

Abstract
Remodeling of synaptic networks through an activity-dependent formation or elimination of synaptic connections is believed to contribute to information processing and long-term memory. Recent work showed that enhanced synaptic activation, including induction of long-term potentiation and sensory stimulation, promote a rapid growth of dendritic filopodia and the formation of new spines or new types of synapses. Here, we investigated whether calcium/calmodulin-dependent protein kinase II (CaMKII), an enzyme implicated in the control of synaptic efficacy, also participated in these mechanisms. We show that the intracellular application of autophosphorylated CaMKII reproduced these morphological changes and triggered filopodia growth and spine formation. In addition, we find that activation of endogenous kinase through the inhibition of phosphatases or the application of calmodulin in the cell produced similar effects. Conversely, blockade of CaMKII activity prevented the synaptic enhancement, the growth of filopodia and formation of new spines triggered by LTP induction, and a short anoxia/hypoglycemia. Together, these results support the interpretation that CaMKII contributes to the control of activity-dependent structural plasticity.
Keywords: hippocampus, LTP (long-term potentiation), phosphorylation, synapse, synaptogenesis, confocal microscopy
Introduction
Work by several laboratories showed that synaptic activation and induction of long-term potentiation (LTP) promote forms of structural plasticity and trigger the growth of dendritic filopodia and formation of new spines or new types of synapses (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Yuste and Bonhoeffer, 2001). Similar changes were also described after potassium depolarization (Goldin et al., 2001), application of a short anoxia/hypoglycemia (Jourdain et al., 2002), and more recently, after sensory stimulation of barrel cortex in living, adult mice (Trachtenberg et al., 2003). Although the functional implication of this structural plasticity remains unclear, it is believed that it could represent a possible long-term adaptation contributing to information processing by brain networks. Therefore, an important question is to understand the molecular mechanisms that underlie this structural plasticity and how they relate to those affecting synaptic efficacy. Among the molecules shown to play a critical role in LTP mechanisms is the enzyme calcium/calmodulin dependent protein kinase II (CaMKII; Hudmon and Schulman, 2002; Lisman et al., 2002). This protein is highly enriched in postsynaptic densities, it is stably activated through an autophosphorylation process, and numerous observations indicate that it contributes to the increase in synaptic efficacy (Fukunaga et al., 1993; Lledo et al., 1995; Wang and Kelly, 1995; Barria et al., 1997; Giese et al., 1998; Lisman et al., 2002). According to the most recent models of LTP, CaMKII, together with other protein kinases, could affect the functioning of postsynaptic AMPA receptors by direct phosphorylation (McGlade McCulloh et al., 1993; Barria et al., 1997) or regulate the synaptic targeting or trafficking of AMPA receptors at the membrane (Shi et al., 1999; Poncer et al., 2002; Song and Huganir, 2003). In view of this central role in the control of synaptic efficacy and a possible involvement in neurite outgrowth (Goshima et al., 1993), we examined whether the enzyme contributed to the dynamics of filopodia growth and spine formation associated with LTP induction. Using hippocampal organotypic slice cultures, we provide evidence that CaMKII is a key regulator of activity-induced structural plasticity.
Materials and Methods
Slice cultures. Hippocampal organotypic slice cultures were prepared from 7-d-old rats and maintained 10-15 d in culture as described previously (Stoppini et al., 1991). Electrophysiology and confocal imaging experiments were performed with slice cultures placed on an infrapatch setup (Luigs & Neumann, Ratingen, Germany) under continuous perfusion (4 ml/min) with a medium containing the following (in mm): 124 NaCl, 1.6 KCl, 2.5 CaCl2, 1.5 MgCl2, 24 NaHCO3, 1.2 KH2PO4, 10 glucose, and 2 ascorbic acid, pH 7.4, at 33°C. Anoxia/hypoglycemia was produced by replacing oxygen with nitrogen and switching to a medium containing sucrose instead of glucose for 5 min. Theta burst stimulation consisted of five trains at 5 Hz, each composed of four pulses at 100 Hz, repeated three times at 10 sec intervals.
