1994) Early phosphorylation of tau in Alzheimer's disease occurs at Ser-202 and is preferentially located within neurites. NeuroReport 5: 2358-2362. [DOI] [PubMed] [Google Scholar]
- Takada K, Becker LE, Chan F ( 1988) Aberrant dendritic development in the human agyric cortex: a quantitative and qualitative Golgi study of two cases. Clin Neuropathol 7: 111-119. [PubMed] [Google Scholar]
- Takahashi M, Iseki E, Kosaka K ( 2000a) Cdk5 and munc-18/p67 co-localization in early stage neurofibrillary tangles-bearing neurons in Alzheimer type dementia brains. J Neurol Sci 172: 63-69. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Iseki E, Kosaka K ( 2000b) Cyclin-dependent kinase 5 (Cdk5) associated with Lewy bodies in diffuse Lewy body disease. Brain Res 862: 253-256. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Yasutake K, Takahashi H, Yokoyama M, Ishiguro K ( 2001) Involvement of cyclin dependent kinase 5 activator p25 on tau phosphorylation in mouse brain. Neurosci Lett 306: 37-40. [DOI] [PubMed] [Google Scholar]
- Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH ( 1995) An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem 270: 26897-26903. [DOI] [PubMed] [Google Scholar]
- Terada M, Yasuda H, Kogawa S, Maeda K, Haneda M, Hidaka H, Kashiwagi A, Kikkawa R ( 1998) Expression and activity of cyclin-dependent kinase 5/p35 in adult rat peripheral nervous system. J Neurochem 71: 2600-2606. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR ( 1987) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51: 503-512. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Takahashi T, Caviness JR VS, Harlow E ( 1993) Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119: 1029-1040. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness Jr VS, Chae T, Harlow E ( 1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419-423. [DOI] [PubMed] [Google Scholar]
- Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F ( 2001) Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis 8: 32-44. [DOI] [PubMed] [Google Scholar]
- Veeranna, Grant P, Pant HC ( 1997) Expression of p67 (Munc-18), Cdk5, P-NFH and syntaxin during development of the rat cerebellum. Dev Neurosci 19: 172-183. [DOI] [PubMed] [Google Scholar]
- Veeranna, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC ( 1998) Mitogen-activated protein kinases (Erk1, 2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci 18: 4008-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Kim E, Davies P ( 1994) Increased production of paired helical filament epitopes in a cell culture system reduces the turnover of tau. J Neurochem 62: 715-723. [DOI] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV, Jope RS ( 2002) Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci USA 99: 7951-7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K ( 1996) Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol (Berl) 92: 232-241. [DOI] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA ( 2002) The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals 11: 270-281. [DOI] [PubMed] [Google Scholar]
- Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH ( 2000) Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26: 633-646. [DOI] [PubMed] [Google Scholar]