

Abstract
Spinal and bulbar muscular atrophy (SBMA) is an inherited motor neuron disease caused by the expansion of the polyglutamine (polyQ) tract within the androgen receptor (AR). The nuclear inclusions consisting of the mutant AR protein are characteristic and combine with many components of ubiquitin–proteasome and molecular chaperone pathways, raising the possibility that misfolding and altered degradation of mutant AR may be involved in the pathogenesis. We have reported that the overexpression of heat shock protein (HSP) chaperones reduces mutant AR aggregation and cell death in a neuronal cell model (Kobayashi et al., 2000). To determine whether increasing the expression level of chaperone improves the phenotype in a mouse model, we cross-bred SBMA transgenic mice with mice overexpressing the inducible form of human HSP70. We demonstrated that high expression of HSP70 markedly ameliorated the motor function of the SBMA model mice. In double-transgenic mice, the nuclear-localized mutant AR protein, particularly that of the large complex form, was significantly reduced. Monomeric mutant AR was also reduced in amount by HSP70 overexpression, suggesting the enhanced degradation of mutant AR. These findings suggest that HSP70 overexpression ameliorates SBMA phenotypes in mice by reducing nuclear-localized mutant AR, probably caused by enhanced mutant AR degradation. Our study may provide the basis for the development of an HSP70-related therapy for SBMA and other polyQ diseases.
Keywords: HSP70, chaperone, polyglutamine, SBMA, transgenic mice, protein degradation
Introduction
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders caused by the expansion of a trinucleotide CAG repeat in the causative genes (Zoghbi and Orr, 2000). To date, nine polyQ diseases have been identified (Ross, 2002). Spinal and bulbar muscular atrophy (SBMA) is a polyQ disease, characterized by proximal muscle atrophy, weakness, contraction fasciculations, and bulbar involvement (Kennedy et al., 1968; Sobue et al., 1989;Takahashi, 2001). In SBMA, a polymorphic CAG repeat with 14–32 CAGs expands to 40–62 CAGs in the first exon of the androgen receptor (AR) gene (Tanaka et al., 1996) and has somatic mosaicism (Tanaka et al., 1999). There is an inverse correlation between the CAG repeat size and the age at onset or the disease severity in SBMA (Doyu et al., 1992; Igarashi et al., 1992; La Spada et al., 1992). In SBMA, nuclear inclusions (NIs) containing mutant AR have been observed in the brainstem motor nuclei, spinal motor neurons, and some visceral organs (Li et al., 1998a,b). Such neuronal inclusions are common pathological features in polyQ diseases and are also colocalized with many components of ubiquitin–proteasome and molecular chaperones (Chai et al., 1999; Huynh et al., 2000; Adachi et al., 2001; Zander et al., 2001; Schmidt et al., 2002), raising the possibility that misfolding and altered degradation of the mutant protein may be involved in the pathogenesis of SBMA as well as other polyQ diseases (Stenoien et al., 1999; Waelter et al., 2001). Furthermore, these chaperones and proteasomes would facilitate refolding or proteolysis of the mutant protein and may play a role in protecting neuronal cells against the toxic properties of the expanded polyQ (Cummings et al., 1998;Kobayashi et al., 2000). We have shown recently that overexpression of heat shock proteins (HSPs) decreases the aggregate formation of truncated AR with the expanded polyQ and markedly prevents cell death in the neuronal cell model of SBMA (Kobayashi et al., 2000; Kobayashi and Sobue, 2001). HSP70 overexpression has been reported to enhance the solubility and degradation of mutant AR (Bailey et al., 2002). HSPs have also been shown to suppress aggregate formation and cellular toxicity in a wide range of polyQ disease models (Cummings et al., 1998; Warrick et al., 1999; Carmichael et al., 2000). Recently, overexpression of the inducible form of rat HSP70 ameliorated neurologic deficits and the neuronal degeneration of spinocerebellar ataxia type 1 (SCA1) transgenic mice, whereas the NIs consisting of mutant ataxin-1 were not reduced (Cummings et al., 2001).
In the present study, we report that overexpression of the inducible form of human HSP70 markedly ameliorated clinical and pathological phenotypes, and that this amelioration was correlated with the reduction of nuclear-localized mutant AR protein complexes in the mouse model of SBMA (Katsuno et al., 2002). Furthermore, the amount of monomeric mutant AR was also significantly reduced in the double-transgenic mice, suggesting that the degradation of mutant AR may have been accelerated by the overexpression of HSP70.
Materials and Methods
Assessment of motor ability. All animal experiments were performed in accordance with the National Institutes of HealthGuide for the Care and Use of Laboratory Animals and were approved by the Nagoya University Animal Experiment Committee. Motor ability was assessed using an Economex Rotarod (Columbus Instruments, Columbus, OH) on a weekly basis as described previously (Adachi et al., 2001). The period for which a mouse could remain on a rotating axle (diameter, 3.6 cm; speed of rotation, 16 rpm) without falling was measured. Three trials were performed, and the longest duration on the rod was recorded for every mouse. The timer was stopped if the mouse fell from the rod or after an arbitrary limit of 180 sec. Cage activity was measured weekly while each mouse was in a transparent acrylic cage (16 × 30 × 14 cm, width × depth × height) within a soundproofed box as described previously (Katsuno et al., 2002). Spontaneous motor activity was measured by means of an animal behavior system (Neuroscience Inc., Tokyo, Japan), which monitored and counted all spontaneous movements, both vertical and horizontal, including locomotion, rearing, head movements, etc. All counts were automatically totaled and recorded in 24 hr.
Immunohistochemistry. We perfused 20 ml of a 4% paraformaldehyde fixative in 0.1 m phosphate buffer, pH 7.4, through the left cardiac ventricle of mice deeply anesthetized with ketamine–xylazine, postfixed tissues in 10% phosphate-buffered formalin, and processed tissues for paraffin embedding. Then we deparaffinized 4-μm-thick tissue sections, dehydrated with alcohol, and treated for antigen retrieval (Katsuno et al., 2002). For the HSP70 immunohistochemical study, the paraffin sections were pretreated with trypsin (Dako, Glostrup, Denmark) for 20 min at 37°C. The tissue sections were blocked with normal animal serum (1:20) and incubated with mouse anti-expanded polyQ (1:10,000) (1C2; Chemicon, Temecula, CA) and goat polyclonal antibody to HSP70 (1:500) (K-20; Santa Cruz Biotechnology, Santa Cruz, CA). Then the sections were incubated with biotinylated anti-species-specific IgG (Vector Laboratories, Burlingame, CA). Immune complexes were visualized using streptavidin–horseradish peroxidase (Dako) and 3,3′-diaminobenzidine (Dojindo, Kumamoto, Japan) substrate. Sections were counterstained with methyl green.
