

Abstract
Cyclin-dependent kinase 5 (Cdk5) is activated on binding of activator proteins p35 and p39. A N-terminally truncated p35, termed p25, is generated through cleavage by the Ca2+-dependent protease calpain after induction of ischemia in rat brain. p25 has been shown to accumulate in brains of patients with Alzheimer's disease and may contribute to A-β peptide-mediated toxicity. Studies from transfected neurons as well as p35 and p25 transgenic mice have indicated that Cdk5, when activated by p25, gains some toxic function compared with p35/Cdk5. It remains unclear, however, whether p25/Cdk5 signaling additionally channels into pathways usually used by p35/Cdk5 and whether p25 is associated with a loss of p35 function. To clarify these issues, we have generated p25-transgenic mice in a p35-null background. We find that low levels of p25 during development induce a partial rescue of the p35−/− phenotype in several brain regions analyzed, including a rescue of cell positioning of a subset of neurons in the neocortex. In accordance with the partial rescue of brain anatomy, phosphorylation of the Cdk5 substrate mouse disabled 1 is partially restored during development. Besides this, p25/Cdk5 fails to phosphorylate other substrates that are normally phosphorylated by p35/Cdk5. Our results show that p25 can substitute for p35/Cdk5 under certain circumstances during development. In addition, they suggest that p25 may have lost some functions of p35.
Keywords: Cdk5, p35, p25, brain, development, phosphorylation, migration, calpain, degeneration
Introduction
Cyclin-dependent kinase 5 (Cdk5) is a Ser-Thr kinase that is activated on binding to p35 or p39, two closely related proteins predominantly expressed in neurons (Dhavan and Tsai, 2001). Cdk5−/− and p35/p39 double knock-out mice die perinatally and display severe neuronal lamination defects in several brain areas suggestive of impaired neuronal migration (Ohshima et al., 1996, 1999; Gilmore et al., 1998; Gilmore and Herrup, 2001; Ko et al., 2001). The phenotype of p35−/− mice is less pronounced but similar to the one of Cdk5−/− mice, but the mice are viable and fertile (Chae et al., 1997; Kwon and Tsai, 1998).
Recently, it has been shown that p35 is cleaved by the Ca2+-dependent Cys protease calpain after induction of ischemia in the brain (Lee et al., 2000; Nath et al., 2000). Calpain cleaves p35 in the N-terminal domain, which results in the generation of a C-terminal truncation product termed p25. p25 contains all elements necessary for Cdk5 catalytic activation and activates Cdk5 in vitro and in vivo (Lew et al., 1994; Ahlijanian et al., 2000; Bian et al., 2002). In addition to ischemia, generation of p25 in the nervous system has been described in patients with Alzheimer's disease, in a mouse model of familial amyotrophic lateral sclerosis, and in a mouse model of Niemann–Pick type C disease (Patrick et al., 1999; Nguyen et al., 2001; Bu et al., 2002; Tseng et al., 2002). Furthermore, neurons challenged with toxic A-β peptide display increased p25 generation, and inhibition of Cdk5 or calpain partially prevents neuronal cell death in this system (Alvarez et al., 1999; Lee et al., 2000). In sum, these data suggest that p25/Cdk5 contributes to neuronal cell death after neurotoxic insults.
Transgenic mice that overexpress p25 (p25-tg) under the neuronal-specific enolase (NSE) promoter show disruption of the axonal cytoskeleton and hyperphosphorylation of the microtubule-associated protein tau and of neurofilament subunits (Ahlijanian et al., 2000). Mice transgenic for p35 do not exhibit a comparable phenotype, arguing that signaling by the p25/Cdk5 complex differs from signaling by p35/Cdk5 (Van den Haute et al., 2001). Indeed, expression of p25/Cdk5 in COS-7 cells leads to hyperphosphorylation of tau, whereas similar levels of p35/Cdk5 fail to do so, suggesting a toxic gain of function by p25/Cdk5 (Patrick et al., 1999). This is further supported byin vitro studies demonstrating that p25/Cdk5 is more effective at phosphorylating tau than p35/Cdk5, and that it might target sites within tau that are only poorly phosphorylated by p35/Cdk5 (Hashiguchi et al., 2002). Although these studies investigated a possible toxic gain of function of p25/Cdk5, they did not address the question of whether further differences might exist between p25/Cdk5 and p35/Cdk5 signaling. In particular, it remains unclear whether p25/Cdk5 can signal in pathways normally used by p35/Cdk5 and whether p25/Cdk5 has lost some functions of p35/Cdk5 signaling.
To address this question, we have generated NSE p25-tg mice in a p35−/− background. This allowed us to compare p25/Cdk5 with p35/Cdk5 signaling in vivo by analysis of the developmental p35−/− phenotype and the phosphorylation of endogenous substrates of Cdk5. On the basis of our results, we suggest that p25/Cdk5 can signal through pathways used by p35/Cdk5 but also may have lost some p35/Cdk5 function.
Materials and Methods
Generation of p25-tg/p35−/− mice. Generation of mice expressing p25 under the control of the NSE promoter and of p35−/− mice has been described previously (Chae et al., 1997;Ahlijanian et al., 2000). To produce p25-tg/p35−/− mice, hemizygous p25-tg mice were mated with p35−/− mice. F1 p25-tg/p35+/− mice were mated again with p35−/− mice to obtain the desired F2 p25-tg/p35−/− offspring as well as p35−/−, p35+/−, and p25-tg/p35+/− control mice. Additional controls were generated by crossing p25-tg mice with p35+/+ mice over two generations to produce F2 p35+/+ and p25-tg/p35+/+ animals. For analysis, littermates were used whenever possible. Genotypes were determined by PCR on genomic tail DNA, as published previously (Kwon and Tsai, 1998; Ahlijanian et al., 2000).
Histochemical and immunohistochemical analysis. Adult mice (6 months–2 years old) were anesthetized by intraperitoneal injections of Avertin (10 ml/kg body weight; Aldrich, Milwaukee, WI). Anesthetized animals were transcardially perfused with 0.1m sodium phosphate buffer, pH 7.4, to remove blood. Next, animals were fixed by perfusion with 70 ml of 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4, or Bouin's fixative (Polysciences, Warrington, PA). Dissected brains were fixed at 4°C either for 20–24 hr in 4% paraformaldehyde solution or for 48 hr in Bouin's fixative. To perform histology, brains were embedded in paraffin, cut into 6- to 7-μm-thick sections, and stained with hematoxylin and eosin or cresyl violet using standard procedures. Three or more animals of each genotype were analyzed. To label fibers within the brain, we used a reduced silver fiber-staining technique according to previously published procedures (Desclin and Escubi, 1975).
Immunohistochemical staining was performed on paraformaldehyde-fixed sections using the avidin–biotin–peroxidase complex technique (Vector Laboratories, Burlingame, CA) with diaminobenzidine as a substrate. Calbindin D-28-K (Sigma, St. Louis, MO; 1:3000) and SMI 32 and SMI 34 (Sternberger Monoclonals Inc., Lutherville, MD; both 1:1000) antibodies were used for staining according to the instructions of the manufacturers. SMI 32 reacts with a nonphosphorylated epitope in the neurofilament H chain. SMI 34 detects a phosphorylated epitope in neurofilament H and to a lesser extent in the neurofilament M chain.
Western blot analysis. Timed pregnant mice were killed, and embryos were collected in ice-cold HBSS and 20 mm HEPES, pH 7.3. The cerebral cortex anlage or the cerebral cortex of adult animals was dissected and lysed in radioimmune precipitation assay buffer [50 mmTris, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1× protease inhibitor mixture (Roche Molecular Biochemicals, Mannheim, Germany), and phosphatase inhibitors 1 mm sodium fluoride, 1 mm sodium orthovanadate, 2 mm imidazole, and 25 mmβ-glycerophosphate]. Lysates were homogenized and spun at 16,000 × g for 5 min. Supernatants were removed, mixed 1:1 with 2× sample buffer, boiled for 5 min, quick-frozen, and stored at –80°C.
