

Abstract
The reinforcing and psychomotor effects of morphine involve opiate stimulation of the dopaminergic system via activation of μ-opioid receptors (μOR). Both μ-opioid and dopamine receptors are members of the G-protein-coupled receptor (GPCR) family of proteins. GPCRs are known to undergo desensitization involving phosphorylation of the receptor and the subsequent binding of βarrestins, which prevents further receptor-G-protein coupling. Mice lacking βarrestin-2 (βarr2) display enhanced sensitivity to morphine in tests of pain perception attributable to impaired desensitization of μOR. However, whether abrogating μOR desensitization affects the reinforcing and psychomotor properties of morphine has remained unexplored. In the present study, we examined this question by assessing the effects of morphine and cocaine on locomotor activity, behavioral sensitization, conditioned place preference, and striatal dopamine release in βarr2 knock-out (βarr2-KO) mice and their wild-type (WT) controls. Cocaine treatment resulted in very similar neurochemical and behavioral responses between the genotypes. However, in the βarr2-KO mice, morphine induced more pronounced increases in striatal extracellular dopamine than in WT mice. Moreover, the rewarding properties of morphine in the conditioned place preference test were greater in the βarr2-KO mice when compared with the WT mice. Thus, βarr2 appears to play a more important role in the dopaminergic effects mediated by morphine than those induced by cocaine.
Keywords: morphine, cocaine, dopamine, βarrestin, knock-out mice, locomotor activity, sensitization, microdialysis, conditioned place preference
Introduction
Morphine (Mor) has been used for centuries to alleviate pain; however, its use is limited by a number of adverse consequences, including constipation, respiratory suppression, and the potential development of dependence and addiction after extended periods of administration. The physiological actions of morphine are recognized to be mediated principally through the μ-opioid receptor (μOR). This concept, which is suggested by extensive pharmacological experiments, has been greatly strengthened by the finding that mice lacking the μOR do not experience morphine-mediated antinociception, respiratory suppression, inhibition of gastrointestinal transit, or increased locomotor activity (Matthes et al., 1996; Sora et al., 1997; Kieffer, 1999). Moreover, mice lacking the μOR do not show a preference for morphine over vehicle in studies designed to test drug reinforcement (Matthes et al., 1996; Kieffer, 1999), supporting the pharmacological inhibitor studies demonstrating the importance of the μOR in self-administration paradigms (Mello and Negus, 1996). Therefore, the disruption of μOR regulation could potentially alter the behavioral effects of morphine.
As with other members of the G-protein-coupled receptor (GPCR) superfamily, the μOR is subject to regulation by phosphorylation and subsequent interactions with βarrestins. This process has been shown to be an important step in receptor desensitization (Ferguson et al., 1998; Lefkowitz, 1998; Zhang et al., 1998; Perry and Lefkowitz, 2002). When the βarrestin-2 (βarr2) gene is inactivated in mice by genetic deletion, the antinociceptive effects of morphine are enhanced and prolonged (Bohn et al., 1999, 2002). The antinociceptive behaviors are correlated with enhanced μOR-G-protein coupling, suggesting that the βarr2 molecule acutely regulates the signaling potential of the receptor (Bohn et al., 1999, 2002). Furthermore, tolerance to the antinociceptive effects of morphine is attenuated in βarr2 knock-out KO mice (βarr2-KO) (Bohn et al., 2000). Together, these studies demonstrate that, by eliminating the βarr2 regulatory molecule, the desensitization of the μOR is impaired, leading to dramatically attenuated antinociceptive tolerance.
Alternatively, when the same mice that do not develop morphine tolerance were tested for opiate dependence, they displayed physical withdrawal symptoms to the same extent during acute naltrexone treatment, revealing that, although the βarr2-KO mice did not experience antinociceptive tolerance to morphine, they still became physically dependent on the drug (Bohn et al., 2000). These findings suggest that the means of regulating the μOR may vary in different systems, with βarr2 playing an important role in morphine antinociceptive tolerance but not necessarily in the development of opiate dependence. Therefore, it is interesting to determine whether the βarr2-KO mice reveal differences in other behavioral effects of morphine, particularly those associated with drug reinforcement.
Although the action of morphine on brain circuitry related to reward mechanisms is complex, the substantial contribution of the mesolimbic dopamine (DA) system is well recognized (Di Chiara and Imperato, 1988; Wise and Rompre 1989; White and Kalivas, 1998; De Vries and Shippenberg, 2002). For instance, mice lacking the D2 dopamine receptor do not present morphine-induced locomotor activity, conditioned place preference, and self-administration (Maldonado et al., 1997; Elmer et al., 2002). Morphine and dopamine bind to their respective GPCRs to mediate their physiological responses. Because βarr2 has been shown to regulate dopamine receptors in vitro (Kim et al., 2001) as well as opioid receptors both in vitro and in vivo (Whistler and von Zastrow, 1998; Zhang et al., 1998; Bohn et al., 1999; Oakley et al., 2000), the loss of this regulatory component could potentially impact the opioid and/or dopamine system directly. In an attempt to pharmacologically dissect these two GPCR signaling systems (opioid and dopamine), cocaine (COC) (which exerts its action in part as an indirect agonist of the dopamine receptors) and morphine were used in parallel. By measuring both neurochemical and behavioral parameters of the dopaminergic system, we asked whether differences between the wild-type (WT) and βarr2-KO mice could be seen with either drug.
Materials and Methods
Animals and drugs
WT, heterozygous (HT), and KO mice were generated as littermates by crossing heterozygous βarr2 C57BL/6 × 129SvJ mice (which has now been maintained over nine generations) as initially described (Bohn et al., 1999). Mice used in this study were age-matched, 3- to 5-month-old male siblings weighing between 20 and 35 gm, and genotype was determined by PCR analysis of DNA extracted from ear punch tissue. In all experiments, WT littermates served as controls for the βarr2-HT and βarr2-KO mice, and all genotypes were evaluated simultaneously. Before the experiment, all mice are provided food and water ad libitum. Mice were only used once in each experiment as indicated; a new group of mice was used for each dose and drug tested. Experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals and with an approved animal protocol from the Duke University Animal Care and Use Committee. Morphine sulfate and cocaine were purchased from Sigma-RBI (St. Louis, MO) and freshly prepared in saline (Sal). All compounds were injected at a volume of 10 μl/gm animal weight as specified.