Confocal imaging. CA1 pyramidal neurons were patched using electrodes filled with a medium containing the following (in mm): 130 K-gluconate, 4 NaCl, 5 EGTA, 20 HEPES, 1 CaCl2, 1 MgCl2, 0.2 Na3GTP, and 2 Na2ATP, pH 7.2-7.4; 290 mOsm, sulforhodamine 0.04-0.06%. Imaging of dendritic spines was performed through a 40× water immersion objective using a Bio-Rad (Hercules, CA) MRC1024 scan head and a Mira 900 laser (Coherent Inc., Santa Clara, CA) set at a 830 nm wavelength. Small dendritic segments (50-250 μm in length), located on secondary and tertiary dendrites, were imaged every 2-5 min over a 1 hr period. Morphological changes (filopodia or new spines) were assessed for an observation period of 60 min. Filopodia were defined as thin, dynamic dendritic protrusions of over 1.5 μm in length. They usually extended rapidly, and then tended to retract, but in all cases remained present until the end of the experiment. All analyses were done blind by two investigators.
CaMKII, purified from rat brain, contained the α and β isoforms as well as the association domain. It was activated by incubation on ice for 10 min at a concentration of 8 μm with 0.5 mm calcium, 3 μm calmodulin, 10 mm Mg acetate, 0.1 mm ATP, and 1 mg/ml BSA and then diluted to a final concentration of 200 nm in the patch pipette medium. After whole-cell access, evoked synaptic responses were continuously monitored and dendritic segments imaged for 90 min. Inactivated CaMKII was prepared in the same way, except that the incubation was performed at 100°C. Calyculin A was diluted in the pipette medium at a final concentration of 150 nm and calmodulin at concentrations of 150 and 450 nm. Two CaMKII inhibitors were used: 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN93) (10 μm), a potent but nonspecific inhibitor, and autocamtide-2-related inhibitory peptide (AIP; 200 nm), a specific CaMKII inhibitor that does not block CaMKI or CaMKIV in in vitro tests (Ishida et al., 1995).
For LTP experiments, the stimulation electrode was placed in the stratum radiatum in the vicinity of the imaged dendritic segment of the recorded cell (20-50 μm). The stimulation intensity was adjusted to evoke 50-150 pA EPSCs. Recordings were started immediately after whole-cell access and, if baseline conditions were stable, high-frequency stimulation was applied after 10 min. Two sets of images were taken before high-frequency stimulation as baseline controls and then continued for 60 min. These experiments were then reproduced in the presence of KN93, applied in the perfusion medium (10 μm). For anoxia/hypoglycemia experiments, a 20 min baseline was observed, anoxia/hypoglycemia was applied for 5 min, and imaging was continued for 60 min. CaMKII was blocked by adding AIP or KN93 to the pipette medium at final concentrations of 200 nm and 40 μm, respectively.
Results
To assess structural plasticity, we used time-lapse two-photon confocal imaging of dendritic segments of CA1 pyramidal cells in hippocampal organotypic slice cultures. Cells were injected with a fluorescent dye (sulforhodamine), and mechanisms of filopodia growth and spine formation were analyzed by repeatedly imaging the same dendritic segment every 2-5 min over a period of at least 60 min. All events (filopodia or new spines) appearing during the imaging period (60 min for all experiments) were quantified, expressed as a function of the length of the dendritic segment under analysis, and averaged across experiments.
As indicated in Figure 1A, analyses of the morphology of dendrites and spines under control conditions revealed few if any changes over time in examined dendritic segments (Fig. 1A). In 16 experiments (2.7 mm of dendritic segments analyzed), only one filopodia and one new spine were found to appear spontaneously during the imaging period (60 min). Overall, this represented <1 new filopodia or spine per millimeter of dendritic segment analyzed or a change affecting <0.3% of all spines. Thus, the frequency of spontaneously occurring new filopodia or new spines was low under control conditions.