For double-labeling immunohistochemistry, sections were preincubated with normal horse serum diluted in 0.02 m PBS buffer, pH 7.4, containing bovine serum albumin. The sections were then incubated with goat anti-HSP70 antibody (1:500) (K-20; Santa Cruz) at 4°C overnight, washed with 0.02 m PBS buffer, incubated with biotinylated horse anti-goat IgG, stained with streptavidin–alkaline phosphatase, and visualized with fast red. 1C2 antibody (1:10,000; Chemicon) was subsequently applied to sections at 4°C overnight. After being washed, the sections were incubated with horseradish peroxidase-labeled donkey anti-mouse Ig F(ab′)2 (Amersham Biosciences, Buckinghamshire, UK), which had been demonstrated to cross-react with neither goat nor horse sera, and visualized with 3,3′-diaminobenzidine. For double-immunofluorescence staining of the spinal cord, sections were blocked with 5% normal horse serum and then sequentially incubated with K-20 antibody (1:500; Santa Cruz Biotechnology) and 1C2 antibody (1:10,000; Chemicon) at 4°C overnight. After incubation with biotinylated horse anti-goat IgG (Vector Laboratories) for 8 hr at 4°C, the sections were incubated with Alexa-488-conjugated streptavidin (1:400; Molecular Probes, Leiden, The Netherlands) and Alexa-568-conjugated goat anti-mouse IgG (1:1300; Molecular Probes), which had been demonstrated to cross-react with neither goat nor horse sera, for 8 hr at 4°C. The sections were then examined and photographed under a confocal laser scanning microscope (MRC 1024; Bio-Rad, Hercules, CA).
As for the immunohistochemistry of SBMA patients, nine patients with clinicopathologically and genetically confirmed SBMA (age, 51–84 years; mean, 64.3) and three non-neurological controls (age, 51–76 years; mean, 64.0) served as the subjects of the present study. Paraffin-embedded sections of the spinal cord and brain were obtained and examined in the same way as for the transgenic mice.
Quantification of 1C2-positive cells in the spinal cord and muscle. For the assessment of 1C2-positive cells, 4-μm-thick coronal sections of the thoracic spinal cord and gastrocnemius muscle stained by 1C2 antibody (1:10,000; Chemicon) were prepared, and the number of 1C2-positive cells for one mouse was counted using a light microscope with a computer-assisted image analyzer (Luzex FS; Nikon, Tokyo, Japan). For the assessment of 1C2-positive cells in the ventral horn of the spinal cord, 50 consecutive transverse sections of the thoracic spinal cord were prepared, and the 1C2-positive cells present within the ventral horn on every fifth section were counted as described previously (Terao et al., 1996; Adachi et al., 2001). Populations of 1C2-positive cells were expressed as the number per square millimeter. For the assessment of 1C2-positive cells in the muscle, 1C2-positive cells were calculated from counts of >500 fibers in randomly selected areas and were expressed as the number per 100 muscle fibers.
Western blots. We exsanguinated mice under ketamine–xylazine anesthesia and snap-froze their tissues with powdered CO2 in acetone. Frozen tissue (0.1 gm wet weight) was homogenized in 1000 μ1 of lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mmNaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS with 1 mm PMSF and aprotinin at 6 μg/ml). Homogenates were spun at 2500 × g for 15 min at 4°C. The protein concentration of the supernatant was determined using detergent-compatible protein assay (Bio-Rad). Each lane on a 5–20% SDS-PAGE gel was loaded with protein (200 μg for the spinal cord and 80 μg for the muscle from the supernatant fraction), which was transferred to Hybond-P membranes (Amersham Biosciences) using 25 mm Tris, 192 mm glycine, 0.1% SDS, and 10% methanol as transfer buffer. Kaleidoscope prestained standards were used as size markers (Bio-Rad). Proteins were then transferred to Hybond-P membranes, which were subsequently blocked in 5% milk in TBS containing 0.05% Tween 20 and incubated with appropriate primary antibodies using standard techniques. Primary antibodies were used at the following concentrations: rabbit anti-AR antibody (1:1000 N-20; Santa Cruz Biotechnology); mouse anti-HSP70/heat shock cognate 70 (HSC70) antibody (1:5000, W-27; Santa Cruz Biotechnology). We performed second antibody probing and detection using the ECL+plus kit (Amersham Biosciences). The HRP-conjugated secondary antibodies used were anti-rabbit Ig F(ab′)2 and anti-mouse Ig F(ab′)2 (1:5000; Amersham Biosciences). Nuclear and cytoplasmic fractions were extracted with a NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit according to the protocol of the manufacturer (Pierce, Rockford, IL). Each lane on a 5–20% SDS-PAGE gel was loaded with 200 μg of protein for the spinal cord and 80 μg for the muscle from each fraction. Immunoprecipitation was performed using 1 mg of the total protein lysate, 10 μl of protein G–Sepharose (Amersham Biosciences), and 5 μl of anti-AR antibody (N-20; Santa Cruz Biotechnology). Protein was eluted from beads by boiling for 3 min in 10 μl of elution buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 60 μl/ml 2-mercaptoethanol, and 10% glycerol). The elutes were loaded on SDS–polyacrylamide gels. Blots were sequentially probed with goat anti-HSP70 antibody (K-20; Santa Cruz Biotechnology).
The signal intensity was analyzed using the NIH Image program (version 1.62). Relative signal intensity was computed as the signal intensity of each sample divided by that of the AR-97Q/HSP70(−/−) mice.
Filter-trap assay. Filtration of proteins through a 0.2 μm cellulose acetate membrane (Sartorius AG, Goettingen, Germany) was performed using a slot-blot apparatus (Bio-Rad). The membranes were washed three times with TBS buffer and supported by three pieces of filter paper (Bio-Rad). We also put 0.45 μm nitrocellulose membrane (Bio-Rad) under the cellulose acetate membrane to capture the monomeric AR protein passing through this membrane. Samples of protein (200 μg) for the spinal cord and for the muscle (80 μg) were prepared in a final volume of 200 μl in lysis buffer, loaded, and gently vacuumed. Membranes were washed three times with TBS containing 0.05% Tween 20. Slot-blots were probed as described for Western blots.
Statistical analysis. We analyzed data using the unpairedt test and log-rank test from Statview software version 5 (Hulinks, Tokyo, Japan).
Results
Nondeleterious effects of HSP70 overexpression and generation of double-transgenic mice
Because HSP70s have a wide variety of functions, we examined whether the overexpression of HSP70 under the control of the human β-actin promoter has deleterious effects on phenotypes in mice (Plumier et al., 1995). Motor function in the mice with HSP70 overexpression was not affected; a Rotarod task until 40 weeks revealed no impairment in either hemizygous or homozygous transgenic mice overexpressing HSP70 (data not shown). Histological examination at 40 weeks of age did not show any detectable effect on the neuronal cell morphology and population and on the muscular structure in the overexpression of human HSP70 alone (data not shown). These studies indicated that the overexpression of human HSP70 alone does not impair neuronal development and motor function.
To determine whether the overexpression of human HSP70 could ameliorate the disease phenotype of the SBMA transgenic mouse model, we crossed the mice expressing full-length human AR with 97-polyQ tract (AR-97Q mice, 4–6 line) (Katsuno et al., 2002) with mice that overexpress human HSP70 under the control of the human β-actin promoter (Plumier et al., 1995). The SBMA model (AR-97Q mice) shows small body size, short lifespan, progressive muscle atrophy and weakness, and reduced cage activity (Katsuno et al., 2002). Because the phenotypes of these SBMA transgenic mice are markedly pronounced in male transgenic mice similarly to SBMA patients (Katsuno et al., 2002), we used male transgenic mice in this study. We generated the AR-97Q/HSP70(tg/tg) mice as homozygotes and the AR-97Q/HSP70(tg/−) mice as hemizygotes, as well as the AR-97Q/HSP70(−/−) mice as a control transgenic mouse line. The SBMA transgene expression was at the hemizygous level in all AR-97Q/HSP70 double-transgenics.
