

Abstract
Antipsychotic drugs require days of treatment to begin to produce therapeutic effects. We report that in vivo treatment with the antipsychotic drug haloperidol acts with a delay of days to slow spontaneous repetitive firing by isolated midbrain dopamine neurons. The decreased excitability is caused by an increased number of functional A-type K+ channels without any change in gating properties. Upregulation of dopamine neuron Kv4.3 mRNA accounts for this effect, demonstrating a role for channel gene expression in this delayed drug action. The resultant long-term dampening of dopamine neuron excitability may serve to tone down the dopamine system.
Keywords: dopamine neuron, pacemaker activity, voltage clamp, haloperidol, A-type K+ channel, mRNA
Introduction
Antipsychotic drugs act on the dopamine (DA) system. This was first revealed from the relationship between D2 receptor antagonism and the efficacy of many antipsychotic drugs (Creese et al., 1976; Seeman et al., 1976). A role for this antagonism is supported by the finding of excess D2 DA receptor occupancy in schizophrenics (Abi-Dargham et al., 2000). Furthermore, chronic treatment with the antipsychotic drug haloperidol reduces synaptic DA release (Ichikawa and Meltzer, 1990, 1991), suggesting a presynaptic action on DA neurons. Yet, simple blockade of D2 receptors (Seeman et al., 1975, 1976; Creese et al., 1976) present on DA neurons and their synaptic targets cannot directly account for delayed antipsychotic drug action. First, the therapeutic effect of antipsychotic drugs develops with a delay of days to weeks (Johnstone et al., 1978; Kandel et al., 2000; Konradi and Heckers, 2001), in contrast to rapid receptor inhibition. Furthermore, increasing administration of an antipsychotic drug over its optimal dose does not accelerate the slow onset of clinical action (Baldessarini et al., 1988). Hence, D2 receptor blockade likely initiates a chain of events that later results in inhibition of DA neuron function.
The basis of delayed DA neuron inhibition by antipsychotic drugs has been controversial. In vivo recordings led to the suggestion that antipsychotic drugs such as haloperidol eventually inhibit DA neurons by synaptically driven depolarization inactivation (Chiodo and Bunney, 1983; White and Wang, 1983a, b; Grace et al., 1997). However, silencing of DA neurons in vivo by antipsychotic drugs is only seen in the presence of general anesthetics (Mereu et al., 1994, 1995; Melis et al., 1998). Thus, a long-term antipsychotic drug action on physiological DA neuron activity has not been identified.
In vivo studies have not tested the hypothesis that antipsychotic drugs change intrinsic DA neuron excitability. DA neurons display intrinsic pacemaker activity that is generated by the interplay of a number of voltage-gated ion channels (Grace and Bunney, 1984; Silva et al., 1990; Hainsworth et al., 1991). Altering the activity of these channels could have a large impact on the drive to fire spontaneous action potentials and the sensitivity to synaptic inputs (Levitan and Levitan, 1988; Marder et al., 1996; Häusser et al., 2000). Resultant changes in electrical activity would alter DA release (Cragg et al., 2000). Thus, a change in DA neuron intrinsic activity could be an important feature of antipsychotic drug action. Yet, nothing is known about the chronic effect of antipsychotic drugs on DA neuron pacemaker activity.
In this study, we tested the hypothesis that the antipsychotic drug haloperidol remodels DA neuron excitability by long-term regulation of voltage-gated ion channels. An experimental approach that avoids the confounding effect of anesthetics shows that in vivo haloperidol treatment acts with a delay of days to slow DA neuron intrinsic pacemaker activity. We then identified the mechanism responsible for this long-term change.
Materials and Methods
Drug treatment and cell preparation. Haloperidol (Research Biochemicals, Natick, MA) at a dose of 2 mg/kg or vehicle (20 mm tartaric acid and 10% ethanol) was injected intraperiteoneally into Sprague Dawley rats every other day starting from postnatal day 6 (P6) to P10. Each treatment group included five to six animals. Animal weight did not change with haloperidol treatment. Midbrain DA neurons were acutely dissociated and labeled on P11 or P12 using a previously described method (Cardozo and Bean, 1995). The cells were plated on glass coverslips and kept in the dark at room temperature (20-24°C) until they were viewed through a 40×/340 UV Olympus (Tokyo, Japan) objective using 340 nm light for excitation and a 420 nm long-pass filter for emission fluorescence.
Electrophysiology. Whole-cell recordings were performed in current-clamp and voltage-clamp modes. Leak subtraction was performed by p/5 protocols, and 70% series resistance compensation was used. Electrodes were filled with a solution containing the following (in mm): 130 KCl, 4 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES, 2 ATP, pH 7.4. The bath solution contained the following (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH 7.4. Data are expressed as mean ± SEM. Statistical significance was assessed by one-way ANOVA with Dunnett's post-test for electrophysiology results.