Samples were analyzed by SDS-PAGE in 8% polyacrylamide gels for postsynaptic density 95 (PSD-95) protein, in 10% gels for mouse disabled 1 (mDab1), NUDE-like (NUDEL), and tau proteins, and in 12% gels for p25, p35, and p39 proteins. Proteins were transferred to an Immobilon-P transfer membrane (Millipore, Bedford, MA). Blots were subjected to protein detection by incubation with various antibodies diluted in Tris-buffered saline (in mm: 25 Tris, 137 NaCl, and 2.7 KCl, pH 7.4) containing 0.2% Tween 20 and 5% nonfat dry milk. Bound antibodies were visualized using enhanced chemiluminescence solutions (PerkinElmer Life Sciences, Boston, MA). Affinity-purified rabbit polyclonal antibodies to p35 and p39 have been described previously (Tsai et al., 1994; Humbert et al., 2000) and were used at 1 μg/ml. Mouse monoclonal antibody (clone AC-40) to actin was purchased from Sigma and used at a 1:2000 dilution. Mouse monoclonal antibody to mDab1 was a kind gift from Dr. A. M. Goffinet (University Louvain Medical School, Brussels, Belgium) and was used at a 1:250 dilution. Phospho-epitope-specific antibody to mDab1 phosphorylated at Ser-491 was generated by injection of a keyhole limpet hemocyanin (KLH)-coupled peptide with the sequence PAPRQS(phosphoS)PSKSSC into rabbits (Keshvara et al., 2002). Rabbit serum was affinity-purified by a phospho-peptide column and proven to specifically recognize mDab1 phosphorylated at Ser-491 and was used at a dilution of 1:1000. Rabbit antibody to NUDEL (serum 211) was described previously and was used at 0.2 μg/ml (Niethammer et al., 2000). Phospho-epitope-specific antibody to Ser-231 of NUDEL was made by injection of the KLH-coupled peptide TENTFP(phosphoS)PKAIPC into rabbits. Rabbit serum was first affinity-purified by a column containing resin-bound peptide, which was not phosphorylated at Ser-231. The flow-through was further purified by a phospho-peptide column and proven to specifically recognize NUDEL phosphorylated at Ser-231 and was used at a dilution of 1:500. Mouse monoclonal antibody to PSD-95 (clone K28/43) was obtained from Upstate Biotechnology (Lake Placid, NY) and used at a 1:10,000 dilution. Affinity-purified rabbit antibody specific to PSD-95 phosphorylated at Thr-19 and Ser-25 was generated by injection of a KLH-coupled peptide with the sequence QDED(phosphoT)PPLEH(phosphoS)PAHC into rabbits. Rabbit serum was first affinity-purified by a column containing resin-bound peptide, which was not phosphorylated at Thr-19 and Ser-25. The flow-through was further purified by a phospho-peptide column and proven to specifically recognize PSD-95 phosphorylated at Thr-19 and Ser-25. The antibody was used at a 1:50 dilution. Mouse monoclonal antibody recognizing total tau (clone TAU-5, reacts with phosporylated and nonphosphorylated forms) was purchased from Neomarkers and was used at a 1:250 dilution. Rabbit polyclonal antibody against phosphorylated Ser-202 of tau was obtained from Biosource (Camarillo, CA) and was used at a 1:1000 dilution. Mouse monoclonal antibody PHF-1 was a kind gift from Dr. P. Davies (Albert Einstein College of Medicine, New York, NY) and was used at a 1:100 dilution. PHF-1 detects tau phosporylated at Ser-396 and Ser-404.
Densitometric analysis. Quantitation of optical density of Western blot signals was performed on scanned films by NIH Image 1.62 analysis software. Values for phospho-epitope-specific signals were divided by values for total protein signals. To allow for comparison between different litters, ratios obtained from p35−/− or p25-tg/p35−/− animals were normalized to the average ratio derived from p35+/− control animals (which was arbitrarily set to 1) within a single litter. Normalized ratios from p35−/− and p25-tg/p35−/− mice were then used to perform statistical analysis (n = 3–5 for each genotype and each developmental stage; unpaired, two-tailed t test).
Results
Partial restoration of cytoarchitecture in the cerebral cortex of p25-tg/p35−/− mice
The mature cerebral cortex is characterized by organization of neurons into readily distinguishable layers. During development, neurons born in the ventricular zone migrate radially toward the pial surface to settle in their final position within the cerebral wall. To establish this pattern, later-born neurons migrate past their predecessors, resulting in a so-called “inside-out” layer formation (Bayer and Altman, 1991).
To assess whether p25 can substitute for p35 in vivo, we crossed p25-tg mice with p35−/− mice. We then compared the morphology of the cerebral cortex among wild-type (wt), p35−/−, p25-tg, and p25-tg/p35−/− mice after cresyl violet staining (Fig.1A). The wt cerebral cortex shows morphological organization into six layers, including the large pyramidal neurons of layer V. In contrast, p35−/− mice display an apparent loss of the laminar structure within the cerebral cortex, with most large pyramidal neurons normally found in layer V located just beneath layer I. This phenotype has been explained by the inability of later-born neurons to migrate past earlier-born ones, resulting in an inverted layering order within the cerebral cortex (Chae et al., 1997; Kwon and Tsai, 1998; Kwon et al., 2000). In p25-tg/p35−/− mice, most large pyramidal neurons are located in a position close to the one found in the wt cortex. The neurons are located superficial to their normal position, corresponding approximately to the location where layer IV neurons normally reside. In addition, the superficial aspect of the cerebral cortex acquires some laminar structure, although it remains less organized than in wt mice. Control p25-tg mice exhibit the same cortical organization as wt mice (Fig. 1A) and display normal gross brain morphology (Ahlijanian et al., 2000). In summary, expression of the p25-tg in a p35-null background results in a partial rescue of the developmental p35−/− lamination phenotype in the cerebral cortex.
Fig. 1.