Human HSP70 overexpression ameliorates motor phenotypes of SBMA transgenic mice
To determine whether HSP70 overexpression has an ameliorative effect on the motor phenotypes, we performed the Rotarod task and the measurement of locomotor cage activity by infrared sensor system with the double-transgenic mice (Fig.1A,B). The AR-97Q/HSP70(−/−) mice showed motor impairment on the Rotarod task as early as 9 weeks after birth; by 12 weeks and 25 weeks of age they began to show significant impairment compared with AR-97Q/HSP70(tg/−) mice (p< 0.05) and AR-97QHSP70(tg/tg) mice (p< 0.001), respectively. (Fig. 1A). Although both the AR-97Q/HSP70(tg/tg) and AR-97Q/HSP70(tg/−) mice performed better than the AR-97Q/HSP70(−/−) mice, the AR-97Q/HSP70(tg/tg) mice were on the rod longer than the AR-97Q/HSP70(tg/−) mice during the trial. The locomotor cage activity of the AR-97Q/HSP70(−/−) mice was also significantly decreased at 21 weeks in comparison with the other two double-transgenics (p < 0.05) (Fig.1B). No lines were distinguishable in terms of body weight at birth. The AR-97Q/HSP70(−/−)mice lost weight significantly earlier than the AR-97Q/HSP70(tg/tg) mice (p < 0.01) (Fig. 1C). The survival rate was significantly more prolonged in the AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice than in the AR-97Q/HSP70(−/−) mice (p < 0.01 and p < 0.005, respectively) (Fig. 1D). The affected AR-97Q/HSP70(−/−) mice exhibited motor weakness, with dragging of the legs or short steps, whereas the AR-97Q/HSP70(tg/tg) mice showed almost normal ambulation and the AR-97Q/HSP70(tg/−) mice only somewhat short steps (Fig. 1E). The AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice showed significantly longer steps than the AR-97Q/HSP70(−/−) mice (Fig.1F). Although both the AR-97Q/HSP70(tg/tg) and AR-97Q/HSP70(tg/−) mice showed ameliorated phenotypic expressions, the AR-97Q/HSP70(tg/tg) mice were better than the AR-97Q/HSP70(tg/−) mice in most of the parameters, suggesting that the improved motor phenotype depended on the HSP70 expression level rather than on the genetic background.
Fig. 1.
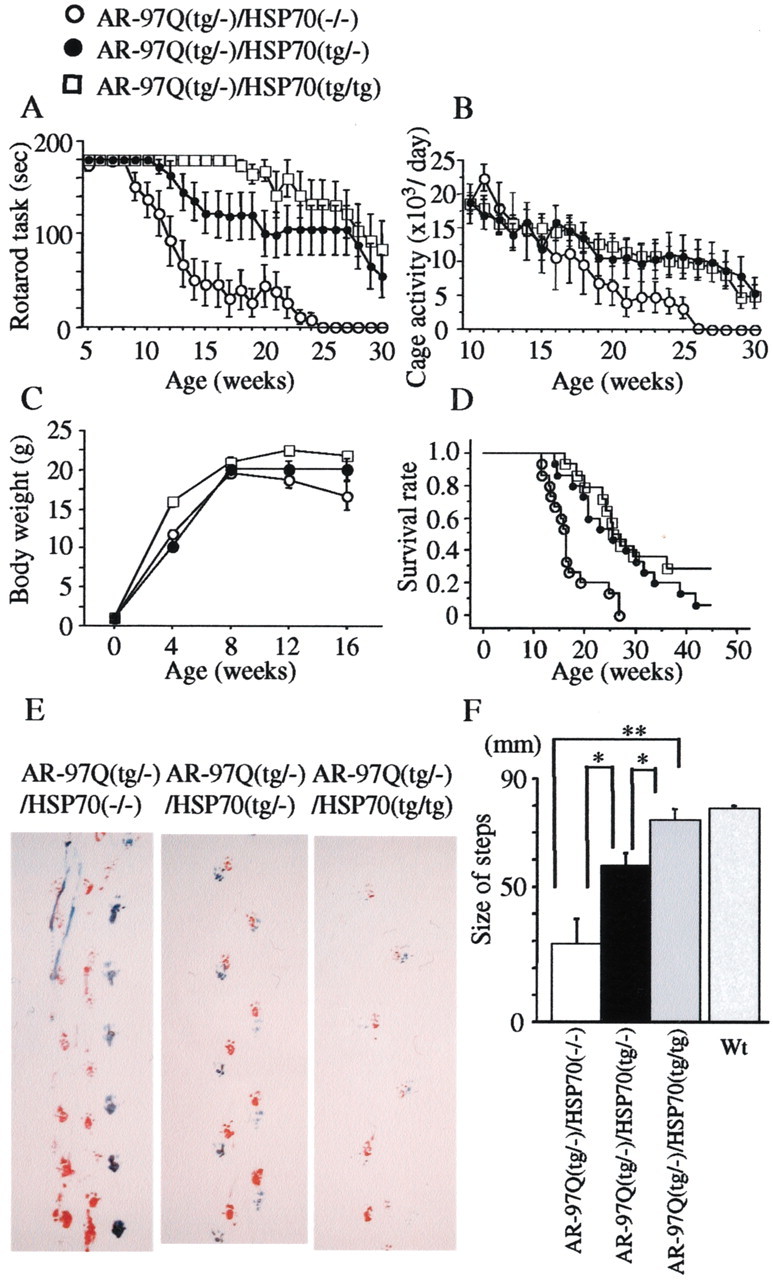
Effects of human HSP70 overexpression on the symptomatic phenotypes of male AR-97Q mice. Rotarod task (A; n = 10), cage activity (B; n = 10), body weight (C; n = 12), and survival rate (D; n = 14) of the AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice. All parameters were significantly different among AR-97Q/HSP70(−/−)mice, AR-97Q/HSP70(tg/tg), and AR-97Q/HSP70(tg/−) mice (p < 0.001, p < 0.05,p < 0.05, and p < 0.005, respectively). AR-97Q mice overexpressing human HSP70 lasted longer on the Rotarod and showed higher cage activity than AR-97Q/HSP70(−/−) mice. The AR-97Q/HSP70(−/−) mice lost weight earlier than the other two double-transgenics. Survival was prolonged in AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice compared with AR-97Q/HSP70(−/−) mice. E, Footprints of representative 16-week-old AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice. Front paws are indicated in red and hindpaws in blue. AR-97Q/HSP70(−/−) mice exhibit motor weakness, with dragging of the legs; AR-97Q/HSP70(tg/tg) mice walk almost normally; and AR-97Q/HSP70(tg/−) mice walk with somewhat short steps. F, The size of steps was measured in 16-week-old AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice (n = 4), respectively. Each column shows an average of steps of the hindpaw. AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice walked with significantly longer steps than AR-97Q/HSP70(−/−) mice. *p < 0.05; **p < 0.01. Error bars indicate SD. Wt, Wild type.