Figure 1.

Growth of filopodia and formation of new spines induced by intracellular injection of activated CaMKII. A, Confocal microscopic images of dendritic spines taken 15 and 60 min after intracellular injection of the fluorescent dye sulforhodamine in a control experiment. B, Growth of two filopodia (arrows) and the formation of three new spines (asterisks) in a cell injected with activated CaMKII. C, Absence of changes in a cell injected with heat-inactivated CaMKII. Scale bars, 2 μm. D, Changes in EPSC slope measured as a function of time in seven cells injected with activated CaMKII (black circles) and five cells injected with the heat-inactivated enzyme (open circles). Data are means ± SEM. E, Quantitative analysis of the growth of filopodia (black columns) and formation of new spines (open columns) expressed as events per 100 μm of dendritic segment and per 60 min under control condition (n = 16), after the injection of activated (n = 14) or heat-inactivated (n = 9) CaMKII (*p < 0.01; t test).
To investigate the role of CaMKII in these mechanisms, we tested the effects of intracellularly injecting an autophosphorylated form of the enzyme in CA1 pyramidal cells (200 nm; see Materials and Methods). To verify that the injected enzyme was functional, we assessed its effect on the size of evoked synaptic responses. Previous work has shown that this procedure results in a potentiation of synaptic transmission that occludes LTP (Lledo et al., 1995). As illustrated in Figure 1D, cells injected with the activated kinase showed a marked potentiation of evoked EPSCs. Time-lapse analyses of dendritic morphology revealed that injected cells also displayed a markedly enhanced structural plasticity (Fig. 1B). In 13 of 14 experiments (1.93 mm of dendritic segments analyzed), we could detect either the growth of filopodia (10 cases) or the appearance of new spines (10 cases), and in many experiments, we could detect the two events simultaneously. Compared with control conditions, the spontaneous occurrence of filopodia and new spines was enhanced by more than an order of magnitude (Fig. 1E) (p < 0.01, t test; p < 0.05, Mann-Whitney U test). The number of new events so detected represented an increase of ∼2.7% of the total number of spines. Filopodia, in these experiments, appeared mostly 40-60 min after whole-cell access (mean onset time, 48.3 ± 3.9 min), whereas the formation of the new spines was delayed (mean onset time, 56.6 ± 2.5 min).
To verify that these morphological changes were caused by the injection of activated CaMKII, we performed experiments with a heat-inactivated form of the enzyme. As illustrated in Figure 1C,D, injection of the heat-inactivated kinase produced no increase in the size of evoked EPSCs and no evidence of structural plasticity. In nine experiments (1.25 mm of dendritic segments analyzed), only one case of new filopodia and no new spines were detected (Fig. 1E) (p < 0.02; t test).
We then investigated whether the activation of endogenous CaMKII was sufficient to produce these effects. For this, pyramidal cells were injected either with calyculin A (150 nm), a phosphatase inhibitor that markedly enhances autophosphorylation of the endogenous enzyme (Fukunaga et al., 1993), or calmodulin (150 or 450 nm), a direct activator of CaMKII (Hudmon and Schulman, 2002). As illustrated in Figure 2, these conditions also resulted in a marked increase in structural plasticity. In 13 experiments with calyculin A (analysis of 2.0 mm of dendritic segments), 14 filopodia and 13 new spines were observed, resulting in averaged changes comparable with those produced by CaMKII. Similarly, nine experiments with 150 nm calmodulin (1.7 mm analyzed) and six experiments with 450 nm (0.8 mm analyzed) yielded comparable increases in both filopodia growth and newly formed spines (12 filopodia and 12 new spines detected) (Fig. 2B). We verified that the effect of calmodulin was attributable to the activation of endogenous CaMKII by coinjecting calmodulin together with AIP (200 nm), a specific and potent inhibitor of CaMKII (Ishida et al., 1995). In six experiments (1.2 mm analyzed), this procedure completely prevented induction of structural plasticity by calmodulin (p < 0.05; t test).