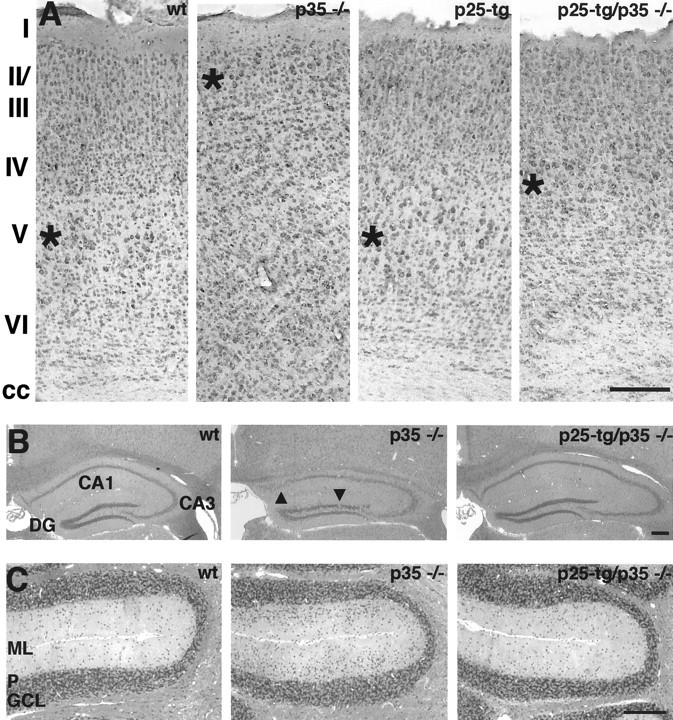
Partial rescue of cytoarchitecture in the brain of p25-tg/p35−/− mice. A, Cerebral cortex. Coronal sections of paraformaldehyde-fixed and paraffin-embedded brains from 6-month-old mice were stained with cresyl violet. Images are taken from the motor cortex area at the level of the anterior commissure. Genotypes are as indicated. The wt cortex displays distinct neuronal cell layers marked I–VI. Large pyramidal neurons are located in layer V, whose center is marked by anasterisk. In the p35−/− cortex, large pyramidal neurons are located in ectopic superficial positions (asterisk). Cell layering is severely disturbed. The cortex of p25-tg mice appears normal. In p25-tg/p35−/− mice, large pyramidal neurons are located in a layer (asterisk) that resides in a slightly superficial position compared with wild-type animals. The superficial aspect of the cortex displays some restoration of layer formation. Note the loss of corpus callosum (cc) fiber tract in p35−/− cortex. The corpus callosum is restored in p25-tg/p35−/− mice but appears disorganized. Medial is to the left; dorsal is at the top.B, C, Restoration of morphology in the hippocampus (B) and the cerebellum (C) of p25-tg/p35−/− mice. Coronal sections of Bouin's-fixed and paraffin-embedded brains from 9-month-old mice were stained with hematoxylin and eosin.B, The wt hippocampus displays the normal configuration of densely packed CA1, CA3, and dentate gyrus (DG) cell layers. The hippocampus of p35−/− mice shows loose packing and occasional disruptions of neuronal cell layers (arrowheads) and is oddly shaped. In p25-tg/p35−/− mice, neuronal layering and overall shape of the hippocampus appear to be largely normal. Medial is to the left; dorsal is at the top. C, The cerebellum of wild-type mice displays characteristic laminar organization into a cell-sparse molecular layer (ML), a Purkinje cell layer (P), and a granule cell layer (GCL). The molecular layer in the p35−/− cerebellum shows an increased density of cells with a granule cell-like morphology. In p25-tg/p35−/− mice, the cell density in the molecular layer is strongly reduced compared with p35−/− mice but seems still slightly elevated when compared with wild-type mice. Medial is at thetop; dorsal is to the left. Scale bars, 200 μm.
Restoration of morphology in the hippocampus and the cerebellum of p25-tg/p35−/− mice
In addition to the cerebral cortex, other brain regions whose laminar organization is affected in p35−/− mice include the hippocampus and the cerebellum (Chae et al., 1997; Ohshima et al., 2001). The hippocampus of p35−/− mice is characterized by loose packing and occasional interruption of CA1 and CA3 neuronal layers as well as the granule cell layer of the dentate gyrus (Fig.1B). In contrast, p25-tg/p35−/− mice display a hippocampal formation, which seems almost indistinguishable from that of the wt hippocampus. The only abnormality we noticed was a slight decrease in the density of neurons at the border between CA1 and CA3 layers. The hippocampus of control p25-tg mice is indistinguishable from that of wt mice (data not shown).
The main feature of the cerebellum of p35−/− mice is a molecular layer that is filled with cells resembling granular cells (Fig.1C). Expression of the p25 transgene in p35-null mice leads to a pronounced reduction of ectopic cells in the molecular layer, although their density still seems slightly increased in comparison with the wt cerebellum. The cerebellum of control p25-tg mice appears normal (data not shown). Taken together, expression of p25 in the p35−/− mice results in alleviation of the developmental lamination defects in the hippocampus and the cerebellum. This provides evidence that p25/Cdk5 can substitute to a significant extent for p35/Cdk-mediated signaling in these brain structures.
Analysis of p25-tg/p35−/− mice cortices by neuronal subtype-specific markers
As indicated by histochemical staining (Fig.1A), the laminar configuration of the cerebral cortex becomes partially restored in p25-tg/p35−/− mice compared with p35-null mice. To investigate this in more detail and not solely based on morphological criteria, we performed immunohistochemistry with antibodies against calbindin and neurofilament chains to identify neuronal subpopulations. Calbindin staining identifies lightly labeled pyramidal neurons of layers II and III as well as a darkly marked fraction of GABAergic interneurons scattered throughout the adult cerebral cortex (Fig.2A,A′,A") (Hendry et al., 1989; Van Brederode et al., 1990; Stuhmer et al., 2002). In p35−/− mice, superficial layers are devoid of lightly stained pyramidal neurons in dorsomedial parts of the cortex (Fig.2B). Instead, layer II and III late-born neurons remain in deep layers of the cortex, in accordance with the inverted layering phenotype described in p35−/− mice. Abnormal layering in p35-null mice is more pronounced in dorsomedial parts compared with ventrolateral parts of the cortex (Chae et al., 1997). Indeed, the abnormal distribution of calbindin-expressing neurons is more obvious in dorsomedial versus ventrolateral parts of the cortex (Fig.2B). Notably, in p25-tg/p35−/− mice, calbindin-positive neurons resume their normal position ventral to layer I (Fig. 2C). This finding suggests that p25/Cdk5 can signal in the same manner as p35/Cdk5 normally signals regarding cell positioning during development. In the cingulate cortex and medial motorcortex, the rescue seems almost complete, whereas more laterally calbindin-positive neurons are still found in ectopic positions in deep cortical layers, although they are less abundant compared with p35−/− mice.
Fig. 2.

Analysis of p25-tg/p35−/− cortices by neuronal subtype-specific markers. Coronal sections of paraformaldehyde-fixed brains were stained with anti-calbindin antibody (A–C) or anti-neurofilament antibody SMI 32 (D–F). Genotypes are as indicated.A′–C′, Superficial layers of the medial motorcortex.A"–C", Corresponding deep layers of the medial motorcortex. C′", Deep layers of lateral the motorcortex. A, In the cerebral cortex of 9-month-old wt mice, calbindin staining labels mostly layer II and III neurons.A′, Magnification of the wt superficial motor cortex area reveals lightly stained pyramidal neurons (arrow) as well as darkly stained presumptive inhibitory neurons (arrowhead). A", Magnification of corresponding deep cortical layers shows few darkly labeled nonpyramidal inhibitory neurons. B, In p35−/− mice, calbindin-positive neurons fail to populate their normal position but localize to deep cortical areas (asterisks). More laterally, however, calbindin-positive neurons can be found in normal positions (lateral of arrow). B′, Magnification of an area comparable with A′ displays few darkly stained neurons but no pyramidal neurons usually found in this location. Instead, they are found in deep cortical layers (B") along with some inhibitory neurons.C, In p25-tg/p35−/− mice, the distribution of calbindin-positive neurons is partially restored. Notably, the localization of calbindin-positive neurons in the cingulate cortex and in the medial motorcortex (arrowheads) appears primarily normal (compare A,C). More laterally, the superficial layer of calbindin-positive neurons seems thinned compared with wild-type animals, and the staining reveals neurons remaining in deep cortical positions (asterisk).C′, Enlargement of superficial layers in the medial motorcortex demonstrates calbindin-positive pyramidal neurons (arrow) in their normal position but not in deep cortical layers (C"), which display only a few inhibitory neurons (arrowhead). C′", Unlike deep layers in the medial motorcortex (C"), corresponding layers in the lateral motorcortex still contain lightly stained pyramidal neurons (arrow). D, Staining of wild-type brains with SMI 32 labels mostly large pyramidal neurons in layer V of the motorcortex. E, In p35−/− mice, the staining identifies large pyramidal neurons scattered abnormally in superficial layers of the motorcortex. Note aberrant fiber fascicles coursing through the cortex. F, In p25-tg/p35−/− mice, the staining labels a discrete layer of large pyramidal neurons. Unlike in p35−/− mice, areas just ventral to layer I are mostly devoid of ectopic SMI 32-positive neurons. The stained layer is somewhat superficially positioned compared with wild-type mice. For all images, medial is to the left; dorsal is at thetop. Scale bars, 200 μm. in p35−/− mice compared with p35+/− animals, as determined by a phospho-epitope-specific antibody targeting Ser-231. No significant changes are detected between p35−/− mice and compound mice, indicating failure of p25/Cdk5 to phosphorylate NUDEL.D, Western blot results obtained with anti-tau antibody PHF-1 to reveal phosphorylation at Ser-396 and Ser-404 and with TAU-5 antibody to detect total tau protein. Quantification of lysates from P0 cortices shows that tau phosphorylation, unlike phosphorylation of mDab1, PSD-95, and NUDEL, appears unchanged between p35+/− and p35−/− mice. No significant differences in tau phosphorylation are detected between p35−/− and compound mice. Statistical analysis was performed by unpaired, two-tailed t test (n ≥ 3; *p < 0.05; **p < 0.01). Errors bars indicate SEM.ko, p35−/− animals; tgko, p25-tg/p35−/− animals.
