

Abstract
Although the cause of neuronal death in Parkinson's disease (PD) is mainly unknown, growing evidence suggests that both apoptotic and non-apoptotic death may occur in PD. Using primary cultures of mesencephalic dopaminergic neurons and the MN9D dopaminergic neuronal cell line, we attempted to evaluate specifically the existence of the mitochondrial apoptotic pathway, focusing on the mitochondrial release of cytochrome c to the activation of the caspases after 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenylpyridinium (MPP+) treatment. Both immunofluorescent labeling and immunoblot analysis indicated mitochondrial release of cytochrome c into the cytosol after 6-OHDA or MPP+ treatment. However, the appearance of activated caspase-3 immunoreactivity in tyrosine hydroxylase (TH)-positive neurons was detected only after 6-OHDA. Immunoblot and biochemical analysis also confirmed that activation of both caspase-9 and caspase-3 was induced by 6-OHDA, but not by MPP+. Consequently, cotreatment with a caspase inhibitor (zVAD-fmk) or with an antioxidant (N-acetylcysteine) not only deterred 6-OHDA-induced loss of TH-positive neurons but also abolished the appearance of activated caspase-3 in TH-positive neurons. In contrast, the same treatment did not spare MPP+-treated TH-positive neurons. Interestingly, a reconstitution assay indicated that the addition of ATP to the cytosolic fraction obtained from MPP+-treated cells was sufficient to activate both caspase-9 and caspase-3. Taken together, our results indicate that distinct mechanisms underlie neurotoxin-induced cell death. They also suggest that, after mitochondrial release of cytochrome c in dopaminergic neurons after neurotoxin treatment, intracellular levels of ATP may constitute a critical factor in determining whether a neuron will die by a caspase-dependent or -independent pathway.
Keywords: cytochrome c, caspase, 6-hydroxydopamine, MPP+, dopaminergic neuron, ATP
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by progressive loss of dopaminergic (DA) neurons of the substantia nigra pars compacta. Although recent advances have been made in defining biochemical events in the pathogenesis of PD, evidence is accumulating that either favors or argues against apoptosis as a sequela in postmortem parkinsonian brains (Mochizuki et al., 1996; Anglade et al., 1997; Kosel et al., 1997; Tompkins et al., 1997; Banati et al., 1998; Burke and Kholodilov, 1998; Kingsbury et al., 1998; Wullner et al., 1999; Hartmann et al., 2000, 2001; Tatton, 2000). Evidence implicating apoptosis in PD also has remained controversial in studies that used experimental models generated by such neurotoxins as 1-methyl-1,2,3,6-tetrahydropyridine (MPTP). For example, several studies have suggested that apoptosis may reflect the primary mechanisms associated with DA neuronal death (Hartley et al., 1994; Mochizuki et al., 1994; Tatton and Kish, 1997; Dodel et al., 1998; Viswanath et al., 2001; Bilsland et al., 2002). In contrast, several other reports still conflict with regard to the mode of cell death (Jackson-Lewis et al., 1995; Jeon et al., 1995; Lotharius et al., 1999; Soldner et al., 1999). For example, Lotharius et al. (1999) have found no evidence of phosphatidylserine externalization, an early apoptotic marker, after treatment of primary cultures of DA neurons with 1-methyl-4-phenylpyridinium (MPP+) and have reported that a caspase inhibitor does not block MPP+-mediated neurotoxicity.
Previously, we have presented the possibility that 6-hydroxydopamine (6-OHDA) and MPP+ act on distinct cell death pathways in a dopaminergic neuronal cell line, MN9D (Oh et al., 1995; Choi et al., 1999a,b; Kim et al., 2001). More specifically, reactive oxygen species (ROS) and/or an ROS-mediated signal play(s) an essential role in 6-OHDA-induced apoptosis in MN9D cells. MPP+-mediated cell death is not blocked by caspase inhibitors and is accompanied by a severe mitochondrial swelling. Metabolic abnormalities with the activation of calpain seem to be involved primarily in MPP+-induced MN9D cell death (Choi et al., 2001; Kim et al., 2001). Overexpression of Bcl-2 spares MPP+-treated MN9D cells significantly by blocking at cellular loci that are distinct from those known for anti-caspase activity (Choi et al., 2001; Kim et al., 2001). When these data and those from the other reports listed above are all taken into consideration, it can be concluded that the mechanism by which these neurotoxins kill DA neuronal cells still remains equivocal and is yet to be thoroughly resolved.
In the present study, therefore, we attempted to examine specifically the mitochondrial apoptotic pathway from the point of mitochondrial release of cytochrome c to activation of capsase-9 and caspase-3 after treatment with MPP+ or 6-OHDA. For the experiments we primarily used cultures of mesencephalic DA neurons derived from embryonic day 14 (E14) rat embryos; the MN9D cells were used for some biochemical analyses. We present data suggesting that (1) both caspase-dependent and -independent pathways exist and (2) the levels of intracellular ATP may play a critical role for deciding the route to caspase-dependent or -independent cell death in in vitro culture models of PD.
Materials and Methods
Cell cultures and drug treatments. To prepare primary cultures of DA neurons, the ventral mesencephalon was removed from 14 d gestation Sprague Dawley rat embryo (Daehan Biolink, Daejon, Korea). Tissues were incubated with 0.01% trypsin in HBSS for 10 min at 37°C and triturated with a constricted Pasteur pipette. DA neurons were plated at 1.0 × 10 5 cells per 8-mm-diameter Aclar embedding film (Electron Microscopy Sciences, Fort Washington, PA) precoated with 100 μg/ml poly-d-lysine (Sigma, St. Louis, MO) and 4 μg/ml laminin (Invitrogen, San Diego, CA). DA neurons were maintained at 37°C in a humidified 5% CO2 atmosphere in MEM supplemented with 10% FBS (Invitrogen), 2 mm glutamine, and 6.0 gm/l glucose. At 5 or 6 d in vitro (DIV) the DA neurons were washed with MEM and treated with various experimental reagents for the time periods indicated. Reagents used were 6-OHDA (Sigma), MPP + (Sigma), staurosporine, N-acetylcysteine, and N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk; Enzyme Systems Products, Livermore, CA). MN9D cells that can synthesize, release, and take up dopamine were established from embryonic mesencephalic DA neurons by a somatic fusion with N18TG2 neuroblastoma cells (Choi et al., 1991, 1992; Tang et al., 1994). As previously described in our laboratory (Choi et al., 2001), briefly, the MN9D cells were plated on 25 μg/ml poly-d-lysine-precoated culture dishes or plates (Corning Glass Works, Corning, NY), maintained in DMEM supplemented with 10% FBS in an incubator with an atmosphere of 10% CO2 at 37°C, and switched to serum-free N2 medium (Bottenstein and Sato, 1979) containing various experimental reagents.
Immunocytochemistry and fluorescent stains for mitochondria. For a double immunolabeling of tyrosine hydroxylase (TH; a rate-limiting enzyme of dopamine synthesis) and cytochrome c or activated caspase-3, DA neurons treated with 20 μm 6-OHDA or 3 μm MPP + were fixed with 4% paraformaldehyde for 20 min at room temperature (RT) and blocked for 1 hr in PBS containing 5% normal goat serum and 0.1% Triton X-100. Then the DA neurons were incubated with the primary antibody in PBS containing 1% normal goat serum and 0.1% Triton X-100 at 4°C overnight. Primary antibodies used were a rabbit polyclonal anti-TH (1:500; Pel-Freez, Rogers, AR), a mouse monoclonal anti-TH (1:7500; Pel-Freez), a mouse monoclonal anti-cytochrome c (1:200; PharMingen, San Diego, CA), and a rabbit polyclonal antibody that recognizes activated caspase-3 (CM1, 1:200; PharMingen). After washes with PBS the DA neurons were incubated at RT for 1 hr with Alexa Fluor 568 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG (1:200; Molecular Probes, Eugene, OR) for a double localization of TH and cytochrome c or with Texas Red-conjugated goat anti-mouse IgG and Oregon green-conjugated goat anti-rabbit IgG (1:200; Molecular Probes) for a double localization of TH and activated caspase-3 in PBS containing 1% normal goat serum and 0.1% Triton X-100. As a negative control the DA neurons were incubated with the secondary antibody only. Cells were examined under an Axiovert 100 microscope equipped with an epifluorescence unit and Axiocam Digital Camera (Zeiss, Zena, Germany) or a Radiance 2100 laser-scanning confocal microscope (Bio-Rad, Hercules, CA). Activated caspase-3-positive neurons among TH-positive neurons were scored by examining 16 consecutive fields (20× objective) across the Aclar embedding film. Approximately 190–210 TH-positive neurons were counted in the untreated controls. For immunofluorescent localization of cytochrome c and activated caspase-3 in MN9D cells the cultures were fixed and blocked as described above. Cultures then were incubated with a mouse monoclonal anti-cytochrome c or a rabbit polyclonal anti-activated caspase-3. This was followed by Alexa Fluor 488 goat anti-mouse IgG or Oregon green-conjugated goat anti-rabbit IgG. To label mitochondria, we treated MN9D cells plated on Aclar embedding film with 6-OHDA or MPP + for the time periods indicated and incubated them with freshly prepared 25 nm MitoTracker Orange CMTMRos (a mitochondrion-selective probe; Molecular Probes) in N2 medium for 20 min at 37°C in an incubator with an atmosphere of 10% CO2. Then the cells were washed with N2 medium, mounted on a glass slide, and examined under a confocal microscope.
