

Abstract
The underlying mechanisms of neuropathic pain are poorly understood, and existing treatments are mostly ineffective. We recently demonstrated that antisense mediated “knock-down” of the sodium channel isoform, NaV1.8, reverses neuropathic pain behavior after L5/L6 spinal nerve ligation (SNL), implicating a critical functional role of NaV1.8 in the neuropathic state. Here we have investigated mechanisms through which NaV1.8 contributes to the expression of experimental neuropathic pain. NaV1.8 does not appear to contribute to neuropathic pain through an action in injured afferents because the channel is functionally downregulated in the cell bodies of injured neurons and does not redistribute to injured terminals. Although there was little change in NaV1.8 protein or functional channels in the cell bodies of uninjured neurons in L4 ganglia, there was a striking increase in NaV1.8 immunoreactivity along the sciatic nerve. The distribution of NaV1.8 reflected predominantly the presence of functional channels in unmyelinated axons. The C-fiber component of the sciatic nerve compound action potential (CAP) was resistant (>40%) to 100 μm TTX after SNL, whereas both A- and C-fiber components of sciatic nerve CAP were blocked (>90%) by 100 μm TTX in sham-operated rats or the contralateral sciatic nerve of SNL rats. Attenuating expression of NaV1.8 with antisense oligodeoxynucleotides prevented the redistribution of NaV1.8 in the sciatic nerve and reversed neuropathic pain. These observations suggest that aberrant activity in uninjured C-fibers is a necessary component of pain associated with partial nerve injury. They also suggest that blocking NaV1.8 would be an effective treatment of neuropathic pain.
Keywords: voltage-gated Na+, tetrodotoxin resistant, nerve injury, peripheral nerve, dorsal root ganglion, nociceptor
Introduction
Voltage-gated Na+ channels (VGSCs) are critical for the initiation and propagation of action potentials in neurons. These channels are also involved in the dynamic regulation of neuronal excitability: they are targets of modulation, and changes in the biophysical properties and expression of voltage-gated Na+ channels have a profound impact on neuronal excitability (Cantrell and Catterall, 2001). Peripheral nerve injury results in an increase in the excitability of both injured afferents and their uninjured neighbors, which appears to be critical for the expression of neuropathic pain. That changes in the biophysical properties and expression of VGSCs may contribute to the nerve injury-induced increase in neuronal excitability, and therefore neuropathic pain, is suggested by the observation that the most effective treatments for neuropathic pain involve compounds that block VGSCs (Waxman et al., 1999). At least one specific VGSC, the tetrodotoxin-resistant (TTX-R) channel NaV1.8 [formerly peripheral nerve Na+ channel type 3 (Sangameswaran et al., 1996) and sensory neuron specific (Akopian et al., 1996)], is likely to contribute to neuropathic pain because of its unique biophysical properties and expression pattern (Gold, 2000). We have demonstrated recently that the intrathecal (IT) application of an antisense (AS) oligodeoxynucleotide (ODN) to NaV1.8 reversibly and selectively decreases NaV1.8 protein and functional channels in sensory neurons (Lai et al., 2002). Importantly, full expression of NaV1.8 is necessary for the expression of neuropathic pain behavior. In this study we have sought to determine how NaV1.8 contributes to neuropathic pain.
Theoretical predictions and empirical data suggest that an increase in Na+ current through VGSCs results in an increase in excitability. There are several ways in which such an increase may occur, including a change in biophysical properties, an increase in expression, and a redistribution of channels. Therefore, we hypothesized that at least one of these mechanisms underlies the contribution of NaV1.8 to neuropathic pain.
Our results show that this channel is primarily redistributed to the axons of uninjured unmyelinated afferents enabling TTX-R conduction. We also tested the hypothesis that such a redistribution of axonal NaV1.8, and the resultant increase in TTX-R current, is critical to noxious thermal hyperalgesia and tactile hypersensitivity. Intrathecal administration of NaV1.8 antisense ODN, but not a mismatch (MM) control ODN, reverses nerve injury-induced pain behavior as well as the redistribution of NaV1.8 channels to the uninjured axons.
Preliminary results have been published previously in abstract form (Gold et al., 2001).
Materials and Methods
Animals. Adult male Sprague Dawley rats (150–300 gm; Harlan Sprague Dawley, Madison, WI) were used for the experiments. All surgical procedures used in this study were reviewed and approved by the University of Maryland Baltimore and the University of Arizona Institutional Animal Care and Use Committees. Animal care and handling were in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Animal model of neuropathic pain. The surgical procedure for L5/L6 spinal nerve ligation was performed according to that described previously (Kim and Chung, 1992). Sham-operated control rats were prepared in an identical manner except that the L5/L6 nerve roots were not ligated. The behavior of the rats was monitored carefully for any visual indication of motor disorders or change in weight or general health.
Evaluation of tactile and thermal sensitivity. Mechanical threshold was determined by measuring the paw withdrawal threshold to probing with a series of calibrated (0.4–15 gm) von Frey filaments (Chaplan et al., 1994). Thermal threshold was determined by measuring paw withdrawal latencies to a radiant heat source applied to the plantar surface of the affected paw of nerve-injured or sham-operated rats (Hargreaves et al., 1988). Baseline mechanical and thermal sensitivity were determined for all experimental subjects before sham or L5/L6 spinal nerve ligation (SNL) surgery. Testing was performed daily subsequent to the surgery. Statistical analysis of the data involved ANOVA followed by Fisher's least significant difference test. The baseline latency to paw withdrawal in response to von Frey probing or thermal stimulus was determined for each animal before surgery and thereafter monitored once daily.
Oligodeoxynucleotides that target Nav1.8. An antisense sequence that is complementary to nucleotides 107–129 of the coding region of the rat NaV1.8 (“antisense”: 5′-TCCTCTGTGCTTGGTTCTGGCCT-3′) and a corresponding mismatch sequence (“mismatch”: 5′-TCCTTCGTGCTGTGTTCGTGCCT-3′) were synthesized as phosphodiester ODNs (Midland Certified Reagent, Midland, TX). The sequence specificity and uptake of these ODNs into DRG cells, and the conditions under which the antisense ODN elicits a “knock-down” of NaV1.8, have been characterized in a previous study (Lai et al., 2002). The ODNs were reconstituted in nuclease-free ultrapure water to a final concentration of 9 μg/μl.
Administration of ODNs. Chronically indwelling intrathecal catheters were implanted into rats according to the method described byYaksh and Rudy (1976) for ODN administration. The animals were allowed to recover from the implantation surgery for 3–5 d before any experimentation, and they were monitored daily after surgery for signs of motor deficiency. Intrathecal injections of ODNs or saline were made in a volume of 5 μl followed by a 9 μl saline to flush the catheter. The ODNs were administered twice daily at 45 μg per injection. Injection was initiated 5 d after sham or SNL surgery, when tactile hypersensitivity and thermal hyperalgesia were established in the SNL rats. The animals were given either the antisense or mismatch ODN for up to 5 d.
Retrograde labeling. To identify sensory neurons in L4 and L5 DRG giving rise to axons comprising the sciatic nerve, we injected the retrograde tracer DiIC18 (DiI) into the sciatic nerve at mid thigh. The sciatic nerve was exposed with blunt dissection at mid thigh level at least 10 d before any subsequent manipulation. DiI was dissolved in DMSO (170 mg/ml) and then diluted 1:10 in saline (Eckert et al., 1998). Two microliters of tracer were injected slowly into the nerve via a 30 gauge needle with the aid of a dissecting microscope. The surgical incision was closed with sutures, and animals were monitored daily after surgery for signs of nerve injury. The labeling procedure had no detectable influence on mechanical or thermal nociceptive threshold (data not shown).