Interestingly, darkly stained neurons with nonpyramidal morphology remain scattered throughout the cortex in p35−/− mice (Fig. 2B,B′,B"), suggesting that their positioning within the cortex is unaffected by loss of p35. A similar result in the positioning of calbindin-positive and GABAergic neurons was observed in Cdk5−/− ↔ wild-type chimeric mice (Gilmore and Herrup, 2001). Therefore, our observation supports the conclusion that positioning of GABAergic interneurons is unaffected in mice with compromised Cdk5 activity (Gilmore and Herrup, 2001).
Antibodies to neurofilament heavy chain (SMI 32) label large pyramidal neurons in layers III and V in the adult mammalian cortex (Campbell and Morrison, 1989; Graus-Porta et al., 2001). In the motorcortex area, the staining identifies mostly large pyramidal neurons of layer V in the wild-type mouse brain (Fig. 2D). In the p35−/− cortex, staining is detected in large neurons that are scattered throughout an area normally occupied by layer II and III neurons (Fig.2E). In contrast, p25-tg/p35−/− mice display a discrete layer of SMI 32-positive neurons (Fig. 2F). The position of this layer is significantly rescued in that the neurons are slightly superficial to what would be expected in the wild-type brain. Opposite to p35−/− mice, very few SMI 32-positive neurons are found in locations just ventral to layer I. These results confirm the observations made after histochemical staining of the cortex (Fig.1A). In summary, p25/Cdk5 expression is sufficient for a subset of calbindin-positive neurons to reach their normal position within the cortex of the compound mice. Moreover, positioning of SMI 32-positive neurons appears rescued to some extent in p25-tg/p35−/− mice (see Discussion).
Partial rescue of the p35−/− corpus callosum phenotype in the compound mice
In addition to lamination abnormalities, p35−/− mice display a prominent disruption of corpus callosum morphology (Chae et al., 1997;Kwon et al., 1999). In contrast, Reeler mice, which show an inverted layering phenotype, have normal corpus callosum morphology (Caviness, 1976; Terashima et al., 1985; Aoki et al., 2001). This suggests that the aberrant corpus callosum morphology observed in p35-null mice is a primary defect and not secondary because of malpositioning of neurons (Kwon et al., 1999). Using a modified silver-staining technique to visualize neuronal projections, we investigated whether expression of p25 affects the formation of fiber tracts in p35-null mice. In p35−/− mice, fibers defasciculate from the corpus callosum adjacent to the midline, resulting in a markedly reduced thickness of the corpus callosum when compared with wt mice (Fig. 3A,B). Furthermore, aberrant fiber fascicles course through the neocortex. In p25-tg/p35−/− mice, the corpus callosum morphology is restored adjacent to the midline (Fig. 3C,D). Moving laterally, it is apparent that the corpus callosum is still thinned and less compact, although the defect is reduced compared with p35-null mice. Moreover, no aberrant fiber fascicles are found in superficial layers of the neocortex, indicative of a restoration of normal cell layering in the cortex (Caviness, 1976; Caviness and Yorke, 1976). These results show that the organization of fiber tracts associated with the neocortex can be partially rescued by p25/Cdk5 signaling in p35−/− mice, indicating that p25 can substitute for p35 in developmental contexts other than cell-positioning defects.
Fig. 3.
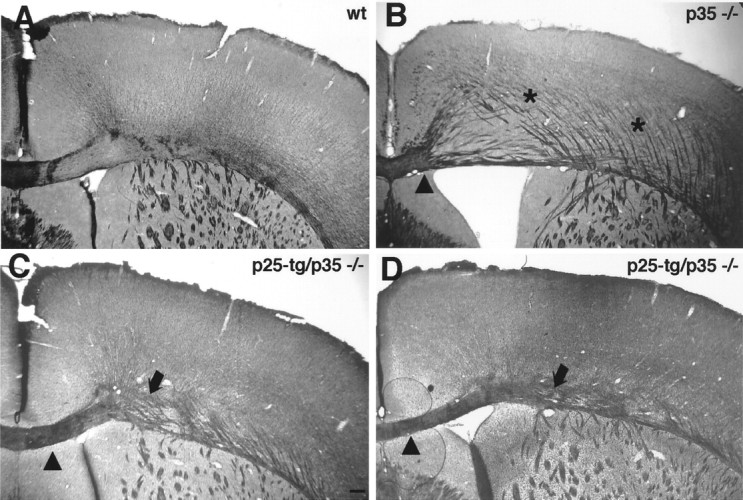
Partial rescue of p35−/− corpus callosum phenotype in p25-tg/p35−/− mice. Coronal sections of paraformaldehyde-fixed brains from 6-month-old mice were stained with a reduced silver technique to reveal fiber tracts. A, In the wt brain, the corpus callosum is tightly fasciculated.B, p35−/− mice display a reduced corpus callosum except for the part close to the midline (medial ofarrowhead). In addition, thick aberrant fiber fascicles course through the cortex, which are not seen in wild-type mice.C, D, In p25-tg/p35−/− mice, corpus callosum morphology is significantly restored (compare areas just lateral to arrowheads in B–D). However, the corpus callosum remains loosely organized in more lateral aspects of the brain (arrows). Unlike in the p35−/− cortex, aberrant fiber fascicles are not prominent in superficial layers.C, Anterior part of the corpus callosum;D, more posterior part of the corpus callosum. Medial is to the left; dorsal is at the top. Scale bar, 200 μm.
Increased levels of p39 do not account for the rescue observed in p25-tg/p35−/− mice
The phenotypic rescue of our compound mice suggested that Cdk5 signaling was partially restored during brain development. p39, the other major activator of Cdk5 in the brain, is upregulated in p35−/− mice, indicating that levels of p39 are regulated by p35 (Ko et al., 2001). To exclude the possibility that the phenotypic rescue we observed is simply a result of increased p39 levels, we investigated whether p39 levels are altered by expression of the p25 transgene during development. As control mice, we used p35+/− mice, which are phenotypically normal (Chae et al., 1997). Throughout development, levels of p39 in the cerebral cortex are increased in p35−/− mice in comparison with p35 heterozygous mice, in accordance with previously described results (Fig.4A–D) (Ko et al., 2001). The levels of p39 in p25-tg/p35−/− mice are very similar to those in p35-null animals (Fig. 4A–D). Therefore, expression of p25 did not alter p39 levels. In summary, our results suggest that the phenotypic rescue in the compound mice is a direct consequence of p25/Cdk5 signaling rather than an indirect effect attributable to increased p39/Cdk5 signaling.
Fig. 4.

