

Abstract
We have investigated the potential of human pluripotent cells to restore function in rats paralyzed with a virus-induced motor neuronopathy. Cells derived from embryonic germ cells, termed embryoid body-derived (EBD) cells, introduced into the CSF were distributed extensively over the rostrocaudal length of the spinal cord and migrated into the spinal cord parenchyma in paralyzed, but not uninjured, animals. Some of the transplanted human cells expressed the neuroglial progenitor marker nestin, whereas others expressed immunohistochemical markers characteristic of astrocytes or mature neurons. Rare transplanted cells developed immunoreactivity to choline acetyltransferase (ChAT) and sent axons into the sciatic nerve as detected by retrograde labeling. Paralyzed animals transplanted with EBD cells partially recovered motor function 12 and 24 weeks after transplantation, whereas control animals remained paralyzed. Semi-quantitative analysis revealed that the efficiency of neuronal differentiation and extension of neurites could not account for the functional recovery. Rather, transplanted EBD cells protected host neurons from death and facilitated reafferentation of motor neuron cell bodies. In vitro, EBD cells secrete transforming growth factor-α (TGF-α) and brain-derived neurotrophic factor (BDNF). Neutralizing antibodies to TGF-α and to BDNF abrogated the ability of EBD-conditioned media to sustain motor neuron survival in culture, whereas neutralizing antibodies to BDNF eliminated the axonal outgrowth from spinal organotypics observed with direct coculture of EBD cells. We conclude that cells derived from human pluripotent stem cells have the capacity to restore neurologic function in animals with diffuse motor neuron disease via enhancement of host neuron survival and function.
Keywords: motor neuron disease, Sindbis virus, stem cells, embryonic germ cell, trophic factors, embryoid body
Introduction
Pluripotent stem cells represent an attractive strategy for treating neurologic diseases, and early successes in animal models have generated optimism for their use to restore or maintain function in humans. These cells have been shown to replace damaged or lost neural cells (Liu et al., 1999a; Yandava et al., 1999; Kondziolka et al., 2000), provide enzymatic machinery to correct metabolic defects (Snyder et al., 1995; Lacorazza et al., 1996), deliver trophic or tropic support to host neural cells (Liu et al., 1999a), provide cellular bridges between neural populations disconnected by disease such as spinal cord injury (Onifer et al., 1997; McDonald et al., 1999), and deliver toxic substances to combat neoplastic processes (Benedetti et al., 2000).
Human pluripotent stem cells can be derived from the inner cell mass of blastocysts (embryonic stem cells) or from the primordial germ cells of the gonadal ridge (embryonic germ cells; EG cells) (Shamblott et al., 1998). EG cells differentiate by forming complex three-dimensional cell aggregates termed embryoid bodies (EBs), which may contain pluripotent cells and cells in various stages of differentiation. Cultured cell lines derived from EBs, embryoid body-derived cells (EBDs), have been expanded in semi-selective growth environments, proliferate robustly with a normal karyotype, and have been characterized for expression patterns (Shamblott et al., 2001). These cell lines express a wide range of lineage-specific markers, and some have neurally biased expression patterns. One of the cell lines (SDEC) that express relatively high levels of nestin and other neural markers was chosen for the current study.
Neuroadapted Sindbis virus (NSV) is a neuronotropic, single-stranded RNA virus that specifically targets motor neurons in the spinal cord, causing hindlimb paralysis in mice (Jackson et al., 1988; Griffin et al., 1994; Kerr et al., 2002). Although NSV is cleared from animals within 7–10 d after infection, it triggers progressive motor neuron death, resulting in permanent hindlimb paralysis. Here we have adapted this model of motor neuron degeneration to rats and have investigated whether human EBD cells could restore function to paralyzed animals with virus-induced motor neuron disease. Transplantation of EBD cells shortly after the onset of paralysis resulted in partial restoration of function at 3 and 6 months, whereas control animals remained paralyzed. Although EBD cells acquired the immunohistochemical features of mature neurons, they did so inefficiently, and the recovery of function was independent of the reconstitution of motor neurons. Rather, EBD cells protected host motor neurons from death and enhanced the recovery of synaptic complement on motor neuron soma, likely because of the secretion of trophic factors including transforming growth factor-α (TGF-α) and brain-derived neurotrophic factor (BDNF).
Materials and Methods
Sindbis virus. NSV was passaged into 3- to 4-week-old Lewis rats via intracranial inoculation to generate a strain of Sindbis virus (rat-adapted NSV, raNSV) that caused paralysis in rats. Rats were inoculated initially with 1000 plaque-forming units (PFU) of NSV while subsequent animals were inoculated with 1000 PFU isolated from brain lysate of the previous passage. Initial titers were ∼1–2 × 10 4 PFU/100 mg of tissue. After passage 4 the viral titers increased to ∼10 6 PFU/100 mg of tissue, and the animals developed an encephalomyelitis characterized by hindlimb paralysis. By passage 6 virtually all rats developed some degree of paralysis, although mortality from the encephalomyelitis remained low at ∼5%. Virus was cleared in large part from these animals by day 8–9 (maximal titers of 10 1 PFU/100 mg of tissue). Viral lysate was generated from passage 6 rats and was used for all subsequent transplantation experiments.
Quantitation of neuronal death. Animals were anesthetized by intraperitoneal injection of 2 ml of 2.5% Avertin and were killed by intracardiac perfusion of saline, followed by fresh 4% paraformaldehyde (PFA) in PBS. The spinal cord and all lumbar roots and associated nerves were dissected en bloc. For ventral root counting, tracing the sciatic nerve into the spinal roots identified ventral roots L3, L4, and L5. A 3 mm segment of the L4 ventral root was isolated midway between the spinal cord and dorsal root ganglion, postfixed in 5% glutaraldehyde for 24 hr, and stored in PBS. Ventral roots from three animals at each time point were examined. These root segments were osmicated and embedded in plastic; the 1 μm sections were stained with toluidine blue. The numbers of axons within each root segment were determined by standard stereology techniques at light level; all profiles with a distinct ring of compact myelin were counted by using a Bioquant Morphometric system (R & M Biometrics, Memphis, TN)
Human embryonic germ-derived culture. Human pluripotent embryonic germ (EG) culture SD was generated from primordial germ cells as described (Shamblott et al., 1998). Embryoid bodies formed in the presence of leukemia inhibitory factor were disaggregated with Collagenase/dispase (Roche, Indianapolis, IN), and the constituent cells (EBD cells) were grown in medium containing 5% serum, bFGF (basic fibroblast growth factor), EGF (epidermal growth factor), VEGF (vascular endothelial growth factor), and IGF1 (insulin-like growth factor 1) (Clonetics EGM2MV, Cambrex, East Rutherford, NJ) and a collagen 1I biomatrix (the bold letters indicate the derivation of the name of the particular EBD culture used in the present study, SDEC). Like many of the EBD cultures grown in this environment, SDEC has a high proliferative capacity (>70 population doublings) with a normal, genetically unmodified karyotype. SDEC was selected from a collection of ∼100 human EBD lines and cultures on the basis of the relatively high levels in vitro of mRNA, protein expression of neural precursor and neuronal and glial markers such as nestin, and neurofilament light chain as well as its high proliferative capacity (Shetty et al., 1994; Shamblott et al., 2001). Then the cells were washed four times in PBS, trypsinized, washed an additional three times, and resuspended in PBS at a cellular density of 30,000 cells/μl.
Behavioral analysis and statistical analyses. The 21 point Basso, Beattie, and Bresnahan (BBB) scale was used as an outcome measure for hindlimb recovery in treated rats. Because of the nonparametric nature of the data (as determined by using tests of normality and skewed distribution of each treatment group at each time point), nonparametric tests were used to analyze the BBB scores. Nonparametric equivalent tests of ANOVA and repeated measures ANOVA were used to increase the robustness of the results. The Kruskal—Wallis test was performed to analyze BBB scores across groups at each time point, and Friedman's nonparametric repeated measures comparison was used to analyze differences in BBB scores across time within a treatment group. These tests were used because they make no assumptions about the distribution of the data such as normality. Change in time to righting at 12 and 24 weeks between the control and EBD groups also was analyzed with the Kruskal—Wallis test. Bonferroni correction was used also to test for statistical significance at the 5% level after adjustment that was based on the number of groups that were compared.