Immunoblot analysis. After treatment with 100 μm 6-OHDA or 50 μm MPP + the MN9D cells were washed with ice-cold PBS and lysed in a buffer containing (in mm) 50 Tris, pH 7.0, 2 EDTA, and 2 PMSF plus 1% Triton X-100, 50 μg/ml aprotinin, and 50 μg/ml leupeptin for 10 min on ice. Cells were homogenized in a Dounce homogenizer on ice, followed by centrifugation at 13,000 × g for 30 min at 4°C. For the detection of mitochondrial release of cytochrome c, cellular fractionation was performed as previously described with minor modifications (Gross et al., 1998). Protein contents were measured with a Bio-Rad protein assay kit. Protein (50–75 μg) was separated on a 10% SDS-PAGE, blotted onto pre-wetted polyvinylidene difluoride (PVDF) nitrocellulose filters, and processed for immunoblot analysis as described (Choi et al., 2001). Primary antibodies used were a rabbit polyclonal anti-activated caspase-9 (1:1000; Cell Signaling, Beverly, MA), a rabbit polyclonal anti-activated caspase-3 (1:1,000; Cell Signaling), a mouse monoclonal anti-cytochrome c (1:3000; PharMingen), a mouse monoclonal anti-cytochrome oxidase subunit IV (1:1000; Molecular Probes), and a rabbit polyclonal anti-IκB (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). Specific bands were detected by enhanced chemiluminescence (ECL; Amersham Biosciences, Piscataway, NJ).
TH immunoreactive neurons, soma size, and dopamine uptake assay. To determine the number of surviving TH-positive cells after neurotoxin treatment in the presence or absence of various inhibitors, we fixed, blocked, and incubated DA neurons with a mouse monoclonal anti-TH as described above. After being washed with PBS, the DA neurons were incubated with biotinylated goat anti-mouse IgG at RT for 1 hr, followed by alkaline phosphatase-conjugated avidin–biotin complex (1:200; Vector Laboratories, Burlingame, CA) for 30 min at RT. Staining was visualized in 0.1 m Tris, pH 8.2, containing 0.2 mg/ml naphthol-AS-MX and 1.0 mg/ml fast red-TR. Manual counts from each condition present in each 8 mm Aclar embedding film were made of all of the TH-positive cells having neurites twice the length of the soma. A total 300–400 TH-positive neurons were counted in the untreated control 8 mm film. After cells were immunostained with anti-TH antibody and subsequently were scanned with a confocal microscope, we measured the greatest soma diameter in ∼60–80 TH-positive neurons. For measuring [3H]DA uptake after neurotoxin treatment, we rinsed the DA neurons with Krebs–Ringer solution, incubated them in Krebs–Ringer solution containing 1 mm ascorbate, 2 mm β-alanine, and 100 μm pargyline at 37°C for 5 min in a humidified 5% CO2 atmosphere, and further incubated them with 25 nm [3H]dopamine (45 Ci/mmol; Amersham Biosciences) for 15 min. Control DA neurons were treated with the above medium in the presence of a dopamine uptake blocker, 10 μm mazindol (Sigma). Cells then were washed with ice-cold Krebs–Ringer solution and lysed with 0.5 m NaOH. Radioactivity was quantified by a liquid scintillation counter (Beckman, Fullerton, CA).
In vitro reconstitution assay. After treatment of MN9D cells with 50 μm MPP + or 1 μm staurosporine for the time periods indicated, cytosolic fractions were prepared as described (Gross et al., 1998), and an in vitro reconstitution assay of caspase activity was performed as previously described with some modifications (Zou et al., 1999). Briefly, cytosolic fractions (50 μg) were incubated with 25 μm acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC for caspase-3-like activity) or 40 μm acetyl-Leu-Glu-His-Asp-7-amino-4-trifluoromethylcoumarin (Ac-LEHD-AFC for caspase-9-like activity; Calbiochem–Novabiochem, La Jolla, CA) for 2 hr at 30°C in a reaction buffer [containing (in mm)20 HEPES-KOH, pH 7.5, 10 KCl, 1.5 MgCl2, 1 EDTA, 1 EGTA, and 1 dithiothreitol] with or without 1 mm dATP or ATP. Fluorogenic caspase activity was measured with excitation at 360 nm and emission at 430 nm (for Ac-DEVD-AMC) or with excitation at 400 nm and emission at 505 nm (for Ac-LEHD-AFC) with a fluorescence plate reader FL 600 (Bio-Tek Instruments, Winooski, VT). Aliquots of reaction mixtures were subjected to immunoblot analysis for activated caspase-3.
Statistics. Data are given as the mean ± SEM. Significance of difference between control and samples treated with various drugs was determined by one-way ANOVA and post hoc Student's t test. Values of p < 0.01 or 0.05 were taken as statistically significant.
Results
Cytotoxic effects of 6-OHDA and MPP+ on primary cultures of DA neurons
In the primary cultures derived from the ventral mesencephalon of E14 rat embryos, up to ∼80% of the total populations were neurons. DA neurons routinely comprised 5.8% of the total population of cells as defined by immunocytochemical staining for TH at 5 DIV (n = 6). In these cultures 4.2% of the total cells were positive for glial fibrillary acidic protein (GFAP) and thus were astrocytes (n = 6). In addition, GABA-positive neurons were present in approximately equivalent numbers to DA neurons, and virtually no OX42-positive microglia were found. Using these cultures, we first characterized the cytotoxic effect of 6-OHDA and MPP+ on DA neurons by determining the number of TH-positive neurons and the rate of [3H]DA uptake. From a dose–response study with various concentrations ranging from 0.5 to 40 μm of both 6-OHDA and MPP+, ∼30–40% of TH-positive neurons survived at 20 μm 6-OHDA for 24 hr or at 3 μm MPP+ for 48 hr. Typically, at these concentrations a time-dependent decrease in the number of TH-positive neurons and in the rate of [3H]DA uptake was observed (Fig. 1A,B). Interestingly, a marked decrease in [3H]DA uptake was observed in cells treated with 3 μm MPP+ for 12 hr, although virtually no TH-positive neurons were dying at this time point. Although this is speculative at this point, the state of phosphorylation of the dopamine transporter by such protein kinases as protein kinase C (Vaughan et al., 1997) may be regulated rapidly by MPP + without significantly affecting the cell death of DA neurons. A recovery period after neurotoxin treatment may be necessary to ensure a correlation between decrease in dopamine uptake and loss of TH-positive neurons. Photomicrographs demonstrating the morphology of TH-immunoreactive neurons in untreated control and neurotoxin-treated groups are shown in Figure 1C. Most of TH-immunoreactive cells in untreated control display multiple processes. After neurotoxin treatment the dying neurons appear to retract processes. Interestingly, DA neurons exposed to MPP+ exhibit a marked reduction in the size of their soma by 30–40%, whereas DA neurons treated with 6-OHDA display virtually no reduction in soma size (Fig. 1D).