Locomotor activity
Activity was measured in an Omnitech Digiscan activity monitor (20 × 20 cm2; Accuscan Instruments, Columbus, OH). Locomotor activity was measured at 5 min intervals, and cumulative counts were taken for data analysis; data were analyzed for the total number of beam breaks seen in 5 min increments, a measure that assesses all movements, including running and turning behaviors (Wang et al., 1997; Xu et al., 2000). To evaluate the effects of morphine or cocaine on locomotor behavior, mice were placed in activity monitor for a 60 min habituation period. Immediately after the 60 min habituation period, mice were injected with drug or saline and placed back into the monitor, and locomotor activity was recorded for the following 120 min. In all acute experiments, each animal received only a single injection of each drug. In morphine sensitization experiments, mice were first exposed to the activity boxes for 30 min, 4 d before the test. On day 1, mice were habituated to the activity box for 30 min, morphine (10 mg/kg, s.c.) was then given, and activity was recorded for 2 hr. The same group of mice was administered morphine (10 mg/kg, s.c.) once per day for the next 6 consecutive days in home cages for a total of 7 d of chronic daily morphine treatment. On the eighth day, the mice were not handled or treated. On day 9, the mice were treated the same as on day 1; after a 30 min habituation, morphine (10 mg/kg, s.c.) was administered, and activity was monitored for 2 hr. The cocaine sensitization protocol was performed in a similar manner as morphine. After a 30 min habituation period, mice received cocaine (20 mg/kg, i.p.) on day 1, and locomotor activity was immediately assessed. Mice then received four additional daily injections of cocaine (20 mg/kg, i.p.) in home cages, were not handled or treated on the sixth day, and received another dose of cocaine (20 mg/kg, i.p.) on the seventh day, at which time activity was again assessed (Wang et al., 1997; Xu et al., 2000).
In vivo microdialysis
Microdialysis was performed as described previously (Wang et al., 1997; Xu et al., 2000). Anesthetized mice were placed in a stereotaxic frame, and dialysis probes (2 mm membrane length, 0.24 mm outer diameter, Cuprophane, 6 kDa cutoff; model CMA-11; CMA Microdialysis, Solna, Sweden) were implanted into the right striatum. The stereotaxic coordinates for implantation of microdialysis probes were as follows: anteroposterior, 0.0 mm; dorsoventral, -4.4 mm; lateral, 2.5 mm relative to bregma (Franklin and Paxinos, 1997). Placement of the probe was verified by histological examination subsequent to the experiments. After surgery, animals were returned to their home cages with access to food and water ad libitum. Twenty-four hours after surgery, the dialysis probe was connected to a syringe pump and perfused at 1 μl/min with artificial CSF (in mm): 150 Na+, 3.0 K+, 1.4 Ca2+, 31.0 PO4-, 0.8 Mg2+, and 155 Cl- (ESA, Bedford, MA). After a 1 hr equilibration period, the perfusates were collected every 20 min to a tube containing 2 μl of 1 m HClO4. At least four control samples were taken before drugs were administered systemically. Dialysates were analyzed for levels of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) using HPLC with electrochemical detection. DA and metabolites were separated on a microbore reverse-phase column (5 μm, 1 × 150 mm; model C-18, Unijet; BAS, West Lafayette, IN) with a mobile phase consisting of 0.03 m citrate-phosphate buffer with 2.1 mm octyl sodium sulfate, 0.1 mm EDTA, 10 mm NaCl, and 17% methanol, pH 3.6, at a flow rate of 80 μl/min and detected by a 6 mm glass carbon electrode (Unijet; BAS) set at +0.8 V. The volume of injection was 5 μl. Under these conditions, the basal levels of dopamine, DOPAC, and HVA in the dialysates were collected from each genotype before treatment (saline, morphine, or cocaine) and were as follows: (1) dopamine, 52 ± 6.6 fmol/20 μl; 68 ± 17 fmol/20 μl; (2) DOPAC, 5.9 ± 0.6 pmol/20 μl; 7.2 ± 1.2 pmol/20 μl; and (3) HVA, 9.3 ± 1.0 pmol/20 μl; 9.4 ± 1.0 pmol/20 μl for WT (n = 19) and βarr2-KO (n = 15) mice, respectively.
Conditioned place preference
The commercially designed place preference chambers (model ENV-3013) for mice were purchased from Med Associates (St. Albans, VT). Each chamber was housed in a sound-attenuating box, and individual fans provided ventilation. The chambers consisted of three distinct compartments, separated by optional manual guillotine doors and illuminated with a separate light in the laminate top of each compartment. The location of the mouse was determined by automated data collection using photobeam strips that recorded the time spent in each of the three compartments. The experiments were conducted in a designated mouse testing room in which no other activities took place during the test times.
Each of the three compartments in the place preference chamber was distinct. The center compartment was 2.85 × 5 × 5 inches, with all gray walls and floor. The two “choice” compartments were 6.6 × 5 × 5 inches. One choice compartment was black with a stainless steel grid rod floor, and the other compartment was white with a stainless steel mesh floor. Each compartment also contained a removable, stainless steel waste pan. A small amount of corncob bedding was added to the waste pan under the mesh floor of one side, and an intact “Orange Spice” Bigelow tea bag was included along with the corncob bedding in the waste pan under the grid floor of the other compartment. The place preference procedure consisted of three phases: preconditioning, conditioning, and postconditioning.
Preconditioning. During the preconditioning phase of the experiment, mice were place into the center gray compartment, and the doors between the black and white compartments were open. The time spent in each of the compartments was recorded for a 30 min period. If a mouse spent more than 60% of the 30 min preconditioning period in either the black or white chamber or more time in the gray chamber than in one of the choice chambers, it was eliminated from further experimentation. This resulted in ∼15-25% elimination in each treatment group. The conditioning phase commenced the following day.
Conditioning phase. Mice were treated with morphine or cocaine at the designated doses on days 1, 3, and 5 and saline on days 2, 4, and 6 of the conditioning phase. Because the initial criteria required that no mice show a marked preference for either side on the preconditioning day, the drug was randomly paired with alternating compartments such that, in any group, half of the mice received drug in the black compartment and the other half received drug in the white compartment. After drug or saline administration, mice were placed in the appropriate compartment. Testing occurred on the day after the 6 d conditioning phase.
Postconditioning test. Mice were handled on the test day in the same manner as on the preconditioning day and received no drug or saline administrations. With the chamber doors open, the mouse movements were automatically recorded for a total of 30 min. Data were analyzed for the first 10, 15, and 20 min of the preconditioning and postconditioning phases of the experiment by subtracting the time spent in the drug-assigned chamber in the preconditioning period from the time spent in that chamber in the postconditioning period.