Electrophysiological analysis. Surgical procedures and dissociation protocol were similar to those described previously (Lai et al., 2002). Isolated neurons were studied between 2 and 7 hr after removal from the animal. Voltage-clamp recordings were performed using a Heka EPC9 (Heka Electonik, Lambrecht/Pfaz, Germany). Currents were low-pass filtered at 5–10 kHz with a four-pole Bessel filter and digitally sampled at 25–100 kHz. Capacity transients were cancelled and series resistance was compensated (>80%); a P/4 protocol was used for leak subtraction. Patch pipettes (0.7–3 MΩ) were filled with (in mm): 100 Cs methansulfonate, 40 tetraethylammonium-Cl, 5 NaCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, 2 Mg-ATP, 1 Li-GTP; pH was adjusted to 7.2 with TRIS-base, and osmolarity was adjusted to 310 mOsm with sucrose. The bath solution used to record whole-cell Na+ currents in isolation contained (in mm): 35 NaCl, 30 tetraethylammonium-Cl, 65 choline-Cl, 0.1 CaCl2, 5 MgCl2, 10 HEPES, 10 glucose; pH was adjusted to 7.4 with Tris-base, and osmolarity was adjusted to 325 mOsm with sucrose. TTX-R INa was isolated from TTX-sensitive Na+ currents (TTX-SINa) with a 500 msec prepulse to −40 mV (Gold et al., 1996, 1998).
After formation of a tight seal (>5 GΩ) and compensation of pipette capacitance with amplifier circuitry, whole-cell access was established. Four hyperpolarizing pulses (10 msec, 20 mV from −60 mV) were recorded for use in the determination of the cell capacitance. Whole-cell capacitance and series resistance was compensated with the amplifier circuitry. The holding potential was then changed to −80 mV. Voltage-gated Na+ currents were evoked after a 500 msec voltage step to either −120 or −40 mV. Current evoked from −40 mV was considered to be TTX-R current (Gold et al., 1996), whereas the difference between the current evoked from −120 and −40 mV was considered to be TTX-SINa. Application of TTX (1 μm) to five cells confirmed that these voltage-clamp protocols were sufficient to isolate the two currents (data not shown). Current–voltage (I–V) relationship data were collected for an I–Vcurve every 2 min. Membrane potential was held at −80 mV. Current was evoked after a 500 msec prepulse to either −120 or −40 mV with a 15 msec step to potentials between −60 and +45 mV in 5 mV increments. Steady-state availability was assessed for TTX-RINa with a 1 sec prepulse varying between −100 and +10 mV followed by a voltage step to 0 mV. Conductance–voltage (G–V) curves were constructed from I–V curves by dividing the peak evoked current by the driving force on the current, such thatG = I/(Vm− Vrev), whereVm is the potential at which current was evoked and Vrev is the reversal potential for the current determined by extrapolating the linear portion of the I–V curve through 0 current. We have demonstrated previously that the approach used for the determination of Vrev in sensory neurons is valid under the recording conditions used in the present study because the I–V curve for voltage-gated Na+ currents in sensory neurons is linear though 0 pA (Gold et al., 2002). Activation and steady-state availability data were fitted with a Boltzmann equation of the formG = Gmax/1 + exp[(V0.5 −Vm)/k], whereG = observed conductance,Gmax = the fitted maximal conductance,V0.5 = the potential for half activation or availability, Vm = command potential, and k = the slope factor. Recovery from inactivation (or repriming) was assessed at −80 mV with a two-pulse protocol. The conditioning and test pulses were to +10 and −25 mV for TTX-R and TTX-S currents, respectively. Recovery data were fitted with an exponential equation of the form Y =f0*(1 − exp(−x/τ1)) + (1 −f0)*(1 − exp(−x/τ2)), wheref0 is the fraction of recovery accounted for by the first time constant, x is the interpulse duration, τ1 is the first time constant, and τ2 is the second time constant. The density TTX-R and TTX-S current was determined by dividing theGmax by the cell capacitance. Two groups of rats were studied: one group received SNL of L5/L6 spinal nerve (n = 7), and the other group received a sham-operation (n = 4). Neurons from both injured (L5/L6) and uninjured (L4) ganglia were studied from each rat. Ten to 20 neurons were studied from each rat.
Suction electrode recording. Sciatic nerves were dissected and immediately submerged in ice-cold (4°C) Locke solution of the following composition (in mm): 136 NaCl, 5.6 KCl, 14.3 NaHCO3, 1.2 NaH2PO4, 2.2 CaCl2, 1.2 MgCl2, 11 dextrose, equilibrated continuously with 95% O2, 5% CO2, pH 7.2–7.4. Nerves were trimmed of excess connective tissue, and blood vessels were then pinned to the Sylgard (Dow Corning, Midland MI)-coated floor of a recording chamber (∼0.25 ml volume). Nerves were superfused (3–5 ml/min) with oxygenated Locke solution at room temperature, 22–24°C. Locke solution was delivered via a gravity-driven perfusion system and exited the recording chamber through a hole in the floor of the chamber. TTX was added to the recording chamber via a manifold connected to the inflow line.
Compound action potentials (CAPs) were recorded with a glass suction electrode connected to the input stage of an AC-coupled differential preamplifier (0.1–1 kHz; WPI, Sarasoto, FL; model DAM-5A). Data were filtered at 2 kHz and sampled at 10 kHz. The sciatic nerve was threaded through a grease (Dow Corning)-lined notch into a mineral oil-containing compartment and placed onto two platinum-stimulating electrodes. CAPs were evoked with supramaximal electrical pulses that were 0.1–0.5 msec in duration. CAPs were elicited at 0.2 Hz. Voltage data were digitized via a TL1 A/D converter (Axon Instruments, Union City, CA) and stored on a Pentium PC. Six CAPs were averaged before saving. Data acquisition and storage were controlled via pClamp6.1 (Axon Instruments).
CAP data were analyzed with Clampfit 8.2 (Axon Instruments). The A-fiber deflection of the CAP (A-wave) was easily distinguished from that associated with the C-fiber deflection (C-wave) because of the time delay between the arrival of the two waves at the recording electrode.
A one-way ANOVA test with a Tukey post hoc test was used to assess the presence of statistically significant differences between groups; p < 0.05 was considered statistically significant. Values presented are mean ± SEM.
Western transblot analysis. A section of the sciatic nerve ∼1 cm proximal to the trifurcation was isolated from each of six rats and pooled. The tissues were homogenized (with glass/glass homogenizer) in a total of 1.5 ml of ice-cold 10 mm PBS/100 μm phenylmethylsulfonylfluoride/50 μg/ml bacitracin/30 μm bestatin/10 μm captopril. Membranes were pelleted by high-speed centrifugation (42,700 × g for 60 min at 4°C). Membrane proteins were then solubilized by resuspending the pellet with 0.4 ml of ice-cold 10 mm PBS, pH 7.4, containing 2% Triton X-100, 4% SDS, and protease inhibitor mixture as above. Protein content in the extracts was determined by the Lowry method. Direct comparisons between the sciatic nerve extracts from sham control or days 2, 3, 5, and 7 after SNL injury were performed by processing and analyzing the samples strictly in parallel under identical conditions. Sample loading was standardized by protein content. Two lots of extracts were prepared for each of the treatments and analyzed independently. Two independent Western analyses were performed for each sample (a total of four independent analyses per time point). Extracts were separated by SDS-PAGE and transblotted onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were preblocked with Tris-buffered saline with 0.05% Tween 20 (TBST), 3% nonfat dried milk, 2% goat serum, and 2% mouse serum. Immunostaining of NaV1.8 was performed using a polyclonal antiserum raised in rabbit against a synthetic peptide derived from rat NaV1.8 [epitope: L808–L828 (C terminal of IIS5/outer loop 3); 1:1000 in TBST/3% nonfat dried milk (NFDM)/1% goat serum]. This antiserum predominantly labels a single band at ∼200 kDa in extracts from DRG, trigeminal ganglia, and dorsal horn of the spinal cord (presumably contributed by central projections of the primary afferent), but not from whole brain or nontransfected HEK293 cells. The antiserum also labels primary cultures of rat DRG cells and HEK293 cells that have been transfected with NaV1.8 by immunocytochemical methods. Immunostaining of transient receptor potential cation channel V1 (formerly VR1 for vanilloid receptor 1) (TRPV1) was performed using an affinity-purified, rabbit polyclonal antiserum raised against a synthetic peptide derived from rat TRPV1 [epitope: E824–K838 (C-terminal); 2 μg/ml in the same dilution buffer as for antiserum to NaV1.8]. This antiserum predominantly labels a band at ∼100 kDa, which comigrates with the single band that is detected in extracts from HEK293 cells that had been transfected with the cloned TRPV1 cDNA from rat (Amgen, Thousand Oaks, CA). The blots were washed twice in TBST/3% NFDM and twice in TBST/1% NFDM, followed by incubation with a secondary antibody: HRP-conjugated anti-rabbit IgG goat antiserum (80 ng/ml in TBST/1% NFDM/1% goat serum; Jackson Immunoresearch, West Grove, PA), washed twice in TBST/1% NFDM and twice in TBST, and processed for chemiluminescence detection (ECL, Amersham, Piscataway, NJ). The chemiluminescent images of transblots were scanned at 600 dots per inch.