Expression of Cdk5 activators during development. Western blot analysis of lysates obtained from cerebral cortex at different embryonic and postnatal stages (as indicated) is shown.A–D, p39 levels in littermates of each stage. Genotypes are indicated. Throughout development, p39 levels are elevated in p35−/− and p25-tg/p35−/− mice compared with p35+/− control animals. p39 levels in p25-tg/p35−/− mice are not increased compared with p35−/− animals, indicating that the partial rescue of the p35-null phenotype is not attributable to upregulation of p39 by p25.E, P25/35 levels in p25-tg/p35+/− mice and p25 levels in p25-tg/p35−/− mice. p35 levels strongly increase between E10.5 and E12.5 and continue to increase throughout embryonic development. p25 levels are lower compared with p35 and reach higher levels only from E16 onward. Similar results regarding p25 levels are obtained in p25-tg/p35+/− and p25-tg/p35−/− mice.
Reduced expression of the p25 transgene compared with endogenous p35 during development
The partial rescue of the p35−/− phenotype in the compound mice indicates increased p25/Cdk5 signaling during brain development. Given the incompleteness of the rescue, however, we were interested in exploring whether differences might exist between the levels of p25 expressed from the transgene and endogenous p35 during development of the cerebral cortex. To allow for direct comparison of p25 and p35 levels in the same animals, we performed a detailed Western blot analysis in p25-tg/p35+/− mice throughout embryonic development. Levels of p35 and p25 are low during early [embryonic day 10.5 (E10.5)] cortex development compared with levels of the cytoskeletal protein actin (Fig. 4E). The identity of weak p25 and p35 bands was confirmed in control animals lacking either p25 (data not shown) or p35 (Fig. 4E). p35 levels increase substantially between E10.5 and E12.5, whereas p25 levels remain low. During further development, p35 levels increase slowly until postnatal day 0 (P0), the last age analyzed. Notably, although p25 levels steadily increase until P0, they remain reduced compared with p35 over the entire time frame covered. Determination of p25 levels at E12.5, E16, and P0 in the compound mice reveals results similar to those in p25-tg/p35+/− animals. Taken together, transgenic p25 levels are reduced compared with endogenous p35 levels during embryonic development of the cerebral cortex.
Phosphorylation of endogenous Cdk5 substrates during development
To explore differences in p25/Cdk5 to p35/Cdk5 signaling in vivo, we analyzed the phosphorylation of known endogenous substrates of Cdk5 during development. The partial rescue of the p35−/− phenotype in p25-tg/p35−/− mice suggested that physiological substrates of p35/Cdk5 were phosphorylated by p25/Cdk5 during development. Therefore, we chose to analyze the phosphorylation of mDab1 at Ser-491, a residue that shows diminished phosphorylation in the Cdk5−/− brain at E16.5 and P0 (Keshvara et al., 2002). Western blot analysis conducted with an antibody that specifically detects mDab1 phosphorylated at Ser-491 reveals decreased phosphorylation in the E16 cortex of p35−/− mice compared with p35+/− control animals (Fig. 5A). Densitometric quantification of signals for Ser-491 phosphorylation (relative to total mDab1) demonstrates a decrease by ∼60% in p35-null mice compared with p35+/− animals, which is consistent with results obtained in Cdk5−/− animals (Keshvara et al., 2002). In p25-tg/p35−/− animals, Ser-491 phosphorylation is significantly increased over that in p35−/− animals at E16; however, it is still decreased compared with p35+/− control animals. In P0 animals, phosphorylation of Ser-491 remains reduced by ∼60% in p35−/− mice compared with p35+/− mice. Again, phosphorylation of Ser-491 is significantly increased in the compound mice compared with p35−/− animals. Additionally, levels of phosphorylation at P0 are higher compared with E16, and this might reflect increased expression of p25 at later stages (Fig. 4E). Taken together, these results show that expression of p25 can partially restore phosphorylation of an endogenous substrate of p35/Cdk5 during cortex development.
Fig. 5.
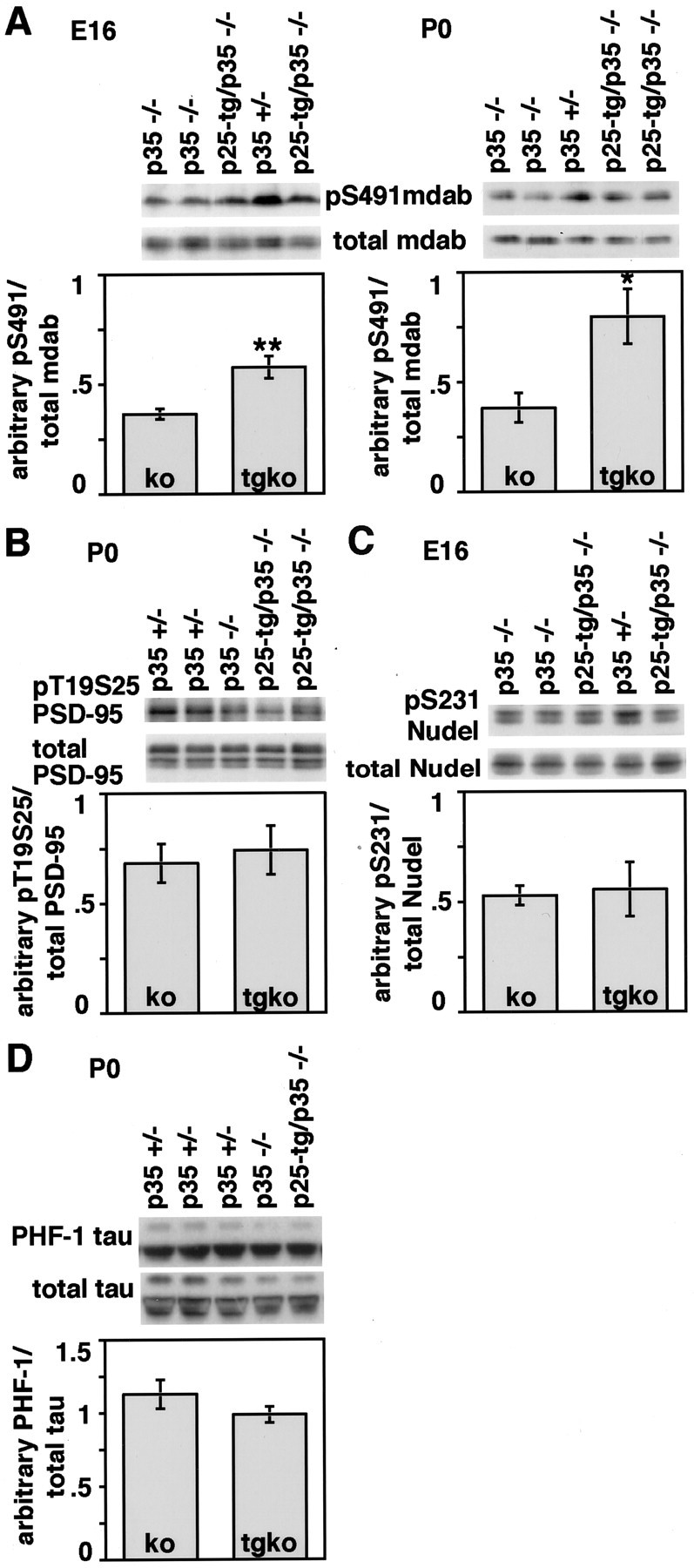
Phosphorylation of endogenous Cdk5 substrates during development. Western blot analysis of lysates obtained from cerebral cortex of littermate mice is shown. A, E16 and P0 Western blot results using a phospo-epitope-specific antibody to Ser-491 of mDab1 and an antibody recognizing total mDab1. Densitometric quantification of Western blot results reveals that at both developmental stages, phosphorylation at Ser-491 relative to total mDab1 levels is reduced by ∼60% in p35−/− compared with control p35+/− animals (for which levels were arbitrarily set to 1). However, phosphorylation is significantly increased in p25-tg/p35−/− mice compared with p35-null mice. B, Western blot results using a phospo-epitope-specific antibody to Thr-19 and Ser-25 of PSD-95 and an antibody recognizing total PSD-95. Phosphorylation of PSD-95 is slightly decreased in p35−/− compared with p35+/− cortices at P0. Quantification shows that phosphorylation levels are not significantly different between p35−/− and p25-tg/p35−/− mice. C, Western blot analysis of Ser-231 phosphorylation of NUDEL and total NUDEL in the cerebral cortex of E16 mice. Phosphorylation is reduced by ∼50%
Another endogenous substrate of Cdk5 is PSD-95. Phosphorylation of PSD-95 is decreased at P0 in p35−/− and p35/p39 double knock-out mice, as determined by a phospho-epitope-specific antibody that recognizes residues Thr-19 and Ser-25 (M. Morabito, M. Sheng, and L.-H. Tsai, unpublished observations). To explore whether expression of p25 in p35−/− mice can restore phosphorylation of PSD-95, we investigated the phosphorylation of PSD-95 in the compound mice compared with p35-null animals. In the cortex of p35−/− mice, phosphorylation is reduced by ∼30% compared with p35+/− mice at P0 (Fig. 5B). Unlike in the case of mDab1, phosphorylation remains unaltered in the compound mice, indicating a failure of p25/Cdk5 to phosphorylate PSD-95 in vivo during development.