Hindlimb grip strength testing was done by using a Chatillon CE digital force gauge (model DFIS 2, Ametek, Paoli, PA). Readings were taken in T-peak and measured in pounds of force. The animals were supported by the examiner under their forelimbs. Then the hindlimbs were lowered to the grip bar until the animal attempted to hold onto the bar. The animal was moved away from the bar slowly and was force measured if the animal exerted active force against that movement. Each animal was given three trials per examining period. The mean digital force gauge (DFG) values were compared with the Kruskal—Wallis test among BHK, human fibroblast-transplanted, and EBD-transplanted animals at 12 and 24 weeks after transplantation. Change in time to righting at 12 and 24 weeks between the control and EBD groups also was analyzed with the Kruskal—Wallis test. Bonferroni correction was used to test for statistical significance at the 5% level after adjustment that was based on the number of groups that were compared.
Animal care. At 8 d after raNSV infection the rats were anesthetized, and a lumbar cannula was inserted caudal to the L6 nerve root. A laminotomy was performed, and a heparinized 26 gauge cannula was inserted with the tip positioned 5 mm rostral to the laminotomy. The cannula was secured with Loctite bone cement (Henkel Loctite, Louisville, KY). Cells were administered in a final volume of 10 μl in PBS, followed by 5 μl of PBS to wash the cells through the cannula. All rats were given cyclosporine (Calbiochem, San Diego, CA) at 10 mg/kg per day on the day before surgery/transplantation and then every day after surgery.
Immunohistochemistry. For quantitation of EBD engraftment five NSV-paralyzed and five uninfected animals that had been transplanted were killed as described above at 1 month after transplantation. After perfusion with 4% PFA, spinal columns were dissected and postfixed for 1–2 hr in 4% PFA in PBS. Spinal cords were cryoprotected, and every 50th 25 μm section spanning the entire spinal column was collected on glass slides. Slides were permeabilized in 0.1–0.4% Triton X-100, blocked with normal serum, and then incubated overnight with primary antibodies. Presented photomicrographs in Figures 1, 2, 3 and 5 are from the lumbar enlargement of the spinal cord. Quantitation of surviving human EBD cells and colocalization of human cells with neural antigens were performed with human-specific antibodies [polyclonal anti-SOD1 (Pardo et al., 1995), anti-nuclear matrix protein 1 (Numa; NA08, Oncogene Research Products, San Diego, CA), or anti-human nuclei (mAb 1281, Chemicon, Temecula, CA)] and a variety of other antibodies to neural antigens [CNPase (clone 11-5B, Sigma, St. Louis, MO), β3-tubulin (Research Diagnostics, Flanders, NJ), glial fibrillary acidic protein (GFAP; Ab5804, Chemicon), choline acetyltransferase (ChAT; Ab5042P, Chemicon), and MAP2 (Ab5622, Chemicon)]. Staining with synaptophysin (mAb 5358, Chemicon) in conjunction with Nissl red (1:100; Molecular Probes, Eugene, OR) was performed at 6 months after transplantation. The possibility of cell fusion was assessed by dual immunohistochemistry with human-specific SOD-1 antibody and MO-1 antibody (ATCC, Manassas, VA), which are specific for rat neurons. Bromodeoxyuridine (BrdU) staining was performed with a sheep polyclonal antibody (Research Diagnostics). Tissue was fixed in cold methanol for 30 min and immersed in 0.007N NaOH for 10–15 sec, followed by permeabilization/blocking as above. Unbiased counting was used to score the number and distribution of Numa + cells in the spinal cord with an optical dissector procedure (Sterio, 1984; Kuhn et al., 1997; West, 1999; Benraiss et al., 2001). We sampled 60 sections per animal for both experimentals and controls. For each, every 50th section was stained at 25 μm intervals. The first section of each axial series was chosen randomly from the most caudal end of the spinal cord.
Figure 1.
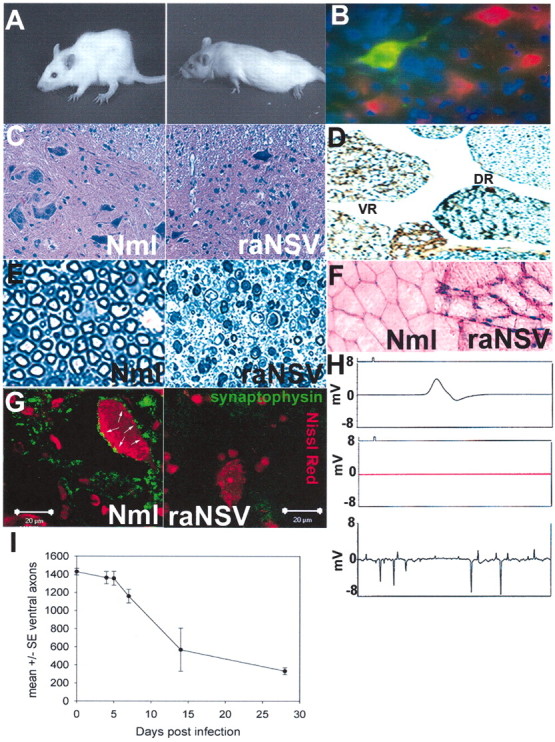
Neuroadapted Sindbis virus causes progressive lower motor neuron injury in rats. A, Progressive kyphoscoliosis (left) and then hindlimb paralysis (right) develop in rats after intracranial infection of 1000 PFU of rat-adapted Sindbis virus (raNSV). B—E, Effect of raNSV on motor neurons. Rats were given intracranial infections of NSV encoding green fluorescent protein (raNSVGFP). At 3 d later the spinal cord sections were costained with Nissl red and Hoechst 33342. B, Note the prominent green fluorescence in spinal motor neurons. C, Lumbar enlargement of spinal cord 28 d after raNSV infection. Note a significant reduction in motor neurons in paralyzed rat (right) as compared with nonparalyzed rat (left). D, Silver-stained sections of lumbar spinal cord 28 d after raNSV infection. Note the selective depletion of axons in the ventral root (VR) in a paralyzed rat and preservation of dorsal roots(DR).E, Toluidine blue-stained sections (1 μm) of the L4 ventral root from uninfected rat (left) and 28 d after infection (right) show prominent Wallerian degeneration. F, Muscle biopsy of paralyzed (right) and control (left) rat. Note neurogenic atrophy with angulated and degenerating fibers in the paralyzed rat, with marked variation in fiber size. G, Loss of presynaptic input to surviving motor neurons after raNSV infection. Note the reduction in synaptophysin contacts (green; arrows reflect synaptic contacts) onto motor neurons 28 d after infection (right) as compared with uninfected motor neurons. H, Electrophysiologic recordings of nonparalyzed and paralyzed rats. Shown are normal compound motor unit potentials in nonparalyzed rat (5.12 mV; top), absent motor action potential in severely paralyzed rat (middle), and positive sharp waves on EMG in raNSV-paralyzed rat (bottom). I, Temporal course of motor neuron loss after raNSV infection as defined by quantitation of ventral L4 root axons. Stereologic examination of semithin sections was performed. Sections from three animals were examined at each time point. Data are presented as the mean at each time point ± SE, with the y-axis representing number of ventral axons.
Figure 2.