Immunohistochemistry. Three rats per experimental group were used for tissue analysis. Animals were deeply anesthetized with ketamine/xylazine (100 mg/kg, i.p.) and perfused intracardially with 500 ml of 0.1 m PBS followed by 500 ml of a solution of 10% formalin in 0.1 m PBS. A section of the sciatic nerve proximal to the trifurcation was isolated from the ipsilateral side of sham or SNL rats, postfixed with 10% formalin for 2 hr at 4°C, and then transferred to 30% sucrose in 0.1m PBS for 24 hr at 4°C. The ligated L5 spinal nerves were isolated from SNL rats 7 d after the surgery and processed in the same manner. Frozen longitudinal sections (20 μm) were incubated with a rabbit anti-NaV1.8 antiserum (1:1000 in PBS with 10% normal goat serum) for 48 hr at 4°C, followed by a Cy3-conjugated goat anti-rabbit IgG (1:500 in PBS with 2% normal serum) for 2 hr at room temperature (Jackson Labs). The specificity of the primary antibody has been reported previously (Novakovic et al., 1998; Lai et al., 2002). To facilitate direct comparison between treatment groups, tissue sections from the treatment groups were always processed and labeled at the same time and under identical conditions.
Results
Behavioral testing
Mechanical and thermal threshold were determined before and 7 d after SNL or sham surgery. SNL but not sham surgery resulted in a dramatic decrease in the mechanical force necessary to evoke a withdrawal. Before experimental manipulations, mechanical threshold was 15 ± 00 gm, whereas after surgery, mechanical threshold was 3.1 ± 0.2 gm. An SNL-induced decrease in thermal threshold was also observed: the latencies for withdrawal were 20 ± 1.0 and 12.0 ± 0.5 sec before and after surgery, respectively (p < 0.05). These results are similar to our previous observations (Lai et al., 2002) as well as those of others (Kim and Chung, 1992; Ringkamp et al., 1999).
DRG recording
The SNL model enables the study of both injured and uninjured neurons from the same animal. All of the neurons in the L5 DRG are injured after ligation of the L5 spinal nerve, whereas all of the neurons in the L4 DRG are uninjured. Furthermore, because behavioral testing was performed on a dermatome receiving innervation via the sciatic nerve, we characterize the impact of SNL on DRG neurons giving rise to the sciatic nerve. By injecting the retrograde tracer DiI into the sciatic nerve before SNL, it is possible to study both the injured and uninjured neurons that give rise to the sciatic nerve after SNL. We refer to the labeled L5 DRG neurons as injured neurons and the labeled L4 DRG neurons as their uninjured neighbors. Because the impact of SNL on functional voltage-gated Na+ channels present in uninjured and injured DRG neurons has not been described previously, we recorded from the cell bodies of DRG neurons in short-term culture (<7 hr). DRG cells from 11 rats were studied, including 4 sham-operated rats and 7 SNL rats. Neurons from both L4 and L5 ganglia were studied from each animal. DiI-labeled DRG neurons were readily detected in vitro after dissociation of DRG. A voltage protocol was used to study TTX-resistant Na+ current in isolation from TTX-sensitive currents (see Materials and Methods). The identical currents were observed by adding 1 μmTTX to the bath solution to isolate TTX-SINa from TTX-RINa (data not shown). TTX-RINa observed in the present study was similar to that described previously in DRG neurons (Lai et al., 2002). Peak inward current was evoked at ∼0 mV, and the current had a high threshold for activation and inactivation and a relatively slow rate of inactivation during a sustained depolarizing voltage step (Fig.1). TTX-RINa density was significantly reduced in injured DRG neurons compared with uninjured neurons or neurons from sham-operated rats (p < 0.01) (Fig. 1, Table1). In addition to the decrease in TTX-R conductance, there were two additional changes observed in the biophysical properties of TTX-R INa in injured neurons. First, there was an 8 mV depolarizing shift in theV1/2 of inactivation. This change reflected a parallel shift in the availability curve of the current because there was no change in the slope factor (Table 1). Second, there was a slowing in the recovery from inactivation. The primary reason for the slowing was a significant decrease in the fraction of recovery that occurred over the fast time constant (τ1) (Table 1). There was no detectable difference between uninjured L4 DRG neurons from SNL rats and those in L4 or L5 of sham-operated animals with respect to either the density (Fig. 1) or biophysical properties of TTX-R INa. Therefore, data from uninjured L4 DRG neurons from SNL rats and those from L4 and L5 DRG neurons from sham-operated rats were pooled for the comparisons shown in Table 1.
Fig. 1.
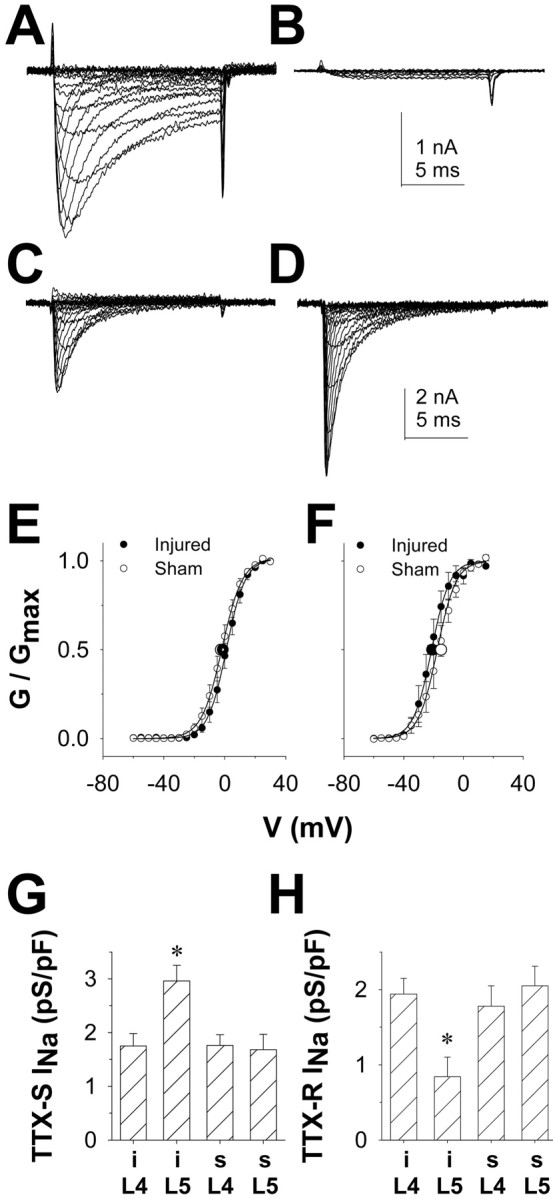
SNL-induced changes in VGSCs in injured DRG neurons. A, TTX-R INa evoked from a typical uninjured DRG neuron with 5 mV voltage steps between −60 and +45 mV after a 500 msec voltage step to +50 mV.B, TTX-R INa evoked as inA, from a typical injured DRG neuron. C, TTX-S INa was evoked in a typical uninjured DRG neuron with a voltage-clamp protocol similar to that used to evoke TTX-R INa and digitally isolated from TTX-RINa as described in Materials and Methods.D, TTX-S INa was evoked in a typical injured DRG neuron as described in C.E, TTX-R INaconductance–voltage data from 8 injured DRG neurons and 10 uninjured DRG neurons (L5 ganglia from sham-treated rats) were normalized with respect to maximal conductance (Gmax), pooled, and plotted as mean ± SEM. Data were fitted with a modified Boltzmann function.Larger symbols reflect mean ± SEM of fitted values obtained for the voltage of half activation (V0.5). F, TTX-SINa conductance–voltage data from 10 injured and 10 uninjured DRG neurons were normalized, plotted, and fitted as described for E. G, SNL results in a significant increase in TTX-S current density in injured (i) DRG neurons compared with uninjured (s, sham) sciatic DRG neurons. H, SNL results in a significant decrease in the density of TTX-RINa in injured (i) sciatic DRG neurons compared with that of uninjured (s, sham) sciatic DRG neurons.