NUDEL, a lissencephaly 1-interacting protein, displays diminished phosphorylation in mice with compromised Cdk5 function (Niethammer et al., 2000; Ko et al., 2001). One of the phosphorylation sites targeted by Cdk5 is Ser-231 (Niethammer et al., 2000). Indeed, phosphorylation of NUDEL at Ser-231 is reduced by ∼50% in the cortex of p35−/− mice compared with control p35+/− mice at E16 (Fig. 5C). Similar to the results obtained by analysis of PSD-95 phosphorylation, we did not detect a significant increase in Ser-231 phosphorylation of NUDEL in p25-tg/p35−/− mice compared with p35−/− mice.
p25/Cdk5 phosphorylates tau protein at multiple sites in vitro and in confined regions of the adult mouse brain that display high levels of p25 (Ahlijanian et al., 2000; Lund et al., 2001). At P0, however, tau phosphorylation at Ser-199, Ser-202, and Thr-205 (AT8 antibody) and Ser-396 and Ser-404 (PHF-1 antibody) does not differ between wt and p35/p39 double knock-out mice brain lysates (Ko et al., 2001). In support of this, we did not observe changes in tau phosphorylation at the PHF-1 epitope between p35+/− control mice and p35 knock-out animals at P0 (Fig. 5D). Additionally, we did not detect significant differences in tau phosphorylation between p35−/− and the compound animals. The same results were obtained with an antibody that specifically recognizes tau phosphorylation at Ser-202 (data not shown). Similarly, Ahlijanian et al. (2000) did not observe changes in tau phosphorylation at the AT8 and PHF-1 epitopes in adult mouse brain lysates between wt and NSE p25-tg animals. It is possible that hyperphosphorylation of tau in these animals only occurs in restricted regions of the brain, or alternatively that p25/Cdk5 levels need to reach a certain threshold to achieve widespread hyperphosphorylation of tau (see below).
In summary, we observe differences in the phosphorylation of endogenous p35/Cdk5 substrates in the developing cortex of p35−/− mice compared with compound animals. On the one hand, phosphorylation of mDab1 becomes partially restored in p25-tg/p35−/− mice; on the other, phosphorylation of the endogenous substrates PSD-95 and NUDEL seems unaffected. These results indicate that p25/Cdk5 can signal to pathways normally used by p35/Cdk5, but that some of the signaling of p35/Cdk5 may be lost.
Pathogenic changes of the brain are detected in aged NSE p25-tg mice
Four-month-old NSE p25-tg mice display a number of pathogenic changes in certain brain regions, including hyperphosphorylation of neurofilament subunits and tau, increased number of argyophilic neuronal somata, and disruption of the axonal cytoskeleton (Ahlijanian et al., 2000). Our p25-tg mice did not show increased immunoreactivity for the phosphorylation-dependent neurofilament antibody SMI 34 at 9 months of age or appreciable differences after silver staining at 6 months of age compared with wt animals (data not shown). This may suggest that alteration of the genetic background from FVB/N (p25-tg mice) to C57bl6 (p35−/− mice) attenuates the phenotype of p25-tg mice. Indeed, we observed degeneration of various brain regions, including cerebral cortex and spinal cord, in 2-year-old p25-tg and p25-tg/p35−/− animals, whereas 6-month-old (data not shown) and 9-month-old animals appeared histologically normal (Fig.6). FVB/N p25-tg mice display approximately a 1:1 ratio of p25 to endogenous p35 in 4-month-old cortices (Ahlijanian et al., 2000). To investigate whether the discrepancies in the phenotype of FVB/N p25-tg mice to our mice might be a result of differences in p25 levels, we tested for p25 levels in the cortices of aged transgenic mice (Fig. 6D). The ratio of p25 to p35 is relatively low in p25-tg/p35+/− animals at P0. At 7 months of age, the ratio is slightly increased, but in 28-month-old mice, p25 levels reach p35 levels. Therefore, low levels of p25 in our young adult mice may account for the lack of pathogenic changes in these animals compared with FVB/N p25-tg mice. In summary, the absence of pathogenic changes in 6- and 9-month-old mice that carry the p25 transgene indicates that the analysis of brain morphology in the compound mice is not confounded by early toxicity of p25. Deregulation of Cdk5 by moderate levels of p25, however, does eventually result in remarkable pathogenic changes in aged brains.
Fig. 6.

Aged p25-tg mice display brain degeneration.A, B, Coronal sections of paraformaldehyde-fixed mouse brains stained with hematoxylin and eosin. The depicted area corresponds to deep layers of the lateral motorcortex at the level of the anterior commissure. A, In 9-month-old mice, no obvious morphological degeneration is apparent in transgenic mice compared with nontransgenic control mice (genotypes as indicated). B, In 2-year-old mice, a spongiform degeneration in the cortex is evident in p25-tg and p25-tg/p35−/− mice compared with corresponding control animals. The spongiform degeneration is characterized by large holes in the tissue, which often contain amorphous material. C, Cross sections of cervical spinal cord in 2-year-old animals. The anterior horn of p25-tg mice displays a degeneration compared with wild-type animals. For all images, dorsal is at the top; medial is to theleft. Scale bars, 200 μm. D, p25 levels increase with age. Western blot analysis of p25 and p35 levels in the cerebral cortex of p25-tg/p35+/− mice is shown. At P0, levels of p25 are relatively low compared with endogenous p35; however, they increase slightly in 7-month-old (7mo) mice. In 28-month-old (28mo) animals, p25 levels are similar to p35 levels. The single lane to the right is a longer exposure of the first P0 lane (compare with Fig.4E).
Discussion
To address the question of whether p25/Cdk5 signaling differs from p35/Cdk5 signaling in vivo, we have generated NSE p25-tg mice in a p35-null background. We find that expression of low levels of p25 during development alleviates several aspects of the p35−/− phenotype independently of p39 levels. Consistent with the phenotypic rescue, phosphorylation of the endogenous Cdk5 substrate mDab1 is partially restored in compound animals during development. Our results provide evidence that p25/Cdk5 signals through pathways normally used by p35/Cdk5. Furthermore, analysis of the phosphorylation of other endogenous Cdk5 substrates in the compound mice indicates that p25/Cdk5 and p35/Cdk5 signaling may differ to some extent in vivo.