Statistical analyses
Data presented as responses repeatedly measured over time were analyzed using a two-way ANOVA for repeated measures using the SAS statistical software (version 6.11; SAS Institute, Cary, NC). Dose-effect curves were compared using standard two-way ANOVA, and individual doses were compared via Bonferroni post hoc analysis (GraphPad Software, San Diego, CA). Sensitization data were analyzed by a three-way ANOVA for repeated measures. Degrees of freedom for corresponding factor and error are shown for the ANOVA analyses. A Mann-Whitney U test (two-tailed) was used to compare genotypes in the microdialysis assays in which the data has been presented in a normalized percentage basis (Morari et al., 1998). Comparisons of drug effects are made after drug administration (after habituation periods on locomotor activity curves).
Results
As an initial screen for potential alterations in dopamine functions, locomotor activity was measured in the βarr2-KO mice in the novel environment of the test chamber. During a 1 hr habituation period, although both genotypes became less active over time (Fig. 1A) (F(11,1022) = 32.89; p < 0.0001), the βarr2-KO mice demonstrated significantly less horizontal activity compared with WT mice (Fig. 1A) (F(1,93) = 8.26, p < 0.005; no interaction of factors was detected, F(11,1022 = 0.223, p < 0.9952). A similar trend was also observed when activity was analyzed for distance traveled or vertical activity (data not shown). Morphine (10 mg/kg, s.c.) was administered to mice of both genotypes at a dose that has been shown to increase activity in mice (Oliverio et al., 1975; Brase et al., 1977; Miner 1997). Morphine produced marked increases in locomotor activity in all three genotypes (F(23,483) = 27.94; p < 0.0001). At this dose, the WT mice appear to be more activated than the βarr2-KO or heterozygotes, although this did not prove to be significantly different (for genotype, F(2,21) = 2.62, p = 0.0965; interaction genotype × time, F(46,483) = 1.70, p < 0.01). To further investigate whether there was a difference in morphine-induced locomotor activity between genotypes, several doses were tested (Fig. 1C). Increases in the number of beam breaks (as accumulated during a 120 min period) after morphine injection were significantly lower in βarr2-KO mice than in their WT counterparts overall (Fig. 1C) (for genotype, F(1,95) = 70.22, p < 0.001; for dose, F(4,95) = 36.07, p < 0.0001; interaction genotype × dose, F(4,95) = 9.49, p < 0.0001). Although it is possible that the lower basal activity of the βarr2-KO mice may contribute to the decreased responsiveness to morphine, it is unlikely that this alone can completely explain the extent of the differences in activity after morphine administration measured in these mice. This observation was surprising in the context of our previous studies in which morphine produced a greater degree of antinociception in βarr2-KO mice than in their WT littermate controls because, in the locomotor activity test, the degree of activation is actually less in the knock-out mice.
Figure 1.

Locomotor activity after acute morphine treatment. A, Basal locomotor activity in WT and βarr2-KO mice during a 1 hr habituation period (for time, F(11,1022) = 32.89, p < 0.0001; for genotype, F(1,93) = 8.26, p < 0.005; n = 46-49). B, Effects of an acute dose of morphine. Morphine (10 mg/kg, s.c.) was administered to wild-type (n = 8), heterozygotes (n = 8), and βarr2-KO (n = 8) mice after a 1 hr habituation period. Locomotor activity was assessed as the number of infrared beam breaks in 5 min intervals. The values were averaged across mice, and the means ± SEM are shown here. Morphine-induced locomotor activity in all three groups, yet the genotypes did not differ significantly at this dose (for genotype, F(23,483) = 27.94, p < 0.0001; for dose, F(2,21) = 2.62, p = 0.0965). C, Morphine dose-response curve for locomotor activity. The number of beam breaks was summed over the 120 min test period, and the mean ± SEM after morphine or saline (10 μl/g, s.c.) administration is presented. The sum of the beam breaks in B is included in this graph, representing the 10 mg/kg dose. Overall, morphine produced greater increases in locomotor activity in WT mice than in the βarr2-KO mice (for genotype, F(1,95) = 70.22, p < 0.001; for dose, F(4,95) = 36.07, p<0.0001; n = 8-16). Bonferroni post hoc analysis revealed significant differences between the genotypes at the doses of 5, 20, and 40 mg/kg subcutaneous morphine (WT vs KO; *p < 0.05; **p < 0.001).
At the same time, a moderate dose of cocaine (20 mg/kg, i.p.) produced similar profiles of locomotor activation in both genotypes (Fig. 2A) (for genotype, F(1,23) = 0.96, p = 0.3371; for time, F(17,391) = 12.40, p < 0.0001; interaction genotype × time, F(17,391) = 0.15, p = 1.0000). Although the βarr2-KO mice appear to be slightly less activated by cocaine than their WT littermates in Figure 2A, this profile of activity closely resembles that seen in the basal state (Fig. 1A) in which the βarr2-KO mice are generally less active. In the dose-response curve, both genotypes displayed similar response profiles to different doses (10, 20, and 30 mg/kg, i.p.) of cocaine tested (Fig. 2B) (for genotype, F(1,62) = 0.08, p = 0.7723; for dose, F(3,62) = 22.88, p < 0.0001; interaction genotype × dose: F(3,62) = 0.37, p = 0.7738). Together, these data indicate that morphine produces markedly differential effects on locomotor activity in the knock-out versus WT mice, although no such obvious differences were observed with cocaine.
Figure 2.