Expression levels of HSP70 in double-transgenic mice
We examined whether the AR-97Q/HSP70 double-transgenic mice express increased levels of the HSP70 protein in the spinal cord and skeletal muscle. Immunohistochemical studies of double-transgenic mice stained with the specific antibody for HSP70 confirmed that spinal neurons and muscular cells expressed the HSP70 (Fig.2A–C). The HSP70 was diffusely distributed to the nuclei and occasionally formed various-sized NIs (Fig. 2A–C). Glial cells also showed diffuse nuclear staining and NIs of HSP70 protein (data not shown). Western blot analysis revealed that the HSP70 expression level was fivefold greater in the AR-97Q/HSP70(tg/−) mice and 10-fold greater in the AR-97Q/HSP70(tg/tg) mice than endogenous HSP70 in the AR-97Q/HSP70(−/−) mice in the spinal cord and muscle (Fig. 2D). The AR-97Q transgene expression did not alter HSP70 expression levels in the spinal cord of the wild-type and HSP70(tg/tg) mice, whereas the AR-97Q transgene expression increased HSP70 levels in the muscle, suggesting that the stress-induced response is different between the spinal cord and the skeletal muscle (Fig.2D). Absence of the stress-induced response was also demonstrated in the nervous system of the other polyQ disease model mice (Jana et al., 2000; Cummings et al., 2001). The nuclear fraction of the spinal cord and muscle surely contained an increased amount of HSP70 in the double-transgenic mice. The amount of HSP70 in the nuclear fraction was most abundant in the AR-97Q/HSP70(tg/tg) mice (Fig.2E). The increased HSP70 was coimmunoprecipitated with mutant AR, suggesting that HSP70 directly binds to the mutant AR protein (Fig. 2F).
Fig. 2.
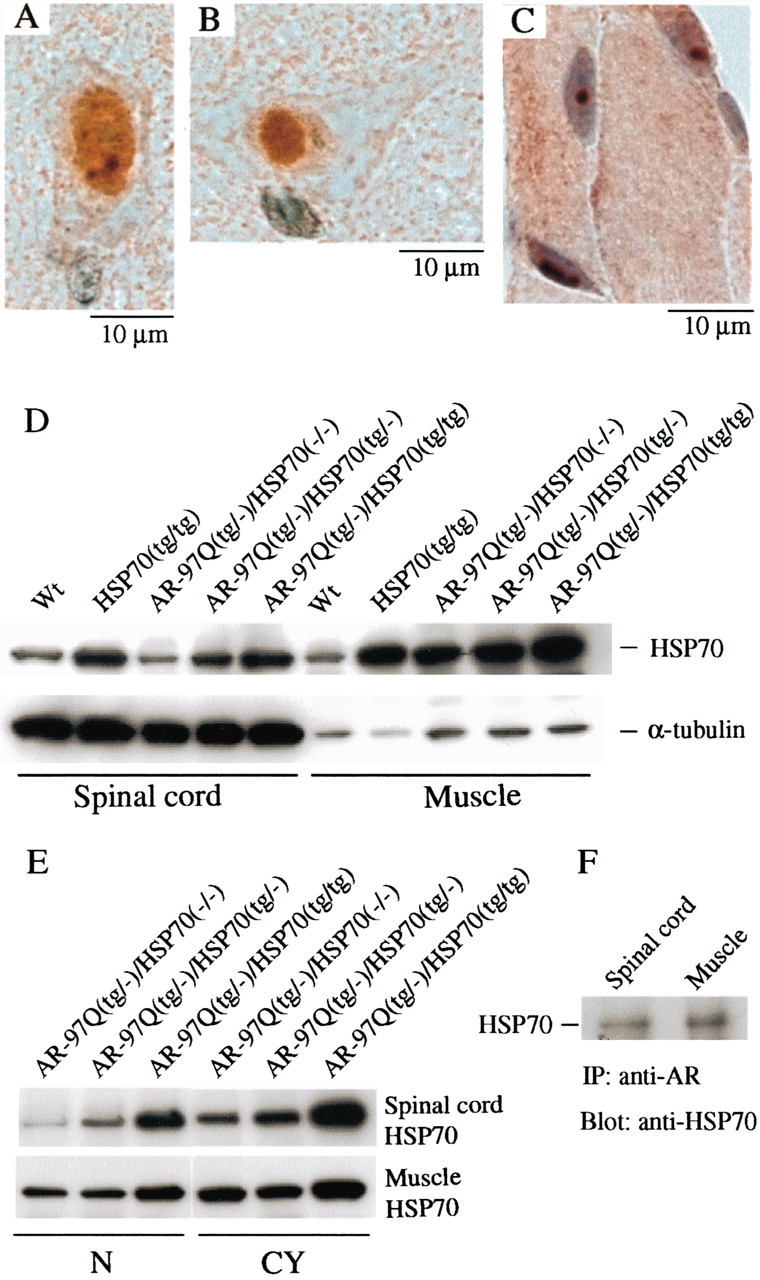
Increased HSP70 expression in double-transgenic mice. A–C, Immunohistochemical study from the 16-week-old AR-97Q/HSP70(tg/−) mice in the spinal anterior horn and skeletal muscle stained with the antibody specific for the HSP70. The immunoreactivity of HSP70 was localized to the nuclei with intense and diffuse staining, and small NIs were present in the anterior horn cell (A). A large nuclear inclusion was also present in the anterior horn cell (B). Skeletal muscle showed diffuse nuclear staining and NIs (C). D, E, Western blot analysis of total spinal cord and muscle protein lysate immunolabeled with an antibody against HSP70. AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice express higher levels of HSP70 than wild-type (Wt) and AR-97Q/HSP70(−/−) mice (D). The HSP70 expression level is fivefold higher in AR-97Q/HSP70(tg/−) mice and 10-fold higher in the AR-97Q/HSP70(tg/tg) mice than endogenous HSP70 in AR-97Q/HSP70(−/−) mice in the spinal cord and muscle, respectively (D). The AR-97Q transgene expression did not alter HSP70 levels in the spinal cord, whereas the AR-97Q transgene expression gained the respective HSP70 levels in the muscle (D, E). Therefore, the AR-97Q transgene expression in the double-transgenics alters HSP70 levels in the muscle but not in the spinal cord. E, Western blots of nuclear and cytoplasmic extracts immunolabeled with an antibody against HSP70. HSP70 localized in the nucleus (N) as well as in the cytoplasm (CY) in the spinal cord and muscle of all lines examined. AR-97Q/HSP70(tg/tg) mice expressed the largest amount of HSP70 in both extracts.F, Immunoprecipitation (IP) Western blots for HSP70. Soluble fractions were collected from the spinal cord and muscle, and equal protein concentrations were immunoprecipitated with an antibody to the N-terminal portion of AR and immunoblotted for HSP70. Coimmunoprecipitation of the HSP70 chaperone and the polyQ-expanded mutant AR was detected.
Colocalization of HSP70 with mutant AR in the nuclei
We evaluated the colocalization of HSP70 and mutant AR in the AR-97Q/HSP70 double-transgenic mice. We performed double-labeling immunohistochemistry and immunofluorescence double-staining with two primary antibodies: goat anti-HSP70 and mouse anti-expanded polyQ (1C2). These double-immunostaining studies revealed that HSP70 (Fig.3A,C) and mutant AR (Fig.3B,D) present diffusely in the nuclei and colocalize each other (Fig. 3B,E) in the spinal anterior horn neurons of the AR-97Q/HSP70(tg/tg) mice. We also determined that such diffuse staining of HSP70 in the nuclei was also present in the spinal neurons of SBMA patients (Fig.3F,J). Immunofluorescence double-staining with anti-HSP70 and anti-expanded polyQ antibodies revealed that the endogenous HSP70 (Fig. 3G,K) and mutant AR (Fig.3H,L) were colocalized on the NI (Fig. 3I) and diffusely in the nuclei (Fig. 3M) in the spinal cord neurons of SBMA patients, suggesting that the endogenous HSP70 preferentially coexists with mutant AR and exerts its function in the nuclei of SBMA patients as well.