Figure 1.
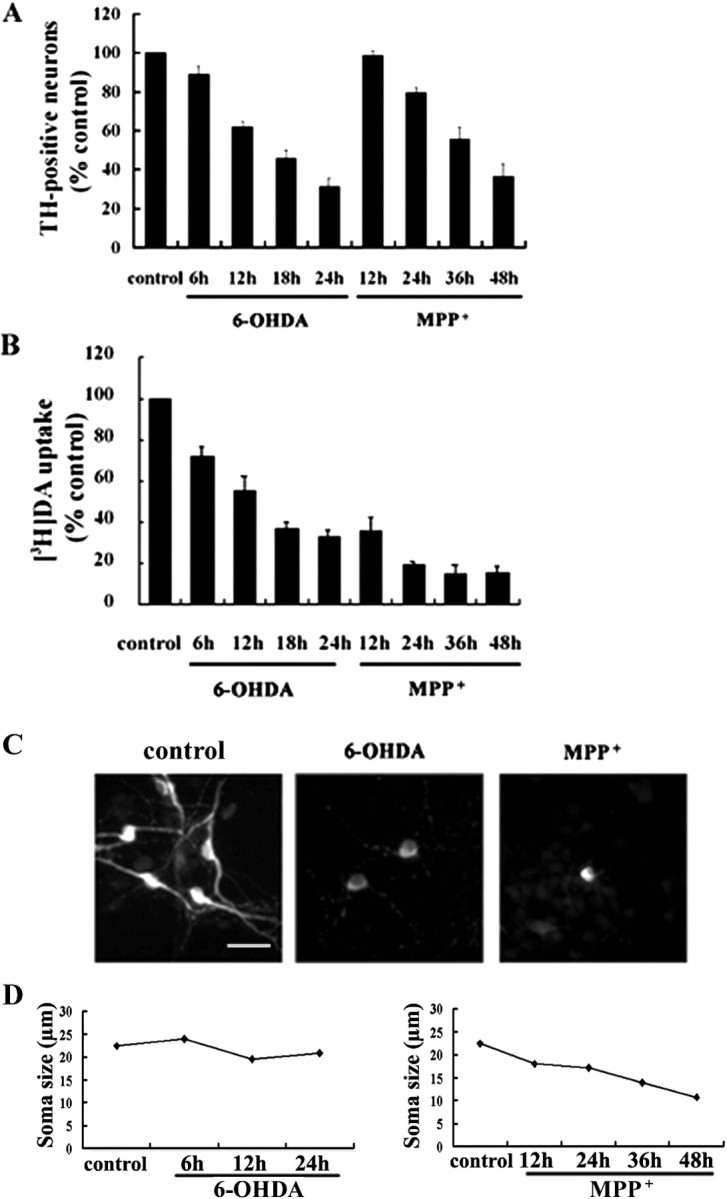
Effects of neurotoxins on loss of tyrosine hydroxylase (TH)-positive neurons, the rate of [3H]DA uptake, and size of soma in primary cultures of DA neurons. Cultures derived from the mesencephalon of E14 rat embryos were treated with 20 μm 6-OHDA or 3 μm MPP + for the time periods indicated. A, Cultures then were immunostained for TH, and the number of surviving TH-positive neurons was counted manually as described in Materials and Methods. B, Similarly, cultures were processed for a [3H]DA uptake assay. Values from each treatment were expressed as a percentage over the untreated control (100%). Data represent the mean ± SEM from at least three independent experiments. C, Photomicrographs of immunofluorescent labeling of TH are representative of untreated control, 6-OHDA-treated, or MPP +-treated cultures, respectively. Scale bar, 50 μm. D, Size of soma was measured as described in Materials and Methods after treatment of cells with neurotoxin for the indicated time periods. Values represent the mean ± SEM of two independent experiments.
To determine whether caspase mediates neurotoxin-induced death, we tested the protective effect of a broad-spectrum caspase inhibitor, zVAD-fmk, in DA neurons treated with 20 μm 6-OHDA or 3 μm MPP+. As shown in Figure 2A, 50 μm zVAD-fmk treatment significantly alleviated the loss of TH-positive neurons induced by 6-OHDA, but not by MPP+. Concentrations of zVAD-fmk ranging from 25 to 200 μm did not spare TH-positive neurons exposed to MPP+. Interestingly, zVAD-fmk significantly did not prevent a decrease in the rate of [3H]DA uptake induced by 6-OHDA (Fig. 2B). In accordance with our previous report that used MN9D cells (Choi et al., 1999b), 0.5 mmN-acetylcysteine (NAC) prevented the loss of TH-positive neurons and preserved [3H]DA uptake after 6-OHDA treatment. In contrast, the compound did not exert a protective role in the death of DA neurons after MPP+ or in the loss of [3H]DA uptake (Fig. 2A,B). None of these inhibitors blocked the MPP+-mediated loss of TH-positive neurons and decreased dopamine uptake regardless of the duration of the MPP+ treatment (ranging from 6 to 48 hr).
Figure 2.

6-OHDA-mediated, but not MPP +-mediated, neuronal death is attenuated by caspase inhibitor or antioxidant. Primary cultures of mesencephalic DA neurons were treated with 20 μm 6-OHDA for 24 hr or with 3 μm MPP + for 12 or 48 hr in the presence or absence of 50 μm zVAD-fmk (Z-VAD) or 0.5 mmN-acetylcysteine (NAC). A, The number of TH-positive neurons and the rate of [3H]DA uptake (B) were measured. Values from each treatment were expressed as a percentage over the untreated control (100%). Data represent the mean ± SEM from at least three independent experiments. Significance of difference was determined by ANOVA with post hoc Student's t test (*p < 0.01).
Colocalization of cytochrome c with TH-positive neurons after neurotoxin treatment
Cytochrome c plays a critical role in the initiation of the mitochondrial apoptotic pathway that activates procaspase-9 (Liu et al., 1996; Li et al., 1997; Zou et al., 1999). Activated caspase-9 then cleaves and activates downstream effector caspases. To distinguish further whether the death of DA neurons triggered by 6-OHDA and MPP+ is mediated by the mitochondrial apoptotic pathway, we attempted to visualize the profile of cytochrome c release in TH-positive neurons after 20 μm 6-OHDA or 3 μm MPP+ treatment for the various time periods indicated. After double-immunofluorescent labeling of cytochrome c and TH the distribution pattern of cytochrome c was imaged in neurons immunoreactive to TH with a confocal microscope. In untreated control cultures the immunostaining of cytochrome c displayed a punctate pattern and was distributed throughout the cytosol in the cell body and neurites in TH-positive neurons (Fig. 3). Clustering and fusion of cytochrome c immunoreactivity were induced markedly in a time-dependent manner after 6-OHDA treatment (Fig. 3, second and third panels). Although two separate patterns of staining distribution, namely, diffused or clustered, have been demonstrated previously for cytochrome c in cells that had undergone mitochondrial release of cytochrome c into the cytosol during tumor necrosis factor-induced apoptosis (Qin et al., 2001), we did not observe any sign of diffused staining pattern of cytochrome c in TH-positive neurons after treatment with 6-OHDA for up to 18 hr. When DA neurons were treated with a well known apoptotic inducer, staurosporine (1 μm), the clustered pattern of staining also prevailed here too (Fig. 3, fourth panel). Similarly, the clustered pattern of cytochrome c staining was apparent in primary cultures of striatal neurons during staurosporine-induced cell death (data not shown). Cotreatment with 0.5 mm NAC, but not with 50 μm zVAD-fmk, significantly inhibited a clustered staining pattern of cytochrome c in DA neurons after 6-OHDA treatment (Fig. 3, two bottom panels).
Figure 3.