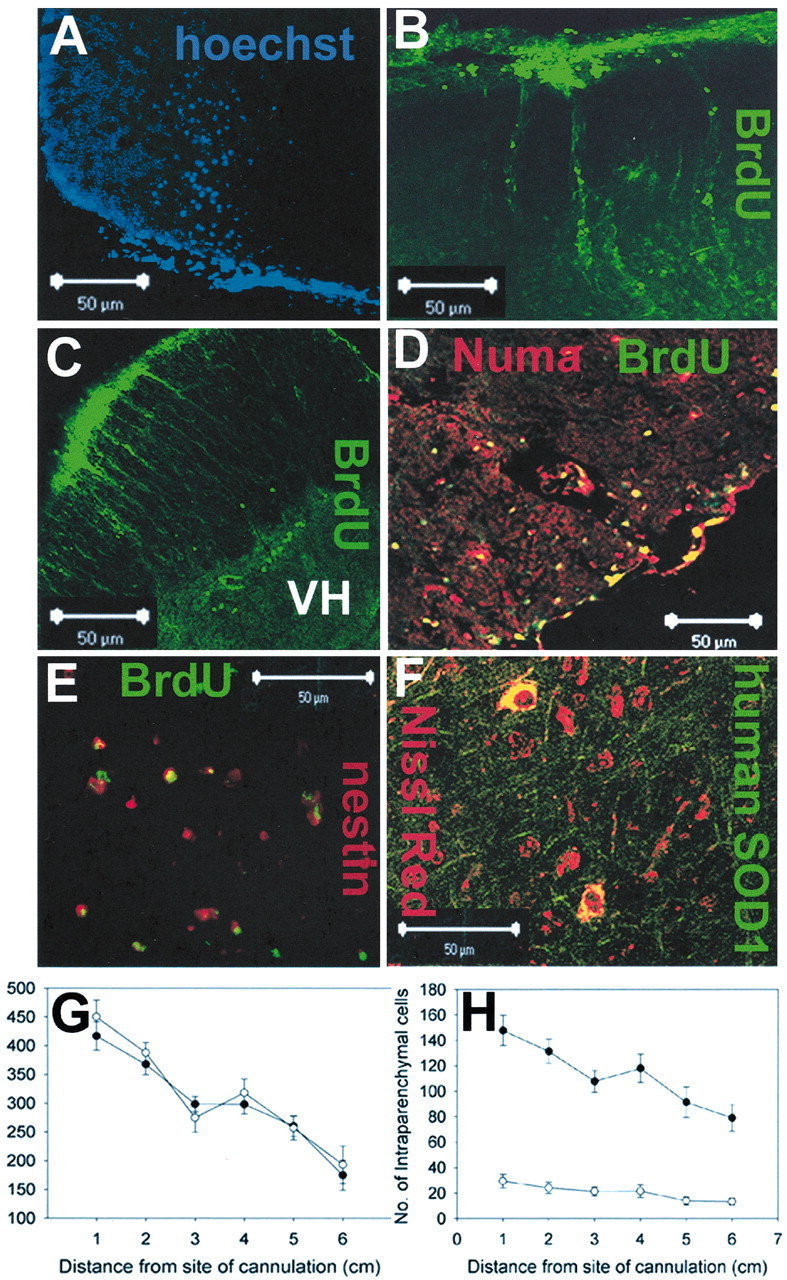
EBD cells introduced into the lumbar CSF migrate into the spinal cord and widely distribute over the length of the spinal cord. A—F, Histologic analysis of rats 4 weeks after implantation revealed intraparenchymal engraftment of EBD cells with multiple distinct markers. Cells prelabeled with Hoechst (A) or BrdU (B—E) showed migration into the spinal cord from the subarachnoid space. Immunohistochemical analysis of spinal cords with antisera specific to human Numa (D) or human SOD1 (F) confirmed migration into the spinal cord. G, Quantitation of the rostrocaudal distribution of all surviving cells (meningeal and intraspinal) at 4 weeks after implantation relative to the cannula placement. Every 50th axial spinal cord section was examined for the number of cells that exhibited human nuclear matrix antigen positivity. Five nonparalyzed EBD-transplanted animals and five paralyzed EBD-transplanted animals were analyzed. EBD cells were detected at all levels of the neuraxis; EBD cell survival was similar in raNSV-paralyzed (filled circles) and in nonparalyzed animals (open circles). H, When only intraparenchymal EBD cells (Numa +) were counted, a greater migration of cells into the spinal cord was seen in paralyzed animals (filled circles) as compared with nonparalyzed (open circles) animals (p = 0.001). In both graphs, data are presented at each distance from the cannulation as mean ± SE.
Figure 3.

Some transplanted EBD cells acquire immunohistochemical markers of neural cells. At 28 d after implantation spinal cord sections were generated and examined by immunohistochemistry to determine whether the implanted EBD cells expressed neuronal markers such as GFAP (A), β3-tubulin (B), MAP2 (C), or ChAT (D). Orthogonal images confirm the colocalization of GFAP, β3-tubulin, and ChAT with human-specific nuclear matrix antigen staining. E,F, Rare retrograde labeling of prelabeled EBD cells from the sciatic nerve. E, Rare Hoechst-prelabeled EBD cells (blue) in the lumbar gray matter that were retrogradely labeled from the sciatic nerve (green) and were strongly Nissl+ (red). Retrograde labeling (green) of EBD cells in the field (top left) reveals two large and one smaller cell in the ventral gray matter. Nissl red staining (top right) reveals four Nissl+ cells in the field, including the three that were retrogradely labeled. Hoechst staining (bottomleft) reveals that one of the retrogradely labeled Nissl+ cells is EBD in origin, whereas several additional EBD cells were not retrogradely labeled and one additional EBD cell acquired strong Nissl+ staining (but did not retrogradely label). The asterisk denotes an EBD-derived cell that retrogradely labeled the sciatic nerve. F, Numa+ EBD cells (red) that retrogradely label from the sciatic nerve (green) with orthogonal views confirming colocalization. Note the nonretrogradely labeled EBD cell (red only, top right) and the host cell that retrogradely labeled (green only). Control animals that had a second sciatic nerve transection more proximal to the original site to prevent axonal transport of the chemical never exhibited any intraspinal label.
Figure 5.
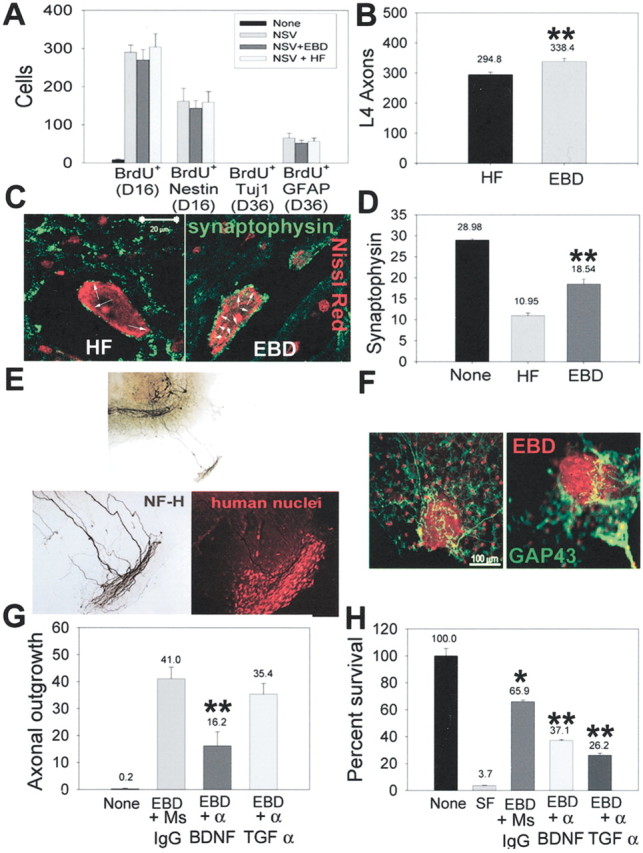
EBD transplantation facilitates host motor neuron survival and axonal reafferentation of host motor neurons. A, EBD transplantation did not increase the proliferation or differentiation of endogenous spinal neural stem cells. Rats were given BrdU for 3d from days 12–15 after infection and then were killed at either day 16 or day 36 after infection. Immunohistochemical analysis determined the frequency of BrdU+ cells per section or the colocalization of BrdU with nestin, β-tubulin, or GFAP. Although raNSV induced neural stem cell proliferation within the spinal cord, no new neurons could be detected at day 36 with any of the transplantation paradigms. B, Quantitation of the number of L4 ventral root axons at 6 months after transplantation shows a significant increase in surviving axons in transplanted animals as compared with control-transplanted animals. L4 nerve roots from 15 animals in each group were analyzed; the comparisons were made with a Student's t test (p < 0.03). C, Representative photomicrographs of the synaptophysin complement on motor neurons at 6 months after transplantation in HF-transplanted (left) and EBD-transplanted (right) rats. D, The number of synaptophysin contacts per motor neuron cell body was enhanced in transplanted animals at 6 months: 50 motor neurons were counted in each of five animals per group. Motor neurons in HF-transplanted rats had 10.95 ± 0.65 synaptophysin contacts per soma, whereas EBD-transplanted rats had 18.54 ± 1.14 synaptophysin contacts per soma. Comparisons between HF- and EBD-transplanted animals were performed by the Mann—Whitney test (p < 0.02). E, F, EBD cells exhibit tropic properties for host axons in a spinal cord organotypic model. EBD cells were placed outside the organotypic margin and cultured for an additional 2 weeks. E, In the presence of EBD cells the host axons were stimulated to send out processes from the organotypic margin for up to 1.2 mm after 14 d. The bottom panels represent a higher power view of axonal growth (left) into the cluster of human nuclei-labeled EBD cells (right). F, Stimulation of GAP 43 expression (green) by host axons in response to EBD cells (red). G, Inhibition of EBD-stimulated axonal growth in organotypic cultures by neutralizing antibodies to BDNF, but not TGF-α. Spinal cord organotypic cultures were cocultured with EBD cells as above, with neutralizing antibodies to BDNF, TGF-α, or control antibody. The scores are depicted as the number of Smi32+ axons that have grown out of the margin of the organotypic culture per slice (p = 0.007). H, Conditioned medium from EBD cells allowed for survival of cultured motor neurons in the absence of exogenous growth factors (compare serum-free medium, lane two, with EBD-conditioned medium, lane 3) (*p = 0.001 compared with lane 2). The addition of neutralizing antibodies to either BDNF or TGF-α partially abolished the ability of EBD-conditioned medium to support motor neuron survival. **p < 0.001 compared with lane 3.