Fig. 3.
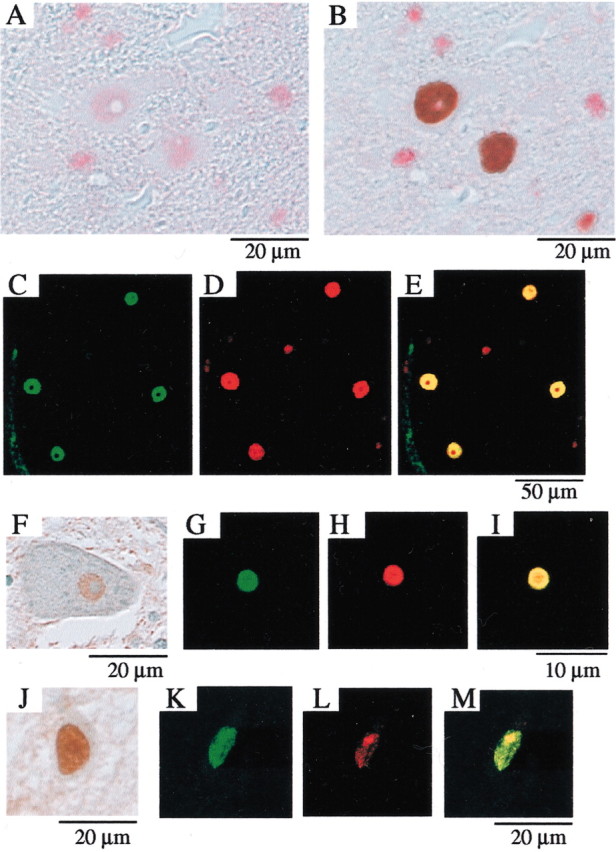
Colocalization of the nuclear-localized HSP70 chaperone with mutant AR. Immunohistochemical analysis for the antibody specific to the HSP70 as well as to the expanded polyQ stretch (immunostained with a monoclonal antibody, 1C2) in the spinal cords of 16-week-old AR-97Q/HSP70(tg/tg) mice (A–E) and SBMA patients (F–M). Double-labeling immunohistochemistry revealed diffuse nuclear staining for goat anti-HSP70 (A) and expanded polyQ (B), suggesting that HSP70 and mutant AR are colocalized in the spinal motor neurons of AR-97Q/HSP70(tg/tg) mice. Immunofluorescence double-staining with antibodies against HSP70 and the expanded polyQ also revealed that HSP70 and mutant AR are colocalized as shown in HSP70 (C, green), expanded-polyQ (D, red), and an overlay of the two signals (E, yellow). Diffuse staining of neuronal nuclei for HSP70 is also observed in the spinal neurons (F, J) of SBMA patients. Immunofluorescence double-staining with anti-HSP70 (green) and anti-expanded polyQ (red) antibodies revealed that the HSP70 (G) and mutant AR (H) are colocalized on the NI (shown inyellow inI) in the spinal anterior horn cell. The diffuse nuclear colocalization of HSP70 (K) and mutant AR (H) was also observed in the SBMA posterior horn cell (M). This cell also has an NI (L, M).
Overexpression of HSP70 decreases the nuclear-localized mutant AR
An immunohistochemical study for mutant AR using 1C2 antibody showed a marked reduction in diffuse nuclear staining and NIs in the AR-97Q/HSP70(tg/−) or the AR-97Q/HSP70(tg/tg) mice compared with the AR-97Q/HSP70(−/−) mice in the spinal motor neurons (Fig.4A–C) and muscles (Fig. 4D–F). The AR-97Q/HSP70(−/−) mice showed intense and frequent 1C2 staining in the nuclei (Fig. 4A,D), whereas the 1C2 staining was infrequent in the AR-97Q/HSP70(tg/−) mice (Fig.4B,E) and much less frequent in the AR-97Q/HSP70(tg/tg) mice (Fig.4C,F). Quantitative assessment of diffuse nuclear staining for 1C2 in the spinal motor neurons (Fig. 4G) and muscles (Fig. 4H) revealed significantly more positive cells in the AR-97Q/HSP70(−/−)mice than in the AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice. However, the 1C2-positive cell populations were not statistically different in the AR-97Q/HSP70(tg/−) and the AR-97Q/HSP70(tg/tg) mice. The neuronal cell population in the spinal ventral horn in the AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice was not significantly decreased compared with that in the wild-type mice (data not shown).
Fig. 4.

HSP70 decreases nuclear-localized mutant AR in double-transgenic mice. Immunohistochemical study of the spinal anterior horn (A–C) and muscle (D–F) of AR-97Q/HSP70(−/−)and AR-97Q/HSP70 double-transgenic mice stained with a monoclonal antibody (1C2) against abnormally expanded polyQ (16 weeks old). AR-97Q/HSP70(−/−) mice have intense and frequent staining for 1C2 in the nucleus (A, D). AR-97Q/HSP70(tg/−) (B, E), and AR-97Q/HSP70(tg/tg) (C, F) mice exhibit low levels of 1C2 staining in the nucleus. G, H, Quantitative assessment of diffuse nuclear staining for 1C2 in the spinal ventral horn (G) and muscle (H). Positively stained nuclei were estimated by counting in the thoracic spinal ventral horn and muscle using six transgenic mice (16 weeks of age). There are significantly more 1C2-positive cells in AR-97Q/HSP70(−/−) mice than in AR-97Q/HSP70(tg/−) mice or AR-97Q/HSP70(tg/tg) mice in both tissues. Results are expressed as means ± SD for six mice. The differences in 1C2-positive cell populations are not statistically significant between AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice. *p < 0.05; **p < 0.01; ***p < 0.001.
Overexpression of HSP70 decreases the high-molecular-weight mutant AR protein and monomeric mutant AR protein
Western blot analysis showed that the high-molecular-weight form of mutant AR protein complexes was retained in the stacking gel as well as a band of monomeric mutant AR monomer in the spinal cord and muscle of the transgenic mice (Fig. 5). The mutant AR within the stacking gel was diminished in the AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice compared with the AR-97Q/HSP70(−/−) mice (Fig.5A,B). In addition, the AR-97Q/HSP70(−/−) mice had more monomeric mutant AR protein than the AR-97Q/HSP70(tg/−) or AR-97Q/HSP70(tg/tg) mice (Fig.5A,B). The mutant AR protein within the stacking gel was found primarily in the nuclear fraction (Fig. 5C). The mutant AR within the stacking gel and monomeric form of the nuclear fraction in the spinal cord and muscle were also decreased in the AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice (Fig.5C). These observations suggested that the overexpression of HSP70 markedly decreases not only the high-molecular-weight mutant AR protein present primarily in the nuclear fraction but also the monomeric mutant AR protein.
Fig. 5.