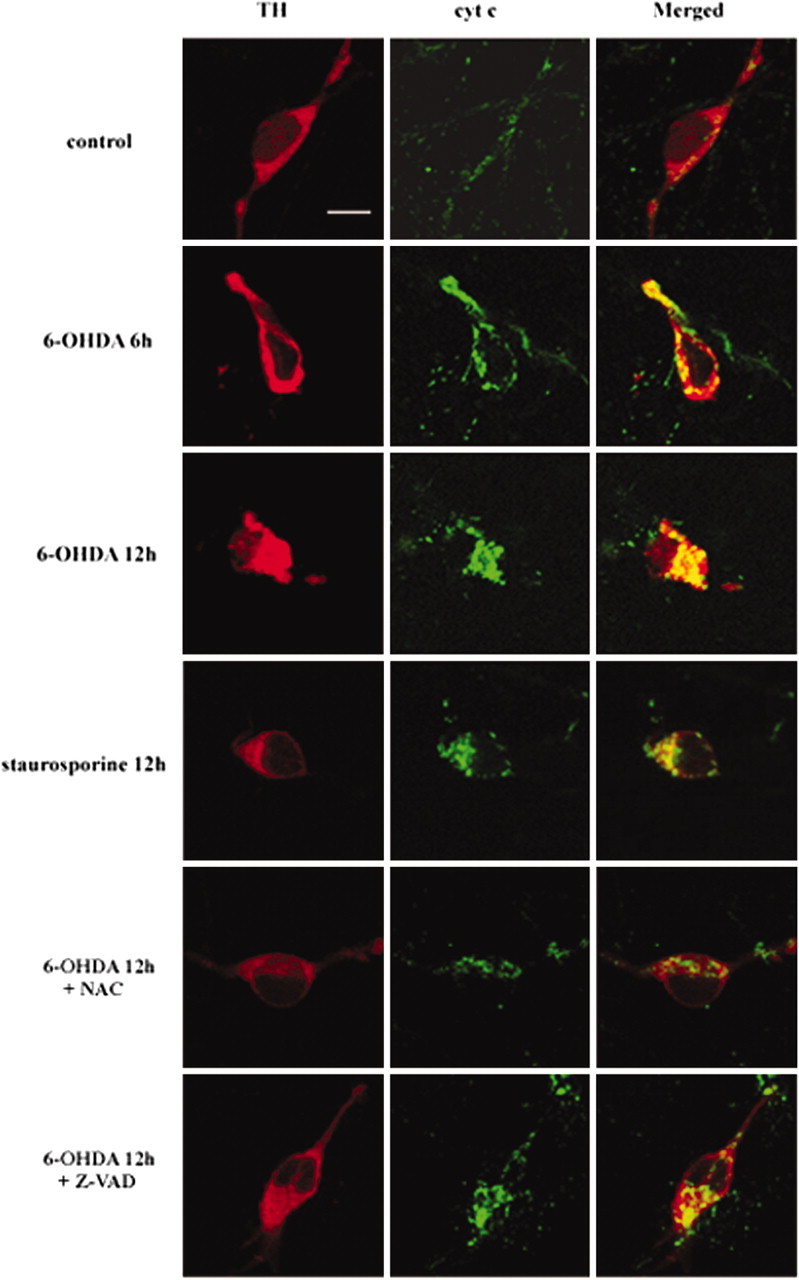
Colocalization of cytochrome c with TH-positive neurons after 6-OHDA treatment. Primary cultures of mesencephalic DA neurons were treated with 20 μm 6-OHDA or 1 μm staurosporine for the time periods indicated in the presence or absence of 50 μm zVAD-fmk (Z-VAD) or 0.5 mmN-acetylcysteine (NAC). Cultures were double immunolabeled with a rabbit polyclonal anti-TH and a mouse monoclonal anti-cytochrome c (cyt c) and were followed by incubation with Alexa Fluor 568 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG as described in Materials and Methods. Cultures then were examined by confocal microscopy. Scale bar, 10 μm.
Surprisingly, a modest clustered pattern of staining for cytochrome c also was observed in TH-positive neurons after exposure to 3 μm MPP + for 12 hr (Fig. 4, top panel). The clustered pattern was much more pronounced after 24–48 hr (Fig. 4, three bottom panels). Clustered staining of cytochrome c was distributed in a “patchy” manner in MPP +-treated DA neurons. The staining pattern of cytochrome c from randomly selected areas was examined under a confocal microscope. Results indicated that after 6-OHDA (n = 40) and MPP + treatment (n = 45) 65.0 and 48.9%, respectively, of TH-positive DA neurons demonstrated a clustered staining pattern of cytochrome c.
Figure 4.
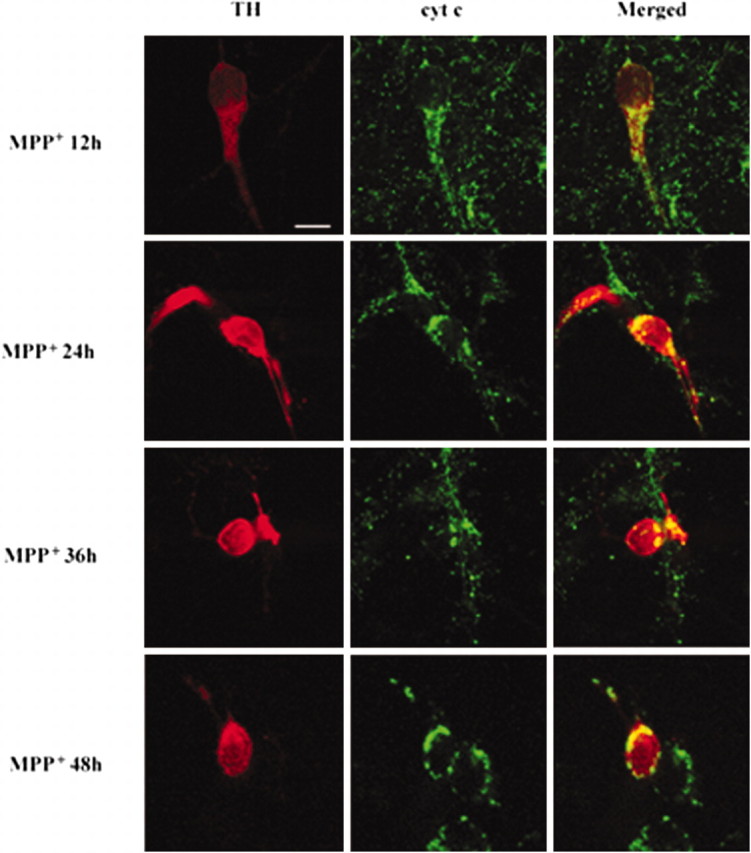
Colocalization of cytochrome c with TH-positive neurons after MPP + treatment. Primary cultures of mesencephalic DA neurons were treated with 3 μm MPP + for the time periods indicated. Cultures were double immunolabeled, using a rabbit polyclonal anti-TH and a mouse monoclonal anti-cytochrome c (cyt c), and were examined with confocal microscopy as described in Figure 3. Scale bar, 10 μm.
Release of mitochondrial cytochrome c into the cytosol in MN9D cells after neurotoxin treatment
Although cytochrome c release from primary cultured DA neurons after drug treatment is still not conclusive at this point, we tried to evaluate whether the clustered staining pattern of cytochrome c in primary cultured DA neurons is indicative of mitochondrial release of cytochrome c. MN9D cells were treated with 100 μm 6-OHDA or 50 μm MPP+ for various time periods and subjected to immunocytochemical localization of cytochrome c. As previously reported by us (Oh et al., 1995; Choi et al., 1999a,1999b), ∼60–70% of MN9D cells were dying after treatment with 100 μm 6-OHDA for 24 hr or with 50 μm MPP+ for 48 hr. Therefore, these doses of drugs were selected to compare the staining pattern of cytochrome c between DA neurons and MN9D cells. As shown in Figure 5B, treatment of MN9D cells with 100 μm 6-OHDA and 50 μm MPP+ induced a quite similar clustered pattern of cytochrome c staining when compared with that observed in primary cultured DA neurons. A similar clustered pattern of cytochrome c staining also was observed in MN9D cells treated with staurosporine (data not shown). Interestingly, the distribution pattern of mitochondria labeled by a mitochondrion-selective probe, MitoTracker Orange CMTMRos, demonstrated that mitochondria are dispersed regardless of treatment (Fig. 5A), suggesting that the clustered pattern of cytochrome c staining both in primary cultures of DA neurons and MN9D cells may indicate redistribution of cytochrome c to the cytoplasm rather than compaction of mitochondria per se after drug treatment. From immunoblot analysis the cytochrome c appeared in the cytosolic fraction as early as 3–6 hr and continued to increase subsequently after 100 μm 6-OHDA treatment (Fig. 5C). None of these cytosolic fractions was positive for cytochrome oxidase subunit IV, a marker protein for the mitochondrial fraction. At 1 μm staurosporine a similar pattern of cytochrome c release into the cytosolic fraction was observed in MN9D cells (data not shown)
Figure 5.