Figure 2.
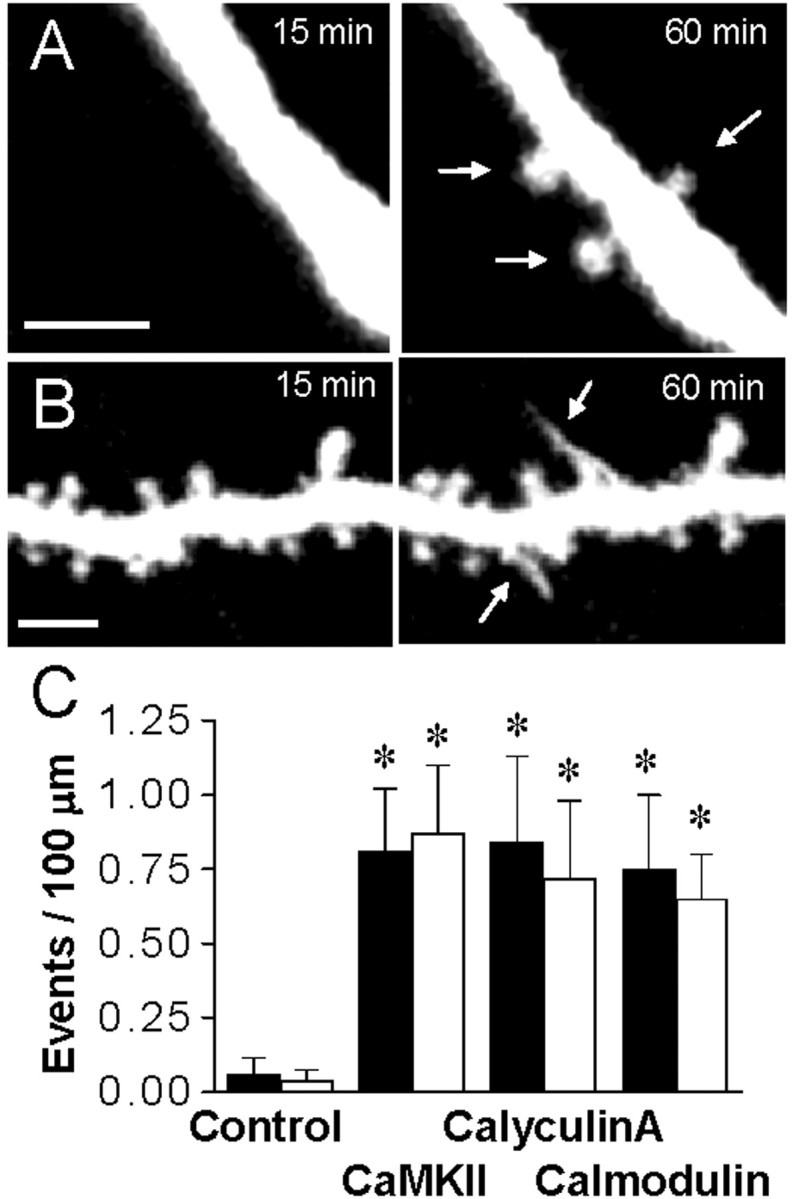
Structural plasticity is induced by the activation of endogenous CaMKII. A, Formation of three new spines (arrows) in a cell injected with the phosphatase inhibitor calyculin A (150 nm). B, Growth of two filopodia in a cell injected with calmodulin (150 nm; bars: 2 μm). C, Growth of filopodia (black columns) and formation of new spines (open columns), expressed as events per 100 μm of dendritic segment and per 60 min, under control conditions (n = 16), after the intracellular injection of activated CaMKII (n = 14), or the phosphatase inhibitor calyculin A (150 nm; n = 13) or calmodulin (150 and 450 nm; n = 15). Data are means ± SEM. *p < 0.01; t test.