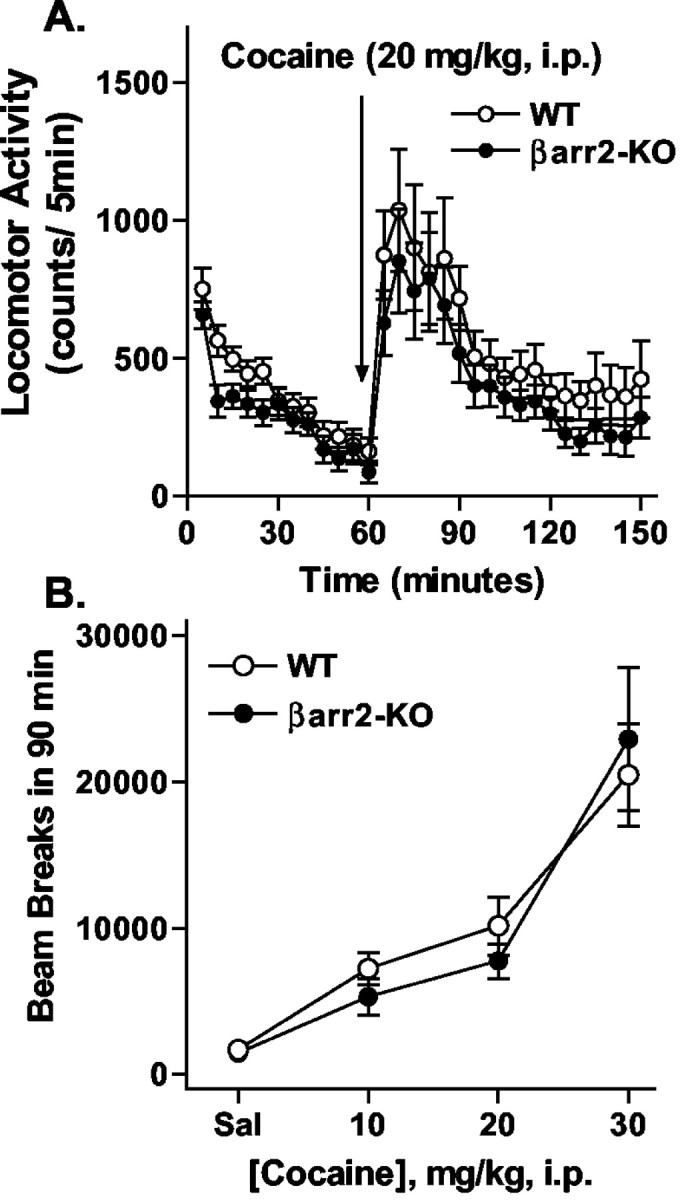
Locomotor activity after acute cocaine treatment. A, Cocaine (20 mg/kg, i.p.) was administered to WT (n = 15) and βarr2-KO (n = 12) mice after a 1 hr habituation period. Locomotor activity was assessed as described in Figure 1. Although cocaine produced marked increases in locomotor activity in both genotypes, WT mice displayed slightly higher activity than the βarr2-KO mice after morphine (for time, F(17,391) = 12.40, p < 0.0001; for genotype, F(1,23) = 0.96, p = 0.3371). B, Dose-response curve for cocaine-induced locomotor activity. Data represent the sum of the activity after each dose over a 90 min period. There were no significant differences between the genotypes (F(1,62) = 0.08; p = 0.7723; n = 7-15).
Locomotor activation can be further enhanced after chronic administration of the drug in a process known as locomotor sensitization or “reverse tolerance” (White and Kalivas, 1998; Robinson and Berridge, 2000; De Vries and Shippenberg, 2002). Although the increases in acute locomotor activity are believed to be a result of increased extracellular dopamine levels, sensitization appears to predominantly involve enhanced sensitivity of dopamine receptor signaling but may also involve alterations in the control of extracellular dopamine dynamics (Koob and Nestler 1997; White and Kalivas, 1998; Chefer and Shippenberg, 2002). This phenomenon is believed to be reflective of long-term cellular plasticity associated with drug addiction (Nestler, 2001). The effects of morphine and cocaine in behavioral sensitization paradigms were assessed. Because the mice were habituated after 30 min (Fig. 1A) and the morphine effect was most prominent in the first 90 min (Fig. 1B), the duration of these test times were shortened accordingly in the sensitization experiment to decrease the overall time spent in the activity box because repeated measurements were to be made on different days. After 6 d of chronic morphine treatment, both genotypes reveal an enhanced increase in locomotor activity compared with their response on the first day of treatment, indicating that both genotypes become sensitized to repeated morphine treatment, and the degree of sensitization was similar in both genotypes (Fig. 3) (for sensitization, F(1,36) = 5.68, p < 0.05; for genotype, F(1,36) = 3.78, p = 0.06; interaction genotype × sensitization, F(1,36) = 0.08, p = 0.7766). In the cocaine sensitization experiment, both genotypes also reveal greater locomotor activity after the repeated administrations of cocaine, yet no difference between the genotypes was determined (Fig. 4) (for sensitization, F(1,39) = 7.67, p < 0.01; for genotype, F(1,39) = 0.00, p = 0.9474; interaction genotype × sensitization, F(1,39) = 0.02, p = 0.8811).
Figure 3.

Locomotor sensitization to morphine. Locomotor activity was initially assessed in WT and βarr2-KO mice during a 30 min habituation period and for 90 min after an acute dose of morphine (10 mg/kg, s.c.). After this observation, mice received one daily dose of morphine (10 mg/kg, s.c.) for 6 additional days in their home cage. After 1 d of rest, the mice were then challenged again with the same dose of morphine (10 mg/kg, s.c.), and locomotor activity was assessed (WT-S, βarr2-KO-S). Locomotor activity was increased significantly after this regimen in both WT and βarr2-KO mice, indicating that sensitization to morphine developed in both genotypes (for sensitization, F(1,36) = 5.68, p < 0.05; for genotype, F(1,36) = 3.78, p = 0.06; n = 10 of each genotype.)
Figure 4.

Locomotor sensitization to cocaine. Locomotor activity was initially assessed in WT and βarr2-KO mice during a 30 min habituation period and for 90 min after an acute dose of cocaine (20 mg/kg, i.p.). Mice then received one daily dose of cocaine (20 mg/kg, i.p.) for 4 additional days in their home cage. After 1 d of rest, the mice were then challenged again with the same dose of cocaine (20 mg/kg, i.p.), and locomotor activity was again assessed (WT-S, βarr2-KO-S). Data represent the sum of the number of beam breaks recorded during 60 min after drug administration. Both groups of mice became sensitized to cocaine to a similar extent (for time, F(1,39) = 7.67, p < 0.01; for genotype, F(1,39) = 0.00, p = 0.9474; n = 7-14.)