Quantitative real-time single-cell PCR (TaqMan). Patch-pipette electrodes were pulled from baked capillary glass and filled with 6 μl of DEPC water and 1 μl (10 U) of RNaseOUT RNase inhibitor (Invitrogen, San Diego, CA). To reduce cell-to-cell variation, five individual cells were aspirated into one patch pipette and ejected into a tube. One microliter (150 ng) of Random hexamers (Invitrogen) and DEPC water were added to a total of 15 μl. The mixture was heated at 70°C for 5 min. After cooling on ice, 2 μl of Sensiscript Buffer and 2 μl of deoxy NTPs were added and then incubated at room temperature for 10 min. Single-stranded cDNA was synthesized from the cellular mRNA by adding 1 μl of Sensiscript reverse transcriptase (RT) (Qiagen, Valencia, CA) and incubating at 37°C for 1 hr. Final volume was 20 μl. The reaction was terminated by heating the mixture to 70°C for 15 min. Single-stranded cDNA (2 μl) from the RT reaction was used as template in a 50 μl PCR reaction. Each reaction was done in triplicate. TaqMan 2× Master Mix (25 μl; Applied Biosystems, Foster City, CA), 900 nm each of the forward and reverse primer, and 250 nm of the probe were used. The sequences of primers and probes for TaqMan experiments, which are the rat homologs of those reported by Liss et al. (2001), were as follows: Kv4.3-F 5′-CTCTCTGGTTCTGATGGTAGATCCT-3′ (AF334791, 1167 bp), Kv4.3-R 5′-AAGCGGCGTCCTGGTCATT-3′ (1246 bp), and Kv4.3-probe 5′-/56-FAM/CCAGTCCCCGTCATAGTCACCAACTTTAGC/36-TAM/-3′ (1192 bp). Real-time PCR was performed in the ABI PRISM 7700 Sequence Detector system and analyzed with the ABI PRISM 1.7 Sequence Detector software. After defining a baseline (normalized background fluorescence of cycles 3-15) in a linear plot of relative fluorescence (Rf) against the PCR cycle number, PCR cycle was detected where the exponentially increasing Rf crosses a manually set detection threshold (Ct) line, defined in a logarithmic plot of Rf values against PCR. A one-tailed Mann-Whitney test was used for TaqMan results.
Results
To test whether chronic treatment with an antipsychotic drug affects DA neuron intrinsic excitability, rats were treated with haloperidol or vehicle, and then DA neurons were acutely dissociated and identified by uptake of the fluorescent marker 5,7-dihydroxytryptamine (Silva et al., 1988; Cardozo and Bean, 1995). Dissociation, identification, and patch-clamp recording of DA neurons were done in the absence of haloperidol to ensure that only persistent effects of the drug could be detected.
Whole-cell recording showed that chronic in vivo haloperidol treatment slows DA neuron intrinsic pacemaker activity. Acutely isolated identified DA neurons were large and ovoid (Fig. 1A) and spontaneously fired long-duration action potentials followed by marked afterhyperpolarizations (Figs. 1B, 2A; Table 1). Likewise, a membrane potential sag induced by hyperpolarizing current injection indicative of pacemaker current (Ih) expected for DA neurons (Silva et al., 1990; Hainsworth et al., 1991) could be detected (Fig. 1B). Spontaneous rhythmic action potential activity was measured from isolated DA neurons from animals previously treated with vehicle or haloperidol for 5 or 6 d. As can be seen from representative membrane potential recordings, chronic haloperidol slowed spontaneous pacemaker firing (Fig. 2A,B). Interspike interval (ISI) histograms show that the doubling of the mean reflects a rightward shift in the peak and a widening of the distribution (Fig. 2C,D). This latter change, which can be quantified as an increase in the interspike interval coefficient of variance (CVISI), is indicative of more irregular firing. Data from independent experiments show that these effects are reproducible (Fig. 2E,F,G). Chronic haloperidol is known to promote irregular firing of DA neurons recorded in anesthetized animals (White and Wang, 1983a). This had been interpreted to be a precursor to synaptically driven depolarization inactivation. However, the response to chronic haloperidol treatment evident in acutely isolated DA neurons cannot be attributed to anesthesia or synaptic activity and thus must reflect a persistent change in intrinsic excitability.
Figure 1.

Identification of acutely dissociated DA neurons. A, Fluorescence micrograph of an isolated DA neuron labeled with 20 μm 5,7-DHT (top). Fluorescence and white light micrograph (middle), and white light micrograph (bottom) of the same field of view. Note the fluorescent-labeled multipolar dopamine neuron (arrow 1) and the unlabeled nondopamine neuron (arrow 2) in the top and middle panels. Scale bar, 10 μm. B, Electrophysiological properties of an isolated DA neuron. Responses to three consecutive current injections (0, -40, and -80 pA) from a holding potential of -77 mV (indicated by broken line) are shown.
Figure 2.

Chronic in vivo haloperidol treatment slows DA neuron intrinsic pacemaker activity. A, B, Whole-cell current-clamp recording from a single DA neuron from rats treated with vehicle (A) and haloperidol (B) for 5 or 6 d (chronic Hal). Spontaneous firing activity was recorded without current injection. Lower traces show pacemaker activity on an expanded time scale. Vertical bars, 25 mV; horizontal bars, 20 sec (top) or 2 sec (bottom). C, Histogram of interspike interval from the trace recorded in experiment A. D, Histogram of interspike interval from the trace in experiment B (bin width, 25 msec). E, The mean interspike interval. F, The coefficient of variation of interspike interval from independent DA neurons (vehicle, n = 4; chronic Hal, n = 4). Chronic treatment with haloperidol increased the mean value of ISI and CVISI, respectively. **p < 0.01; *p < 0.05. G, Cumulative histograms for ISI data combined from vehicle and chronic Hal DA neurons (p < 0.001; Kolmogorov-Smirnov test). Note the rightward shift and decreased slope after chronic haloperidol treatment.
Table 1.
Action potential properties of DA neurons after chronic haloperidol treatment
|
|
Half-width (msec) |
First peak (mV) |
Second peak (mV) |
Threshold (mV) |
AHP (mV) |
|---|---|---|---|---|---|
| Without TEA | |||||
| Control | 3.2 ± 1.4 | 33.1 ± 3.1 | 30.5 ± 2.1 | −44.5 ± 0.9 | −56.2 ± 1.5 |
| Vehicle | 3.3 ± 0.9 | 32.1 ± 3.4 | 32.4 ± 4.6 | −45.0 ± 1.4 | −57.9 ± 2.1 |
| Haloperidol | 4.3 ± 1.3 | 35.4 ± 3.2 | 33.6 ± 3.5 | −46.1 ± 1.2 | −58.9 ± 1.6 |
| With TEA | |||||
| Control | 185 ± 62 | 21.0 ± 1.7 | 20.7 ± 2.4 | −43.2 ± 2.4 | −45.2 ± 2.6 |
| Vehicle | 155 ± 88 | 27.5 ± 2.6 | 25.7 ± 5.7 | −38.9 ± 4.6 | −48.4 ± 2.2 |
| Haloperidol
|
126 ± 55
|
27.9 ± 1.5
|
24.6 ± 4.5
|
−40.5 ± 3.2
|
−53.7 ± 1.6
|
Half-width, The time between two peaks of first derivative; first peak, peak of the first spike; second peak, peak of the second spike; threshold, the first point on the rising phase of the spike where the rate of rise exceeded 20 mV/msec; AHP, afterhyperpolarization. n = 5-6.