EBD cells were colocalized with neural antigens by using two-color confocal imaging with a Zeiss LSM510 microscope (Oberkochen, Germany). Images were acquired in both red and green emission channels by using an argon—krypton laser with single-channel, line-switching mode. Images were acquired as series of single 0.9 μm optical sections and viewed as z-dimension reconstructions with orthogonal views in the x—z and y—z planes.
For retrograde labeling the animals were anesthetized, and one sciatic nerve was transected at the location at which the L4—L5 roots join. Alexa Fluor-conjugated dextran (Molecular Probes) crystals (∼10 mg) were placed at the proximal stump (∼15 mm from the spinal cord). Animals were allowed to recover for 48 hr and then were killed.
Organotypic spinal cord culture. Organotypic spinal cord cultures were prepared from lumbar spinal cord slices obtained from 8-d-old rat pups (Rothstein et al., 1993). Briefly, cords were harvested under sterile conditions and cut into 350 μm sections. Sections were transferred to Millipore (Bedford, MA) Millicell-CM porous membranes and maintained in an incubation medium (50% MEM, 25 mm HEPES, 25% heat-inactivated horse serum, and 25% HBSS supplemented with 25.6 mg/ml d-glucose and 2 mm glutamine at a final pH of 7.2). For immunohistochemistry the organotypic cultures were fixed with 4% PFA in 0.1 m phosphate buffer, pH 7.4, for 30 min at room temperature. After being blocked with 5% normal horse serum in 0.2% Triton X-100 for 1 hr, the cultures were incubated overnight at 4°C with the following primary antibodies: human nuclei (Chemicon), SMI-32 (Sternberger Monoclonals, Lutherville, MD), and growth-associated protein 43 (GAP43; Chemicon). For immunofluorescence the sections were incubated for 1 hr in secondary antibody (Texas Red anti-mouse or fluorescein anti-rabbit; Vector Laboratories, Burlingame, CA); after this the cultures were washed and mounted with Gel/Mount solution (Biomeda, Foster City, CA). The SMI-32 antibody was used with a biotinylated anti-mouse secondary antibody (Vector Laboratories), and after incubation with ABC reagents (Vector Laboratories) the diaminobenzidine reaction was used for color development.
Endogenous neurogenesis. Rats were administered 100 mg/kg BrdU (Sigma) intraperitoneally every hour for 36 hr from days 12–15 after raNSV infection. Animals then were killed at day 16 or 36 for analysis of BrdU + (day 16), BrdU +/nestin + (day 16), BrdU +/Tuj1 + (day 36), or BrdU +/GFAP + cells (day 36) as above.
Motor neuron culture. Motor neurons were cultured as previously described (Vandenberghe et al., 1998). In brief, ventral spinal cords were dissected from 14-d-old Wistar rat embryos in HBSS, cut into 1 mm pieces, and digested in 0.05% trypsin in HBSS for 15 min at 37°C. A motor neuron-enriched neuronal population was purified from the ventral spinal cord by centrifugation on a 6.5% metrizamide cushion and then immunopanned by using the 192 mouse IgG attached to a dish previously coated with anti-mouse antibodies. The motor neuron-enriched suspension was cultured on a glial feeder layer that had been preestablished on 18-mm-round glass coverslips coated with poly-l-ornithine and laminin. The culture medium consisted of L15 supplemented with 0.2% sodium bicarbonate, 3.6 mg/ml glucose, 20 nm progesterone, 5 μg/ml insulin, 0.1 mm putrescine, 0.1 mg/ml conalbumin, 30 nm sodium selenite, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2% horse serum. The cultures were kept in a 7% CO2-humidified incubator at 37°C.
Then 12 hr later the L15 medium with sodium bicarbonate, progesterone, and glucose or the same medium previously conditioned on confluent EBD cells for 24 hr was added to the motor neuron culture. There was no deleterious effect of culturing EBD cells in this medium for periods of time <36 hr. At 24 hr after the addition of the conditioned medium the cells were fixed in 4% paraformaldehyde and immunolabeled with anti-ChAT antibody. The number of ChAT + cells in 20 high power fields was scored.
Enzyme-linked immunosorbent assay. BDNF, glial cell line-derived neurotrophic factor (GDNF), neurotrophin (NT) 3, NT4, and nerve growth factor (NGF) secretion by EBD cells was assessed in EBD-conditioned medium by using E-max ImmunoAssay System Kits for these neurotrophic factors and by following the manufacturer's recommendations (Promega, Madison, WI) in an antibody sandwich format. In all cases 10 5 EBD cells were placed in a 60 mm tissue culture dish with 1 ml of medium. Medium was added and 24 hr later was removed for ELISA analysis. TGF-α ELISA was performed according to the manufacturer's instructions (Calbiochem) with slight modifications (Geng et al., 2000).
Neutralizing antibodies. Anti-TGF-α antibody (TGF-A clone; Research Diagnostics), anti-BDNF (AB-1, GF35L; Novagen), and control mouse IgG (Sigma) were used at 0.5 μg/ml.
Results
Rat model of motor neuron degeneration
We developed a model in rats characterized by widespread lower motor neuron loss. NSV is a neuronotropic virus that causes an acute encephalomyelitis in mice when inoculated intracranially (Jackson et al., 1987; Lustig et al., 1988). Animals develop progressive weakness and atrophy with extensive spinal motor neuron loss, resulting in permanent paralysis (Jackson et al., 1987; Griffin et al., 1994; Kerr et al., 2002). By serial passage of NSV into rat brains, we generated an NSV derivative (raNSV) that induced a similar pathology in infected rats. After intracranial inoculation of raNSV the rats developed progressive kyphoscoliosis (Fig. 1A, left), indicating thoracic muscle weakness and hindlimb paralysis beginning 4–6 d after infection (Fig. 1A, right). raNSV has a relative tropism for motor neurons, as shown by infection with an NSV clone encoding green fluorescent protein (Fig. 1B), which explains in part the selective loss of this neuronal population. At 10 d after infection with raNSV the surviving animals typically have reached the maximal degree of paralysis and in large part have cleared virus from the spinal cord (viral titers <101 PFU/100 mg of tissue; our unpublished data).
At 28 d after raNSV infection a marked loss of ventral motor neurons was observed in the lumbar enlargement (compare section from nonparalyzed rat, left, with section from paralyzed rat, right; Fig. 1C). Ventral nerve roots (VR) showed fulminant degeneration of motor axons (Fig. 1D), whereas dorsal nerve roots (DR) remained mainly intact, indicating the relative specificity of motor neuron death after raNSV infection. Further analysis of the L4 ventral roots revealed Wallerian degeneration, with progressive loss of motor axons at 28 d after infection (Fig. 1E, right) as compared with motor roots from uninfected animals (Fig. 1E, left). Muscle biopsy of paralyzed animals revealed severe neurogenic atrophy with angulated and degenerating fibers (compare nonparalyzed rat, left, with paralyzed rat, right; Fig. 1F) consistent with a lower motor neuron injury. Even surviving motor neurons in the lumbar spinal cord had a markedly diminished synaptic complement on their soma at 28 d after infection, as indicated by diminished synaptophysin staining (compare uninfected, left, with raNSV-infected, right; Fig. 1G). Thus the paralysis of animals may be attributable to both motor neuron loss and to reactive deafferentation of remaining neurons in response to the infection. Electrophysiologic evaluation revealed a marked reduction of compound motor action potential amplitude in the intrinsic foot muscles of paralyzed animals (compare 5.12 mV in a normal animal with no recordable motor action potential in completely paralyzed rats; Fig. 1H) and abnormal fibrillation activity on needle electromyography, further supporting an acute neurogenic etiology for the paralysis (Fig. 1H, bottom). Examination of the temporal features of neural injury revealed progressive loss of axons in the L4 ventral nerve root 2 mm from the spinal cord out to 28 d after infection. There was rapid loss of ventral axons after infection, with ∼50% loss by 8 d after infection (Fig. 1I) and 80% loss by 28 d after infection. Comparable data were found by quantitative analysis of surviving motor neurons in the lumbar enlargement (data not shown), confirming that proximal L4 ventral axon number is an appropriate surrogate marker for neuronal death in this model. Acute motor neuron death during raNSV infection is mediated in part by excitotoxic injury even of noninfected neurons (data not shown), although mechanisms of progressive motor neuron death after viral clearance are unclear. Therefore, raNSV triggers fulminant and progressive motor neuron injury in rat spinal cords, resulting in hindlimb paralysis.