Altered signaling of p25/Cdk5 compared with p35/Cdk5
Previous studies on p25/Cdk5-mediated signaling have indicated that it differs from p35/Cdk5 signal transduction. Foremost, p25-tg mice display breakdown of the axonal cytoskeleton, hyperphosphorylation of tau and neurofilament, as well as behavioral abnormalities (Ahlijanian et al., 2000; Bian et al., 2002), whereas p35-tg mice do not (Van den Haute et al., 2001). This gain of toxic function might be explained by altered subcellular localization of p25 compared with p35, which results in mislocalization of active Cdk5 within the cell (Patrick et al., 1999; Kusakawa et al., 2000; Lee et al., 2000). Furthermore, in vitro studies on the phosphorylation of tau have indicated that p25/Cdk5 could gain some toxic function by targeting phosphorylation sites within substrates that are less efficiently targeted by p35/Cdk5 (Hashiguchi et al., 2002).
Although p25/Cdk5 appears to gain some toxic function, little is known regarding further differences between p25/Cdk and p35/Cdk5. More specifically, it is unknown whether p25/Cdk5 would phosphorylate the same substrates p35/Cdk normally targets. p25 has been shown to be a more stable protein than p35 (Patrick et al., 1998, 1999). Accordingly, accumulation of p25 could result in increased phosphorylation of endogenous p35/Cdk5 substrates and might contribute to disturbed Cdk5 signaling. Analysis of the signaling of p25/Cdk5 requires a system that allows distinguishing unequivocally between p25/Cdk5 and p35/Cdk5 and is therefore best performed in a system that lacks p35. Our results from the p25-tg/p35−/− mice indicate that p25/Cdk5 can substitute for p35/Cdk5 at a cellular level in vivo. This is best illustrated by the normal positioning of a subset of calbindin-positive neurons as well as by the almost normal appearance of the hippocampus and the cerebellum in the compound mouse. Therefore, in addition to a toxic gain of function by p25, p25/Cdk5 must use signaling pathways in the compound mice that are normally used by p35 during development. Consistent with this, we observed increased Ser-491 phosphorylation of the endogenous Cdk5 substrate mDab1 in the compound mice compared with p35−/− mice during development. Interestingly, a study by Hashiguchi et al. (2002) demonstrates that p25/Cdk5 can phosphorylate all sites of a substrate that are targeted by p35/Cdk5, arguing that phosphorylation sites overlap in vitro. Our results regarding mDab1 phosphorylation now suggest that p25/Cdk5 can target the same sites of a substrate that are normally phosphorylated by p35/Cdk5 in vivo.
Despite these findings, our data indicate that p25/Cdk5 cannot execute all aspects of p35/Cdk5 signaling. First, p25/Cdk5 fails to phosphorylate the endogenous p35/Cdk5 substrates PSD-95 and NUDEL during development, although it is able to phosphorylate mDab1. Furthermore, we observed a slight increase of p39 levels in p35−/− mice throughout development. p39 levels are similar in p25-tg/p35−/− animals compared with p35−/− mice, which indicates that p25 cannot compensate for the loss of p35 with regard to regulation of p39 levels. Taken together, our in vivo results implement new aspects of p25/Cdk5-mediated signaling compared with p35/Cdk5, aside from a previously anticipated toxic gain of function. On the one hand, p25/Cdk5 signals through pathways that are normally used by p35/Cdk5; on the other, some aspects of p35/Cdk5 signaling seem to remain lost in the presence of p25/Cdk5.
Substrates of Cdk5 during development
Although several endogenous substrates of p35/Cdk5 have been identified during nervous system development (Niethammer et al., 2000;Keshvara et al., 2002), little is known regarding the functions of these phosphorylation events. mDab1 is an essential adaptor protein in the reelin pathway that regulates neuronal positioning in the cerebral cortex (Rice and Curran, 2001). Phosphorylation of Ser-491 of mDab1 is strongly diminished in the Cdk5−/− brain (Keshvara et al., 2002). As such, two signaling pathways that control cell positioning might cross talk via mDab1 (Keshvara et al., 2002). Our finding that increased Ser-491 phosphorylation of mDab1 in the compound mice correlates with a partial rescue of the p35−/− cell-positioning defect does not contradict this hypothesis. Any further conclusions regarding the function of Ser-491 phosphorylation of mDab1 remain speculative. Unlike mDab1, NUDEL and PSD-95 did not show increased phosphorylation in the compound mice compared with p35−/− animals, suggesting that phosphorylation might not be necessary for the phenotypic rescue. The residual phosphorylation of both substrates in the p35−/− cortex, however, seems higher than that of mDab1. One possible explanation may be that the p39/Cdk5 kinase has more impact on the phosphorylation of NUDEL and PSD-95 than of mDab1. Given such uncertainties, we think that conclusions with regard to the lack of phosphorylation and the observed phenotype would be premature.
Reduced levels of p25 compared with p35 during development
Evaluation of p25/Cdk5 signaling during development ideally requires expression of p25 in the same cells, at the same levels, and at the same time as endogenous p35. Expression of p35 and p39 is primarily neuronal (Lew et al., 1994; Tsai et al., 1994; Tang et al., 1995), and analysis of Cdk5 chimeric mice has shown that Cdk5 acts cell-autonomously in neurons (Ohshima et al., 1999). Expression of transgenes from the NSE promoter is restricted to differentiating and adult neurons (Forss-Petter et al., 1990). Consistent with previously published results obtained from NSE promoter-driven transgenes (Bernard et al., 1998), p25 can be detected early in cortical development (E10.5), when expression is comparable with endogenous p35 levels. During further embryonic development, as expression levels of p35 increase, the levels of p25 remain low relative to those of p35. Despite this expression profile, some p25/Cdk5 signaling is induced during development, resulting in alleviation of several aspects of the p35-null brain phenotype in p25-tg/p35−/− animals. The rescue is not complete and results in an intermediate phenotype. Regarding cell positioning, calbindin-positive neurons segregate into two fractions, with one fraction being rescued and the other one appearing unaffected. Given the relatively low expression levels of the transgene compared with endogenous p35, some calbindin-positive neurons might not attain sufficiently high expression of p25 to allow migration to proper target areas. Alternatively, Cdk5 signaling might differ within calbindin-positive neurons, rendering a subpopulation of them receptive to p25/Cdk5-mediated signaling, whereas in another subpopulation, p25/Cdk5 cannot substitute for p35/Cdk5. More instructive is the finding that some calbindin-positive neurons resume normal positions within the NSE p25-tg/p35−/− cortex. This result is a proof of principle that p25 can substitute for p35, at least in those neurons, and it shows that p25/Cdk5 signaling must use pathways that p35/Cdk5 is engaged in.
Layering in the cerebral cortex of p25-tg/p35−/− mice
A fraction of calbindin-positive pyramidal neurons in the motorcortex area resides in its normal position in layers II and III of the compound mice. In contrast, SMI 32-positive large pyramidal neurons are found superficial to their normal position. The mislocalization of SMI 32-positive neurons in the cortex of the compound mice might be explained in part by the inability of some calbindin-positive neurons to reach superficial positions. This would result in a thinning of layers II and III, which we indeed find in the compound mice. Accordingly, if one assumes that the position of SMI 32-positive neurons is rescued in the compound mice, they should be found in slightly superficial locations compared with wt mice, which is what we observe. In other words, one could consider the position of SMI 32-positive neurons as normal in relation to other layers of a wild-type-like but thinned cortex, which resides superficial to a mass of neurons that failed to reach normal positions. This view is supported by the presence of a thin layer of small granule cells, which resemble layer IV neurons, just above the layer of SMI 32-positive (layer V) neurons in the compound mice (Fig. 1A). Taken together, p25/Cdk5 signaling seems to reestablish accurate positioning of presumptive layer V, some layer IV, and a fraction of layer II and III neurons. Hence, expression of p25 appears to be sufficient to substitute for p35 in a subset of neurons destined for layers II–V, which are the layers primarily affected in Cdk5-deficient mice (Gilmore and Herrup, 2001).