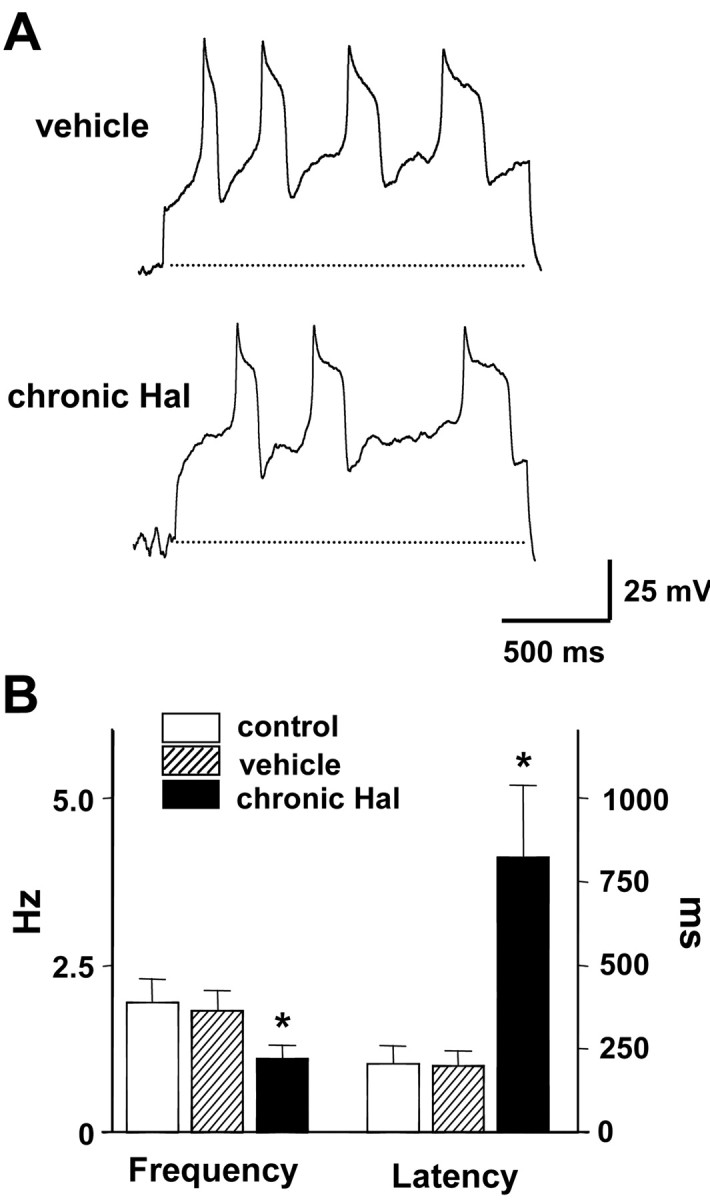
Current-clamp experiments showed that the slowed pacemaker activity is not attributable to depolarization inactivation. A sustained inward current can produce tonic depolarization that inactivates voltage-gated Na+ channels so that action potential firing is inhibited. Such a depolarizing conductance, which would have to be intrinsic in isolated cells, would reduce the latency to the first action potential after releasing clamped DA neurons from a negative potential (i.e., -80 mV). However, the decreased intrinsic firing of DA neurons elicited by chronic haloperidol was associated with an increase in the latency to the first action potential (Fig. 3A,B). Hence, the slowing of intrinsic pacemaker activity is not attributable to depolarization inactivation but appears to reflect dampening of excitability. Note that a 1 d in vivo treatment with haloperidol did not induce this effect (Fig. 3C,D). Hence, haloperidol acts over a period of days to slow DA neuron pacemaking.
Figure 3.

Chronic in vivo haloperidol treatment dampens DA neuron excitability. A, Whole-cell current-clamp recordings from single DA neurons from rats treated with vehicle or haloperidol for 5 or 6 d. Spontaneous firing activity was recorded when clamped current was released to 0 pA from a holding potential of -80 mV. B, Chronic haloperidol treatment decreases spontaneous spike frequency and increases first spike latency and interspike interval between first and second spikes. (control, n = 5; vehicle, n = 6; haloperidol, n = 6). Broken lines indicate -80 mV. C, Whole-cell current-clamp recordings from single DA neurons from rats treated 1 d with vehicle or haloperidol. D, One day haloperidol treatment does not affect spike frequency, first spike latency, and first interspike interval (vehicle, n = 5; haloperidol, n = 6). *p < 0.05.
Current-clamp recordings delimited possible targets of chronic haloperidol. First, the action potential amplitude, duration, and afterhyperpolarization did not change significantly with prolonged in vivo haloperidol exposure (Table 1, top). Therefore, currents that dominate the afterhyperpolarization (i.e., calcium-activated potassium current), as well as the spike waveform, were not obviously altered. Furthermore, the increased latency to the first spike and the slowed firing rate were evident in the presence of 50 mm tetraethylammonium (TEA), whereas action potentials lengthened ∼50-fold by TEA were not significantly affected by chronic haloperidol (Fig. 4; Table 1, bottom). This suggests that TEA-sensitive potassium channels and the calcium and calcium-activated currents that shape TEA-broadened spikes do not mediate the drug effect. Therefore, we hypothesized that a subthreshold voltage-gated calcium-independent current that is especially important for pacemaking activity is regulated by chronic haloperidol.