Transplantation with human embryonic germ-derived cells
To investigate whether EBD cells have the capacity to restore neurologic function to raNSV-paralyzed rats, we transplanted 300,000 cells into the spinal fluid of rats via a lumbar cannula 8 d after NSV infection. At 4 weeks after transplantation Hoechstand BrdU-prelabeled EBD cells were found both adherent to the meninges and within the spinal cord (Fig. 2A—E) in transplanted animals, whereas no signal was seen in tissue from animals given prelabeled, killed EBD cells (data not shown). The ability of transplanted EBD cells to survive and migrate into the spinal cord was confirmed by finding cells immunoreactive to human-specific nuclear matrix antigens (Numa; Fig. 2D) and human-specific superoxide dismutase 1 (SOD1; Fig. 2F).
To examine the rostrocaudal distribution of EBD cells, we sampled human-specific cells (defined by immunoreactivity to human-specific nuclear matrix antigen) in every 50th spinal cord section at 4 weeks after transplantation. The number of surviving human Numa+ cells per rat that was adherent to the meninges or within the spinal cord was determined and grouped in 1 cm intervals (Fig. 2G,H, Table 1). Surviving EBD cells were distributed widely over the rostrocaudal length of the spinal cord, with nearly one-half of the cells (48%) 3 cm or greater from the cannulation site. When all surviving human cells were counted (either adherent to the meninges or parenchymal), rostrocaudal distribution and total cellular survival were similar in nonparalyzed (open circles) and raNSV-paralyzed animals (filled circles; Fig. 2G, Table 1). However, the number and percentage of intraparenchymal cells were reduced significantly in nonparalyzed animals (673 ± 61 vs 122 ± 22 per animal; 37 vs 6.5% of the surviving cells; p = 0.001; Fig. 2H, Table 1), suggesting that migration into the spinal cord depended on local cues in paralyzed animals. We estimate that ∼90,000 EBD cells survived to 1 month after transplantation (30% of transplanted cells). In paralyzed animals 37% of cells engrafted within the spinal cord parenchyma, and, therefore, an estimated 33,650 EBD cells survived within the spinal cord at 1 month after transplantation. These studies thus show that EBD cells administered via the lumbar CSF can be found over a wide area of the rostrocaudal axis of the spinal cord and that they can migrate into the spinal cords of rats with acute motor neuron injury.
Table 1.
Distribution of EBD cells after transplantation
|
|
Number (mean ± SEM) of EBD cells per animal |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adherent to meninges
|
Within spinal cord
|
Total cells (either meninges or within spinal cord)
|
|||||||||
| Sections of spinal cord rostral to cannula
|
No NSV
|
+ NSV
|
No NSV
|
+ NSV
|
No NSV
|
+ NSV
|
|||||
| 0-1 cm | 421 ± 25.4 | 269 ± 14.5 | 29 ± 5.4 | 148 ± 11.9 | 450.2 ± 29.3 | 416.8 ± 24.7 | |||||
| 1-2 cm | 364 ± 16.9 | 236 ± 12.8 | 24 ± 4.4 | 131 ± 9.6 | 387.8 ± 18.2 | 367.8 ± 18.4 | |||||
| 2-3 cm | 253 ± 23.8 | 191 ± 7.1 | 21 ± 3.6 | 108 ± 8.5 | 274.8 ± 25.4 | 298.4 ± 13.3 | |||||
| 3-4 cm | 297 ± 21.2 | 180 ± 10.7 | 21 ± 5.0 | 118 ± 11.2 | 318.4 ± 23.9 | 298 ± 16.7 | |||||
| 4-5 cm | 243 ± 18.9 | 169 ± 11.6 | 14 ± 3.2 | 91 ± 12 | 256.8 ± 20.8 | 260.2 ± 18.2 | |||||
| 5-6 cm | 180 ± 30.9 | 96 ± 18.4 | 13 ± 2.7 | 79 ± 10.6 | 193.2 ± 32.6 | 174.8 ± 26 | |||||
| Total
|
1758 ± 124
|
1141 ± 61
|
122.4 ± 21.5
|
673.4 ± 61.0
|
1880 ± 137
|
1814 ± 107
|
|||||
Expression of neuronal markers by EBD cells
To determine the ability of EBD cells to acquire immunohistochemical markers of neural cells, we performed dual-color microscopy of spinal cord sections from transplanted animals at 4 weeks after transplantation. Some of the EBD cells expressed markers characteristic of astrocytes (Fig. 3A), neurons (Fig. 3B,C), or, rarely, cholinergic neurons (ChAT; Fig. 3D). The colocalization of EBD cells with neural antigens was confirmed by generating 0.9 μm z-stack series with orthogonal x—z and y—z images (Fig. 3A,B,D,F). None of the EBD cells expressed the rat-specific epitope identified by the MO-1 antibody (Urakami and Chiu, 1990; Chen et al., 1995) (data not shown), indicating that cell fusion is not associated with the adoption of neural phenotypes (Terada et al., 2002; Ying et al., 2002). Because the frequency of neuronal differentiation was low and not related to the functional recovery (see below), we have not characterized the expression pattern of ChAT+ human-derived neurons further. Therefore, this differentiation may reflect differentiation toward spinal motor neuron or, alternatively, to other cholinergic neuronal phenotypes. To investigate whether engrafted EBD cells sent out processes from the ventral horn into the periphery, we examined retrograde labeling of neurons in the lumbosacral spinal cord of a subset of animals. One sciatic nerve in each animal was transected at the site at which the L4—L5 contributions merge (15 mm from the spinal cord). Crystals of a fluorescently conjugated leptin (Alexa Fluor dextran conjugate) were placed at the proximal stump of the sciatic nerve; then the animal was allowed to recover for 48 hr. This reagent has minimal intercellular transfer and therefore labels only primary axons and cell bodies (Schmued et al., 1990; Vercelli et al., 2000). Animals then were killed and examined for the presence of retrogradely labeled cells within the lumbosacral spinal cord. In transplanted animals the vast majority of prelabeled EBD cells remained small with no retrograde labeling, although, rarely, we detected one to two large EBD cells that retrogradely labeled (Fig. 3E,F). Control animals had a second sciatic nerve transection more proximal to the original site to prevent axonal transport of the chemical and did not exhibit any intraspinal label. In both presented panels (Fig. 3E,F) retrogradely labeled and nonretrogradely labeled EBD cells are seen, confirming the specificity of the label. Interestingly, we did not detect retrograde labeling of EBD cells in regions with no remaining host motor neurons, and the rare retrograde labeling of EBD cells occurred in a relatively spared region with some preservation of host motor neurons in the area (Fig. 3E,F), suggesting that remaining host neurons influence the ability of EBD cells to extend processes.
Examination of the immunohistochemical features of surviving EBD cells at 4 weeks after transplantation revealed that ∼30% of cells immunoreactive for human nuclear antigen (Numa) cells also expressed GFAP (Table 2). Of the surviving human cells 4.41% expressed β3-tubulin, 0.06% expressed ChAT, and 0.36% of cells expressed the oligodendrocyte marker O4+. Approximately 60% of surviving EBD cells expressed none of these markers and thus presumably remained in a somewhat undifferentiated state. Because virtually none of the surviving EBD cells that remained adherent to the meninges expressed neural antigens, we estimate that ∼10,085 human GFAP+ cells, 1483 human β3-tubulin+ cells, and 20 human choline acetyltransferase cells existed in the transplanted spinal cords at 1 month after transplantation.
Table 2.
Differentiation of intraparenchymal cells
|
|
Number of intraparenchymal cells expressing neural markers |
||||||
|---|---|---|---|---|---|---|---|
| NSV (n = 3367 SC cells)
|
No NSV (n = 612 SC cells)
|
||||||
| Immunohistochemical marker
|
Mean ± SE/animal
|
%
|
Mean ± SE/animal
|
%
|
|||
| β3-tubulin+ | 29.8 ± 2.96 | 4.41 | 2.6 ± 0.98 | 2.1 | |||
| ChAT+ | 0.8 ± 0.2 | 0.12 | 0 | 0 | |||
| GFAP+ | 201.8 ± 10.9 | 29.97 | 24.2 ± 2.35 | 19.8 | |||
| CNPase+
|
2.4 ± 1.12
|
0.36
|
0
|
0
|
|||
Functional recovery of paralyzed rats
To determine whether EBD cell engraftment within the spinal cords of paralyzed rats resulted in any functional recovery, we transplanted 15 rats with EBD cells (filled triangles), baby hamster kidney (BHK) cells (open triangles), or 15 human fibroblast (HF) cells (filled circles; Fig. 4A). All animals received the immunosuppressant FK506 daily for the duration of the experiment. After transplantation the animals were coded, housed in mixed cages (control and experimental animals), and scored according to the BBB locomotor score (Basso et al., 1995) by two raters who were blinded to the treatment regimens. The code for the transplantation regimen was not broken until 24 weeks after transplantation. Baseline BBB scores were similar in the three groups (2.05 ± 0.2 for EBD-, 2.24 ± 0.42 for BHK-, and 2.1 ± 0.34 for HF-transplanted animals). None of the three groups of animals exhibited any significant recovery at 4 or 8 weeks after transplantation. By 12 weeks, however, animals in the EBD group showed significant improvement in hindlimb locomotion as compared with control-transplanted animals (6.12 ± 0.79 for EBD vs 2.14 ± 0.36 for BHK and 2.07 ± 0.29 for HF; p = 0.004). There was slight additional recovery at 24 weeks exclusively in the EBD-transplanted group (6.86 ± 0.68 for EBD, 2.11 ± 0.2 for BHK, and 2.01 ± 0.23 for HF-transplanted animals; p = 0.0001).