Release of mitochondrial cytochrome c into the cytosolic fractions after neurotoxin treatment. MN9D dopaminergic neuronal cells treated with 100 μm 6-OHDA for 12hr or with 50 μm MPP + for 36 hr were processed for labeling with a mitochondrion-selective probe Mito-Tracker Orange CMTMRos (A) and immunofluorescent localization of cytochrome c (cyt c; B) as described in Materials and Methods. Scalebar, 10 μm. MN9D dopaminergic neuronal cells were treated with 100 μm 6-OHDA (C) or with 50 μm MPP + (D) for the time periods indicated. Cells then were homogenized with a Dounce homogenizer and processed to obtain the cytosolic fractions as described previously (Gross et al., 1998). Cytosolic proteins (75 μg) were separated by 10% SDS-PAGE and transferred to PVDF nitrocellulose filters. Blots were immunolabeled with a mouse monoclonal anti-cyt c. Duplicate blots were immunolabeled with a mouse monoclonal anti-cytochrome c oxidase subunit IV (COX IV) to confirm the absence of the mitochondrial contamination. Similarly, the above blots were reprobed with anti-IκB antibody to confirm relatively equal protein loading in each lane. Specific bands were detected by enhanced chemiluminescence.
Slight but detectable levels of cytochrome c in the cytosolic fractions also were observed as early as 12 hr after 50 μm MPP + (Fig. 5D). Thereafter, marked mitochondrial release of cytochrome c into the cytosolic fraction was observed later than 30 hr after MPP + treatment. Again, none of these cytosolic fractions was contaminated with cytochrome c oxidase subunit IV, indicating that cytochrome c indeed was released into the cytosol after 6-OHDA or MPP + treatment in our culture models.
Colocalization of activated caspase-3 with TH-positive neurons after neurotoxin treatment
A double-immunofluorescent localization of activated caspase-3 and TH was performed in DA neurons after 6-OHDA or MPP+ treatment. In untreated control cultures there was virtually no coexpression of activated caspase-3 in TH-positive neurons (Fig. 6A). In DA neurons treated with 20 μm 6-OHDA for 12 or 24 hr, however, colocalization of activated caspase-3 was apparent in 3.8 or 11.9% of TH-positive neurons, respectively (Fig. 6A,B). This number is in agreement with the findings of others, suggesting that only a small subset of DA neurons is undergoing caspase activation at any given time period (Hartmann et al., 2000, 2001; Sherer et al., 2002). However, the possibility that there may be caspase-dependent and caspase-independent mechanisms in play during 6-OHDA-induced cell death cannot be ruled out. Although virtually no expression of activated caspase-3 in non-TH-expressing populations was detected before drug treatment, ∼3.1–7.8% of non-TH-expressing populations were positive for activated caspase-3 after 6-OHDA treatment for 12 and 24 hr, respectively (n = 2). Cotreatment with 50 μm zVAD-fmk or 0.5 mmN-acetylcysteine in large part abolished 6-OHDA-induced activation of caspase-3 in TH-positive neurons (2.1% for zVAD-fmk; 2.0% for N-acetylcysteine; Fig. 6B).
Figure 6.
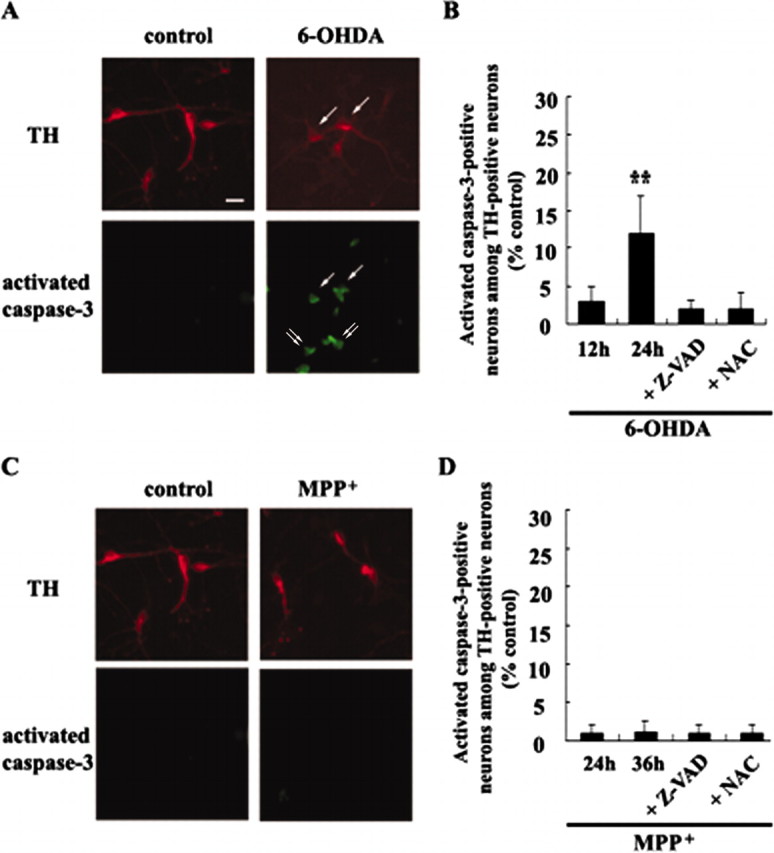
Colocalization of activated caspase-3 with TH-positive neurons after neurotoxin treatment. Primary cultures of mesencephalic DA neurons were treated with 20 μm 6-OHDA (A, B) or with 3 μm MPP + (C, D) for the time periods indicated in the presence or absence of 50 μm zVAD-fmk (Z-VAD) or 0.5 mmN-acetylcysteine (NAC). Cultures were double immunolabeled with a mouse monoclonal anti-TH and a rabbit polyclonal anti-activated caspase-3, followed by incubation with Texas Red-conjugated goat anti-mouse IgG and Oregon green-conjugated goat anti-rabbit IgG as described in Materials and Methods. Cultures then were examined with fluorescent microscopy. Shown are typical photomicrographs after 20 μm 6-OHDA for 24 hr (A) or 3 μm MPP + for 36 hr (C). Arrows indicate neurons that are positive for both activated caspase-3 and TH. Double arrows indicate non-TH-expressing cells that are positive for activated caspase-3. Scale bar, 20 μm. B, D, Neurons that were positive for activated caspase-3 among TH-positive neurons were counted as described in Materials and Methods. Data represent the mean ± SEM from three independent experiments (**p < 0.05; ANOVA with post hoc Student's t test).
However, virtually no TH-positive or TH-negative cells displayed immunoreactivity for activated caspase-3 in mesencephalic cultures treated with 3 μm MPP + for various time periods (< 0.8%; Fig. 6C,D). In addition, we were unable to detect any activated caspase-3-positive DA neurons that had been treated with 3 μm MPP + for up to 48 hr. Similarly, virtually no activated caspase-3-positive cells were observed even after treatment with a wide concentration range of MPP + (0.5–20 μm).
Lack of caspase activation in MN9D cells after MPP+ treatment
Because MPP+ induced mitochondrial release of cytochrome c in MN9D cells (Fig. 5B), we attempted further to examine whether caspases indeed are not activated in MN9D cells after MPP+ treatment. In MN9D cells treated with 100 μm 6-OHDA, activated caspase-9 and caspase-3 appeared (Fig. 7A). In MN9D cells treated with 50 μm MPP+, neither activated caspase-9 nor caspase-3 was detected for up to 48 hr; at this time point the majority of cells (>75%) was dead (Fig. 7A). Immunocytochemistry indicates that ∼25–30% of MN9D cells were positive for activated caspase-3 after 100 μm 6-OHDA for 24 hr (Fig. 7B). In contrast, none of the MN9D cells treated with 50 μm MPP+ for up to 48 hr showed immunoreactivity for activated caspase-3. Failure to activate caspase was observed at a wide concentration range of MPP+ (25–200 μm) as determined by fluorogenic caspase substrate assay, immunoblot analysis for activated caspase-3, -6, -8, and -9, and in a cotreatment study with various types of caspase inhibitors (data not shown). This further supports our data from primary cultured DA neurons that a caspase-dependent apoptotic pathway does not underlie MPP+-mediated death.
