

Summary
Immune‐checkpoint blockade antibodies have been approved for the treatment of cancer. However, poorly immunogenic tumours are less responsive to such therapies. Agonistic anti‐Toll‐like receptor 4 (TLR4) monoclonal antibodies (mAbs) activate only cell‐surface TLR4; in contrast, lipopolysaccharide (LPS) activates both TLR4 and intracellular inflammatory caspases. In this study, we investigated the adjuvant activity of an anti‐TLR4 mAb in T‐cell‐mediated antitumour immunity. The anti‐TLR4 mAb induced the activation of antigen‐specific T‐cells in adoptive transfer studies. The growth of ovalbumin (OVA)‐expressing tumours was significantly suppressed by administration of OVA and the anti‐TLR4 mAb in combination, but not individually. The antitumour effect of anti‐PD‐1 mAb was enhanced in mice administered with OVA plus the anti‐TLR4 mAb. The OVA‐specific IFN‐γ‐producing CD8 T‐cells were induced by administration of OVA and the anti‐TLR4 mAb. The suppression of tumour growth was diminished by depletion of CD8, but not CD4, T‐cells. The inflammatory response to the anti‐TLR4 mAb was of significantly lesser magnitude than that to LPS, as assessed by NF‐κB activation and production of TNF‐α, IL‐6 and IL‐1β. Administration of LPS (at a dose that elicited levels of proinflammatory cytokines comparable to those by the anti‐TLR4 mAb) plus OVA induced no or less‐marked activation of OVA‐specific T‐cells and failed to suppress tumour growth in mice. In conclusion, the agonistic anti‐TLR4 mAb induces potent CD8 T‐cell‐dependent antitumour immunity and an inflammatory response of lesser magnitude than does LPS. The agonistic anti‐TLR4 mAb has potential as an adjuvant for use in vaccines against cancer.
Keywords: antibodies, immunotherapy, inflammasome, Toll‐like receptors, tumour immunology
Abbreviations
- Ab
antibody
- Ag
antigen
- APC
allophycocyanin
- ANOVA
analysis of variance
- BMM
bone marrow‐derived macrophage
- CFSE
5‐ (and 6‐)carboxyfluorescein diacetate succinimidyl ester
- LPS
lipopolysaccharide
- OVA
ovalbumin
- PE
phycoerythrin
- PRR
pathogen recognition receptor
- TLR
Toll‐like receptor
Introduction
Cancer immunotherapy in addition to surgery, radiotherapy and chemotherapy has to date failed to show clinical efficacy.1 However, immune‐checkpoint blockade is reportedly efficacious in patients with cancer,2, 3, 4, 5 and inhibitory antibodies (Abs) against PD‐1, PD‐L1 and CTLA‐4 are approved for the treatment of a growing list of cancers.6, 7 However, only about 30% of patients with cancer benefit from immune‐checkpoint blockade; the exact proportion depends on the cancer type.6, 8 The efficacy of immune‐checkpoint blockade is reportedly associated with the immunogenicity of the tumour.4, 9, 10, 11 Tumours with low immunogenicity tend to be less responsive to immune‐checkpoint blockade therapy because of the small number of tumour‐infiltrating T‐cells.4, 5, 10, 12 Thus, patients with low‐immunogenicity tumours are unlikely to benefit from immune‐checkpoint‐blocking Abs because antitumour immunity is not stimulated and/or due to the immune‐evasion mechanism(s) of the tumours.6, 13, 14 Therefore, boosting the host immune response to the tumour would enhance the cytotoxic effect of immune‐checkpoint blockade therapy.13, 15, 16
In contrast to immune‐checkpoint blockade, cancer vaccines comprising tumour‐associated or ‐specific antigens (Ags) prime and/or activate antitumour immune cells, such as cytotoxic T‐cells.15, 16 However, administration of a vaccine containing only a protein or peptide Ag does not stimulate antitumour immunity.17 Ag presentation under non‐inflammatory conditions fails to elicit an adaptive immune response and can induce tolerance to the Ag.18, 19 The activation of innate immunity is required for the induction of Ag‐specific acquired immunity.20, 21 Immune adjuvants have been reported to enhance the immune responses elicited by cancer vaccines.15, 21