HSP70 decreases nuclear-localized mutant AR protein complexes as well as monomeric mutant AR. A, B, Western blot analysis of total tissue homogenates from the spinal cord and muscle of AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice (16 weeks of age) immunolabeled by an antibody against AR (N-20). The mutant AR appearing within the stacking gel and monomeric mutant AR were diminished in AR-97Q/HSP70(tg/−) and AR-97Q/HSP70(tg/tg) mice compared with AR-97Q/HSP70(−/−) mice (A, B). Values of mutant AR were normalized to endogenous α-tubulin and expressed as the ratio to those of AR-97Q/HSP70(−/−) mice (B). Values are expressed as means ± SD for three mice.C, Western blot analysis of nuclear (N) and cytoplasmic (CY) fractions from the spinal cord and muscle of AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice (16 weeks of age) immunolabeled by N-20. Mutant AR protein within the stacking gel was found primarily in the nuclear fraction. The mutant AR within the stacking gel of the nuclear fraction also significantly decreased in the spinal cord and muscle of AR-97Q/HSP70(tg/tg)mice. R.S.I., Relative signal intensity.
We next performed a filter-trap assay for the quantitative analysis of the large molecular aggregated and soluble monomeric form of the mutant AR protein (Wanker et al., 1999). Only the larger-sized mutant AR protein was retained on the cellulose acetate membrane (pore diameter, 0.2 μm), whereas the nitrocellulose membrane captured proteins of all sizes (Fig.6A). We also put the nitrocellulose membrane under the cellulose acetate membrane to capture the soluble monomeric AR protein passing through this membrane (Fig.6B). Values were normalized to endogenous α-tubulin using the nitrocellulose membrane. Using this approach, we analyzed the ability of the HSP70 to decrease the large aggregated or soluble monomeric mutant AR protein. Overexpression of HSP70 resulted in a significant decrease in large aggregated as well as soluble monomeric mutant AR protein in a dose-dependent manner (Fig.6A–C). The endogenous AR protein was not retained on the cellulose acetate membrane in wild-type mice (data not shown). These results indicate that the HSP70 decreases not only the mutant AR protein complexes in the large aggregated form but also the soluble monomeric mutant AR as observed on Western blot analysis. These observations also suggested that overexpression of HSP70 enhanced the function of the ubiquitin–proteasome pathway and subsequently accelerated the degradation of monomeric mutant AR protein.
Fig. 6.
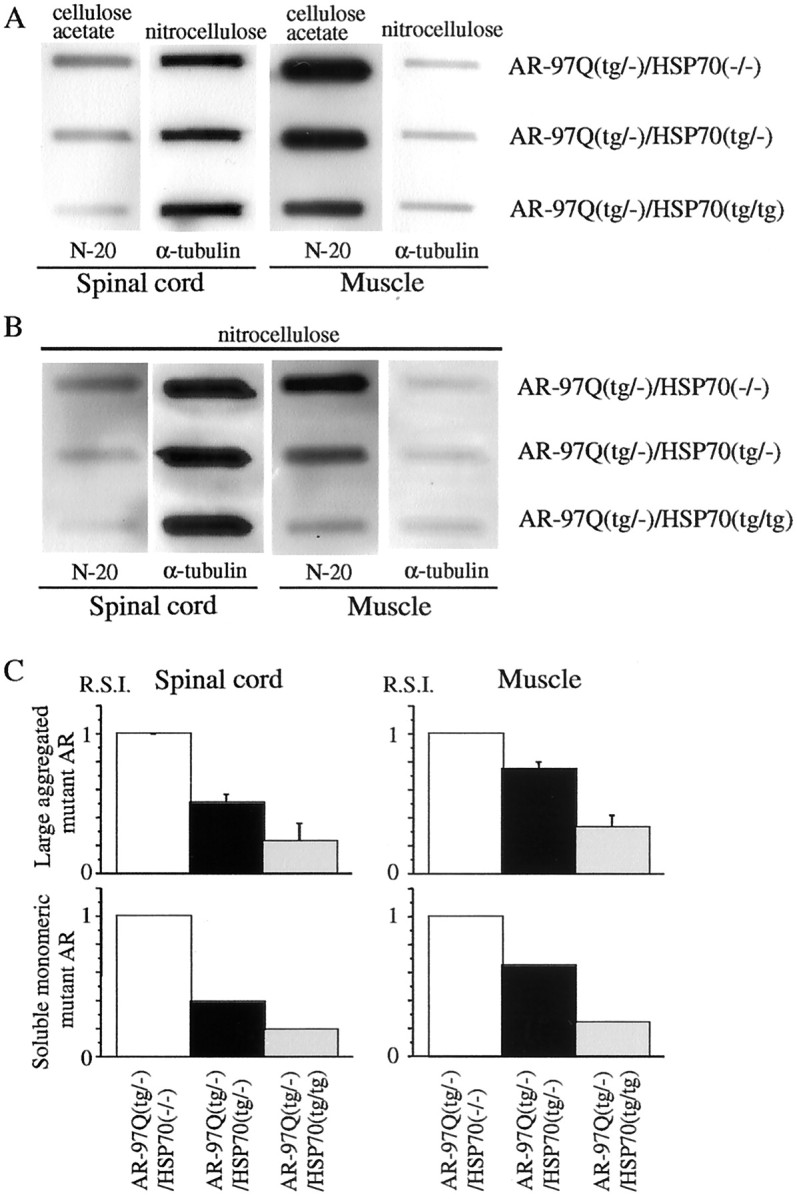
HSP70 decreases large aggregated mutant AR protein and soluble monomeric mutant AR protein. A–C, Filter-trap assay of total tissue homogenates from the spinal cord and muscle of AR-97Q/HSP70(−/−), AR-97Q/HSP70(tg/−), and AR-97Q/HSP70(tg/tg) mice (16 weeks of age) immunolabeled by an antibody against AR (N-20). Large aggregated mutant AR complexes were trapped by the cellulose acetate membrane (A), and soluble monomeric mutant AR passing through the cellulose acetate membrane was trapped by the nitrocellulose membrane beneath the cellulose acetate membrane (B). Endogenous α-tubulin using the nitrocellulose membrane was also shown (A, B). The normalized value of large aggregated mutant AR and soluble monomeric mutant AR against endogenous α-tubulin is shown in C. Relative values against those of AR-97Q/HSP70(−/−)mice were expressed as means ± SD for three mice or a mean of two mice (C). The trapped AR protein was reduced in the spinal cord and muscle of AR-97Q/HSP70(tg/−)and AR-97Q/HSP70(tg/tg) mice in both membranes (A, B). This reduction was most evident in AR-97Q/HSP70(tg/tg) mice (A–C), suggesting that the overexpression of HSP70 resulted in a significant, dose-dependent decrease in large aggregated and soluble monomeric mutant AR protein.R.S.I., Relative signal intensity.
Discussion
We generated a transgenic mouse model carrying a full-length AR containing 97 CAGs (Katsuno et al., 2002). This model showed progressive muscular atrophy and weakness as well as diffuse nuclear staining and NIs consisting of the mutant AR. These phenotypes were very pronounced in male transgenic mice, similar to those in SBMA (Katsuno et al., 2002). Here we demonstrate that the overexpression of human HSP70 exerts dose-dependent therapeutic effects on motor dysfunction in this mouse model. Mutant AR and HSP70 colocalized diffusely to the nuclei and to the NIs in the neurons and muscles of the AR-97Q/HSP70 double-transgenic mice. The overexpression of HSP70 served to decrease the nuclear-localized mutant AR protein complexes in large aggregated form in the double-transgenic mice. Monomeric mutant AR was also significantly reduced by HSP70 overexpression, suggesting that it could accelerate the turnover of mutant AR.