Table 1.
Biophysical properties of voltage-gated Na+currents in DRG neurons supplying the sciatic nerve after SNL
| Property | Injured TTX-S INa | Uninjured TTX-S INa | Injured TTX-RINa | Uninjured TTX-RINa |
|---|---|---|---|---|
| Capacitance (pF) | 62.5 ± 4.6 (17) | 48.9 ± 3.2 (64) | 61.0 ± 4.6 (14) | 48.8 ± 3.0 (73) |
| Density (pS/pF) | 2.71 ± 0.36* (10) | 1.72 ± 0.13 (42) | 0.84 ± 0.281-160 (13) | 1.95 ± 0.14 (55) |
| Activation | ||||
| Gmax (pS) | 187.7 ± 28.71-160 (10) | 82.8 ± 11.5 (42) | 34.0 ± 11.21-160 (13) | 81.3 ± 6.7 (52) |
| Slope (mV) | 5.0 ± 0.4 (10) | 5.7 ± 0.3 (42) | 5.6 ± 0.4 (13) | 5.2 ± 0.2 (52) |
| V1/2(mV) | −21.5 ± 2.0 (10) | −18.4 ± 1.1 (42) | −2.6 ± 1.6 (13) | −2.6 ± 0.7 (52) |
| Inactivation | ||||
| Imax (nA) | 4.9 ± 0.91-160 (16) | 2.4 ± 0.4 (28) | 1.2 ± 0.31-160 (8) | 3.0 ± 0.5 (7) |
| Slope (mV) | 8.8 ± 0.7 (16) | 9.0 ± 0.4 (24) | 5.5 ± 0.4 (8) | 4.8 ± 0.3 (7) |
| V1/2(mV) | −63.9 ± 1.5 (16) | −61.4 ± 1.7 (24) | −17.1 ± 0.81-160 (8) | −25.1 ± 1.2 (7) |
| Repriming | ||||
| F τ1 | 0.86 ± 0.03 (8) | 0.78 ± 0.03 (21) | 0.57 ± 0.08* (10) | 0.82 ± 0.03 (8) |
| τ1 (msec) | 11.8 ± 3.2 (8) | 18.1 ± 2.9 (21) | 3.33 ± 1.44 (10) | 1.42 ± 0.19 (8) |
| Decay rate (msec) @ 0 mV | 0.98 ± 0.09 (8) | 1.3 ± 0.07 (55) | 14.0 ± 3.2 (8) | 9.8 ± 0.9 (50) |
p < 0.05; statistical comparisons made between injured and uninjured neurons for each property.
F1-160: p < 0.01; statistical comparisons made between injured and uninjured neurons for each property. Repriming, Rate of recovery from inactivation assessed with a two-pulse protocol. Repriming was determined at −80 mV. F τ1, Fraction of repriming that was accounted for by the time constant τ1.
In contrast to the decrease in TTX-RINa in labeled L5 DRG neurons from SNL rats, we observed a significant increase in TTX-SINa in this same population of DRG neurons (p < 0.05) (Fig. 1). There was no significant difference between injured and uninjured DRG neurons with respect to the biophysical properties of TTX-SINa present. The currents were similar with respect to the voltage-dependence of activation, inactivation, and recovery from inactivation (Table 1). There did appear to be two populations of DRG neurons with respect to TTX-SINa rate of recovery from inactivation. One population had currents that recovered rapidly (τ of recovery <10 msec), whereas the other had currents that recovered more slowly (τ > 20 msec). There were more injured (6 of 8) than uninjured (12 of 21) neurons with rapidly repriming currents; however, the difference between these two groups was not significantly different (p > 0.05; χ2 test).
Our electrophysiological results with TTX-RINa in both injured and uninjured DRG neurons are consistent with previous results suggesting that there is a decrease in NaV1.8 mRNA (Boucher et al., 2000) and protein (Decosterd et al., 2002; Lai et al., 2002) in injured DRG neurons but no change in NaV1.8 protein levels in the uninjured L4 (Decosterd et al., 2002; Lai et al., 2002). We also evaluated the presence of NaV1.8 protein in the sciatic nerve after SNL. Immunohistochemical staining revealed a dramatic increase in NaV1.8-like immunoreactivity in the sciatic nerve 7 d after SNL of the L5 and L6 spinal roots (Fig. 2). That we could detect an increase in NaV1.8 staining in the sciatic nerve at day 7 was striking in light of the fact that 7 d after SNL, 40–50% of the axons arising from the injured L5 spinal root should have degenerated (Terada et al., 1998). The apparent redistribution of NaV1.8 in the uninjured fibers in the sciatic nerve was in stark contrast to the low level of NaV1.8-like immunoreactivity accumulated on the proximal side of the L5 ligation (Fig. 2C). Consistent with this immunohistochemical evidence, Western transblot analysis showed a significant increase in NaV1.8 labeling in the sciatic nerve with time. The upregulation was evident by day 2 and persisted to day 7, the longest time point studied (Fig.2D). Concurrent analysis of TRPV1 immunoreactivity in these samples showed that TRPV1 levels declined rapidly and were very weak or nondetectable by day 2 after injury (Fig.2D). Together these data suggest that the observed changes in the levels of the proteins over time are specific and not caused by artifact such as protein loading.
Fig. 2.