Figure 4.
TEA does not influence the effect of chronic haloperidol on DA pacemaker activity. A, Whole-cell current-clamp recordings in the presence of 50 mm TEA from DA neurons of rats treated chronically with vehicle or haloperidol (chronic Hal). Decreased firing frequency and increased latency by chronic haloperidol is preserved after acute perfusion of 50 mm TEA. Broken lines indicate -80 mV. B, Summary of 50 mm TEA effects on spike frequency and first spike latency of DA neuron spontaneous activity (control, n = 4; vehicle, n = 5; chronic Hal, n = 5). *p < 0.05.
Whole-cell voltage-clamp experiments identified a fast inactivating voltage-gated current in acutely dissociated DA neurons that is sensitive to in vivo haloperidol treatment. First, a conditioning pulse was used to isolate transient current as the difference between the total and noninactivating currents (Fig. 5A, insets). The peak amplitude of this transient current increased in DA neurons from rats chronically treated with haloperidol (Fig. 5A). Second, we examined the effect of chronic in vivo haloperidol treatment by recording currents in the presence of TEA to block noninactivating K+ currents (Thompson, 1977). As shown in Figure 5B, chronic haloperidol increased the peak amplitude of the TEA-resistant transient current.
Figure 5.

Chronic haloperidol treatment increases Kv4 A-type K+ current in DA neurons. A, Whole-cell voltage-clamp recordings and mean current density of transient K+ currents from DA neurons of rats treated chronically with vehicle or haloperidol (chronic Hal). Left, Middle, Transient currents were obtained by subtracting noninactivating currents from total outward currents (vehicle, 21.2 pF; chronic Hal, 22.3 pF). Inset, Total outward currents were evoked by depolarizing steps (-20 to 60 mV) from a holding potential of -90 in 20 mV steps. Noninactivating currents were evoked by the same pulse protocol after a prepulse to -40 mV for 500 msec. Right, Mean transient K+ current density from each group. Transient K+ current density was obtained by dividing peak transient K+ currents at 60 mV by cell capacitance (control, n = 6; vehicle, n = 9; chronic Hal, n = 10). Transient current density is increased in DA neurons from a haloperidol-treated rat compared with DA neurons from a vehicle-treated rat. Horizontal bars, 500 msec; vertical bars, 2.5 nA. B, Whole-cell voltage-clamp recordings and mean current density of K+ currents in the presence of 50 mm TEA. Left, Middle, Transient currents in DA neurons (vehicle, 26.8 pF; chronic Hal, 27.3 pF) were activated by a pulse to 20 mV from a holding potential of -90 mV. Right, Summary from eight cells in each cell group. **p < 0.01. C, D, Block of transient current in representative DA neurons from rats chronically treated with haloperidol by 4-AP (C) and HpTx3 (D). Currents were recorded at 20 mV (C) and 0 mV (D) from a holding potential of -90 mV.
Pharmacological experiments demonstrated that the regulated current is caused by A-type K+ channels. Figure 5C illustrates that local application of 4-aminopyridine (4-AP; 4 mm) completely blocked the transient current in DA neurons from haloperidol-treated rats (n = 5). Consistent with A-type currents (Castle and Slawsky, 1993; Tseng et al., 1996), inactivation slowed in the presence of 4-AP to produce a “crossover” with the control current traces. Furthermore, like A-type currents (Connor and Stevens, 1971; Thompson, 1977), the increased transient current isolated with the prepulse method was not sensitive to TEA; DA neuron transient current after haloperidol treatment was reduced <5% by 50 mm TEA (data not shown). Finally, A-type K+ current encoded by Kv4 family subunits is selectively inhibited by heteropodatoxin3 (HpTx3) (Sanguinetti et al., 1997). HpTx3 acutely blocked the transient current after prolonged haloperidol treatment in a dose-dependent manner (Fig. 5D). Comparable with the known sensitivity of A-type K+ current in DA neurons (Liss et al., 2001), 100 nm HpTx3 inhibited 53.0 ± 5.4% (n = 5) of the peak current at 0 mV. Therefore, chronic in vivo haloperidol treatment specifically increases Kv4 A-type K+ current in midbrain DA neurons.
This effect of chronic haloperidol is specific. Voltage-gated Na+ current (INa), hyperpolarization-activated cation current (Ih), and delayed rectifier K+ current (IDR) were not changed by chronic in vivo haloperidol treatment (Fig. 6A--C). We also tested whether A-type K+ channel activity developmentally changed during the treatment period. No significant change in current density was found over the week in which experiments were conducted (Fig. 6D). Thus, the effect of chronic haloperidol is specific and occurs on a flat baseline of A-type channel activity.
Figure 6.

Specific effect of chronic in vivo haloperidol treatment. A, Mean voltage-dependent Na+ current density from DA neurons. Current density was obtained by dividing peak Na+ currents at 0 mV by cell capacitance (control, n = 5; vehicle, n = 6; chronic Hal, n = 6). B, Mean hyperpolarization-activated cation current density from DA neurons. Current density was obtained by dividing peak Ih currents at the end of the pulse (-100 mV) by cell capacitance (control, n = 5; vehicle, n = 5; chronic Hal, n = 6). C, Mean delayed rectifier K+ current density from DA neurons. Current density was obtained by dividing K+ currents at the end of the pulse (60 mV) by cell capacitance (control, n = 29; vehicle, n = 25; chronic Hal, n = 30). D, Mean A-type K+ current density from DA neurons does not change during the time period studied here. Current density was obtained by the same protocol described in Figure 5 (P5, n = 6; P10, n = 4; P11, n = 6; P12, n = 6).