Figure 4.

Functional recovery of EBD-transplanted rats at various intervals. In all, 15 paralyzed rats in each group were implanted with 300,000 EBD cells, baby hamster kidney (BHK) cells, or human fibroblast (HF) cells and were scored for recovery by two independent raters who were blinded to the treatment groups. A, BHK-transplanted (open triangles) and HF-transplanted (filled circles) rats exhibited no recovery of locomotor function at 4, 8, 12, or 24 weeks. EBD-transplanted rats showed a significant functional improvement as compared with BHK- or HF-transplanted animals at 12 and 24 weeks. Asterisks denote significant recovery of EBD-transplanted animals (p = 0.004 at 12 weeks; p = 0.0001 at 24 weeks). B, Measurement of hindlimb strength via a digital force gauge revealed improved grip strength in transplanted animals (filled triangles), whereas HF-transplanted (filled circles) and BHK-transplanted (open triangles) animals did not. By 6 months after transplantation the EBD-transplanted animals had recovered ∼40% of their pre-paralysis strength, whereas there had been no recovery in the other groups. Asterisks denote significant improvement in hindlimb strength (p < 0.001 at 12 and 24 weeks). C, Measurement of righting time as an independent and objective measure of motor recovery. All 15 animals transplanted with either EBD or HF cells were tested for the time required to right themselves when placed in a supine position. Two separate trials were conducted, and the total time required for both trials was summed at the time of transplantation and again at 24 weeks after transplantation. The data are presented as a Δ score, reflecting an individual animal's change from its baseline. Positive numbers reflect improved performance on this task, whereas negative numbers reflect worse performance. In all, 11 of 15 EBD-transplanted animals (back row) had an improvement of ≥3 sec, whereas only 1 of 15 HF-transplanted animals did.
As an additional objective measure of functional recovery, the animals were evaluated blindly for hindlimb grip strength. raNSV induced a decrease in hindlimb strength from a mean of 0.27 ± 0.03 lb of pressure to 0.01 ± 0.00 lb of pressure by the time of transplantation (Fig. 4B). By 12 and 24 weeks after transplantation BHK-transplanted (open triangles) and HF-transplanted (filled circles) animals exhibited no return of hindlimb grip strength, whereas EBD-transplanted rats (filled triangles) had significant improvement in hindlimb strength (open circles; 0.09 ± 0.025 at 12 weeks and 0.11 ± 0.27 at 24 weeks; p < 0.001).
Additionally, the animals in both groups were investigated for their ability to right themselves from a supine position as another marker for torso and limb strength (Fig. 4C). Animals were given two blinded trials at each time point, and the sum time required for righting was noted. Then they were retested at 24 weeks after transplantation, and the change in time to righting was recorded. In the HF-treated animals there was little difference in the time required for righting between 0 and 24 weeks (1 of 15 animals performed better at 24 weeks by ≥3 sec; mean Δ score, -0.6 sec). However, at 24 weeks 11 of 15 EBD-transplanted animals performed better at this task compared with their baseline (mean Δ score, 6.93 ± 1.17 sec; p = 0.0001). Therefore, by three measures of functional recovery the transplanted animals partially regained hindlimb function, whereas nontransplanted animals did not.
Possible mechanisms of EBD-induced motor recovery
Because it was clear that the functional recovery of animals was not attributable to structural reconstitution of motor neurons by EBD cells, we examined alternative mechanisms to explain this. Specifically, we investigated the possibility that transplantation of EBD cells altered the local microenvironment and enhanced survival and connectivity of surviving host cells. Initially, we considered the possibility that transplantation of EBD cells initiated endogenous neurogenesis within the spinal cord (Fig. 5A). We pulsed animals with BrdU at 12–15 d after infection and then killed the animals at day 16 or 36 after infection to determine the proliferation of cells within the spinal cord. Similar to that seen in traumatic spinal cord injury in adult rodents (Weiss et al., 1996; Kehl et al., 1997; Johansson et al., 1999; Horner et al., 2000; Shihabuddin et al., 2000; Yamamoto et al., 2001), paralysis from raNSV infection resulted in the proliferation of neural progenitors within the spinal cord (increase from 0 BrdU+/nestin+ cells in nonparalyzed animals to 162 ± 34 cells per section in paralyzed animals; Fig. 5A). Transplantation of EBD cells at 8 d after infection altered neither the number of endogenous neural progenitors (BrdU+/nestin+) present at 16 d after infection nor the number of reactive astrocytes at 36 d after infection (BrdU+/GFAP+; Fig. 5A). Further, we did not observe the generation of BrdU+/TUJ1+ at 36 d after infection in any animals. Therefore, transplantation of EBD cells did not modulate the endogenous neural progenitor response to viral injury. Interestingly, few of the EBD cells became BrdU+ within the spinal cord (data not shown), indicating that they left the cell cycle shortly after transplantation.
Examination of spinal cords in EBD- and HF-transplanted animals at 6 months after transplantation revealed two significant differences: enhanced host motor neuron survival and reafferentation of motor neuron soma in EBD-transplanted animals. We counted L4 ventral axons in all transplanted animals at 6 months after transplantation as a surrogate marker for motor neuron survival. We found a significantly increased number of surviving ventral axons in EBD-transplanted animals as compared with HF-transplanted animals (338.33 ± 11 vs 294 ± 8.73; p < 0.03; Fig. 5B), suggesting that EBD transplantation halted or slowed the progressive motor neuron death triggered by raNSV. Although we cannot exclude a baseline inequality in motor neuron survival between the groups, the observation that they had identical baseline clinical measures of paralysis (Fig. 4) suggests that this is unlikely.
Because it remained unclear that this finding could account for the functional recovery that was seen, we examined whether EBD transplantation facilitated reafferentation of surviving motor neurons (Fig. 5C,D). Indeed, we found that spinal motor neurons from EBD-transplanted rats had markedly increased synaptophysin contacts as compared with HF-transplanted animals (10.95 ± 0.65 vs 18.54 ± 1.14; p < 0.02; Fig. 5D), suggesting that host spinal cord axons had reestablished synaptic contact with them. It is likely, therefore, that EBD transplantation facilitated a partial locomotor recovery via modulation of the local microenvironment, resulting in diminished motor neuron loss and enhanced axonal growth onto surviving motor neurons.
We found that EBD cells had a similar ability to stimulate axonal growth and facilitate motor neuron survival in vitro (see below) and examined the possibility that secretion of trophic factors mediates these effects. We plated 105 EBD cells in 1 ml of medium and examined the presence of various neurotrophic factors in the medium 24 hr later. EBD cells undergo 0.6 population doublings every day (data not shown), and there were 1.6 × 105 cells present at the time of supernatant removal and testing. EBD cells secrete significant amounts of BDNF (332 ± 26 pg/ml) and TGF-α (140 ± 16.35 ng/ml), but not GDNF, NT3, or neurturin. When we use the mean number of cells present during the 24 hr period, this corresponds to 255 pg of BDNF per 105 cells/d and 107 ng of TGF-α per 105 cells/d.
We found that EBD cells, placed outside the margin of spinal cord organotypic cultures, stimulated extensive axonal projections (Fig. 5E) and upregulation of GAP43 within the axonal growth cones (Fig. 5F). Spinal axons grew up to 1.2 mm from the margin of the culture in 2 weeks after the placement of EBD cells in the culture dish (data not shown) and ultimately invaded and surrounded the cluster of EBD cells (Fig. 5F). Incubation of spinal organotypic/EBD cocultures with neutralizing antibodies to BDNF, but not to TGF-α, diminished the number of axons emerging from the organotypic culture (41.3 ± 7.2 vs 16.2 ± 5.23 with anti-BDNF; p = 0.007; Fig. 5G). This indicates that EBD cells stimulated spinal neuron axonal growth by secreting BDNF and suggests that BDNF may have mediated axonal reafferentation of host motor neurons in transplanted animals in vivo.