Figure 7.

Lack of caspase activation in MN9D cells after MPP + treatment. A, MN9D cells were treated with 100 μm 6-OHDA or with 50 μm MPP + for the time periods indicated. Cellular lysates (50 μg) were subjected to immunoblot analysis with an antibody that recognizes activated caspase-9 or activated caspase-3 as described in Materials and Methods. B, Representative photomicrographs of immunofluorescent localization of activated caspase-3 in MN9D cells after 100 μm 6-OHDA for 24 hr or after 50 μm MPP + for 48 hr. Scale bar, 50 μm.
Role of ATP in reconstitution of caspase activation in MN9D cells
Previously, Leist et al. (1997) have reported that sufficient ATP levels are required for the execution of the final phase of drug-induced apoptosis in human T-cells. Furthermore, dATP-dependent oligomerization of Apaf-1 is an important step in initiating an apoptotic protease cascade (Li et al., 1997). Our previous study in MN9D cells demonstrated that MPP+ induced a marked decrease in intracellular levels of ATP, whereas 6-OHDA did not affect the intracellular level of ATP (Kim et al., 2001). Thus we hypothesized that after treatment with MPP+ decreased levels of intracellular ATP at the time of cytochrome c release into the cytosol may be responsible for the failure to activate caspase. To examine this possibility, we performed a reconstitution reaction to measure caspase activity in the presence or absence of dATP or ATP, using the cytosolic fraction harvested from MN9D cells treated with 50 μm MPP+ for 12–15 hr. Caspase activity measured from the cytosolic fraction of 1 μm staurosporine-treated cell was used as a positive control. As shown in Figure 8, A and B, virtually no differences in the levels of caspase-9 and caspase-3 activity were observed in the cytosolic fraction from MN9D cells treated with MPP+ for 12, 24, and 36 hr as compared with those from untreated control. However, the single addition of either 1 mm dATP or 1 mm ATP to the cytosolic fraction obtained from MN9D cells treated with MPP+ for 15 hr was sufficient to activate both caspase-9 and caspase-3; this was determined by a fluorogenic caspase assay that used Ac-LEHDAFC or Ac-DEVD-AMC as a substrate for caspase-9-like and caspase-3-like activity, respectively. The levels of caspase-9-like activity were quite comparable to those from cells treated with staurosporine, whereas the levels of caspase-3-like activity were somewhat lower than those from staurosporine-treated cells. Immunoblot analysis of aliquots from the same reaction mixture also indicates that activated caspase-3 appeared if the cytosolic fraction of cells treated with MPP + was incubated in the presence of 1 mm ATP (Fig. 8C). The intensity and sizes of bands were quite comparable to those from staurosporine-treated cells. However, the addition of dATP to the cytosolic fraction obtained from MN9D cells treated with MPP + for longer than 36 hr did not induce caspase activation (data not shown), indicating that other unidentified factors also are involved in determining caspase activation after the MPP +-induced release of cytochrome c into the cytosol (Lankiewicz et al., 2000).
Figure 8.
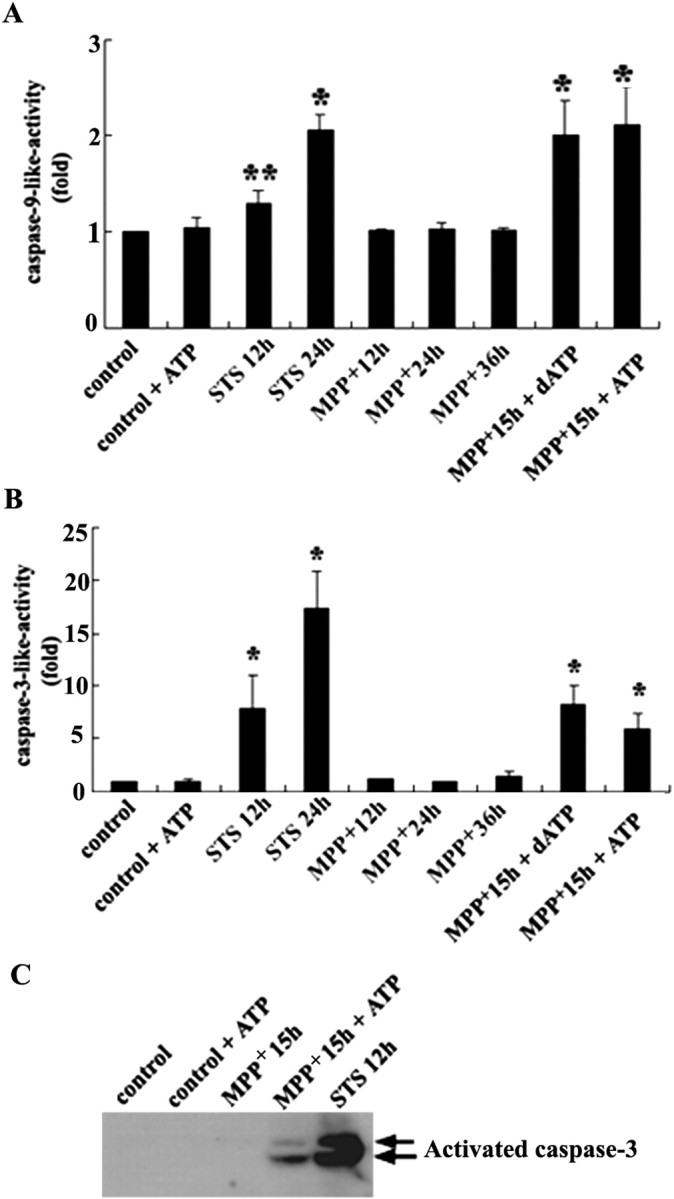
Reconstitution of caspase activation in vitro. MN9D cells were treated with 50 μm MPP + or 1 μm staurosporine (STS) for the time periods indicated. Cytosolic fractions (50 μg) were incubated with 40 μm Ac-LEHD-AFC for caspase-9-like activity (A) or with 25 μm Ac-DEVD-AMC for caspase-3-like activity (B) for 2 hr at 30°C in a reaction buffer with or without 1 mm dATP or ATP. Fluorogenic caspase activity was measured as described in Materials and Methods. Data represent the mean ± SEM from three to four independent experiments (*p < 0.01; **p < 0.05; ANOVA with post hoc Student's t test). C, Aliquots from the same reaction mixtures (MPP + for 15 hr in the presence or absence of 1 mm ATP; STS for 12 hr) were subjected to immunoblot analysis for activated caspase-3.
Discussion
Here we have demonstrated that caspase-dependent and -independent cell death pathways are induced in primary cultures of DA neurons after 6-OHDA or MPP+ treatment, respectively. This was determined by a double immunocytochemical localization of cytochrome c or activated caspase-3 in TH-positive neurons and by a cotreatment study with a caspase inhibitor. In addition, N-acetylcysteine prevents the loss of TH-positive neurons, a decrease in [3H]DA uptake, and the activation of caspase-3 induced by 6-OHDA, but not by MPP+. This is in agreement with our previous demonstration in MN9D cells that the reactive oxygen species-mediated signaling pathway plays a critical role in 6-OHDA-mediated apoptosis, whereas MPP+ acts on distinct cell death pathways (Oh et al., 1995; Choi et al., 1999a,b; Kim et al., 2001). Additionally, we have found that mitochondrial release of cytochrome c into the cytosol does not lead necessarily to the activation of procaspase-9 and procaspase-3. Thus caspase activation is observed only in cells treated with 6-OHDA, but not with MPP+, although cytochrome c is released into the cytosol in both cases. The lack of caspase activation in MPP+-treated cells is confirmed by caspase substrate cleavage assays, immunoblot analysis, and immunofluorescence analysis. Of the many possibilities to explain this phenomenon, decreased levels of intracellular ATP in MPP+-treated cells appear to be responsible for the lack of caspase activation. Taken together, our present data still favor the notion that 6-OHDA and MPP+ produce DA neuronal death by distinct cellular mechanisms.