We then showed that the effect of chronic in vivo haloperidol treatment is not because of a change in voltage-dependent A-type channel gating. Activation kinetics was unaffected because mean time-to-peak versus voltage was similar in all cell groups (Fig. 7A). Furthermore, A-type K+ currents from all experimental groups inactivated with similar mean time constants (Fig. 7B). The voltage dependence of steady state inactivation was measured as the dependence of current amplitude on conditioning prepulse membrane potential (Fig. 7C). Boltzmann function fits of steady state inactivation were virtually identical regardless of treatment (Fig. 7D, left). Similarly, there was no effect of chronic haloperidol on the voltage dependence of activation of A-type K+ current (Fig. 7D, right). A double-pulse protocol with various interpulse intervals showed fast recovery from inactivation of A-type K+ currents (Fig. 7E). All cell groups displayed recovery of inactivation that could be fit with similar single exponential functions (Fig. 7F). Thus, A-type channel gating was not altered by haloperidol. Because membrane surface area measured as capacitance was not affected by the drug treatment (vehicle: 21.1 ± 1.5 pF, n = 18; chronic haloperidol: 22.5 ± 1.1 pF, n = 16), the increase in fast inactivating current with prolonged haloperidol treatment must be because of an increase in the number of functional Kv4 channels.
Figure 7.

Chronic haloperidol treatment does not affect gating of A-type K+ current in DA neurons. A, Mean activation kinetics. B, Mean inactivation kinetics. Averaged activation and inactivation time constants are plotted as a function of the test pulse voltage for each cell group. Triangle, Control; square, vehicle treatment; circle, haloperidol treatment. C, Voltage-dependent inactivation of A-type K+ currents in a DA neuron from a rat treated chronically with haloperidol. Currents were evoked by a test pulse to 20 mV after conditioning prepulses at various voltage levels (from -140 to 0 mV; 500 msec) in the presence of 50 mm TEA. D, Steady state activation and inactivation curves from DA neurons of rats treated chronically with vehicle and haloperidol. For activation curves, the peak amplitudes were measured from transient K+ currents generated by the same protocols described in Figure 5. Membrane conductances (g) at different voltage levels were obtained by dividing the peak transient K+ currents by the current driving force and normalized to conductance (gmax) at 80 mV. The mean data from each group were fitted with Boltzmann functions, which yielded half-activation potentials of control (triangle, 24.8 ± 1.3 mV; n = 7), vehicle (square, 23.8 ± 2.2 mV; n = 7), and haloperidol treatment (circle, 23.6 ± 1.1 mV; n = 8). For inactivation curves, the peak current amplitudes (I) obtained from experiment C were normalized to current amplitude with a -140 mV conditioning prepulse (Imax). The data from each group were described by Boltzmann functions with a half-inactivation potential of control (triangle, -78.7 ± 1.4 mV; n = 5), vehicle (square, -78.0 ± 1.1 mV; n = 7), and haloperidol treatment (circle, -77.2 ± 1.7 mV; n = 7). E, Recovery from inactivation of A-type K+ currents in a DA neuron from a haloperidol-treated animal. Currents were recorded with a double-pulse protocol with various interpulse intervals at -90 mV in the presence of 50 mm TEA. The test pulses were to 20 mV. F, The mean recovery from inactivation in DA neurons of rats treated chronically with vehicle and haloperidol. The data from each group were fitted with a single exponential function with a time constant of control (triangle, 30.3 msec; n = 5), vehicle (square, 32.2 msec; n = 7), and haloperidol treatment (circle, 30.3 msec; n = 7).
The augmentation of A-type K+ current by prolonged in vivo haloperidol treatment is required for the effect on pacemaker activity. The voltage-clamp recording in Figure 5D shows that 100 nm HpTx3 halves A-type K+ current. Therefore, we applied this dose of HpTx3 to DA neurons from rats chronically treated with haloperidol to counter-balance the treatment-induced twofold increase of A-type K+ current. Acutely reestablishing a normal level of A-type current with HpTx3 reversibly accelerated spontaneous spiking (Fig. 8A). Indeed, frequency, first latency, and first interspike interval returned back to values found in vehicle-treated animals (Fig. 8B). This normalizing effect was evident both in anode break and tonic-firing recordings (Fig. 8A,C). Thus, the haloperidol-induced increase of A-type current accounts for the regulation of DA neuron activity.
Figure 8.