We then continued by investigating whether CaMKII phosphorylation activity was required for stimulation-induced structural plasticity. A stimulating electrode was placed in the vicinity (20-50 μm) of the dendritic arbor of a pyramidal cell injected with sulforhodamine. The stimulation intensity was adjusted so as to evoke a synaptic response, and 10 min afterward, whole-cell access LTP was induced by theta burst stimulation and the dendritic segment under analysis was repeatedly imaged. As illustrated in Figure 3, this protocol resulted in a robust LTP, but also in an enhanced structural plasticity. In 10 experiments (1.6 mm analyzed), we could observe 13 cases of filopodia growth and 15 cases of newly formed spines (Fig. 3A). The filopodia were early events, with a mean onset time of 12.5 ± 2.7 min after stimulation, whereas the newly formed spines appeared after a longer delay (32.1 ± 2.6 min). This LTP-associated structural plasticity was dependent on NMDA receptor activation, because the application of theta burst stimulation in the presence of MK801 (40 μm) prevented synaptic potentiation and the structural changes (Fig. 3D) (1.4 mm of dendritic segments analyzed; n = 8; p < 0.05; t test).
Figure 3.
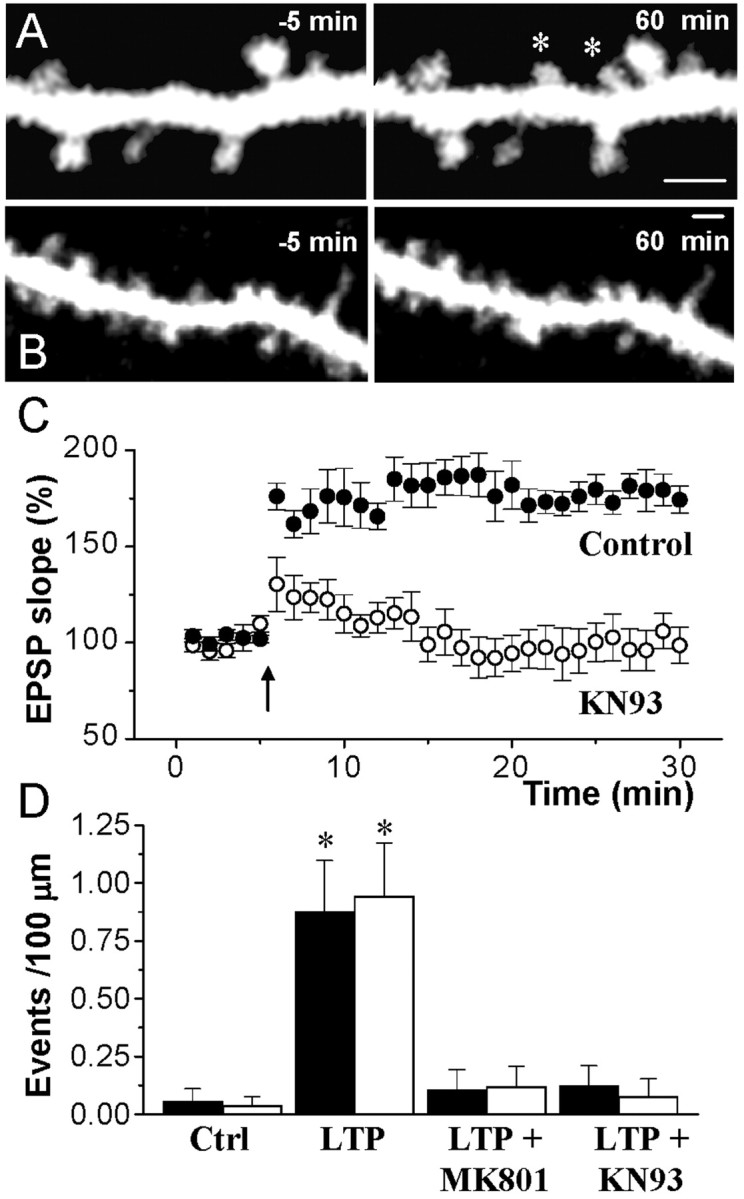
Blockade of LTP-induced filopodia growth and spine formation by inhibition of CaMKII. A, Formation of two new spines (asterisks) induced by theta burst stimulation. B, Application of high-frequency stimulation in the presence of KN93 (10 μm) did not induce the growth of filopodia or the formations of new spines. Scale bars, 2 μm. C, Changes in EPSC amplitude triggered by theta burst stimulation under control conditions (black circles) or in the presence of KN93 (10 μm, open circles). Data are means ± SEM (n = 5, 6). D, Quantitative assessment of filopodia growth (black columns) and spine formation (open columns) observed under control conditions (n = 16), after the application of theta burst stimulation in the absence of drugs (LTP; n = 10), or after stimulation in the presence of MK801 (40 μm; n = 8) or KN93 (10 μm; n = 10). Data are means ± SEM. *p < 0.05; t test.
To assess the involvement of CaMKII, we then reproduced these experiments, but in the presence of the CaMKII inhibitor KN93 (10 μm). As illustrated in Figure 3B,C, this treatment prevented induction of stable LTP, but also the later occurrence of dendritic filopodia and newly formed spines. In 10 experiments (1.6 mm analyzed), only one case of filopodia growth was observed and no examples of newly formed spine were seen. The block of LTP-induced structural plasticity was statistically significant (Fig. 3D) (p < 0.05; t test).