MPTP or its active metabolite MPP+ has been used to induce selective DA neuronal cell death in experimental models of PD. However, several studies focusing on the mode of cell death induced by these neurotoxins have presented contrasting results (for review, see Nicotra and Parvez, 2000; Hartmann and Hirsch, 2001). Although the exact nature of this controversy is not understood, the interpretation of whether neurotoxin elicits apoptosis in bona fide DA neurons should be regarded with caution. For example, most of these studies have used a limited array of criteria such as TUNEL positivity to distinguish between apoptotic and non-apoptotic death and have not confirmed the cell types affected or have not used DA cells. A few studies that used postmortem human brains of PD have demonstrated a caspase-dependent apoptotic process in TH-positive or melanized neurons (Hartmann et al., 2000, 2001; Tatton, 2000). Although it remains to be determined whether (1) non-apoptotic changes exist in DA neurons of the postmortem brain, (2) only a small fraction of DA neurons remaining in the postmortem brain can be totally representative of the death mode of PD, and (3) apoptosis-like changes in the postmortem brain are influenced by perimortem factors, our data from MPP+-treated cells appear less supportive of apoptosis as a sole mode of DA neuronal cell death.
Thus far, several groups have demonstrated the colocalization of activated caspase-3 in TH-positive neurons derived from embryonic mesencephalon after MPP+ treatment (Hartmann et al., 2000; Viswanath et al., 2001; Bilsland et al., 2002). Parameters including culture conditions, severity of insult, and duration of MPP+ treatment may account for the discrepancy between our present data and others. In our study we have not seen any signs of caspase activation in primary cultures of DA neurons treated for up to 3 d with a wide concentration range of MPP+ (0.5–20 μm). Only 0.20–0.75% of both TH-positive and TH-negative cells were positive for activated caspase-3 at these conditions (n = 3). Therefore, severity of insult and duration of MPP+ treatment do not seem to be responsible for the discrepancy. Furthermore, we have not observed MPP+-induced caspase activation even after cultures are maintained in a defined serum-free medium (data not shown). It is interesting to note that a large number of activated caspase-3-positive neurons is present in untreated control cells in other studies, compared with a virtual absence of activated caspase-3-positive cells before and after MPP+ treatment in our case. Although it is highly speculative at present, a certain degree of apoptotic pressure already present even in the absence of MPP+ may prime DA neurons to take an apoptotic pathway over the non-apoptotic pathway in these three reports (Hartmann et al., 2000; Viswanath et al., 2001; Bilsland et al., 2002). In this regard it is interesting to note that the inhibition of caspase in primary cultures of DA neurons treated with MPP+ triggers a switch from apoptosis to necrosis (Hartmann et al., 2001). MPP + may have the potential to induce caspase-dependent and -independent cell death, and the final outcome may be determined by several extrinsic factors placed on cells in defined situations. Likewise, we have observed that the suppression of 6-OHDA-induced apoptosis by cotreatment of zVAD-fmk is not associated with a long-term increase in MN9D cell survival but, rather, seems to redirect from apoptotic death to necrotic form (data not shown). Therefore, it may be possible to assume that blocking of one cell death pathway results in a redirection to a different cell death pathway (Raymond et al., 2003).
Among the several apoptogenic factors released from the mitochondria in response to a wide variety of stimuli, cytochrome c is the best characterized for initiating the mitochondrial apoptotic pathway (Liu et al., 1996, 1997; Zou et al., 1999). Cytosolic cytochrome c and dATP-dependent oligomerization of Apaf-1 allow for recruitment of procaspase-9 into the apoptosome complex, leading to caspase-9 activation, which in turn activates caspase-3. Activated caspase-3 cleaves endogenous apoptotic substrates, completing the mitochondrial apoptotic pathway. In most cases, therefore, mitochondrial release of cytochrome c comprises the critical component of caspase activation in the mitochondrial apoptotic pathway (for review, see Hengartner, 2000). Although cytochrome c release from primary cultured DA neurons after drug treatment is still not conclusive at this point, nevertheless to our knowledge we are the first to demonstrate immunocytochemical localization of cytochrome c in primary cultures of DA neurons after neurotoxin treatment. Although the loss of punctate staining and a move to more diffuse, less intense staining pattern of cytochrome c has been reported in certain cell death paradigms, several other studies have indicated that a clumpy pattern of cytochrome c staining is apparent during cell death and have interpreted this as an indication of mitochondrial release of cytochrome c (Guegan et al., 2001; Jiang et al., 2001; Qin et al., 2001; Sugawara et al., 2002). In the present study the clustered staining pattern of cytochrome c prevails and may be indicative of mitochondrial release of cytochrome c in TH-positive DA neurons after 6-OHDA or MPP+ treatment. Interestingly, clustered staining pattern of cytochrome c in MPP+-treated DA neurons is distributed in a patch-like manner, whereas that in 6-OHDA or staurosporine is distributed mainly evenly throughout the cytosol (compare Figs. 3, 4). We do not know whether this phenomenon is correlated somewhat with the pattern of caspase activation. However, our in vitro reconstitution assay suggesting that intracellular levels of ATP at or around the time of cytochrome c release are important for subsequent caspase activation provides one of several possible explanations of why MPP + does not elicit caspase-dependent cell death even after mitochondrial release of cytochrome c. It would be very interesting to explore this possibility fully by manipulating intracellular levels of ATP in the 6-OHDA- or MPP +-treated cells to see whether it can shift the cell death pathway.
As with the recent findings by others that redistribution of cytochrome c into the cytosol is not sufficient for and does not always result in caspase activation in neurons (Deshmukh and Johnson, 1998; Von Ahsen et al., 2000; Zhou et al., 2000; Torok et al., 2002), the discrepancy between mitochondrial release of cytochrome c and the lack of caspase activation in our MPP+-treated cells also can be explained by several other factors. Among many potential factors, a recent report on calpain draws our attention. This suggested that activation of calpain prevents the entry of NMDA-treated hippocampal neurons into a caspase-dependent apoptosis after mitochondrial release of cytochrome c (Lankiewicz et al., 2000). Our previous study demonstrated that calpain is activated in MN9D cells treated with MPP+, but not with 6-OHDA (Choi et al., 2001). In preliminary studies we consistently have found calpain-cleaved fodrin immunoreactivity in primary cultures of DA neurons treated with MPP+, but not with 6-OHDA. Activation of calpain(s) after MPP+ treatment therefore may prevent the entry of DA neurons into a caspase-dependent cell death by inhibiting the processing of procaspase-9 and caspase-3 into their active subunits. Further studies delineating cellular events associated with MPP +-induced cell death may reveal more definitely a diverse role of cytochrome c and/or the molecular mechanism(s) associated with the failure of cytochrome c-initiated apoptotic pathway in this paradigm.
From a therapeutic point of view the inhibition of caspase activity would be one of the favored methods for rescuing dying DA neurons. However, data from our lab and others imply that the neuroprotective strategy of inhibiting caspase activity may not be sufficient for a full functional recovery of DA neurons in patients with PD. Therefore, the careful examination of the existence of other than caspase-dependent death signaling pathways could contribute to the development of more effective therapeutic concepts that promote both cell survival and functional recovery of DA neurons.
Footnotes
This work was supported by Grant HMP-00-CH-13-0012 from the Ministry of Health and Welfare, in part by the Korea Science and Engineering Foundation through the Brain Disease Research Center at Ajou University, and by Ministry of Science and Technology Grants FPR-02-A-1 and M1-0108-00-0096. We gratefully acknowledge Dr. A. Heller for providing us with the MN9D cell line.