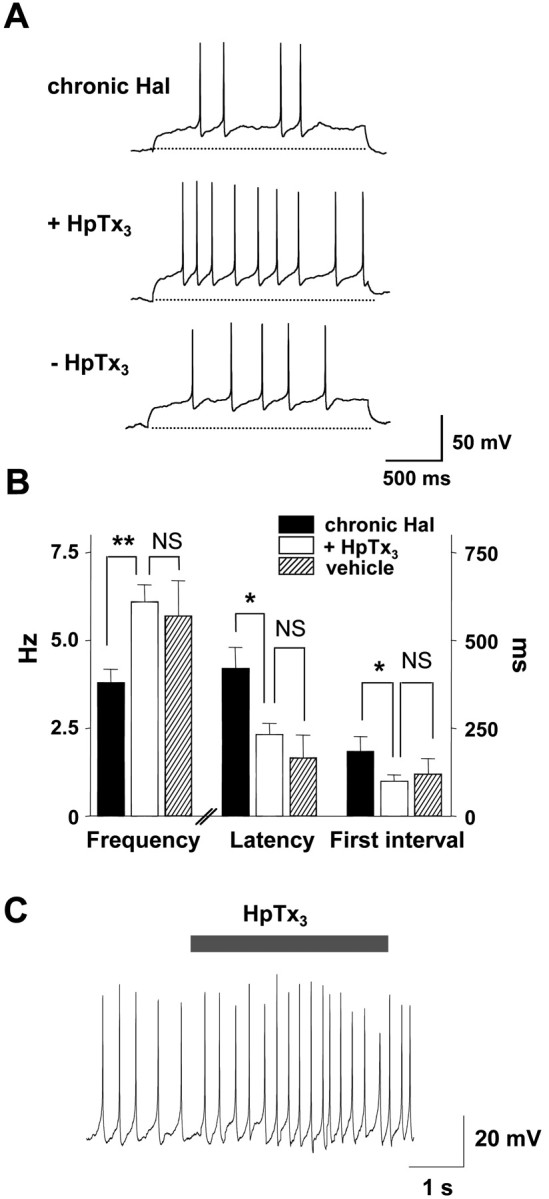
Chronic haloperidol treatment slows intrinsic pacemaker activity by increasing A-type K+ current. A, Whole-cell current-clamp recording showing the effect of 100 nm HpTx3 on spontaneous firing activity of DA neurons from a rat chronically treated with haloperidol. Spontaneous activity was recorded after releasing the current clamp from a holding potential of -80 mV. HpTx3 increases spike frequency and decreases first spike latency and first interspike interval in DA neurons. Broken lines indicate -80 mV. B, Summary of 100 nm HpTx3 effects on spike frequency, first spike latency, and first interspike interval of spontaneous activity in haloperidol-treated DA neurons (chronic Hal, n = 7; +HpTx3, n = 7; -HpTx3, n = 5). Note that acutely reversing the haloperidol effect on A-type current with HpTx3 normalizes pacemaker activity. **p < 0.01; *p < 0.05; NS, no significance. C, The effect of 100 nm HpTx3 on tonic firing activity by a chronic haloperidol-treated DA neuron.
Recently, MAP kinase was shown to mediate the acute regulation of A-type K+ current in DA neurons by glial cell line-derived neurotrophic factor (Yang et al., 2001). If a change in the rapid phosphorylation of channels by MAP kinase is involved in chronic haloperidol action, it would be expected that gating of the channels would be altered and the effect of a MAP kinase inhibitor would change with haloperidol treatment. However, as noted above, no change in gating was found (Fig. 7). Furthermore, application of 100 μm PD098059, a MAP kinase inhibitor, to acutely dissociated DA neurons did not significantly affect A-type K+ currents from vehicle or haloperidol-treated animals; after 3-5 min, the current changed by 0.6 ± 1.3% (n = 7) for vehicle and 7.9 ± 6.3% (n = 6) for chronic haloperidol treatment (p = 0.25). Thus, the change in channel activity induced by chronic haloperidol does not involve acute MAP kinase activity. The kinetics of the chronic haloperidol effect also argues against an acute phosphorylation mechanism. Specifically, in vivo treatment for 1 d with haloperidol did not increase DA neuron A-type K+ current; current density at 60 mV was 451 ± 80 pA/pF (n = 9) for vehicle and 398 ± 71 (n = 9) with haloperidol.
We then identified a Kv4 family member involved in long-term haloperidol action. Specifically, we focused on the Kv4.3 gene, because it encodes for DA neuron A-type K+ current (Liss et al., 2001), and control of Kv4.3 gene expression is very slow in another tissue (Zhang et al., 2001). Because detecting an approximately twofold change in Kv4.3 protein in acutely isolated neurons is not possible with available experimental tools, we tested whether chronic haloperidol increases Kv4.3 mRNA by performing quantitative real-time PCR mRNA measurements. We characterized the Kv4.3 assay over a range of Ct cycle numbers spanning 27-34 by using eight different Kv4.3 double-stranded DNA (dsDNA) concentrations. A Ct value standard curve generated from this experiment yielded a mean slope of -3.25 ± 0.05, which is close to the ideal value of -3.32 (-1/log2) (Fig. 9A). Figure 9B shows examples of triplicate Kv4.3 amplification results generated from samples of five pooled DA neurons from vehicle and haloperidol-treated animals. Drug treatment changed the Ct number by approximately one cycle, indicating an approximately twofold difference in Kv4.3 mRNA in these samples. On average, chronic in vivo haloperidol treatment produced an ∼85% increase in Kv4.3 mRNA in DA neurons (Fig. 9C, left). In contrast, Kv4.3 mRNA in non-DA neurons was not regulated (Fig. 9C, right). The calculated Kv4.3 mRNA expression level from Figure 9C was plotted against A-type current density from Figure 5A to show that the change in channel mRNA was directly proportional to the electrophysiological effect of chronic haloperidol treatment (Fig. 9D). Given the known linear relationship between Kv4.3 mRNA and current in DA neurons (Liss et al., 2001), it is evident that Kv4.3 channels participate in the long-term regulation of DA neuron excitability by chronic haloperidol treatment.
Figure 9.

Chronic haloperidol treatment upregulates Kv4.3 mRNA in DA neurons. A, Standard curve for Kv4.3 mRNA detection. Mean Ct values from triplicates are plotted against their eight different relative concentrations of Kv4.3 dsDNA on a logarithmic scale. B, Representative amplification plots of real-time fluorescent RT-PCR experiments in five DA neurons from haloperidol-treated animals and five DA neurons from vehicle-treated animals. Amplification plots show triplicates from each sample. The horizontal line indicates the threshold of amplification detection at Rf = 0.036. The average Ct values in this example are 30.5 (chronic Hal) and 31.6 (vehicle). C, Summary of normalized Kv4.3 mRNA from DA neurons and non-DA neurons. Each average Ct value derived from triplicate assays was linearized with the equation y = 2(34 - Ct) and normalized to the total mean of DA neurons from vehicle-treated animals (chronic Hal DA, n = 8; vehicle DA, n = 7; chronic Hal non-DA, n = 6; vehicle non-DA, n = 6). *p < 0.05. D, Chronic in vivo treatment with haloperidol proportionally increases DA neuron A-type K+ current density and Kv4.3 mRNA. The solid line is generated from y = x. Superimposed data points on the line are the normalized means of A-type current density and Kv4.3 mRNA.