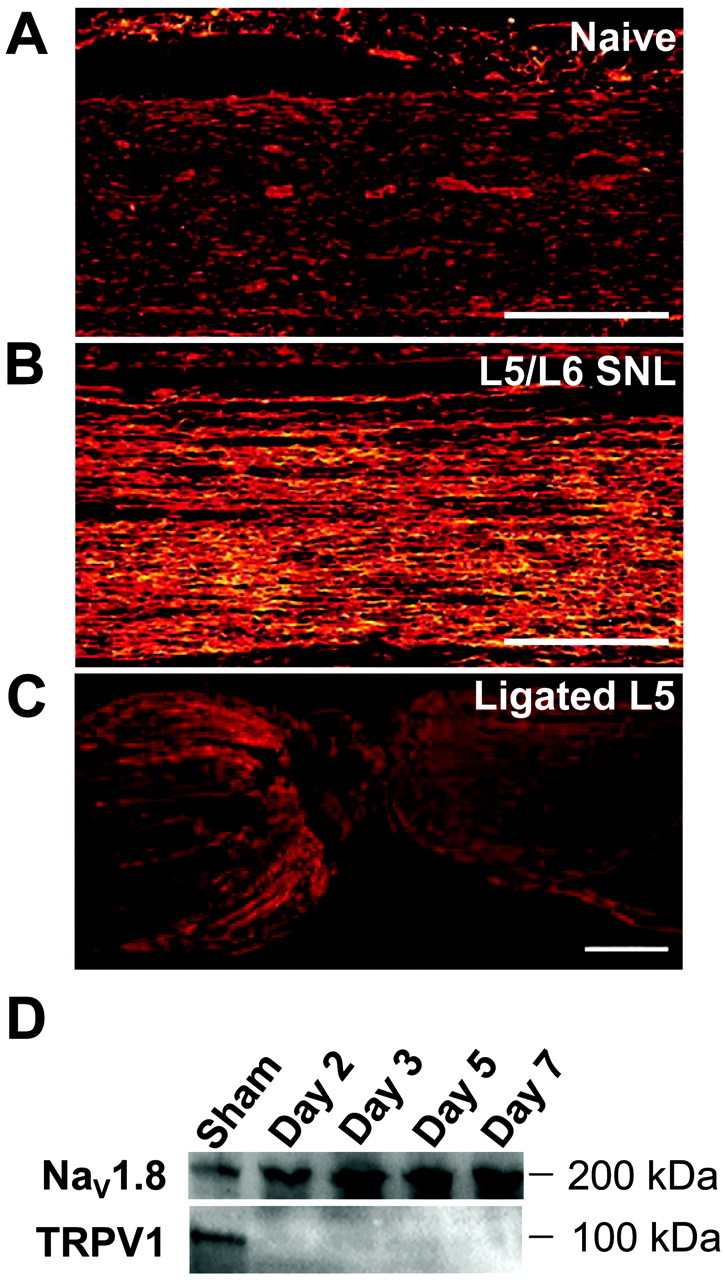
Changes in the expression and distribution of NaV1.8 after SNL by immunohistochemical (A–C) and Western (D) analyses. A, Very little NaV1.8 immunoreactivity is detected in the sciatic nerve of naive rats. B, After SNL, an increase in NaV1.8 immunoreactivity is detected 7 d after nerve injury in the fibers remaining in the sciatic nerve. C, Very little accumulation of NaV1.8 immunoreactivity is found on the proximal side of the ligation site at the L5 spinal root. Scale bars, 400 μm. D, Representative Western transblot analysis of NaV1.8 and TRPV1 in the sciatic nerve of sham-operated rats (Sham) or nerve-injured rats at 2, 3, 5, or 7 d after L5/L6 SNL. A time-related increase in NaV1.8 is concurrent with a loss of TRPV1 immunoreactivity in the same samples. The data are representative of five independent analyses using two separate sets of tissue extracts.
To determine whether the SNL-induced increase in NaV1.8-like immunoreactivity in the sciatic nerves reflects a change in the distribution of functional channels, we examined the sensitivity of action potential conduction in the sciatic nerve to blockade by TTX in nerves from 35 animals, including 14 rats (7 sham and 7 SNL) that did not receive IT catheters and 21 rats that received IT ODN administration (6 SNL + AS, 6 SNL + MM, 4 sham + AS and 5 sham + MM). The sciatic nerve was removed from a point just proximal to the trifurcation at mid thigh to a point 0.5–1 cm proximal to the iliac crest. The average length of nerve studied was 16.5 ± 0.91 mm. This length of nerve was sufficient to enable a clear resolution of the component of the CAP carried by more rapidly conducting myelinated axons (A-wave) and the component carried by the more slowly conducting unmyelinated axons (C-wave) (Fig. 3). To further aid in the resolution of these two components of the CAP, experiments were performed at room temperature (∼22°C). Thus, the conduction velocity calculated for the peak of the A-wave was 15.1 ± 1.27 m/sec, whereas that of the C-wave was 0.41 ± 0.06 m/sec, somewhat slower than the velocity recorded at 37°C.
Fig. 3.
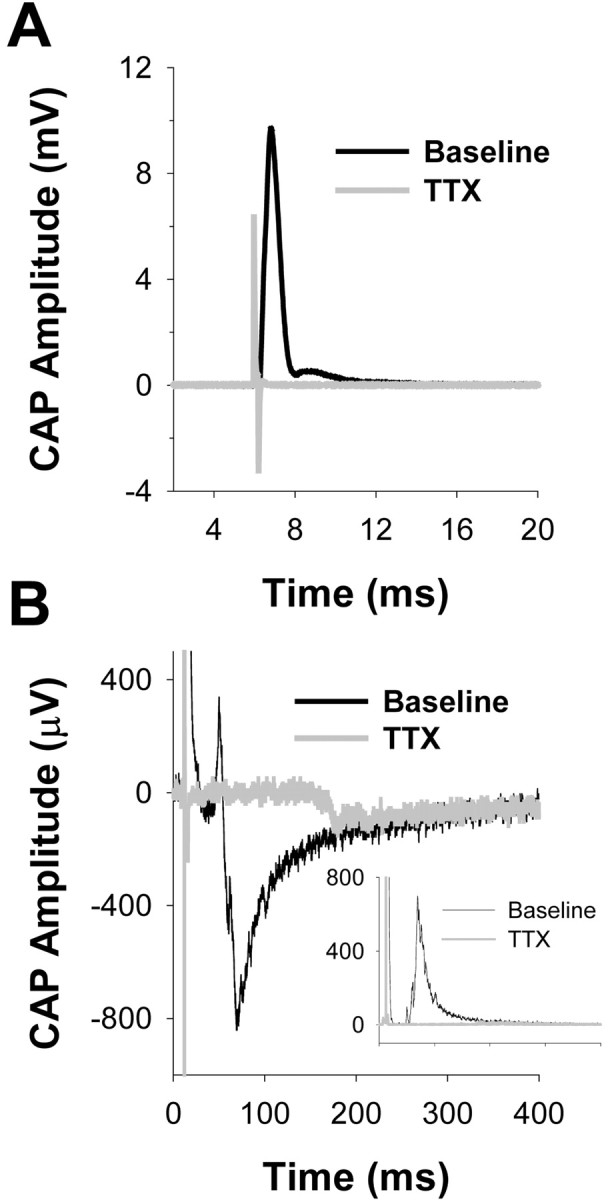
Action potential conduction in naive sciatic nerve is TTX-S. Raw compound action potential (CAP) data for both A- and C-waves before (black traces) and after (gray traces) application of TTX. A, Both Aβ/Aδ and Aδ components of the A-wave are blocked by TTX (100 μm). B, The C-wave is also mostly blocked by TTX. Inset in B is the rectified C-wave before and after application of TTX. Data were analyzed as a change in the area under the rectified waveform.
Application of 100 μm TTX blocked the A-wave in sciatic nerves obtained from both SNL and sham-operated animals: 1.9 ± 0.6% (n = 7) and 2.2 ± 1.1% (n= 7) of A-waves from SNL and sham-operated nerves, respectively, remained after 5 min in the presence of TTX (Figs. 3,4). Similarly, the C-wave in the sciatic nerve from control animals was mostly blocked by TTX: 8.3 ± 1.6% remained after a 5 min exposure. The sciatic nerve contralateral to that receiving the SNL injury was studied in seven rats (four with SNL alone and three from sham-operated animals). Data from these nerves were indistinguishable from those of sham-operated rats in terms of TTX sensitivity but were not included in further analysis. In contrast, 38.8 ± 13.0% of the C-wave remained in sciatic nerves from SNL rats after exposure to TTX (p < 0.05; one-way ANOVA with Tukey post hoc) (Figs. 3, 4). Of note, a small, slowly conducting (∼2 m/sec) TTX-R component of the A-wave became evident after application of TTX to several nerves from SNL-inured animals (Fig. 5A, right panel), suggesting that some of the increase in immunoreactivity observed in the sciatic nerve reflects a redistribution of TTX-R channels in thinly myelinated axons. Because the TTX-R component of the A-wave was not detectable in every nerve studied, we did not quantify changes in the magnitude of this component.