On the basis of our findings, we hypothesize an altered and adverse signaling of Cdk5 under conditions that generate p25. First, p25/Cdk5 gains some toxic function. Second, a buildup of p35/p25 over the normal levels of endogenous p35 could result in hyperphosphorylation of some substrates of p35/Cdk5. Third, p25/Cdk5 could be unable to perform all aspects of normal p35/Cdk5 signaling. Taken together, our findings provide a basis on which changes in Cdk5 signaling under pathological conditions that generate p25 can be investigated further.
Footnotes
This work was supported by National Institutes of Health Grant GM 53049. We are grateful to Jonathan Cruz, Amitabh Gupta, Jane Ko, and Benjamin Samuels for critically reading this manuscript.
Correspondence should be addressed to Dr. Li-Huei Tsai or Dr. Holger Patzke, Department of Pathology and Howard Hughes Medical Institute, 200 Longwood Avenue, Armenise Building, Room 342, Boston, MA 02115. E-mail: li-huei_tsai@hms.harvard.edu.
References
- 1.Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez A, Toro R, Caceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- 3.Aoki T, Setsu T, Okado H, Mikoshiba K, Watanabe Y, Terashima T. Callosal commissural neurons of Dab1 deficient mutant mouse, yotari. Neurosci Res. 2001;41:13–23. doi: 10.1016/s0168-0102(01)00249-8. [DOI] [PubMed] [Google Scholar]
- 4.Bayer SB, Altman J. Neocortical development. Raven; New York: 1991. [Google Scholar]
- 5.Bernard R, Dieni S, Rees S, Bernard O. Physiological and induced neuronal death are not affected in NSE-bax transgenic mice. J Neurosci Res. 1998;52:247–259. doi: 10.1002/(SICI)1097-4547(19980501)52:3<247::AID-JNR1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 6.Bian F, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KKW, Walker LC. Axonopathy, tau abnormalities, and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol. 2002;446:257–266. doi: 10.1002/cne.10186. [DOI] [PubMed] [Google Scholar]
- 7.Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann–Pick type C murine model. J Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell MJ, Morrison JH. Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol. 1989;282:191–205. doi: 10.1002/cne.902820204. [DOI] [PubMed] [Google Scholar]
- 9.Caviness VS., Jr Patterns of cell and fiber distribution in the neocortex of the reeler mutant mouse. J Comp Neurol. 1976;170:435–447. doi: 10.1002/cne.901700404. [DOI] [PubMed] [Google Scholar]
- 10.Caviness VS, Jr, Yorke CH., Jr Interhemispheric neocortical connections of the corpus callosum in the reeler mutant mouse: a study based on anterograde and retrograde methods. J Comp Neurol. 1976;170:449–459. doi: 10.1002/cne.901700405. [DOI] [PubMed] [Google Scholar]
- 11.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 12.Desclin JC, Escubi J. An additional silver impregnation method for demonstration of degenerating nerve cells and processes in the central nervous system. Brain Res. 1975;93:25–39. doi: 10.1016/0006-8993(75)90284-x. [DOI] [PubMed] [Google Scholar]
- 13.Dhavan R, Tsai L-H. A Decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:1–12. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 14.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 15.Gilmore EC, Herrup K. Neocortical cell migration: GABAergic neurons and cells in layers I and VI move in a cyclin-dependent kinase 5-independent manner. J Neurosci. 2001;21:9690–9700. doi: 10.1523/JNEUROSCI.21-24-09690.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 18.Hashiguchi M, Saito T, Hisanaga SI, Hashiguchi T. Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem. 2002;277:44525–44530. doi: 10.1074/jbc.M207426200. [DOI] [PubMed] [Google Scholar]
- 19.Hendry SH, Jones EG, Emson PC, Lawson DE, Heizmann CW, Streit P. Two classes of cortical GABA neurons defined by differential calcium binding protein immunoreactivities. Exp Brain Res. 1989;76:467–472. doi: 10.1007/BF00247904. [DOI] [PubMed] [Google Scholar]
- 20.Humbert S, Dhavan R, Tsai L. p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci. 2000;113:975–983. doi: 10.1242/jcs.113.6.975. [DOI] [PubMed] [Google Scholar]
- 21.Keshvara L, Magdaleno S, Benhayon D, Curran T. Cyclin-dependent kinase 5 phosphorylates disabled 1 independently of Reelin signaling. J Neurosci. 2002;22:4869–4877. doi: 10.1523/JNEUROSCI.22-12-04869.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci. 2001;21:6758–6771. doi: 10.1523/JNEUROSCI.21-17-06758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 24.Kwon YT, Tsai LH. A novel disruption of cortical development in p35(−/−) mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Kwon YT, Tsai LH, Crandall JE. Callosal axon guidance defects in p35(−/−) mice. J Comp Neurol. 1999;415:218–229. doi: 10.1002/(sici)1096-9861(19991213)415:2<218::aid-cne6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 26.Kwon YT, Gupta A, Zhou Y, Nikolic M, Tsai LH. Regulation of N-cadherin-mediated adhesion by the p35-Cdk5 kinase. Curr Biol. 2000;10:363–372. doi: 10.1016/s0960-9822(00)00411-5. [DOI] [PubMed] [Google Scholar]
- 27.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 28.Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 29.Lund ET, McKenna R, Evans DB, Sharma SK, Mathews WR. Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk/p20) using mass spectrometry. J Neurochem. 2001;76:1221–1232. doi: 10.1046/j.1471-4159.2001.00130.x. [DOI] [PubMed] [Google Scholar]
- 30.Nath R, Davis M, Probert AW, Kupina NC, Ren X, Schielke GP, Wang KK. Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun. 2000;274:16–21. doi: 10.1006/bbrc.2000.3070. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- 32.Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee M, Morabito M, Tsai L. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 33.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB. Migration defects of cdk5(−/−) neurons in the developing cerebellum is cell autonomous. J Neurosci. 1999;19:6017–6026. doi: 10.1523/JNEUROSCI.19-14-06017.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohshima T, Ogawa M, Veeranna, Hirasawa M, Longenecker G, Ishiguro K, Pant HC, Brady RO, Kulkarni AB, Mikoshiba K. Synergistic contributions of cyclin-dependent kinase 5/p35 and Reelin/Dab1 to the positioning of cortical neurons in the developing mouse brain. Proc Natl Acad Sci USA. 2001;98:2764–2769. doi: 10.1073/pnas.051628498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–24064. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- 37.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 38.Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 39.Stuhmer T, Puelles L, Ekker M, Rubenstein JL. Expression from a Dlx gene enhancer marks adult mouse cortical GABAergic neurons. Cereb Cortex. 2002;12:75–85. doi: 10.1093/cercor/12.1.75. [DOI] [PubMed] [Google Scholar]
- 40.Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 41.Terashima T, Inoue K, Inoue Y, Mikoshiba K, Tsukada Y. Distribution and morphology of callosal commissural neurons within the motor cortex of normal and reeler mice. J Comp Neurol. 1985;232:83–98. doi: 10.1002/cne.902320108. [DOI] [PubMed] [Google Scholar]
- 42.Tsai LH, Delalle I, Caviness VS, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 43.Tseng HC, Zhou Y, Shen Y, Tsai LH. A survey of Cdk5 activator p35 and p25 levels in Alzheimer's disease brains. FEBS Lett. 2002;523:58–62. doi: 10.1016/s0014-5793(02)02934-4. [DOI] [PubMed] [Google Scholar]
- 44.Van Brederode JF, Mulligan KA, Hendrickson AE. Calcium-binding proteins as markers for subpopulations of GABAergic neurons in monkey striate cortex. J Comp Neurol. 1990;298:1–22. doi: 10.1002/cne.902980102. [DOI] [PubMed] [Google Scholar]
- 45.Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F. Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis. 2001;8:32–44. doi: 10.1006/nbdi.2000.0333. [DOI] [PubMed] [Google Scholar]