Discussion
Antipsychotic drugs such as haloperidol act very slowly to reduce DA release and DA-dependent behavior. Here, we used an experimental approach, which avoids the confounding effect of anesthetics (Mereu et al., 1994, 1995; Melis et al., 1998), to discover that haloperidol acts over a period of many days to slow DA neuron intrinsic pacemaker activity. This is caused by a delayed increase in the number of functional A-type K+ channels. By virtue of their voltage dependence, A-type channels influence repetitive firing, the functional strength of excitatory and inhibitory synapses, spike backpropagation, and synaptic plasticity (Hoffman et al., 1997; Schoppa and Westbrook, 1999; Häusser et al., 2000). Thus, the haloperidol-induced increase in A-type current density will tend to reduce both intrinsically generated and synaptically driven action potential activity.
The simplest interpretation of our results is that increased Kv4.3 A-type channel expression is responsible for the long-term change in DA neuron excitability. First, biophysical measurements show that chronic haloperidol increases the number of functional A-type channels without a change in gating properties (Fig. 7). Second, HpTx3 identifies the channel solely responsible for the change in repetitive firing as a member of the Kv4 family (Fig. 8). Third, Kv4.3 is the only known A-type channel gene expressed in DA neurons (Liss et al., 2001). Fourth, DA neuron pacemaker frequency in the absence of drugs in slices is inversely proportional to Kv4.3 mRNA level (Liss et al., 2001). Fifth, because Kv4.3 mRNA has a long half-life (Zhang et al., 2001), increased Kv4.3 gene expression will change excitability slowly in accordance with the clinical effect of haloperidol. Finally, upregulation of Kv4.3 mRNA quantitatively accounts for the increased A-type channel activity found with chronic haloperidol (Fig. 9). Thus, all of the experimental data are consistent with the conclusion that long-term regulation of DA neuron Kv4.3 channel activity is a novel feature of haloperidol action. Pinpointing this specific gene provides a potential target for drug development.
The long-term control of neuronal excitability is of interest beyond its role in haloperidol action. Because past experiments typically have not been designed to identify persistent changes that occur with such a slow onset, this kind of plasticity may be prevalent. DA neurons are important in motor control, cognitive function, and reward behavior (Brooks, 1995; White, 1996; Spanagel and Weiss, 1999), and thus delayed A-type channel regulation in these cells may be important for control of a variety of physiological behaviors. Similar mechanisms may also be important for the action of addictive drugs that affect dopamine neurons such as amphetamines, cocaine, and nicotine. Finally, remodeling of excitability might occur in neurons that participate in the action of other psychiatric drugs with a slow onset of clinical action such as antidepressants.
It is interesting to relate our findings to previous in vivo results. First, it is notable that the irregular DA neuron firing reflected in interspike interval histograms (Fig. 2C,D) is similar to that reported with chronic haloperidol in vivo (White and Wang, 1983a). Thus, this haloperidol effect reflects a persistent change in intrinsic DA neuron excitability. Second, we did not find any silencing of intrinsic pacemaking. Of course, silencing of DA neurons is also not seen in vivo in the absence of general anesthesia (Mereu et al., 1994, 1995; Melis et al., 1998). However, it is possible that the chronic haloperidol-induced decrease in DA neuron intrinsic excitability acts in concert with general anesthetics to contribute to the silencing of DA neurons in unconscious animals. Finally, altered DA neuron pacemaker activity could be relevant to dopaminergic transmission. In freely moving animals, chronic haloperidol reduces DA release (Ichikawa and Meltzer, 1990, 1991). It has recently been found that schizophrenics have over occupancy of D2 receptors (Abi-Dargham et al., 2000). Thus, the drug-induced reduction in DA release might reestablish a more normal level of D2 receptor activation. Our results raise the possibility that decreased intrinsic firing of DA neurons contributes to decreased DA release. Of course, we cannot be certain that this new delayed mechanism for regulating DA neuron activity in young rats applies to adult human schizophrenics, or that this effect is shared by the classical neuroleptic haloperidol and newer atypical antipsychotic drugs. However, the long-term regulation of intrinsic DA neuron pacemaking should be incorporated into the design and interpretation of future experiments.
Footnotes
This research was supported by a National Alliance for Research on Schizophrenia and Depression-Pfizer Independent Investigator Award and National Institutes of Health Grant HL55312 to E.S.L. Technical expertise was provided by personnel of the Center for Genomic Sciences, a core facility of the Center for Human Genetics and Integrative Biology (University of Pittsburgh). Heteropodatoxin was kindly provided by NPS Pharmaceuticals. We thank Drs. R. H. Kramer, P. Shepard, and W. C. DeGroat for their comments.
Correspondence should be addressed to Edwin S. Levitan, Department of Pharmacology, University of Pittsburgh, Pittsburgh, PA 15261. E-mail: Levitan@server.pharm.pitt.edu.