As an additional test, we also examined the role of CaMKII in the growth processes induced by a short period of anoxia/hypoglycemia (5 min; Jourdain et al., 2002). In 12 experiments performed in this way, we observed 13 cases of filopodia growth and 12 cases of newly formed spines (Fig. 4A,B), changes that were highly significant (1.8 mm analyzed; p < 0.02, t test; p < 0.05, Mann-Whitney U test) (Fig. 4C). In these experiments, the growth of filopodia was again an early event (mean time of onset, 19.8 ± 4.2 min after anoxia/hypoglycemia), whereas the formation of new spines was more delayed (mean time of onset, 33.0 ± 3.3 min). We then reproduced these experiments, but in the presence of KN93 or AIP, two inhibitors of the enzyme, applied intracellularly at concentrations of 40 μm and 200 nm, respectively. Control experiments showed that these conditions blocked LTP and prevented the effects of CaMKII activation by calmodulin (data not shown). When analyzing morphological plasticity, we found, as illustrated in Figure 4B,C, that both the growth of filopodia and the formation of new spines were significantly reduced. In 11 experiments performed with AIP (1.3 mm analyzed), only two cases of filopodia and one newly formed spine were detected (p < 0.05; t test). Comparable results were obtained in seven experiments performed with KN-93 (Fig. 4C) (1.6 mm analyzed; p < 0.05; t test), thereby confirming a role of CaMKII in structural plasticity.
Figure 4.

Blockade of anoxia/hypoglycemia-induced structural plasticity by inhibitors of CaMKII. A, Growth of two filopodia (arrows, top) and formation of two new spines (asterisks, bottom) observed after the application of a brief (5 min) period of anoxia/hypoglycemia. B, Anoxia/hypoglycemia applied in the presence of AIP (200 nm) failed to induce structural plasticity. Scale bars, 2 μm. C, Growth of filopodia (black columns) and formation of new spines (open columns) expressed as events per 100 μm of dendritic segments and per 60 min under control conditions (n = 16), after a short (5 min) period of anoxia/hypoglycemia, (n = 12), or after anoxia/hypoglycemia, but in the presence of KN93 (40 μm; n = 7) or AIP (200 nm; n = 11). Data are means ± SEM. *p < 0.01; t test.