The activation of pathogen recognition receptors (PRRs) in response to pathogens stimulates innate, and subsequently adaptive, immunity.22 Therefore, activators of PRRs show promise as adjuvants.20, 21 Toll‐like receptor 4 (TLR4), the first TLR identified, is highly expressed in Ag‐presenting cells, such as dendritic cells and macrophages.23, 24 Lipopolysaccharide (LPS), a component of the cell walls of Gram‐negative bacteria, is a potent activator of TLR4 and is an endotoxin.25, 26 Upon binding of LPS, TLR4—in conjunction with the accessory molecules MD‐2, CD14 and LPS‐binding protein—activates NF‐κB, leading to the production of proinflammatory cytokines, interferons and lipid mediators.27, 28, 29, 30, 31 Activation of TLR4 in dendritic cells enhances Ag‐specific priming of lymphocytes due to upregulation of MHC and co‐stimulatory molecules.22, 24, 32, 33 Therefore, the efficacy of LPS derivatives as adjuvants in vaccines against infectious diseases and various types of cancer has been evaluated.21, 34 However, the clinical application of LPS is hampered by its potent proinflammatory activity; this problem can be overcome by the development of detoxified LPS derivatives. For instance, monophosphoryl lipid A, the hexa‐acylated di‐glucosamine of which lacks one of the two phosphate groups, was clinically approved as an adjuvant for a vaccine against cervical cancer.35 LPS is recognized intracellularly by the inflammatory caspases 4/5/11 in a TLR4‐independent manner.36, 37 Activation of inflammatory caspases causes activation of the inflammasome, resulting in an excessive inflammatory response.
We produced an agonistic anti‐TLR4 monoclonal antibody (mAb) that binds to and activates cell‐surface TLR4, but not intracellular inflammatory caspases.38, 39 Administration of the agonistic anti‐TLR4 mAb with a protein Ag markedly enhanced the production of Ag‐specific IgG.33 These findings led us to hypothesize that the agonistic anti‐TLR4 mAb would enhance tumour immunogenicity, thereby augmenting tumour‐specific T‐cell responses. In this study, we showed that the agonistic anti‐TLR4 mAb enhances the induction of Ag‐specific T‐cells, resulting in the suppression of tumour growth in mice, but exerts a far less intense inflammatory reaction than did LPS.
Materials and methods
Reagents and Abs
Lipopolysaccharide (Escherichia coli O:111) was purchased from Wako Pure Chemical Industries (Osaka, Japan). The mouse anti‐mouse TLR4/MD‐2 agonistic mAb (UT12)38, 39 was purified from conditioned serum‐free medium (Hybridoma‐SFM; Thermo Fisher Scientific, Waltham, MA) used to culture hybridomas.32 Rat anti‐mouse CD4 (GK1·5), CD8 (YTS 169·4·2·1) and PD‐1 (RMP1‐14) mAbs were purified from ascitic fluid of mice with severe combined immunodeficiency by caprylic acid precipitation followed by diethylaminomethyl ion‐exchange chromatography.26 Pam3CSK4 and low‐molecular‐weight poly(I:C) were from InvivoGen (San Diego, CA). Transfection grade linear polyethylenimine hydrochloride (MW 40 000) was from Polysciences (Warrington, PA). The other primary and secondary Abs were as follows: fluorescein isothiocyanate‐CD4 (GK1·5), allophycocyanin (APC)‐CD4 (GK1·5), APC‐CD8α (53‐6·7), phycoerythrin (PE)‐CD44 (IM7), PE‐CD45·1 (A20), PE‐IFN‐γ (XMG1·2) and horseradish peroxidase‐goat anti‐rat IgG (minimal cross‐reactivity) Ab (BioLegend, San Diego, CA); PE‐H‐2Kb (AF6‐88·5·5·3; eBioscience, San Diego, CA); caspase‐11 (17D9) rat mAb, GAPDH (14C10) rabbit mAb, horseradish peroxidase‐goat anti‐rabbit IgG Ab (Cell Signaling Technology, Danvers, MA); mouse IL‐1β/IL‐1F2 Ab (Clone 166926; R&D systems, Minneapolis, MN); and horseradish peroxidase‐goat anti‐mouse IgG Ab (Jackson ImmunoResearch Laboratories, West Grove, PA). Ovalbumin (OVA) and thiazolyl blue tetrazolium bromide were obtained from Sigma‐Aldrich (St Louis, MO). The OVA257–264 (SIINFEKL) and OVA323–339 (ISQAVHAAHAEINEAGR) peptides were synthesized by MBL (Nagoya, Japan). The 123count eBeads™ counting beads and permeabilization buffer were purchased from Thermo Fisher Scientific. Brefeldin A was obtained from LKT Laboratories (St Paul, MN). The Immobilon‐P membrane was from Millipore (Bedford, MA), and the Chemi‐Lumi One Super chemiluminescence kit was from Nacalai Tesque (Kyoto, Japan).