T.E. Tse's present address: Children's Hospital of Pittsburgh, Pittsburgh, PA 15213.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310859-08$15.00/0
References
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M ( 2000) Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA 97: 8104-9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldessarini RJ, Cohen BM, Teicher MH ( 1988) Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psychiatry 45: 79-91. [DOI] [PubMed] [Google Scholar]
- Brooks DJ ( 1995) The role of the basal ganglia in motor control: contributions from PET. J Neurol Sci 128: 1-13. [DOI] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP ( 1995) Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol 74: 1137-1148. [DOI] [PubMed] [Google Scholar]
- Castle NA, Slawsky MT ( 1993) Characterization of 4-aminopyridine block of the transient outward K+ current in adult rat ventricular myocytes. J Pharmacol Exp Ther 265: 1450-1459. [PubMed] [Google Scholar]
- Chiodo LA, Bunney BS ( 1983) Typical and atypical neuroleptics: differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J Neurosci 3: 1607-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Stevens CF ( 1971) Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol (Lond) 213: 21-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Hille CJ, Greenfield SA ( 2000) Dopamine release and uptake dynamics within nonhuman primate striatum in vitro J Neurosci 20: 8209-8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH ( 1976) Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192: 481-483. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS ( 1984) The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci 4: 2866-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS, Moore H, Todd CL ( 1997) Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 20: 31-37. [DOI] [PubMed] [Google Scholar]
- Hainsworth AH, Röper J, Kapoor R, Ashcroft FM ( 1991) Identification and electrophysiology of isolated pars compacta neurons from guinea-pig substantia nigra. Neuroscience 43: 81-93. [DOI] [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ ( 2000) Diversity and dynamics of dendritic signaling. Science 290: 739-744. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D ( 1997) K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurones. Nature 387: 869-875. [DOI] [PubMed] [Google Scholar]
- Ichikawa J, Meltzer HY ( 1990) Apomorphine does not reverse reduced basal dopamine release in rat striatum and nucleus accumbens after chronic haloperidol treatment. Brain Res 507: 138-142. [DOI] [PubMed] [Google Scholar]
- Ichikawa J, Meltzer HY ( 1991) Differential effects of repeated treatment with haloperidol and clozapine on dopamine release and metabolism in the striatum and the nucleus accumbens. J Pharmacol Exp Ther 256: 348-357. [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Frith CD, Carney MW, Price JS ( 1978) Mechanism of the antipsychotic effect in the treatment of acute schizophrenia. Lancet 1: 848-851. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM ( 2000) Principles of neural science, Ed 4, pp 1205-1206. New York: McGraw-Hill.
- Konradi C, Heckers S ( 2001) Antipsychotic drugs and neuroplasticity: insights into the treatment and neurobiology of schizophrenia. Biol Psychiatry 50: 729-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan ES, Levitan IB ( 1988) Serotonin acting via cyclic AMP enhances both the hyperpolarizing and depolarizing phases of bursting pacemaker activity in the Aplysia neuron R15. J Neurosci 8: 1152-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J ( 2001) Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J 20: 5715-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Abbott LF, Turrigiano GG, Liu Z, Golowasch J ( 1996) Memory from the dynamics of intrinsic membrane currents. Proc Natl Acad Sci USA 93: 13481-13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Mereu G, Lilliu V, Quartu M, Diana M, Gessa GL ( 1998) Haloperidol does not produce dopamine cell depolarization-block in paralyzed, unanesthetized rats. Brain Res 783: 27-32. [DOI] [PubMed] [Google Scholar]
- Mereu G, Lilliu V, Vargiu P, Muntoni AL, Diana M, Gessa GL ( 1994) Failure of chronic haloperidol to induce depolarization inactivation of dopamine neurons in unanesthetized rats. Eur J Pharmacol 264: 449-453. [DOI] [PubMed] [Google Scholar]
- Mereu G, Lilliu V, Vargiu P, Muntoni AL, Diana M, Gessa GL ( 1995) Depolarization inactivation of dopamine neurons: an artifact? J Neurosci 15: 1144-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Johnson JH, Hammerland LG, Kelbaugh PR, Volkmann RA, Saccomano NA, Mueller AL ( 1997) Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol Pharmacol 51: 491-498. [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL ( 1999) Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci 2: 1106-1113. [DOI] [PubMed] [Google Scholar]
- Seeman P, Chau-Wong M, Tedesco J, Wong K ( 1975) Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci USA 72: 4376-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Lee T, Chau-Wong M, Wong K ( 1976) Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 261: 717-719. [DOI] [PubMed] [Google Scholar]
- Silva NL, Mariani AP, Harrison NL, Barker JL ( 1988) 5, 7-Dihydroxytryptamine identifies living dopaminergic neurons in mesencephalic cultures. Proc Natl Acad Sci USA 85: 7346-7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva NL, Pechura CM, Barker JL ( 1990) Postnatal rat nigrostriatal dopaminergic neurons exhibit five types of potassium conductances. J Neurophysiol 64: 262-272. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Weiss F ( 1999) The dopamine hypothesis of reward: past and current status. Trends Neurosci 22: 521-527. [DOI] [PubMed] [Google Scholar]
- Thompson SH ( 1977) Three pharmacologically distinct potassium channels in molluscan neurons. J Physiol (Lond) 265: 465-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GN, Jiang M, Yao JA ( 1996) Reverse use dependence of Kv4.2 blockade by 4-aminopyridine. J Pharmacol Exp Ther 279: 865-876. [PubMed] [Google Scholar]
- White FJ ( 1996) Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci 19: 405-436. [DOI] [PubMed] [Google Scholar]
- White FJ, Wang RY ( 1983a) Comparison of the effects of chronic haloperidol treatment on A9 and A10 dopamine neurons in the rat. Life Sci 32: 983-993. [DOI] [PubMed] [Google Scholar]
- White FJ, Wang RY ( 1983b) Differential effects of classical and atypical antipsychotic drugs on A9 and A10 dopamine neurons. Science 221: 1054-1057. [DOI] [PubMed] [Google Scholar]
- Yang F, Feng L, Zheng F, Johnson SW, Du J, Shen L, Wu CP, Lu B ( 2001) GDNF acutely modulates excitability and A-type K+ channels in midbrain dopaminergic neurons. Nat Neurosci 4: 1071-1078. [DOI] [PubMed] [Google Scholar]
- Zhang TT, Takimoto K, Stewart AF, Zhu C, Levitan ES ( 2001) Independent regulation of cardiac Kv4.3 potassium channel expression by angiotensin II and phenylephrine. Circ Res 88: 476-482. [DOI] [PubMed] [Google Scholar]