Fig. 4.

After SNL, C-fiber conduction in sciatic nerve becomes TTX-R. A, Rectified waveform of C-wave (A-wave is off scale) evoked in an ipsilateral sciatic nerve, 7 d after SNL, before and after application of TTX. B, Pooled data from sciatic nerves ipsilateral to sham (n = 7) or SNL (injury; n = 7) treated animals. The A-waves in both groups of nerves were mostly blocked by TTX. C, Approximately 40–50% of C-wave evoked in the same group of nerves as described in B is resistant to 100 μm TTX after SNL. Asterisk indicates a significant difference with p < 0.05.
Fig. 5.
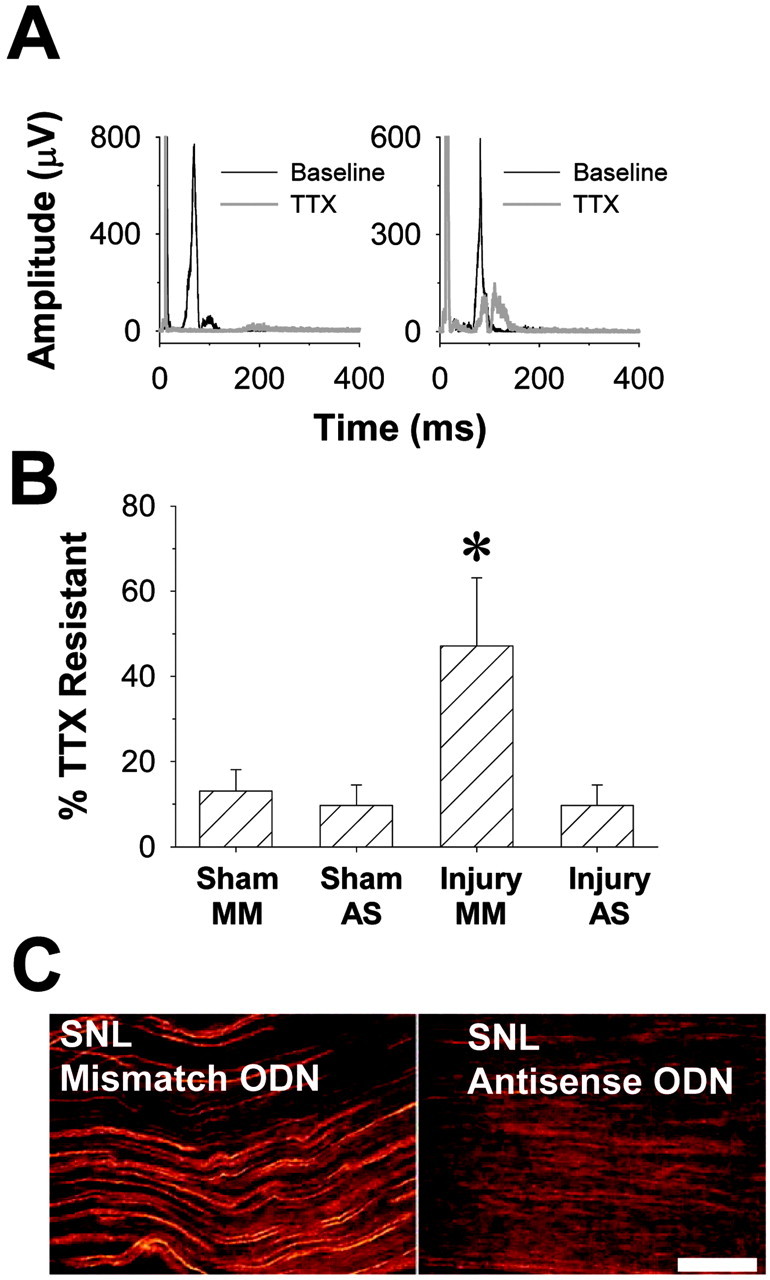
NaV1.8 antisense ODNs reverse SNL-induced changes in TTX sensitivity of C-fiber conduction in the sciatic nerve as well as the redistribution of NaV1.8-like immunoreactivity. A, left panel, NaV1.8 antisense ODN administration eliminates the TTX-R component of the CAP after SNL. Typical data are from the sciatic nerve ipsilateral to the SNL injury of an antisense-treated rat before (black trace) and after (red trace) application of TTX (100 μm). The A-wave is off scale, and the C-wave has been rectified. Right panel, Mismatch ODN administration has no influence on the SNL-induced increase in the TTX-R component of the CAP. Data from the sciatic nerve ipsilateral to the SNL injury of a mismatch ODN-treated rat before (black trace) and after (red trace) application of TTX (100 μm). A small, slowly conducting (∼2 m/sec) TTX-R A-fiber component is visible in this nerve after application of TTX.B, Bar chart showing that TTX-R conduction depends on NaV1.8. Antisense (AS) ODNs have no effect on conduction in sciatic nerves from sham-treated animals but significantly attenuate TTX-R conduction after SNL. Mismatch (MM) ODNs had no detectable effect on TTX-R conduction. Asterisk indicates a significant difference with p < 0.05. C, left panel, NaV1.8-like immunoreactivity (LI) in sciatic nerve after SNL and the administration of MM ODN. Right panel, NaV1.8-LI is eliminated in the sciatic nerve after SNL and the administration of AS ODN. Before tissue harvest, the AS ODN-treated animals exhibited normal paw withdrawal thresholds to both tactile and thermal stimuli, whereas the MM ODN-treated animals were hyperalgesic and showed enhanced tactile sensitivity. Scale bar, 100 μm.
To determine whether the TTX-R component of the C-wave in the sciatic nerve of SNL rats reflects expression of NaV1.8, we used antisense ODNs specific for NaV1.8 (Lai et al., 2002) to knock down this channel protein in vivo. The ODNs were delivered spinally via indwelling catheters. Thus, four additional groups were studied in 2 × 2 design with nerve injury (SNL or sham) as one arm of the design and ODN (antisense or mismatch) as the other arm of the design. We observed that antisense ODN has no significant impact on the percentage of the C-wave that was TTX-R in sham-operated animals (Fig. 5). Mismatch ODN failed to attenuate the SNL-induced increase in percentage of C-wave that was TTX-R (Fig. 5). Importantly, antisense ODN significantly attenuated the SNL-induced increase in the percentage of C-wave that was TTX-R (Fig. 5). The percentage of C-wave that was TTX-R in the sciatic nerves from antisense-treated animals was indistinguishable from that seen in the sham-operated controls.
Finally, to determine whether the increase in NaV1.8-like immunoreactivity in the sciatic nerve reflects the presence of NaV1.8, rats were treated with mismatch or antisense ODNs as for the electrophysiological experiments. Antisense, but not mismatch, ODN treatment resulted in a significant reduction in NaV1.8 labeling in the sciatic nerve ipsilateral to SNL (Fig. 5).
Discussion
Peripheral nerve injury may result in pain behavior that reflects, at least in part, changes in voltage-gated Na+ channels (Gold, 2000). These changes include a decrease in TTX-R INa and an increase in TTX-S INa in injured neurons (Fig. 1). The changes in TTX-RINa are associated with a decrease in NaV1.8 expression (Dib-Hajj et al., 1996; Boucher et al., 2000) and protein (Decosterd et al., 2002), whereas the increase in TTX-S INa is associated with an increase in NaV1.3 expression (Waxman et al., 1994) and protein (Black et al., 1999). Previous observations of nerve injury-induced changes in NaV1.8 led to the suggestion that this channel does not contribute to neuropathic pain (Okuse et al., 1997; Boucher et al., 2000; Decosterd et al., 2002). The observations reported in the present study, including the absence of NaV1.8 redistribution to the site of nerve injury, support the suggestion that this channel does not contribute to neuropathic pain through a role in injured neurons. However, we also observed that nerve injury results in a redistribution of NaV1.8 to the axons of uninjured neurons and that these redistributed channels are functional, enabling TTX-R conduction in predominantly unmyelinated axons. Furthermore, antisense ODN treatment specifically knocks down NaV1.8 and TTX-R conduction in the sciatic nerve, indicating that this α subunit mediates TTX-R conduction. Importantly, the same antisense ODN treatment prevents and reverses experimental neuropathic pain (Porreca et al., 1999; Lai et al., 2002). Together, these data suggest that the NaV1.8 contribution in uninjured primary afferent fibers is critical for the expression of neuropathic pain.