The acute administration of morphine and cocaine increases the extracellular levels of dopamine in several brain regions (Di Chiara and Imperato, 1988; Pontieri et al., 1995; Rocha et al., 1998; He and Shippenberg, 2000). Extracellular levels of dopamine can be directly measured by in vivo microdialysis in which a probe is inserted into the striatum to allow collection of samples in a conscious, freely moving mouse (Wang et al., 1997; Gainetdinov et al., 1999b; Chefer and Shippenberg, 2002). Here, striatal microdialysis was performed to see whether the rise in extracellular dopamine is comparable between the βarr2-KO and WT mice after drug treatment. Samples were collected from the striatum of freely moving mice and assessed for dopamine, and its metabolites DOPAC and HVA, by HPLC after either saline or morphine (10 mg/kg, s.c.) treatment (Fig. 5). As might be expected, morphine induced a significant increase in extracellular dopamine and metabolite levels in both genotypes (Fig. 5) (WT plus Sal vs WT plus Mor: dopamine, DOPAC, and HVA, p < 0.01; βarr2-KO plus Sal vs βarr2-KO plus Mor: dopamine, DOPAC, and HVA, p < 0.01; two-tailed Mann-Whitney U test). Surprisingly, significantly more dopamine was released, particularly in the first hour of treatment after morphine, in the βarr2-KO mice than in the WT littermates (Fig. 5) (p < 0.05; two-tailed Mann-Whitney U test). This effect was also seen in the measurements of the dopamine metabolites DOPAC and HVA, indirect indicators of increased dopaminergic neuron function (Fig. 5) (WT vs βarr2-KO; p < 0.01; two-tailed Mann-Whitney U test). Saline treatment did not reveal differences in dopamine or metabolite levels between the genotypes, nor did the basal levels vary (see Materials and Methods) (Fig. 5).
Figure 5.

Invivo microdialysis after acute morphine. The effects of morphine (10 mg/kg, s.c.) and saline (10 μl/g, s.c.) on extracellular DA, DOPAC, and HVA levels in the striatum of freely moving mice as measured using in vivo microdialysis. Data are presented as the percentage of the average level of dopamine or metabolite measured in at least three samples collected before the drug administration. Analysis of area under curve values for 3 hr after administration (data not shown) revealed a significant effect of morphine on extracellular dopamine and metabolites in both genotypes: WT+Sal vs WT+Mor, dopamine, DOPAC, and HVA, p < 0.01; βarr2-KO+Sal vs βarr2-KO+Mor, dopamine, DOPAC, and HVA, p < 0.01; Mann-Whitney U test (two-tailed). Analysis of the 1 hr period after morphine administration (data not shown) revealed that βarr2-KO mice produced a greater accumulation of dopamine and metabolites: WT (n = 9) vs βarr2-KO (n = 6) after morphine, DA, p < 0.05; DOPAC and HVA, p < 0.01; Mann-Whitney U test (two-tailed). Similar analysis of saline effects (1 or 3 hr period) revealed no significant differences between the genotypes (WT, n = 5; βarr2-KO, n = 5; p > 0.05; two-tailed Mann-Whitney U test).
Cocaine-induced elevation in extracellular dopamine was also assessed by in vivo microdialysis. Cocaine at 20 mg/kg intraperitoneally increased extracellular dopamine over saline-treated controls (WT plus Sal vs WT plus COC, βarr2-KO plus Sal vs βarr2-KO plus Mor: p < 0.01; two-tailed Mann-Whitney U test). This degree of stimulation was to the same extent in both the βarr2-KO and WT mice (Fig. 6). The levels of dopamine metabolites after cocaine did not differ between genotype or with the saline-treated controls (data not shown) as is expected for a transporter blocker (Nomikos et al., 1990). The levels of dopamine after cocaine are similar between the genotypes and correlates well with the similar extent of their locomotor responses to the drug (Fig. 2).
Figure 6.

In vivo microdialysis after acute cocaine. The effects of cocaine (20 mg/kg, i.p.) and saline (10 μl/g, i.p.) on extracellular DA levels in the striatum of freely moving mice as measured using in vivo microdialysis. Data are presented as the percentage of the average level of dopamine measured in at least three samples collected before the drug administration. Analysis of area under curve values for 2 hr after cocaine treatment (data not shown) revealed a significant increase in DA in both genotypes compared with the saline treatment of the respective genotype (p < 0.01; two-tailed Mann-Whitney U test). No significant differences were observed between genotypes after saline (WT, n = 5; βarr2-KO, n = 5) or cocaine (WT, n = 5; KO, n = 4; p > 0.05; two-tailed Mann-Whitney U test).
A conditioned place preference paradigm was used to determine whether the rewarding or reinforcing properties of morphine or cocaine were affected by elimination of βarr2 (Shoaib et al., 1995; Miner, 1997; Martin et al., 2000). In Figure 7A, the preference is shown at three different experimental conditions after 3 mg/kg subcutaneous morphine in which we examined the change in time spent in the drug-paired compartment, in which the preconditioning and postconditioning times were set at 10, 15, or 20 min. By examining the data in this way, it demonstrates that even early (10 min) in the time period of choosing sides, the βarr2-KO mice experienced more preference than the WT mice for the morphine-paired side (Fig. 7A) (for genotype: F(1,11) = 5.53, p < 0.05; for time, F(2,22) = 15.20, p < 0.0001; interaction genotype × time, F(2,22) = 0.49, p = 0.6218). A dose-response analysis was also performed, and, at each of the doses of morphine examined (1, 3, and 6 mg/kg, s.c.), the βarr2-KO mice showed a greater preference for the morphine-paired compartment than their WT littermates (Fig. 7B) (for genotype, F(1,41) = 6.31, p < 0.05; for dose, F(2,41) = 7.05, p < 0.01; interaction genotype × dose, F(2,41) = 0.21, p = 0.8105). Together, these data indicate that the rewarding properties of morphine, as measured in the conditioned place preference procedure, are enhanced in βarr2-KO mice over those observed in the WT controls (Fig. 7)
Figure 7.