To determine whether soluble factors from EBD cells were capable of supporting motor neuron survival, we examined the potential of conditioned medium from EBD cells to allow the survival of cultured motor neurons in a serum-free environment. Conditioned medium from EBD cells was capable of supporting survival of motor neurons without additional growth factor support, whereas virtually 100% of motor neurons incubated with unconditioned L15 medium died within 24 hr (65.9 ± 1.27% survival with EBD conditioned medium vs 3.7 ± 0.176% with unconditioned medium; Fig. 5H)(p = 0.001). Incubation of the motor neuron cultures with conditioned medium and neutralizing antibodies to either BDNF or TGF-α partially abolished this survival effect (37.1 ± 0.72% with anti-BDNF, p < 0.001; 26.2 ± 1.56% with anti-TGF-α, p < 0.001), Therefore, TGF-α and to a lesser degree BDNF secreted from EBD cells supported the survival of cultured motor neurons and may account for the in vivo protection from motor neuron death seen in transplanted animals.
Discussion
In the present study we demonstrate the ability of human pluripotent stem cell derivatives to engraft diffusely throughout the spinal cord and to bring about functional recovery of animals with paralysis because of motor neuron injury. Because of the relatively widespread motor neuron injury induced by raNSV, we adopted a CSF-based delivery system. Thus EBD cells transplanted directly into the lumbar CSF were distributed over the entire rostrocaudal length of the spinal cord and were engrafted over a substantial area. This distribution likely reflects passive dissemination from CSF flow because distribution was identical in nonparalyzed animals. We further show that EBD cells can migrate across the pial layer into the spinal cord parenchyma but do not do so in control animals. Although immortalized stem cells previously have been delivered into the ventricular system and have been shown to migrate into the brain (Snyder et al., 1995; Yandava et al., 1999), these v-myc-transformed cells adopt the migratory behavior more of tumor cells than of native stem cells. There is no real precedent for parenchymal spinal invasion of nontransformed stem cells or progenitors, and these data raise the possibility that functional restoration over a wide region of the neuraxis may be possible with less invasive transplantation paradigms. The lack of parenchymal migration of EBD cells in nonparalyzed animals indicates that the cells can respond to contextual and damage-associated cues provided either by soluble factors or by the extracellular matrix.
However, the present study also shows that 63% of cells remained adherent to the meninges and did not migrate into the spinal cord parenchyma. Although they may have been critical to the functional recovery that was seen, it is also possible that they might stimulate an inflammatory response that could result in some degree of neurologic injury. In initial studies we have not observed any inflammatory response to the EBD cells nor neurologic deterioration in any transplanted animal, but further investigations should clarify the issue. Additionally, we have not seen any human-derived tumors in >100 transplanted animals that have been examined to date.
Our initial expectations were that the neurally biased EBD cells potentially would differentiate into spinal neurons, thus facilitating a motor recovery. However, although neuronal differentiation of transplanted cells did occur, it did not occur with enough efficiency to account for the functional recovery. Additionally, we have no evidence to suggest that transplanted cells, even those that did differentiate into neurons, formed synapses with peripheral muscle. It is surprising, therefore, that a significant functional recovery did occur and suggests alternative mechanisms for plasticity in the adult mammalian spinal cord. We found that EBD transplantation slowed or halted progressive motor neuron degeneration triggered by raNSV infection and facilitated a reconstitution of synaptic contacts on surviving host motor neurons. EBD cells secrete large quantities of TGF-α, a factor known to support the survival of motor neurons (Junier et al., 1998; Boillee et al., 2001) and axonal regrowth after axotomy (Herzog and Otto, 2002). Indeed, in our studies the amount of TGF-α was sufficient to protect cultured motor neurons from death, although it is not yet clear the extent of TGF-α secretion by transplanted cells. Interestingly, although TGF-α has been reported to enhance astrogliosis in the CNS (Rabchevsky et al., 1998), we did not see increased astrocyte numbers or activated astrocyte morphology in transplanted animals. Although TGF-α has been shown to induce endogenous stem cell proliferation (Fallon et al., 2000), we did not find any enhanced neurogenesis in the presence of transplanted cells.
Further examination of the spinal cords revealed a virtually complete loss of synaptic complement on surviving motor neurons in the acute phase and enhanced restoration of synaptic complement in transplanted animals. It is well established via pharmacologic, physiologic, and morphologic studies that synaptic input onto motor neuron cell bodies is reduced dramatically after injury (Purves, 1975; Taxi and Eugene, 1995). This process, termed reactive deafferentation (Titmus and Faber, 1990), results in neurons that have diminished electrical connectivity to other CNS neurons and plays a role in paralysis. Enhanced reestablishment of such contacts by EBD transplantation may allow the remaining motor neurons to move the hindlimbs more effectively than in nontransplanted animals. Interestingly, diminished presynaptic input onto spinal motor neurons has been observed in human spinal cords from patients with amyotrophic lateral sclerosis, spinal muscular atrophy, and poliomyelitis (Sasaki and Maruyama, 1994; Ince et al., 1995; Cruz-Sanchez et al., 1996). It is therefore possible that the clinical disability seen in such patients reflects a summation of neuronal loss and neuronal disconnection caused by deafferentation. The tropic support for this axonal reafferentation may involve the secretion of BDNF by EBD cells. BDNF is a factor both for survival of spinal neurons (Lindsay and Peters, 1984; Barde et al., 1987) and for axonal growth (Yuan et al., 2000; Tobias et al., 2001). We found that EBD cells secrete large quantities of BDNF in vitro and that this amount of BDNF was sufficient to cause robust axonal growth in a spinal cord organotypic model and to promote survival of cultured motor neurons.
In this context EBD cells may be similar to genetically modified non-stem cells, engineered to express neurotrophic factors (Liu et al., 1999b; Tobias et al., 2001). However, unlike fibroblasts, EBD cells are capable of migrating into the spinal cord of paralyzed animals. The ability of EBD cells to migrate and secrete trophic factors differentiates them from other cellular transplants that have the ability either to migrate or to secrete trophic factors, but not both. Although the amount of neurotrophin secreted by the EBD cells was sufficient for potent biological effects in vitro, it is not clear that this is the mechanism for transplantation effect in vivo. We have estimated that ∼9 × 104 EBD cells survive at 1 month after transplantation (either adherent to the meninges or within the spinal cord). If we assume that the rate of neurotrophin secretion is unchanged in vivo, this corresponds to ∼229 pg of BDNF and 96 ng of TGF-α secretion per day in each spinal cord of transplanted animals. Although these levels do not approach those required to see biological effects when neurotrophins are delivered extrinsically to the spinal cord, it remains possible that local delivery by transplanted cells is more effective than extrinsic neurotrophin delivery. EBD cells exist within close contact of host neurons and axons, and, therefore, local concentration of neurotrophins may approach that seen with extrinsic delivery. Additionally, cell-mediated contacts or locally high neurotrophin levels may stimulate additional neurotrophin secretion by host cells.
Footnotes
Project ALS (Amyotrophic Lateral Sclerosis), Families of Spinal Muscular Atrophy, and Andrew's Buddies supported this work. We acknowledge M. Hardwick for scientific advice and P. Talalay for manuscript suggestions.