Mice
C57BL/6N mice were purchased from Japan SLC (Hamamatsu, Japan) and CLEA Japan (Tokyo, Japan). OT‐I (Ly5·1‐congenic) and OT‐II T‐cell receptor‐transgenic mice were gifts from Dr Ishii (Tohoku University, Sendai, Japan), and were used as sources of CD8 and CD4 T‐cells responsive to OVA257–264 and OVA323–339 in the context of H‐2Kb and I‐Ab, respectively.40, 41 An OT‐II mouse strain congenic for Ly5·1 was generated in our laboratory.32 The mice were bred and maintained under specific pathogen‐free conditions according to the Guidelines for Animal Experimentation of Tohoku University. The study protocol was approved by the Institutional Animal Care and Use Committee of Tohoku University.
Cells
EG7 cells42 were purchased from the American Type Culture Collection (Rockville, MD). EL4 and B16F10 cells were purchased from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan). MC38‐transfected cells secreting OVA were generated previously.32 Ba/F3‐transfected cells carrying the mouse TLR4/MD‐2/NF‐κB reporter gene were generated previously,24 and were maintained in RPMI‐1640 medium supplemented with 10% fetal calf serum, penicillin (100 U/ml) and streptomycin (100 μg/ml); Ba/F3 cells were maintained in medium containing murine IL‐3. Bone marrow‐derived macrophages (BMMs) were prepared from the bone marrow cells of C57BL/6N mice using the conditioned medium of L929 cells, as described previously.24 The cells were incubated at 37° in a humidified CO2 incubator. B16F10 cells were transfected with the OVA/pCAGGS3 expression construct32 using Lipofectamine® 2000 reagent and established by G418 selection. The resulting B16F10‐transfected cells express OVA intracellularly in a stable manner due to deletion of the signal sequence comprising amino acids 18–143.
Cell staining and flow cytometry
Cells were stained and subjected to flow cytometry using the CytoFLEX instrument (Beckman Coulter Life Sciences, Brea, CA), as described previously.43 Data were analysed using FlowJo software (Tree Star, Ashland, OR). 123count eBeads™ counting beads were used in some experiments to enumerate cells.
Adoptive transfer
Ly5·1+ OT‐I or OT‐II spleen cells were labelled with 5 μm 5‐ (and 6‐)carboxyfluorescein diacetate succinimidyl ester (CFSE) in phosphate‐buffered saline (PBS) containing 0·1% bovine serum albumin as described previously,44 and 1–2 × 107 labelled cells were injected i.v. into C57BL/6N (Ly5·2+) mice. The following day, the mice were immunized i.p. with OVA and the anti‐TLR4 mAb or LPS in PBS. Two–three days after immunization, the proliferation of spleen OT‐I CD8 and OT‐II CD4 T‐cells was assayed by fluorescence‐activated cell sorting based on the intensity of CFSE fluorescence.