In our SBMA transgenic mouse model, nuclear translocation of mutant AR, which is dependent on the testosterone level, has been demonstrated to be essential for mutant AR-induced neurotoxicity (Katsuno et al., 2002). Reduction of the testosterone level by castration diminished nuclear-localized mutant AR and markedly prevented phenotypic expression in the male transgenic mice, whereas testosterone administration enhanced the nuclear localization of mutant AR and caused significant motor dysfunction in the female transgenic mice (Katsuno et al., 2002). In particular, the large aggregated complexes of the mutant AR protein detected in the stacking gel or slowly migrating species in the Western blot analysis in the nuclear fraction were well correlated with the phenotypic expression in this mouse model (Katsuno et al., 2002). This suggested that oligomeric or polymeric mutant AR large complex molecules positively associated with other molecules would exert the toxicity rather than monomeric mutant AR (Katsuno et al., 2002).
In the present study, we demonstrated that the amount of nuclear-localized mutant AR protein, particularly that of the large complex form present in the stacking gel or trapped by the cellulose acetate membrane, was significantly reduced in the AR-97/HSP70 double-transgenic mice. Thus, the overexpression of HSP70 is suggested to exert its amelioration of the phenotypic expression by diminishing the amount of nuclear-localized mutant AR protein. However, in the previously reported SCA1 transgenic mouse model, NIs of the mutant protein were not apparently decreased in the double-transgenic mice with rat HSP70 overexpression, although the neurological deficit and neuronal degeneration were ameliorated (Cummings et al., 2001). Because the gain of amelioration for phenotypic expression in the model mice of Cummings et al. (2001) was mild even in the double-transgenics with HSP70 homozygotes, the change in the frequency of the NIs would not have been significant enough to detect. In our mouse model, NIs were present only in the small subpopulation of neurons and muscles, particularly in the early phase of phenotypic expression, whereas the 1C2-positive nuclei were abundant (Katsuno et al., 2002). In addition, 1C2-positive neurons are more extensive than those of NI-bearing neurons in the tissues of the autopsied samples from patients with polyQ diseases, and the distribution of 1C2-positive neurons is well correlated with the neurological symptoms (Yamada et al., 2001). These observations suggest that 1C2 staining is a more sensitive histological marker for the detection of the nuclear localization of the mutant protein with an expanded polyQ stretch compared with NIs detected by antibodies for the responsible protein.
The interesting observation in our study was the diminution of monomeric mutant AR in the double-transgenic mice with overexpression of HSP70. Recently, HSP70 overexpression in the cell culture model has revealed enhanced solubility of mutant AR with an expanded polyQ and degradation through the ubiquitin–proteasome system (Bailey et al., 2002). Overexpression of chaperones generally enhances the function of the ubiquitin–proteasome pathway and subsequently accelerates protein degradation (Bukau and Horwich, 1998). The ubiquitin–proteasome pathway, particularly its activity, is known to be related to chaperone expression levels (Bukau and Horwich, 1998). The molecular mechanism for this relationship remains unsolved, but recently CHIP (C terminal of HSC70-interacting protein), U-box-type E3 ubiquitin ligase, has been shown to interact with HSP90 or HSP70 (Connel et al., 2001) and ubiquitylate unfolded proteins trapped by molecular chaperones and degrades them, thus acting as a “quality control E3” (Murata et al., 2001). Furthermore, there is a cofactor of HSC70/HSP70, Bcl-2 associated athanogene 1, which possesses a ubiquitin-like domain and promotes binding of HSC70/HSP70 to the proteolytic complex (Lüders et al., 2000). Although such coupling factors between the HSP70 chaperone system and the protein degradation machinery for mutant AR are unknown at present, if the similar E3 for mutant AR is present, it could ubiquitylate and degrade mutant AR as a result of interacting with HSP70. In this scenario, the overexpression of HSP70 may accelerate E3-dependent capture of mutant AR and its degradation through the proteasome pathway. A remarkable reduction of the monomeric mutant AR in the double-transgenics with HSP70 overexpression can be the reflection of the accelerated degradation of mutant AR through the HSP70-mediated E3–proteasome system. Interaction between mutant AR and HSP70 detected by coimmunoprecipitation and Western blot analysis in the double-transgenic mice would support this view. The overexpression of HSP70 could enhance the degradation of the monomeric mutant AR, presumably through the HSP70-interacting quality control E3 activation, and subsequently it could reduce the amount of nuclear-localized mutant AR, resulting in the amelioration of phenotypic expression induced by mutant AR. To substantiate this, however, one needs to identify the HSP70-interacting E3 ligase, which recognizes mutant AR as a substrate.
Another possibility is that overexpressed HSP70 directly renaturates the misfolded mutant AR and normalizes the interaction of mutant AR with proteins that are essential to maintain the cell function (Hendricks and Hartl, 1993). The overexpression of HSP70 and HSP40 or HSC70 and Drosophila human DNAJ homolog-1 (dHdj-1) changed the distribution and morphologic pattern of NI formation of mutant huntingtin and ataxin-1 (Cummings et al., 1998;Fernandez-Funez et al., 2000; Muchowski et al., 2000). The overexpression of dHdj-1 and HSP70 increased the proportion of the monomeric mutant protein with an expanded polyQ, suggesting that chaperones modulate the biochemical properties of mutant polyQ-bearing protein (Chan et al., 2000). It has been proposed that the disease proteins with an expanded polyQ participate in inappropriate protein–protein interactions that lead to cell dysfunction and eventual cell death (Sherman and Goldberg, 2001). Molecular chaperones can be involved in the conformational modification by stabilizing the unfolded mutant proteins and can facilitate or inhibit the interaction with self or other proteins (Opal and Zoghbi, 2002). To date, a number of proteins that interact with polyQ-bearing disease protein have been cloned, including huntingtin-associated protein (Li et al., 1995), huntingtin-interacting protein (Kalchman et al., 1997), glyceraldehyde-3-phosphate dehydrogenase (Burke et al., 1996), leucine-rich acidic nuclear protein (Matilla et al., 1997), polyglutamine tract-binding protein-1 (Waragai et al., 1999), 130 kDa human TATA-binding protein-associated factor subunit of the human transcription factor TFIID (Shimohata et al., 2000), and cAMP response element-binding protein (CREB) binding protein (CBP) (Nucifora et al., 2001; Zander et al., 2001). CBP has been demonstrated to interact with mutant AR, colocalize in the NIs, and reduce the mutant AR-induced cell toxicity by CBP overexpression in the cell culture model by modifying CBP-dependent transcriptional activity (McCampbell et al., 2000). HSP70 may reduce the toxicity of mutant AR proteins through the inhibition or acceleration of the interaction with these proteins. However, interacting protein involvement in association with mutant AR still needs to be investigated.
The other possible avenue by which HSP70 acts to improve polyQ-induced toxicity is the anti-apoptotic activities of HSP70. HSP70 suppresses apoptosis by inhibiting the c-Jun N-terminal kinase (Gabai et al., 1998) or by inhibiting cytochromec release and caspase-3 activation (Li et al., 2000; Jana et al., 2001). Furthermore, HSP40 and mammalian relative of DNAJ chaperones can inhibit caspase-3 and caspase-9 activation mediated by mutant huntingtin, independent of huntingtin aggregation (Zhou et al., 2001; Chuang et al., 2002). However, the involvement of anti-apoptotic activities of HSPs in protection against mutant AR toxicity through reducing the nuclear-localized mutant AR remains to be elucidated.