Correspondence should be addressed to Dr. Douglas Kerr, Department of Neurology, Pathology 627 C, Johns Hopkins University School of Medicine, 600 North Wolfe Street, Baltimore, MD 21287. E-mail: dkerr@jhmi.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235131-10$15.00/0
References
- Barde YA, Davies AM, Johnson JE, Lindsay RM, Thoenen H ( 1987) Brain-derived neurotrophic factor. Prog Brain Res 71: 185–189. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC ( 1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12: 1–21. [DOI] [PubMed] [Google Scholar]
- Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De Fraja C, Vescovi A, Cattaneo E, Finocchiaro G ( 2000) Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med 6: 447–450. [DOI] [PubMed] [Google Scholar]
- Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA ( 2001) Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J Neurosci 21: 6718–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Cadusseau J, Coulpier M, Grannec G, Junier MP ( 2001) Transforming growth factor α: a promoter of motoneuron survival of potential biological relevance. J Neurosci 21: 7079–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EW, Loera S, Chiu AY ( 1995) Target regulation of a motor neuron-specific epitope. J Neurosci 15: 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Sanchez FF, Moral A, Rossi ML, Quinto L, Castejon C, Tolosa E, de Belleroche J ( 1996) Synaptophysin in spinal anterior horn in aging and ALS: an immunohistological study. J Neural Transm 103: 1317–1329. [DOI] [PubMed] [Google Scholar]
- Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J, Korc M, Endo TL, Duong A, Nguyen G, Karkehabadhi M, Twardzik D, Patel S, Loughlin S ( 2000) In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci USA 97: 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Naylor PH, Dosescu J, Skunca M, Majumdar AP, Moshier JA ( 2000) TGFα is required for full expression of the transformed growth phenotype of NIH 3T3 cells overexpressing ornithine decarboxylase. Carcino-genesis 21: 567–572. [DOI] [PubMed] [Google Scholar]
- Griffin DE, Levine B, Ubol S, Hardwick JM ( 1994) The effects of alphavirus infection on neurons. Ann Neurol [Suppl] 35: S23–S27. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Otto T ( 2002) Administration of transforming growth factor-α enhances anatomical and behavioral recovery following olfactory nerve transection. Neuroscience 113: 569–580. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH ( 2000) Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci 20: 2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince PG, Slade J, Chinnery RM, McKenzie J, Royston C, Roberts GW, Shaw PJ ( 1995) Quantitative study of synaptophysin immunoreactivity of cerebral cortex and spinal cord in motor neuron disease. J Neuropathol Exp Neurol 54: 673–679. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Griffin DE, Johnson RT ( 1987) The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab Invest 56: 418–423. [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Trapp BD, Griffin DE ( 1988) Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest 58: 503–509. [PubMed] [Google Scholar]
- Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J ( 1999) Identification of a neural stem cell in the adult mammalian central nervous system. Cell 96: 25–34. [DOI] [PubMed] [Google Scholar]
- Junier MP, Legendre P, Esguerra CV, Tinel M, Coulpier M, Dreyfus PA, Bahr M ( 1998) Regulation of growth factor gene expression in degenerating motoneurons of the murine mutant wobbler: a cellular patch-sampling/RT-PCR study. Mol Cell Neurosci 12: 168–177. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Fairbanks CA, Laughlin TM, Wilcox GL ( 1997) Neurogenesis in postnatal rat spinal cord: a study in primary culture. Science 276: 586–589. [DOI] [PubMed] [Google Scholar]
- Kerr DA, Larsen T, Cook SH, Fannjiang YR, Choi E, Griffin DE, Hardwick JM, Irani DN ( 2002) BCL-2 and BAX protect adult mice from lethal Sindbis virus infection but do not protect spinal cord motor neurons or prevent paralysis. J Virol 76: 10393–10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondziolka D, Wechsler L, Goldstein S, Meltzer C, Thulborn KR, Gebel J, Jannetta P, DeCesare S, Elder EM, McGrogan M, Reitman MA, Bynum L ( 2000) Transplantation of cultured human neuronal cells for patients with stroke. Neurology 55: 565–569. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH ( 1997) Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci 17: 5820–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacorazza HD, Flax JD, Snyder EY, Jendoubi M ( 1996) Expression of human β-hexosaminidase α-subunit gene (the gene defect of Tay—Sachs disease) in mouse brains upon engraftment of transduced progenitor cells. Nat Med 2: 424–429. [DOI] [PubMed] [Google Scholar]
- Lindsay RM, Peters C ( 1984) Spinal cord contains neurotrophic activity for spinal nerve sensory neurons. Late developmental appearance of a survival factor distinct from nerve growth factor. Neuroscience 12: 45–51. [DOI] [PubMed] [Google Scholar]
- Liu Y, Himes BT, Solowska J, Moul J, Chow SY, Park KI, Tessler A, Murray M, Snyder EY, Fischer I ( 1999a) Intraspinal delivery of neurotrophin-3 using neural stem cells genetically modified by recombinant retrovirus. Exp Neurol 158: 9–26. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kim D, Himes BT, Chow SY, Schallert T, Murray M, Tessler A, Fischer I ( 1999b) Transplants of fibroblasts genetically modified to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci 19: 4370–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH ( 1988) Molecular basis of Sindbis virus neurovirulence in mice. J Virol 62: 2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, Choi DW ( 1999) Transplanted embryonic stem cells survive, differentiate, and promote recovery in injured rat spinal cord. Nat Med 5: 1410–1412. [DOI] [PubMed] [Google Scholar]
- Onifer SM, Cannon AB, Whittemore SR ( 1997) Altered differentiation of CNS neural progenitor cells after transplantation into the injured adult rat spinal cord. Cell Transplant 6: 327–338. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW ( 1995) Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci USA 92: 954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D ( 1975) Functional and structural changes in mammalian sympathetic neurones following interruption of their axons. J Physiol (Lond) 252: 429–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier MP ( 1998) A role for transforming growth factor α as an inducer of astrogliosis. J Neurosci 18: 10541–10552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW ( 1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA 90: 6591–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Maruyama S ( 1994) Decreased synaptophysin immunoreactivity of the anterior horns in motor neuron disease. Acta Neuropathol (Berl) 87: 125–128. [DOI] [PubMed] [Google Scholar]
- Schmued L, Kyriakidis K, Heimer L ( 1990) In vivo anterograde and retrograde axonal transport of the fluorescent rhodamine-dextran-amine, Fluoro-Ruby, within the CNS. Brain Res 526: 127–134. [DOI] [PubMed] [Google Scholar]
- Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ, Blumenthal PD, Huggins GR, Gearhart JD ( 1998) Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci USA 95: 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblott MJ, Axelman J, Littlefield JW, Blumenthal PD, Huggins GR, Cui Y, Cheng L, Gearhart JD ( 2001) Human embryonic germ cell derivatives express a broad range of developmentally distinct markers and proliferate extensively in vitro Proc Natl Acad Sci USA 98: 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Madison RD, Bradley J, Turner DA ( 1994) Quantitative graft integration of fetal hippocampal transplants labeled with 5′ bromodeoxyuridine into normal adult hippocampus. Exp Neurol 126: 205–224. [DOI] [PubMed] [Google Scholar]
- Shihabuddin LS, Horner PJ, Ray J, Gage FH ( 2000) Adult spinal cord stem cells generate neurons after transplantation in the adult dentate gyrus. J Neurosci 20: 8727–8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EY, Taylor RM, Wolfe JH ( 1995) Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature 374: 367–370. [DOI] [PubMed] [Google Scholar]
- Sterio DC ( 1984) The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc 134[Pt 2]: 127–136. [DOI] [PubMed] [Google Scholar]
- Taxi J, Eugene D ( 1995) Effects of axotomy, deafferentation, and reinnervation on sympathetic ganglionic synapses: a comparative study. Int Rev Cytol 159: 195–263. [DOI] [PubMed] [Google Scholar]
- Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW ( 2002) Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416: 542–545. [DOI] [PubMed] [Google Scholar]
- Titmus MJ, Faber DS ( 1990) Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog Neurobiol 35: 1–51. [DOI] [PubMed] [Google Scholar]
- Tobias CA, Dhoot NO, Wheatley MA, Tessler A, Murray M, Fischer I ( 2001) Grafting of encapsulated BDNF-producing fibroblasts into the injured spinal cord without immune suppression in adult rats. J Neurotrauma 18: 287–301. [DOI] [PubMed] [Google Scholar]
- Urakami H, Chiu AY ( 1990) A monoclonal antibody that recognizes somatic motor neurons in the mature rat nervous system. J Neurosci 10: 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe W, Van Den Bosch L, Robberecht W ( 1998) Glial cells potentiate kainate-induced neuronal death in a motoneuron-enriched spinal coculture system. Brain Res 807: 1–10. [DOI] [PubMed] [Google Scholar]
- Vercelli A, Repici M, Garbossa D, Grimaldi A ( 2000) Recent techniques for tracing pathways in the central nervous system of developing and adult mammals. Brain Res Bull 51: 11–28. [DOI] [PubMed] [Google Scholar]
- Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, Reynolds BA ( 1996) Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J Neurosci 16: 7599–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ ( 1999) Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22: 51–61. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Nagao M, Sugimori M, Kosako H, Nakatomi H, Yamamoto N, Takebayashi H, Nabeshima Y, Kitamura T, Weinmaster G, Nakamura K, Nakafuku M ( 2001) Transcription factor expression and Notch-dependent regulation of neural progenitors in the adult rat spinal cord. J Neurosci 21: 9814–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandava BD, Billinghurst LL, Snyder EY ( 1999) “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc Natl Acad Sci USA 96: 7029–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Evans EP, Smith AG ( 2002) Changing potency by spontaneous fusion. Nature 416: 545–548. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Wu W, So KF, Cheung AL, Prevette DM, Oppenheim RW ( 2000) Effects of neurotrophic factors on motoneuron survival following axonal injury in newborn rats. NeuroReport 11: 2237–2241. [DOI] [PubMed] [Google Scholar]