Results from NaV1.8 heterologous expression of studies (Akopian et al., 1996; Sangameswaran et al., 1996), biophysical characterization of TTX-R INa in sensory neurons (Roy and Narahashi, 1992; Elliott and Elliott, 1993), NaV1.8 antisense ODN administration (Khasar et al., 1998; Lai et al., 2002), and NaV1.8 knock-out mice (Akopian et al., 1999) all support the suggestion that this α subunit underlies TTX-R INaas described in the present study. The nerve injury-induced redistribution of NaV1.8 in uninjured axons shown here may reflect changes in several cellular processes, including an increase in transcription, translation, and insertion as well as a decrease in protein degradation and turnover. RT-PCR analysis for NaV1.8 transcripts in the uninjured L4 DRG detected a moderate upregulation (Boucher et al., 2000). Similar analysis in our hands, however, did not reach statistical significance between sham and SNL up to day 7 after SNL (data not shown). Thus, an increase in transcription may not account for the increase in NaV1.8 in uninjured axons. An increase in the half-life of the channel protein is also an unlikely mechanism because the short time course (∼2 d) for onset and recovery from the effects of NaV1.8 antisense ODN treatment suggests that the NaV1.8 turnover rate is relatively high. Rather, channel redistribution may be caused by an increase in the stability of NaV1.8 mRNA or the efficiency of translation, or both, resulting in a net increase in the synthesis of NaV1.8 protein coupled with an increase in the transport of the protein to the axons. Such changes in NaV1.8 synthesis and transport could account for the observed increase in functional protein in the axons in the absence of an increase in functional channels in the cell body (Fig. 1) or dramatic changes in NaV1.8 protein (Decosterd et al., 2002) or mRNA levels (Boucher et al., 2000) in the cell soma.
At present, few data are available regarding the potential mechanism(s) underlying the redistribution of NaV1.8. The association of Na+ channel α subunits with β subunits, in particular the β2 subunit, appears to be primarily responsible for anchoring the channels at specific sites in the plasma membrane (Isom, 2000). It is possible that the redistribution of NaV1.8 follows a redistribution of a subunit such as β2. Other possible mechanisms include the binding of NaV1.8 to annexin II light chain (p11) to facilitate translocation of NaV1.8 to the plasma membrane (Okuse et al., 2002), or interaction with contactin, which was found to target the Na+ channel NaV1.9 to specific sites along unmyelinated axons (Liu et al., 2001).
We observed that application of TTX resulted in a slowing of C-fiber conduction velocity in sciatic nerves from SNL animals (Figs. 4, 5). The simplest explanation for the conduction velocity slowing is a TTX-induced decrease in the number of functional channels contributing to the upstroke of the action potential. This implies that after nerve injury, both TTX-S and TTX-R channels underlie action potential conduction in “uninjured” axons. Importantly, TTX-R channels alone are sufficient to underlie action potential conduction in a significant minority of uninjured axons after SNL.
It is interesting to note that even in sciatic nerves from naive animals, ∼8% of the C-fiber volley was resistant to 100 μm TTX. A similar percentage of TTX-R C-fiber volley remained after NaV1.8 antisense ODN administration in SNL rats, suggesting that expression and distribution of the channel(s) underlying this component are not influenced by nerve injury. Furthermore, that NaV1.8 antisense ODN had no influence on the TTX-R fraction of the C-fiber volley in sciatic nerves from sham-treated animals suggests that NaV1.8 does not underlie this component. It is possible that the residual TTX-R component of the C-fiber volley reflects NaV1.9, because this channel is TTX-R (Dib-Hajj et al., 1998; Tate et al., 1998) and appears to be present in uninjured C-fibers (Fjell et al., 2000).
There are two previous reports of NaV1.8 redistribution after nerve injury. The first used the chronic constriction injury model of nerve injury (Novakovic et al., 1998), and the second used tissue from nerve injury patients (Coward et al., 2000). Although it was not possible to determine whether the redistribution occurred in injured or uninjured axons in either study, our results suggest that these previous investigators described the latter. Interestingly, Coward and colleagues (2000) described time-dependent changes in the distribution of NaV1.8 after nerve injury such that the initial redistribution in axons reversed to baseline levels, whereas at much later time points (many months), NaV1.8-like immunoreactivity was observed in neuromas. Thus, there may be time-dependent changes in the distribution of NaV1.8 such that its contribution to neuropathic pain shifts from an action in uninjured neurons to an action in injured neurons with time after nerve injury. Time-dependent changes in the underlying mechanisms of neuropathic pain have also been described in the CNS where descending pain facilitatory projections (Porreca et al., 2001; Burgess et al., 2002) and the upregulation of spinal dynorphin (Wang et al., 2001) are critical for the maintenance, but not the initiation, of neuropathic pain.
Our observations have several important implications for the underlying mechanisms contributing to neuropathic pain as well as its treatment. First, the redistribution of NaV1.8 to uninjured axons is necessary for neuropathic pain behavior. This conclusion is based on the observations that (1) intrathecal NaV1.8 antisense ODN completely eliminates SNL-induced pain behavior, (2) NaV1.8 antisense ODN completely eliminates the SNL-induced increase in the TTX-R component of the C-fiber volley, and (3) NaV1.8 antisense ODN completely reverses the upregulation of NaV1.8 immunoreactivity in the sciatic nerve after SNL. The role of the uninjured afferents in neuropathic pain is also implicated in recent reports that activity in injured fibers is not sufficient for the manifestation of SNL-induced pain behavior (Li et al., 2000; X. Liu et al., 2000) [but see Sheen and Chung (1993);Yoon et al. (1996); C. Liu et al. (2000)].
Second, activity in uninjured C-fibers is necessary for SNL-induced pain behavior. This conclusion is supported by results obtained in the present study, in which redistribution of functional TTX-R channels appears to be associated predominantly with unmyelinated axons. This conclusion is consistent with the modest increase in spontaneous activity observed in uninjured C-fibers after SNL (Wu et al., 2001). Increased C-fiber input as a basis of nerve injury-induced hypersensitivity is also supported by selective lesion of C-fibers by the TRPV1 agonist, resiniferatoxin, which blocks thermal hyperalgesia after SNL (Ossipov et al., 1999). Interestingly, this treatment did not block tactile hypersensitivity in these animals. Thus, a redistribution of NaV1.8 may also occur in a subpopulation of resiniferatoxin-insensitive C-fibers and in a small population of large-diameter fibers (Lai et al., 2002) that ascend through the dorsal column or synapse with the postsynaptic dorsal column cells (Sun et al., 2001; Ossipov et al., 2002). Of note, recent observations obtained with a mouse deficient in NaV1.8 indicates that these animals develop mechanical and thermal hypersensitivity in response to partial nerve injury identical to wild-type mice (Kerr et al., 2001). This observation has been used to argue that NaV1.8 does not contribute to neuropathic pain behavior. However, a compensatory increase in the expression of TTX-SINa is observed in the NaV1.8 knock-out mouse (Akopian et al., 1999), which may account for the presence of neuropathic pain behavior in these animals. Importantly, compensatory changes in TTX-SINa are not observed in rats after administration of NaV1.8 antisense ODN (Lai et al., 2002).