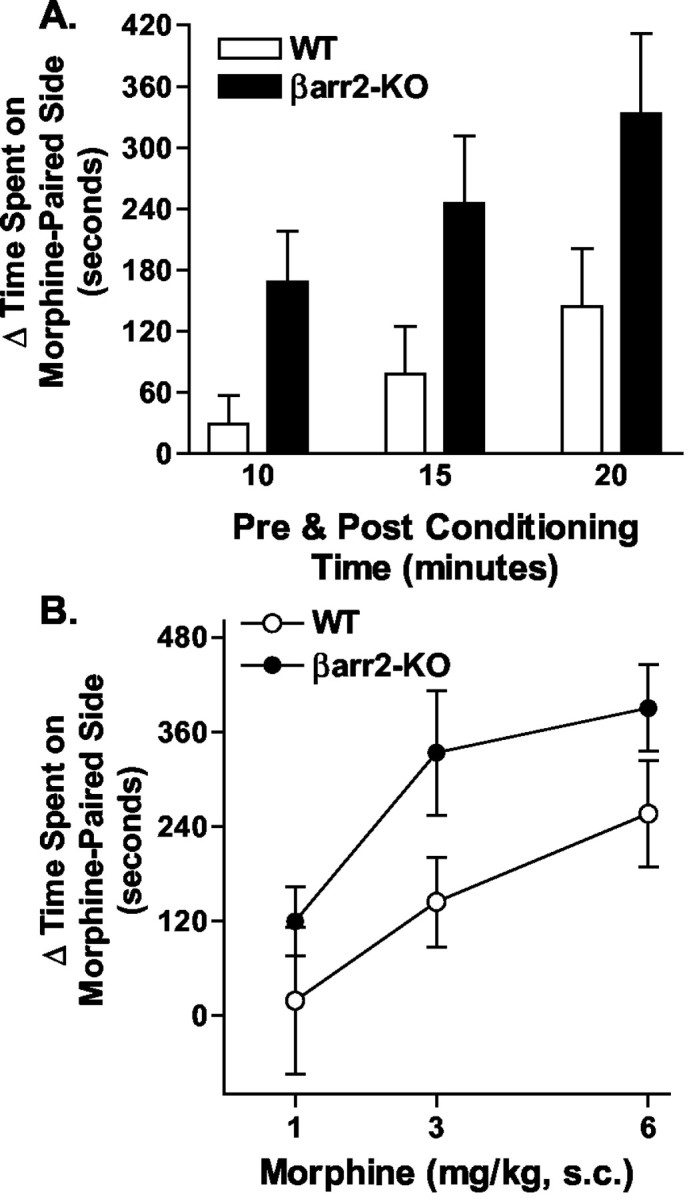
Morphine-induced conditioned place preference. Mice were assessed for the time spent in either the black or white compartment on the preconditioning day. Drug was alternatively paired with either side for both groups of mice as described in Materials and Methods. After the 6 d conditioning regimen (drug on days 1, 3, and 5; saline on days 2, 4, and 6), the time spent in each compartment was assessed in the absence of drug or saline (postconditioning). A, Data are shown as the difference in the time spent in the drug-paired (morphine, 3 mg/kg, s.c.) compartment on the postconditioning day and the preconditioning day when the data are analyzed at 10, 15, or 20 min preconditioning and postconditioning times (mean ± SEM). The βarr2-KO mice spent more time in the drug-paired side than the WT mice when compared over all times (for genotype, F(1,11) = 5.53, p < 0.05; for time, F(2,22) = 15.20, p < 0.0001). B, Dose-response curve at 20 min preconditioning and postconditioning times. Morphine produced a dose-dependent increase in preference for the morphine-paired compartment in both genotypes; however, when comparing genotypes, the effect was significantly greater in the βarr2-KO mice compared with their WT littermates (for genotype, F(1,41) = 6.31, p < 0.05; for dose, F(2,41) = 7.05, p < 0.01; n = 9 per group).
We also examine the conditioned place preference induced by cocaine in each genotype. A dose of 10 mg/kg intraperitoneally, which has been used previously in this type of experiment to induce preference in normal mice (Zhang et al., 2002), induced similar levels of preference in both genotypes, and this remained constant at each of the preconditioning and postconditioning time points examined (Fig. 8) (for genotype, F(1,16) = 0.05, p = 0.8179; for time, F(2,32) = 5.95, p < 0.01; interaction genotype × time, F(2,32) = 0.34, p < 0.01).
Figure 8.

Cocaine-induced conditioned place preference. Conditioning was performed as described in Figure 7A with cocaine (10 mg/kg, i.p.). Data are shown as the difference in the time spent in the drug-paired compartment on the postconditioning day and the preconditioning day when the preconditioning and postconditioning times were set at 10, 15, or 20 min (mean ± SEM). Cocaine (10 mg/kg, i.p.) produced preference for the cocaine-paired compartment in both genotypes that did not differ under any of the conditions (for genotype, F(1,16) = 0.05, p = 0.8179; for time, F(2,32) = 5.95, p < 0.01; n = 9-10 per group).
Discussion
The βarr2-KO mice have proven to be a valuable animal model for studying how regulation of the μOR can lead to differences in behavioral responses to the μOR agonist morphine. Previously, we reported that the anitinociceptive properties of morphine are enhanced in mice lacking βarr2 (Bohn et al., 1999). Here we demonstrate that morphine induces a greater increase in striatal dopamine release after morphine treatment (Fig. 5), which correlates with the increased conditioned place preference (Fig. 6A) in the βarr2-KO mice compared with the WT mice. These observations give additional support to the general principle that the inactivation of components of G-protein-coupled receptor kinase/arrestin-mediated desensitization mechanisms can lead to enhanced receptor signaling, and thereby, enhanced behavioral responses (Bohn et al., 1999, 2002; Gainetdinov et al., 1999a, 2003). However, removal of βarr2 actually leads to less enhancement of locomotor activity after morphine (Fig. 1), although both genotypes experience comparable behavioral sensitization to this drug (Fig. 3) after repeated administration, indicating that not all of the effects of morphine are potentiated in the absence of βarr2 and could reflect differential roles of βarr2 in the regulation of the receptor in different neuronal populations (Bohn et al., 2002).
The activation of both opioid and dopamine receptors plays a critical role in the reinforcing effects of morphine (Koob, 1992; Shippenberg et al., 1993; Maldonado et al., 1997; Kieffer, 1999; Elmer et al., 2002). Morphine and cocaine, as well as many other major drugs of abuse, lead to increased dopamine signaling in mesolimbic brain structures, such as the nucleus accumbens (NAc) (Di Chiara and Imperato, 1988; Pontieri et al., 1995; Shoaib et al., 1995; He and Shippenberg, 2000; Murphy et al., 2001). However, in the case of morphine, stimulation of dopamine pathways is indirect, originating from a disinhibition of GABAergic cells in dopamine cell body regions (within substantia nigra and the ventral tegmental area), leading to increased firing, and consequently, increased dopamine release in terminal regions (within striatum and NAc) (Di Chiara and North, 1992; Johnson and North, 1992). Morphine, however, can also exert a direct effect on dopamine dynamics in the NAc (Spanagel et al., 1992) and exert some effects in a dopamine-independent manner (Kalivas et al., 1983).
Regardless of the mechanism for increasing dopamine signaling, the activation of the dopaminergic system, particularly in the mesolimbic system, is an integral part of the initial response to most major drugs of abuse, including morphine and cocaine, and therefore was the focus of our behavioral and neurochemical studies (Di Chiara and Imperato, 1988; Wise and Rompre, 1989; White and Kalivas, 1998; De Vries and Shippenberg, 2002). Cocaine is known to act by blocking the monoamine transporters and thereby preventing reuptake of monoamines, including dopamine, which leads to elevated levels of neurotransmitter in the extracellular space. In our findings, cocaine promotes similar levels of locomotor activity (Fig. 2), behavioral sensitization (Fig. 4), dopamine response (Fig. 6), and conditioned place preference (Fig. 8) in both βarr2-KO and WT mice, which suggests that the elimination of βarr2 has little effect on the behaviors elicited by cocaine, which may reflect a less critical role of βarr2 in the regulation of dopaminergic signaling.