In summary, the overexpression of HSP70 significantly ameliorates the phenotypes of SBMA transgenic mice by reducing the amount of nuclear-localized mutant AR protein, particularly that of the large complex form. The amount of monomeric mutant AR was also reduced by HSP70 overexpression, suggesting enhanced degradation of mutant AR. A recent study revealed that the ansamycin antibiotic Geldanamycin induced a heat shock response and inhibited aggregation of mutant huntingtin in COS-1 cells (Sittler et al., 2001). Thus, HSP70 overexpression would provide a potential therapeutic avenue for SBMA and other polyQ diseases.
Footnotes
This work was supported by a Center of Excellence grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by grants from the Ministry of Health, Labor, and Welfare of Japan. We thank Sugiko Yokoi for her technical assistance.
Correspondence should be addressed to Dr. Gen Sobue, Department of Neurology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho Showa-ku, Nagoya, 466-8550, Japan. E-mail:sobueg@med.nagoya-u.ac.jp.
References
- 1.Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Do J, Sang C, Kobayashi Y, Doyu M, Sobue G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10:1039–1048. doi: 10.1093/hmg/10.10.1039. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 3.Bukau KT, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 4.Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996;2:347–350. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 5.Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht AR, Rubinsztein DC. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan HYE, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 8.Chuang JZ, Zhou H, Zhu M, Li SH, Li XJ, Sung CH. Characterization of a brain-enriched chaperone, MRJ, that inhibits Huntingtin aggregation and toxicity independently. J Biol Chem. 2002;277:19831–19838. doi: 10.1074/jbc.M109613200. [DOI] [PubMed] [Google Scholar]
- 9.Connel P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 10.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 11.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Overexpression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 12.Doyu M, Sobue G, Mukai E, Kachi T, Yasuda T, Mitsuma T, Takahashi A. Severity of X-linked recessive bulbospinal neuronopathy correlates with size of the tandem CAG repeat in androgen receptor gene. Ann Neurol. 1992;23:707–710. doi: 10.1002/ana.410320517. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 14.Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY. Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett. 1998;438:1–4. doi: 10.1016/s0014-5793(98)01242-3. [DOI] [PubMed] [Google Scholar]
- 15.Hendricks JP, Hartl FU. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem. 1993;62:349–384. doi: 10.1146/annurev.bi.62.070193.002025. [DOI] [PubMed] [Google Scholar]
- 16.Huynh DP, Figueroa K, Hoang N, Pulst SM. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet. 2000;26:44–50. doi: 10.1038/79162. [DOI] [PubMed] [Google Scholar]
- 17.Igarashi S, Tanno Y, Onodera O, Yamazaki M, Sato S, Ishikawa A, Miyatani N, Nagashima M, Ishikawa Y, Sahashi K, Ibi T, Miyatake T, Tsuji S. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology. 1992;42:2300–2302. doi: 10.1212/wnl.42.12.2300. [DOI] [PubMed] [Google Scholar]
- 18.Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 19.Jana NR, Zemskov EA, Wang GH, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–1059. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 20.Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- 21.Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi Y, Sobue G. Protective effect of chaperones on polyglutamine diseases. Brain Res Bull. 2001;56:165–168. doi: 10.1016/s0361-9230(01)00593-7. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- 25.La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, Yee WC, Fischbeck KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–304. doi: 10.1038/ng1292-301. [DOI] [PubMed] [Google Scholar]
- 26.Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem. 2000;275:25665–25671. doi: 10.1074/jbc.M906383199. [DOI] [PubMed] [Google Scholar]
- 27.Li M, Miwa S, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998a;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Fischbeck KH, Sobue G. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol. 1998b;153:695–701. doi: 10.1016/S0002-9440(10)65612-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 30.Lüders J, Demand J, Höhfeld J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J Biol Chem. 2000;275:4613–4617. doi: 10.1074/jbc.275.7.4613. [DOI] [PubMed] [Google Scholar]
- 31.Matilla A, Koshy BT, Cummings CJ, Isobe T, Orr HT, Zoghbi HY. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature. 1997;389:974–978. doi: 10.1038/40159. [DOI] [PubMed] [Google Scholar]
- 32.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 33.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 36.Opal P, Zoghbi HY. The role of chaperones in polyglutamine disease. Trends Mol Med. 2002;8:232–236. doi: 10.1016/s1471-4914(02)02310-9. [DOI] [PubMed] [Google Scholar]
- 37.Plumier JC, Ross BM, Currie RW, Angelidis CE, Kaziaris H, Kollias G, Pagoulatos GN. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, Rechsteiner M, Riess O, Landwehrmeyer GB. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol. 2002;51:302–310. doi: 10.1002/ana.10101. [DOI] [PubMed] [Google Scholar]
- 40.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 41.Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T. Expanded polyglutamine stretches interact with TAFII130,interfering with CREB-dependent transcription. Nat Genet. 2000;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- 42.Sittler A, Lurz R, Lueder G, Priller J, Hayer-Hartl MK, Hartl FU, Lehrach H, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 43.Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy: a clinicopathological study. Brain. 1989;112:209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- 44.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins,proteasome components and SRC-1,and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi A. Hiroshi Kawahara (1858–1918). J Neurol. 2001;248:241–242. doi: 10.1007/s004150170236. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka F, Doyu M, Ito Y, Matsumoto M, Mitsuma T, Abe K, Aoki M, Itoyama Y, Fischbeck KH, Sobue G. Founder effect in spinal and bulbar muscular atrophy (SBMA). Hum Mol Genet. 1996;5:1253–1257. doi: 10.1093/hmg/5.9.1253. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka F, Reeves MF, Ito Y, Matsumoto M, Li M, Miwa S, Inukai A, Yamamoto M, Doyu M, Yoshida M, Hashizume Y, Terao S, Mitsuma T, Sobue G. Tissue-specific somatic mosaicism in spinal and bulbar muscular atrophy (SBMA) is dependent on CAG repeat length and androgen receptor gene expression level. Am J Hum Genet. 1999;65:966–973. doi: 10.1086/302578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terao S, Sobue G, Hashizume Y, Li M, Inagaki T, Mitsuma T. Age-related changes in human spinal ventral horn cells with special reference to the loss of small neurons in the intermediate zone: a quantitative analysis. Acta Neuropathol (Berl) 1996;92:109–114. doi: 10.1007/s004010050497. [DOI] [PubMed] [Google Scholar]
- 49.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 51.Waragai M, Lammers C, Takeuchi S, Imafuku I, Udagawa Y, Kanazawa I, Kawabata M, Mouradian MM, Okazawa H. PQBP-1,a novel polyglutamine tract-binding protein,inhibits transcription activation by Brn-2 and affects cell survival. Hum Mol Genet. 1999;8:977–987. doi: 10.1093/hmg/8.6.977. [DOI] [PubMed] [Google Scholar]
- 52.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 53.Yamada M, Wood JD, Shimohata T, Hayashi S, Tsuji S, Ross CA, Takahashi H. Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol. 2001;49:14–23. doi: 10.1002/1531-8249(200101)49:1<14::aid-ana5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 54.Zander C, Takahashi J, Hachimi KHE, Fujigasaki H, Albanese V, Lebre AS, Stevanin G, Duyckaerts C, Brice A. Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase-3. Hum Mol Genet. 2001;10:2569–2579. doi: 10.1093/hmg/10.22.2569. [DOI] [PubMed] [Google Scholar]
- 55.Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]
- 56.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]