Finally, the redistribution of NaV1.8 does not itself have to be the driving force behind spontaneous activity in uninjured C-fibers. Recent evidence suggests that this activity may arise from peripheral terminals of these fibers (Wu et al., 2001) and reflect, at least in part, the activation of α-adrenergic receptors (Ali et al., 1999). An increase in the expression of TRPV1 (Hudson et al., 2001) and BDNF (Fukuoka et al., 2001) in uninjured afferents may also contribute to the increased spontaneous activity. Importantly, because NaV1.8 appears to enable conduction of action potentials arising from several different transduction processes, pharmacologically blocking this channel and processes underlying its redistribution may be the most efficient way of selectively eliminating the underlying basis for neuropathic pain behavior.
Footnotes
This research was supported by National Institutes of Health Grants NS38771 (J.L.), NS36929 (M.S.G.), and NS22069 (D.W.). We thank Dr. Sonja Novakovic of Roche Bioscience for the gift of the antiserum to NaV1.8 (immunohistochemistry). We also thank Shannon E. Burgess and Wenhong Guo for technical assistance.
Correspondence should be addressed to Dr. Michael S. Gold, Department of Oral and Craniofacial Biological Sciences, Room 5-A-12 Hayden-Harris Hall, University of Maryland, Baltimore Dental School, 666 West Baltimore Street, Baltimore, MD 21201. E-mail:msg001@dental.umaryland.edu.
References
- 1.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 2.Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 3.Ali Z, Ringkamp M, Hartke TV, Chien HF, Flavahan NA, Campbell JN, Meyer RA. Uninjured C-fiber nociceptors develop spontaneous activity and alpha- adrenergic sensitivity following L6 spinal nerve ligation in monkey. J Neurophysiol. 1999;81:455–466. doi: 10.1152/jn.1999.81.2.455. [DOI] [PubMed] [Google Scholar]
- 4.Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol. 1999;82:2776–2785. doi: 10.1152/jn.1999.82.5.2776. [DOI] [PubMed] [Google Scholar]
- 5.Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290:124–127. doi: 10.1126/science.290.5489.124. [DOI] [PubMed] [Google Scholar]
- 6.Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–5136. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- 8.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 9.Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S, Bountra C, Anand P. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. 2000;85:41–50. doi: 10.1016/s0304-3959(99)00251-1. [DOI] [PubMed] [Google Scholar]
- 10.Decosterd I, Ji RR, Abdi S, Tate S, Woolf CJ. The pattern of expression of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain. 2002;96:269–277. doi: 10.1016/S0304-3959(01)00456-0. [DOI] [PubMed] [Google Scholar]
- 11.Dib-Hajj S, Black JA, Felts P, Waxman SG. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA. 1996;93:14950–14954. doi: 10.1073/pnas.93.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckert SP, Taddese A, McCleskey EW. Isolation and culture of rat sensory neurons having distinct sensory modalities. J Neurosci Methods. 1998;77:183–190. doi: 10.1016/s0165-0270(97)00125-8. [DOI] [PubMed] [Google Scholar]
- 14.Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fjell J, Hjelmstrom P, Hormuzdiar W, Milenkovic M, Aglieco F, Tyrrell L, Dib-Hajj S, Waxman SG, Black JA. Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. NeuroReport. 2000;11:199–202. doi: 10.1097/00001756-200001170-00039. [DOI] [PubMed] [Google Scholar]
- 16.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold MS. Sodium channels and pain therapy. Curr Opin Anaesthesiol. 2000;13:565–572. doi: 10.1097/00001503-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 18.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gold MS, Weinreich D, Kim CS, Burgess SE, Porreca F, Lai J. NaV1.8 mediates neuropathic pain via redistribution in the axons of uninjured afferents. Soc Neurosci Abstr. 2001;27:55.56. [Google Scholar]
- 21.Gold MS, Zhang L, Wrigley DL, Traub RJ. Prostaglandin E(2) modulates TTX-R I(Na) in rat colonic sensory neurons. J Neurophysiol. 2002;88:1512–1522. doi: 10.1152/jn.2002.88.3.1512. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 23.Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci. 2001;13:2105–2114. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- 24.Isom LL. I. Cellular and molecular biology of sodium channel beta-subunits: therapeutic implications for pain? Am J Physiol Gastrointest Liver Physiol. 2000;278:G349–G353. doi: 10.1152/ajpgi.2000.278.3.G349. [DOI] [PubMed] [Google Scholar]
- 25.Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. NeuroReport. 2001;12:3077–3080. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]
- 26.Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- 27.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 28.Lai J, Gold MS, Kim CS, Biana D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85:493–502. doi: 10.1016/S0304-3959(00)00250-5. [DOI] [PubMed] [Google Scholar]
- 30.Liu C, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- 31.Liu CJ, Dib-Hajj SD, Black JA, Greenwood J, Lian Z, Waxman SG. Direct interaction with contactin targets voltage-gated sodium channel Nav1.9/NaN to the cell membrane. J Biol Chem. 2001;276:46553–46561. doi: 10.1074/jbc.M108699200. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Eschenfelder S, Blenk KH, Janig W, Habler H. Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain. 2000;84:309–318. doi: 10.1016/s0304-3959(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 33.Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okuse K, Chaplan SR, McMahon SB, Luo ZD, Calcutt NA, Scott BP, Akopian AN, Wood JN. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol Cell Neurosci. 1997;10:196–207. doi: 10.1006/mcne.1997.0657. [DOI] [PubMed] [Google Scholar]
- 35.Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417:653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- 36.Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- 37.Ossipov MH, Zhang E-T, Carvajal C, Gardell LR, Quirion R, Dumont Y, Lai J, Porreca F. Selective mediation of nerve-injury induced tactile hypersensitivity by neuropeptide Y. J Neurosci. 2002;22:9858–9867. doi: 10.1523/JNEUROSCI.22-22-09858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the micro-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ringkamp M, Eschenfelder S, Grethel EJ, Habler HJ, Meyer RA, Janig W, Raja SN. Lumbar sympathectomy failed to reverse mechanical allodynia- and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain. 1999;79:143–153. doi: 10.1016/s0304-3959(98)00186-9. [DOI] [PubMed] [Google Scholar]
- 41.Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci. 1992;12:2104–2111. doi: 10.1523/JNEUROSCI.12-06-02104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodoxtoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 43.Sheen K, Chung JM. Signs of neuropathic pain depend on signals from injured nerve fibers in a rat model. Brain Res. 1993;610:62–68. doi: 10.1016/0006-8993(93)91217-g. [DOI] [PubMed] [Google Scholar]
- 44.Sun H, Ren K, Zhong CM, Ossipov MH, Malan TP, Lai J, Porreca F. Nerve injury-induced tactile allodynia is mediated via ascending spinal dorsal column projections. Pain. 2001;90:105–111. doi: 10.1016/s0304-3959(00)00392-4. [DOI] [PubMed] [Google Scholar]
- 45.Tate S, Benn S, Hick C, Trezise D, John V, Mannion RJ, Costigan M, Plumpton C, Grose D, Gladwell Z, Kendall G, Dale K, Bountra C, Woolf CJ. Two sodium channels contribute to TTX-R sodium current in primary sensory neurons. Nat Neurosci. 1998;1:653–655. doi: 10.1038/3652. [DOI] [PubMed] [Google Scholar]
- 46.Terada M, Yasuda H, Kikkawa R. Delayed Wallerian degeneration and increased neurofilament phosphorylation in sciatic nerves of rats with streptozocin-induced diabetes. J Neurol Sci. 1998;155:23–30. doi: 10.1016/s0022-510x(97)00269-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan TP, Jr, Lai J, Porreca F. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci. 2001;21:1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72:466–470. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, Meyer RA. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci 21 2001. RC140(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yaksh TL, Rudy TA. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- 52.Yoon YW, Na HS, Chung JM. Contributions of injured and intact afferents to neuropathic pain in an experimental rat model. Pain. 1996;64:27–36. doi: 10.1016/0304-3959(95)00096-8. [DOI] [PubMed] [Google Scholar]