Discussion
The present study provides strong evidence implicating CaMKII as an important contributor to activity-induced structural plasticity and spine formation. This conclusion is supported by the observation that the activation of the enzyme was sufficient to trigger filopodia growth and spine formation, whereas the blockade of CaMKII prevented the structural remodeling. Thus, these results are very consistent with the recent report showing that CaMKIIβ is an important regulator of dendritic growth and synapse formation in dissociated cultures (Fink et al., 2003) and indicate that CaMKII is both sufficient and necessary for triggering activity-dependent structural plasticity.
The role of CaMKII in LTP is firmly established, and several recent studies have provided plausible mechanisms by which the kinase could enhance synaptic transmission and account for the potentiation (Lisman et al., 2002; Song and Huganir, 2003). Through direct phosphorylation of AMPA receptors or the machinery responsible for the recycling of receptors, CaMKII, together with other kinases, probably regulates the number and function of receptors expressed at postsynaptic densities (Barria et al., 1997; Shi et al., 1999; Poncer et al., 2002; Song and Huganir, 2003). In addition to this, CaMKII, and specifically CaMKIIβ, appears to have a strong morphogenic activity by regulating dendritic growth, filopodia extension, and synapse formation in cell cultures (Fink et al., 2003). Here, we extend these data by providing evidence that CaMKII directly contributes to the process of activity-induced filopodia growth and spine formation in a mature synaptic network (Yuste and Bonhoeffer, 2001). The role of CaMKII in these mechanisms was examined by injecting the active kinase into cells (both the α and β isoforms; Fink et al., 2003) as well as by using classical activators and inhibitors of the enzyme. Taken separately, these different procedures may very well lack specificity. However, the concordance of the different data as well as the controls performed strongly support the contention that the reported changes reflect a specific involvement of CaMKII.
The two parameters analyzed here, growth of dendritic filopodia and formation of new spines, are likely to reflect a process of synaptogenesis. Filopodia are considered as precursors of spines, and analysis of their dynamics has shown that after a phase of elongation, they usually retract and stabilize as new spine synapses (Fiala et al., 1998). Newly formed spines probably also reflect the creation of new synaptic contacts. As shown in the recent in vivo study by Trachtenberg et al. (2003), the newly formed spines induced by sensory stimulation corresponded to fully morphologically mature synaptic contacts. Therefore, the activation of CaMKII could play a significant role in the mechanisms of synapse turnover and experience-dependent changes in spine density and dendrite morphology observed under various learning conditions (Yuste and Bonhoeffer, 2001).
In this regard, the involvement of CaMKII in two different aspects of synaptic plasticity, changes in synaptic efficacy and synaptogenesis, is particularly remarkable. This supports the interpretation that structural and functional plasticity are linked together and could represent graded or sequential changes controlled by a common triggering pathway (Luscher et al., 2000). Increase in synaptic efficacy, filopodia growth, and spine formation all require NMDA receptor activation, increase in intracellular calcium, and CaMKII activation (Maletic-Savatic et al., 1999; Engert and Bonhoeffer, 1999; Luscher et al., 2000; Segal et al., 2000; Jourdain et al., 2002). However, because of their very different time courses, functional and structural plasticity must also rely on different mechanisms. Synaptic potentiation is immediate after stimulation, whereas filopodia growth occurs after 5-15 min (Maletic-Savatic et al., 1999) and spine formation after 30-60 min (Engert and Bonhoeffer, 1999; Toni et al., 1999; Jourdain et al., 2002). The same sequence of changes was observed here after the intracellular injection of CaMKII, although the phenomenon was delayed by ∼15-20 min, probably the time required for diffusion and full action of the enzyme at synaptic sites. Therefore, an interesting possibility is that CaMKII, mainly the α isoform (Silva et al., 1992), contributes to the rapid changes in synaptic function (Barria et al., 1997; Benke et al., 1998; Poncer et al., 2002; Song and Huganir, 2003), whereas more complex regulations involving the β isoform and the cytoskeleton machinery (Fink et al., 2003) would be implicated in initiating growth processes. Much recent work has emphasized the role of actin polymerization and enzymes of the Rho/Rac family in the control of spine dynamics, turnover and morphology (Matus, 2000; Hering and Sheng, 2001; Star et al., 2002; Penzes et al., 2003; Sin et al., 2003). CaMKII could also interact with these mechanisms, and its capacity to maintain a phosphorylation activity for a prolonged period through autophosphorylation (Lisman et al., 2002) could represent an important way to sustain signaling and thus orchestrate events characterized by different delays. Therefore, the identification of CaMKII as a key contributor to structural plasticity strengthens the importance of phosphorylation mechanisms not only in the control of synaptic function, but also in synapse formation and experience-dependent network remodeling.