Correspondence should be addressed to Dr. Young J. Oh, Department of Biology, Yonsei University College of Science, 134 Shinchon-Dong, Seodaemoon-Gu, Seoul 120-749, Korea. E-mail: yjoh@yonsei.ac.kr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235069-10$15.00/0
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y ( 1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12: 25–31. [PubMed] [Google Scholar]
- Banati RB, Daniel SE, Blunt SB ( 1998) Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson's disease. Mov Disord 13: 221–227. [DOI] [PubMed] [Google Scholar]
- Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, Hefti F, Harper SJ ( 2002) Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci 22: 2637–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH ( 1979) Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA 76: 514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Kholodilov NG ( 1998) Programmed cell death: does it play a role in Parkinson's disease? Ann Neurol 44: S126–S133. [DOI] [PubMed] [Google Scholar]
- Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer BH, Hoffmann PC, Heller A ( 1991) Immortalization of embryonic mesencephalic dopaminergic neurons by somatic fusion. Brain Res 552: 67–76. [DOI] [PubMed] [Google Scholar]
- Choi HK, Won L, Roback JD, Wainer BH, Heller A ( 1992) Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Proc Natl Acad Sci USA 89: 8943–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Canzoniero LM, Sensi SL, O'Malley KL, Gwag BJ, Sohn S, Kim JE, Oh TH, Lee EB, Oh YJ ( 1999a) Characterization of MPP +-induced cell death in a dopaminergic neuronal cell line: role of macromolecule synthesis, cytosolic calcium, caspase, and Bcl-2-related proteins. Exp Neurol 159: 274–282. [DOI] [PubMed] [Google Scholar]
- Choi WS, Yoon SY, Oh TH, Choi EJ, O'Malley KL, Oh YJ ( 1999b) Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP +-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res 57: 86–94. [DOI] [PubMed] [Google Scholar]
- Choi WS, Lee EH, Chung CW, Jung YK, Jin BK, Kim SU, Oh TH, Saido TC, Oh YJ ( 2001) Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J Neurochem 77: 1531–1541. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson Jr EM ( 1998) Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c Neuron 21: 695–705. [DOI] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Bales KR, Ling ZD, Carvey PM, Paul SM ( 1998) Peptide inhibitors of caspase-3-like proteases attenuate 1-methyl-4-phenylpyridinum-induced toxicity of cultured fetal rat mesencephalic dopamine neurons. Neuroscience 86: 701–707. [DOI] [PubMed] [Google Scholar]
- Gross A, Jockel J, Wei MC, Korsmeyer SJ ( 1998) Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction, and apoptosis. EMBO J 17: 3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S ( 2001) Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 21: 6569–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley A, Stone JM, Heron C, Cooper JM, Schapira AHV ( 1994) Complex I inhibitors induce dose-dependent apoptosis in PC12 cells: relevance to Parkinson's disease. J Neurochem 63: 1987–1990. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Hirsch EC ( 2001) Parkinson's disease: the apoptosis hypothesis revisited. Adv Neurol 86: 143–153. [PubMed] [Google Scholar]
- Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC ( 2000) Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci USA 97: 2875–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC ( 2001) Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis. J Neurosci 21: 2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO ( 2000) The biochemistry of apoptosis. Nature 407: 770–776. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S ( 1995) Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 4: 257–269. [DOI] [PubMed] [Google Scholar]
- Jeon BS, Jackson-Lewis V, Burke RE ( 1995) 6-Hydroxydopamine lesion of the rat substantia nigra: time course and morphology of cell death. Neurodegeneration 4: 131–137. [DOI] [PubMed] [Google Scholar]
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH ( 2001) Zn 2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem 276: 47524–47529. [DOI] [PubMed] [Google Scholar]
- Kim HE, Yoon SY, Lee JE, Choi WS, Jin BK, Oh TH, Markelonis GJ, Chun SY, Oh YJ ( 2001) MPP + downregulates mitochondrially encoded gene transcripts and their activities in dopaminergic neuronal cells: protective role of Bcl-2. Biochem Biophys Res Commun 286: 659–665. [DOI] [PubMed] [Google Scholar]
- Kingsbury AE, Mardsen CD, Foster OJ ( 1998) DNA fragmentation in human substantia nigra: apoptosis or perimortem effect? Mov Disord 13: 877–884. [DOI] [PubMed] [Google Scholar]
- Kosel S, Egensperger R, von Eitzen U, Mehraein P, Graeber MB ( 1997) On the question of apoptosis in the parkinsonian substantia nigra. Acta Neuropathol (Berl) 93: 105–108. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH ( 2000) Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem 275: 17064–17071. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P ( 1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 185: 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X ( 1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X ( 1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c Cell 86: 147–157. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Dugan LL, O'Malley KL ( 1999) Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci 19: 1284–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki H, Nakamura N, Nishi K, Mizuno Y ( 1994) Apoptosis is induced by 1-methyl-4-phenylpyridinium ion (MPP +) in ventral mesencephalic–striatal coculture in rat. Neurosci Lett 170: 191–194. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Goto K, Mori H, Mizuno Y ( 1996) Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci 137: 120–123. [DOI] [PubMed] [Google Scholar]
- Nicotra A, Parvez SH ( 2000) Cell death induced by MPTP, a substrate for monoamine oxidase B. Toxicology 153: 157–166. [DOI] [PubMed] [Google Scholar]
- Oh YJ, Wong SC, Moffat M, O'Malley KL ( 1995) Overexpression of Bcl-2 attenuates MPP +, but not 6-ODHA, induced cell death in a dopaminergic neuronal cell line. Neurobiol Dis 2: 157–167. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kikly KK, Sapp E, Kegel KB, Aronin N, DiFiglia M ( 2001) Pro-caspase-8 is predominantly localized in mitochondria and released into cytoplasm upon apoptotic stimulation. J Biol Chem 276: 8079–8086. [DOI] [PubMed] [Google Scholar]
- Raymond MA, Mollica L, Vigneault N, Desormeaux A, Chan JS, Filep JG, Hebert MJ ( 2003) Blockade of the apoptotic machinery by cyclosporin A redirects cell death toward necrosis in arterial endothelial cells: regulation by reactive oxygen species and cathepsin D. FASEB J 17: 515–517. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT ( 2002) An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered α-synuclein metabolism and oxidative damage. J Neurosci 22: 7006–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Weller M, Haid S, Beinroth S, Miller SW, Wullner U, Davis RE, Dichgans J, Klockgether T, Schulz JB ( 1999) MPP + inhibits proliferation of PC12 cells by a p21 WAF1/Cip1-dependent pathway and induces cell death in cells lacking p21 WAF1/Cip1 Exp Cell Res 250: 75–85. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH ( 2002) Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci 22: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Todd RD, Heller A, O'Malley KL ( 1994) Pharmacological and functional characterization of D2, D3, and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J Pharmacol Exp Ther 268: 495–502. [PubMed] [Google Scholar]
- Tatton NA ( 2000) Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp Neurol 166: 29–43. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ ( 1997) In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labeling and acridine orange staining. Neuroscience 77: 1037–1048. [DOI] [PubMed] [Google Scholar]
- Tompkins MM, Basgall EJ, Zamrini E, Hill WD ( 1997) Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol 150: 119–131. [PMC free article] [PubMed] [Google Scholar]
- Torok NJ, Higuchi H, Bronk S, Gores GJ ( 2002) Nitric oxide inhibits apoptosis downstream of cytochrome c release by nitrosylating caspase 9. Cancer Res 62: 1648–1653. [PubMed] [Google Scholar]
- Vaughan RA, Huff RA, Uhl GR, Kuhar MJ ( 1997) Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J Biol Chem 272: 15541–15546. [DOI] [PubMed] [Google Scholar]
- Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK ( 2001) Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci 21: 9519–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Ahsen O, Waterhouse NJ, Kuwana T, Newmeyer DD, Green DR ( 2000) The “harmless” release of cytochrome c Cell Death Differ 7: 1192–1199. [DOI] [PubMed] [Google Scholar]
- Wullner U, Kornhuber J, Weller M, Schulz JB, Loschmann PA, Riederer P, Klockgether T ( 1999) Cell death and apoptosis-regulating proteins in Parkinson's disease—a cautionary note. Acta Neuropathol (Berl) 97: 408–412. [DOI] [PubMed] [Google Scholar]
- Zhou H, Li XM, Meinkoth J, Pittman RN ( 2000) Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol 151: 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X ( 1999) An APAF-1·cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549–11556. [DOI] [PubMed] [Google Scholar]