It is interesting that, although the βarr2-KO mice demonstrate increased striatal dopamine release and enhanced conditioned place preference with morphine, they do not display enhanced locomotor activation. In contrast, the βarr2-KO mice actually display markedly less hyperactivity than their WT controls after each dose of morphine tested (Fig. 1C). To some extent, this might be explained by the decreased basal levels of activity in the βarr2-KO mice; however, the magnitude of the difference after morphine treatment, unlike that observed after cocaine, is great enough to suggest that mechanisms other than those responsible for the difference in the basal activity might be involved. Morphine-induced increases in locomotor behavior are likely dependent not only on μOR-mediated activation of dopamine signaling but also on other neurotransmitter mechanisms because several neurotransmitter systems can modulate dopamine-dependent locomotor activation. For example, a suppression of locomotor activation is observed after direct and indirect dopamine agonists when the serotonergic system is coactivated (Gainetdinov et al., 1999b, 2001). Morphine has been shown to elevate serotonergic transmission (Tao and Auerbach, 1994) in addition to elevating dopamine transmission (Di Chiara and Imperato, 1988). Furthermore, it has been shown that inhibition of the serotonin transporter may counteract morphine hyperactivity (Sills and Fletcher, 1997; Ise et al., 2001). Therefore, one potential mechanism may be related to possible alterations in the μOR-mediated regulation of the serotonergic system or in serotonin receptors per se in the βarr2-KO mice, which might contribute to the reduced locomotor response to morphine even in the presence of high striatal extracellular dopamine.
Historically, the locomotor response has been considered a predictor of whether a drug will be reinforcing (Wise and Bozarth, 1987). Furthermore, locomotor activation and sensitization have both been used in initial screening for potential alterations in opiate and psychostimulant reinforcement mechanisms. However, the decreased activation observed in the βarr2-KO mice after morphine not only did not correspond to the higher elevation of dopamine release in these animals but also contrasted the findings in the conditioned place preference paradigm. In fact, enhanced increases in dopamine levels after acute morphine in the βarr2-KO mice correlate best with the enhanced rewarding properties also observed in these animals in the conditioned place preference procedure (Fig. 7). Our findings support a growing body of literature documenting that locomotor responses to morphine do not necessarily correlate with its dopamine-releasing properties and therefore cannot serve as an indicator of the reinforcing properties of a drug (Maldonado et al., 1997; Spielewoy et al., 2000; Murphy et al., 2001; Rouge-Pont et al., 2002).
For the most part, the differences seen in this study comparing the WT and βarr2-KO mice were only observed when morphine, but not cocaine, was administered. The removal of βarr2 had no apparent physiological effect in response to cocaine, as evidenced by the minimal alterations of the behavioral and neurochemical effects observed in the βarr2-KO mice. In addition, the robust sensitization of locomotor-stimulating effects of both chronic morphine and cocaine indirectly suggest that dopamine receptor-mediated signaling may be unchanged in the βarr2-KO mice. To address the question of why removing the βarr2 regulatory element may not effect all GPCRs, an attractive hypothesis is that the complement of the other isoform of βarrestin, βarrestin-1, is sufficient to regulate the dopamine receptors in the absence of βarr2. It has been shown in transfected cell culture systems that both D1 and D2 dopamine receptors will interact with both types of βarrestins (Oakley et al., 2000; Kim et al., 2001). The ability of the complement of βarrestin-1 to suffice for the regulation of most GPCRs may become more apparent as more agonists are tested in the βarr2-KO mice. For example, treatment with the α2 adrenergic receptor agonist clonidine does not reveal differences in antinociception between WT and βarr2-KO mice (L. M. Bohn, unpublished observations). Moreover, these observations highlight how the specificity of regulation of physiological processes may be established in the organism.
Together, these finding indicate that the loss of βarr2 leads to increases in morphine-induced striatal dopamine release, intact locomotor sensitization to morphine and cocaine, as well as enhanced conditioned place preference when morphine, but not cocaine, is the challenging drug. Considering the enhanced morphine-induced antinociception reported previously in the βarr2-KO mice and its correlation with enhanced μOR receptor signaling, it would appear that the loss of βarr2 leads to greater morphine reinforcement that is related to increases in morphine-induced dopaminergic activation and not necessarily attributable to direct alterations in dopaminergic signaling.
Footnotes
This work was supported by National Institutes of Health-National Institute on Drug Abuse Grants DA-14600 (L.M.B.) and DA-13115 (M.G.C.). M.G.C. and R.J.L. are Investigators of the Howard Hughes Medical Institute. We thank S. Suter and J. T. Camp for care and genotyping of the mouse colony.
Correspondence should be addressed to Dr. Marc G. Caron, Duke University Medical Center, Box 3287, Room 487, Clinical and Research Laboratories, Research Drive, Durham, NC 27710. E-mail: m.caron@cellbio.duke.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310265-09$15.00/0
References
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT ( 1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286: 2495-2498. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG ( 2000) Muopioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408: 720-723. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG ( 2002) Differential mechanisms of morphine antinociceptive tolerance revealed in βarrestin-2 knock-out mice. J Neurosci 22: 10494-10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brase DA, Loh HH, Way EL ( 1977) Comparison of the effects of morphine on locomotor activity, analgesia and primary and protracted physical dependence in six mouse strains. J Pharmacol Exp Ther 201: 368-374. [PubMed] [Google Scholar]
- Chefer VI, Shippenberg TS ( 2002) Changes in basal and cocaine-evoked extracellular dopamine uptake and release in the rat nucleus accumbens during early abstinence from cocaine: quantitative determination under transient conditions. Neuroscience 112: 907-919. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Shippenberg TS ( 2002) Neural systems underlying opiate addiction. J Neurosci 22: 3321-3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A ( 1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, North RA ( 1992) Neurobiology of opiate abuse. Trends Pharmacol Sci 13: 185-193. [DOI] [PubMed] [Google Scholar]
- Elmer GI, Pieper JO, Rubinstein M, Low MJ, Grandy DK, Wise RA ( 2002) Failure of intravenous morphine to serve as an effective instrumental reinforcer in dopamine D2 receptor knock-out mice. J Neurosci 22: RC224(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS, Zhang J, Barak LS, Caron MG ( 1998) Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci 62: 1561-1565. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G ( 1997) The mouse brain in stereotaxic coordinates. San Diego: Academic.
- Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT ( 1999a) Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 24: 1029-1036. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG ( 1999b) Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science 283: 397-401. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Bohn LM, Caron MG ( 2001) Glutamatergic modulation of hyperactivity in mice lacking the dopamine transporter. Proc Natl Acad Sci USA 98: 11047-11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT ( 2003) Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron 38: 291-303. [DOI] [PubMed] [Google Scholar]
- He M, Shippenberg TS ( 2000) Strain differences in basal and cocaine-evoked dopamine dynamics in mouse striatum. J Pharmacol Exp Ther 293: 121-127. [PubMed] [Google Scholar]
- Ise Y, Katayama S, Hirano M, Aoki T, Narita M, Suzuki T ( 2001) Effects of fluvoxamine on morphine-induced inhibition of gastrointestinal transit, antinociception and hyperlocomotion in mice. Neurosci Lett 299: 29-32. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA ( 1992) Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci 12: 483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Widerlov E, Stanley D, Breese G, Prange AJ Jr ( 1983) Enkephalin action on the mesolimbic system: a dopamine-dependent and a dopamine-independent increase in locomotor activity. J Pharmacol Exp Ther 227: 229-237. [PubMed] [Google Scholar]
- Kieffer BL ( 1999) Opioids: first lessons from knockout mice. Trends Pharmacol Sci 20: 19-26. [DOI] [PubMed] [Google Scholar]
- Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG ( 2001) Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem 276: 37409-37414. [DOI] [PubMed] [Google Scholar]
- Koob GF ( 1992) Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci 13: 177-184. [DOI] [PubMed] [Google Scholar]
- Koob GF, Nestler EJ ( 1997) The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci 9: 482-497. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ ( 1998) G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem 273: 18677-18680. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E ( 1997) Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature 388: 586-589. [DOI] [PubMed] [Google Scholar]
- Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O ( 2000) Cocaine, but not morphine, induces conditioned place preference and sensitization to locomotor responses in CB1 knockout mice. Eur J Neurosci 12: 4038-4046. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL ( 1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383: 819-823. [DOI] [PubMed] [Google Scholar]
- Mello NK, Negus SS ( 1996) Preclinical evaluation of pharmacotherapies for treatment of cocaine and opioid abuse using drug self-administration procedures. Neuropsychopharmacology 14: 375-424. [DOI] [PubMed] [Google Scholar]
- Miner LL ( 1997) Cocaine reward and locomotor activity in C57BL/6J and 129/SvJ inbred mice and their F1 cross. Pharmacol Biochem Behav 58: 25-30. [DOI] [PubMed] [Google Scholar]
- Morari M, Sbrenna S, Marti M, O'Connor WT, Bianchi C, Fuxe K, Beani L ( 1998) Evidence for a striatal NMDA receptor modulation of nigral glutamate release. A dual probe microdialysis study in the awake freely moving rat. Eur J Neurosci 10: 1716-1722. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lam HA, Maidment NT ( 2001) A comparison of morphine-induced locomotor activity and mesolimbic dopamine release in C57BL6, 129Sv and DBA2 mice. J Neurochem 79: 626-635. [DOI] [PubMed] [Google Scholar]
- Nestler EJ ( 2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Damsma G, Wenkstern D, Fibiger HC ( 1990) In vivo characterization of locally applied dopamine uptake inhibitors by striatal microdialysis. Synapse 6: 106-112. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS ( 2000) Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275: 17201-17210. [DOI] [PubMed] [Google Scholar]
- Oliverio A, Castellano C, Eleftheriou BE ( 1975) Morphine sensitivity and tolerance: a genetic investigation in the mouse. Psychopharmacologia 42: 219-224. [DOI] [PubMed] [Google Scholar]
- Perry SJ, Lefkowitz RJ ( 2002) Arresting developments in heptahelical receptor signaling and regulation. Trends Cell Biol 12: 130-138. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Di Chiara G ( 1995) Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci USA 92: 12304-12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC ( 2000) The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95 [Suppl 2]: S91-S117. [DOI] [PubMed] [Google Scholar]
- Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG ( 1998) Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci 1: 132-137. [DOI] [PubMed] [Google Scholar]
- Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E ( 2002) Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. J Neurosci 22: 3293-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Bals-Kubik R, Herz A ( 1993) Examination of the neurochemical substrates mediating the motivational effects of opioids: role of the mesolimbic dopamine system and D-1 vs. D-2 dopamine receptors. J Pharmacol Exp Ther 265: 53-59. [PubMed] [Google Scholar]
- Shoaib M, Spanagel R, Stohr T, Shippenberg TS ( 1995) Strain differences in the rewarding and dopamine-releasing effects of morphine in rats. Psychopharmacology (Berl) 117: 240-247. [DOI] [PubMed] [Google Scholar]
- Sills TL, Fletcher PJ ( 1997) Fluoxetine attenuates morphine-induced locomotion and blocks morphine-sensitization. Eur J Pharmacol 337: 161-164. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR ( 1997) Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA 94: 1544-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS ( 1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA 89: 2046-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy C, Gonon F, Roubert C, Fauchey V, Jaber M, Caron MG, Roques BP, Hamon M, Betancur C, Maldonado R, Giros B ( 2000) Increased rewarding properties of morphine in dopamine-transporter knockout mice. Eur J Neurosci 12: 1827-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Auerbach SB ( 1994) Increased extracellular serotonin in rat brain after systemic or intraraphe administration of morphine. J Neurochem 63: 517-524. [DOI] [PubMed] [Google Scholar]
- Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG ( 1997) Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron 19: 1285-1296. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M ( 1998) Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad Sci USA 95: 9914-9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ, Kalivas PW ( 1998) Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend 51: 141-153. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA ( 1987) A psychomotor stimulant theory of addiction. Psychol Rev 94: 469-492. [PubMed] [Google Scholar]
- Wise RA, Rompre PP ( 1989) Brain dopamine and reward. Annu Rev Psychol 40: 191-225. [DOI] [PubMed] [Google Scholar]
- Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang YM, Caron MG ( 2000) Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci 3: 465-471. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG ( 1998) Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA 95: 7157-7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mantsch JR, Schlussman SD, Ho A, Kreek MJ ( 2002) Conditioned place preference after single doses or “binge” cocaine in C57BL/6J and 129/J mice. Pharmacol Biochem Behav 73: 655-662. [DOI] [PubMed] [Google Scholar]