Footnotes
This work was supported by the Swiss National Science Foundation, the Ott Foundation, the Novartis Foundation, and the De Reuter Foundation. We thank M. Moosmayer and L. Parisi for excellent technical support.
Correspondence should be addressed to Dr. Dominique Muller, Centre Médical Universitaire, 1211 Geneva 4, Switzerland. E-mail: Dominique.Muller@medecine.unige.ch.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310645-05$15.00/0
References
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR ( 1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276: 2042-2045. [DOI] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL ( 1998) Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393: 793-797. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T ( 1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM ( 1998) Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci 18: 8900-8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell Jr JE, Schulman H, Meyer T ( 2003) Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39: 283-297. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D ( 1993) Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 268: 7863-7867. [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ ( 1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279: 870-873. [DOI] [PubMed] [Google Scholar]
- Goldin M, Segal M, Avignone E ( 2001) Functional plasticity triggers formation and pruning of dendritic spines in cultured hippocampal networks. J Neurosci 21: 186-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima Y, Ohsako S, Yamauchi T ( 1993) Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. J Neurosci 13: 559-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M ( 2001) Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2: 880-888. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H ( 2002) Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 71: 473-510. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H ( 1995) A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun 212: 806-812. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Nikonenko I, Alberi S, Muller D ( 2002) Remodeling of hippocampal synaptic networks by a brief anoxia-hypoglycemia. J Neurosci 22: 3108-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H ( 2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175-190. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA ( 1995) Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA 92: 11175-11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D ( 2000) Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3: 545-550. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K ( 1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283: 1923-1927. [DOI] [PubMed] [Google Scholar]
- Matus A ( 2000) Actin-based plasticity in dendritic spines. Science 290: 754-758. [DOI] [PubMed] [Google Scholar]
- McGlade McCulloh E, Yamamoto H, Tan SE, Brickey DA, Soderling TR ( 1993) Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 362: 640-642. [DOI] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL ( 2003) Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 37: 263-274. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Esteban JA, Malinow R ( 2002) Multiple mechanisms for the potentiation of AMPA receptor-mediated transmission by α-Ca2+/calmodulin-dependent protein kinase II. J Neurosci 22: 4406-4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal I, Korkotian I, Murphy DD ( 2000) Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci 23: 53-57. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R ( 1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1755-1757. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y ( 1992) Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257: 201-206. [DOI] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT ( 2003) Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature 419: 475-480. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL ( 2003) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25: 578-588. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN ( 2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5: 239-246. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D ( 1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D ( 1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402: 421-425. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GK, Feng G, Sanes JR, Welker E, Svoboda K ( 2003) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420: 788-794. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT ( 1995) Postsynaptic injection of CA2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 15: 443-452. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T ( 2001) Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071-1089. [DOI] [PubMed] [Google Scholar]