Tumour challenge and treatments
EG7, B16F10‐cOVA or parental tumour cells (3 × 105) were injected s.c. into the right flanks of the mice, which were 1 day later injected s.c. with OVA (100 μg), the anti‐TLR4 mAb (3 μg), or both. Tumour volumes were measured three times per week using digital calipers, and were calculated using the following formula: tumour volume (mm3) = (long diameter) × (short diameter)2 × 0·5. Mice were killed when the tumour volume reached > 1500 mm3. In some experiments, LPS was injected instead of the anti‐TLR4 mAb. To deplete CD4 and CD8 T‐cells, mice were injected i.v. with an anti‐CD4 or ‐CD8 mAb (100 μg) on day −3, and i.p. on days −1 and 6. MC38‐OVA tumour cells (5 × 105) were injected s.c. into the right flanks of the mice. One day later, the mice were injected s.c. with OVA plus the anti‐TLR4 mAb or vehicle, and then injected i.p. with the anti‐PD‐1 mAb (RMP1‐14, 100 μg) or vehicle on days 6 and 9.
To analyse IFN‐γ‐secreting T‐cells, mice were immunized i.p. with the anti‐TLR4 mAb (UT12, 3 μg) or OVA (100 μg) in sterile PBS. On day 7, spleen cells (6 × 106) were stimulated with OVA257–264 (1 μg/ml) or OVA323–339 (10 μg/ml) in 2 ml culture medium in a 12‐well plate for 2 hr, and subsequently incubated with brefeldin A (10 μg/ml) for a further 6 hr. Next, the cells were fixed in 4% paraformaldehyde in PBS on ice for 30 min, and intracellularly stained using permeabilization buffer.
To analyse systemic inflammation, the mice were injected i.p. with the anti‐TLR4 mAb (UT12) or LPS in 250 μl PBS, and their plasma was collected at 1 and 3 hr for measuring TNF‐α and IL‐6 levels. For IL‐1β, the mice were primed with i.v. injection of poly(I:C) (200 μg) for 21 hr, and then injected i.p. with the anti‐TLR4 mAb or LPS. Plasma samples were collected at 2 and 6 hr.
Stimulation of cells
Bone marrow‐derived macrophages (5 × 104) were inoculated into 96‐well plates, incubated overnight in 200 μl culture medium, and stimulated with the anti‐TLR4 mAb or LPS for 24 hr to assay TNF‐α and IL‐6 production. For IL‐1β ELISA, BMMs (5 × 104) were incubated overnight in 100 μl culture medium in 96‐well plates and primed with the Pam3CSK4, anti‐TLR4 mAb or LPS for 4 hr. The primed cells were washed once with PBS and transfected with anti‐TLR4 mAb or LPS using 2 μg polyethylenimine diluted in Opti‐MEM in 100 μl culture medium for 20 hr.
For Western blot analyses, BMMs (5 × 105) were plated onto 24‐well plates, incubated overnight in 1 ml culture medium, and stimulated with the anti‐TLR4 mAb or LPS. Additionally, cells (5 × 105) were incubated overnight in 24‐well plates containing 1 ml culture medium and then primed for 4 hr with Pam3CSK4. The primed cells were washed once and transfected with anti‐TLR4 mAb or LPS using 10 μg polyethylenimine in 250 μl fresh culture medium. The culture supernatants and whole‐cell lysates were prepared as described previously,27 and subjected to Western blot.
ELISA
The TNF‐α, IL‐6 and IL‐1β concentrations in culture supernatant and serum were determined using the Mouse TNF‐α ELISA MAX™ Standard, Mouse IL‐6 ELISA Ready‐SET‐Go! kit (eBioscience) and IL‐1β Mouse Uncoated ELISA kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Absorbance values were read using a Multiskan™ FC Microplate Photometer (Thermo Fisher Scientific).
Western blot
Proteins were resolved on sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred onto an Immobilon‐P membrane as described previously.27 Blotted membranes were probed with primary mAbs and horseradish peroxidase‐conjugated secondary Ab described above. The immunoreactive protein was exposed on X‐ray film using a Chemi‐Lumi One Super chemiluminescence kit.
NF‐κB reporter assay
Ba/F3‐transfected cells (2 × 104) carrying the mouse TLR4/MD‐2/NF‐κB reporter gene were stimulated with LPS for 5–6 hr in 96‐well round‐bottom plates, and luciferase activity was assayed as described previously.45
Statistical analyses
The statistical significance of differences between two groups was assessed by Student's t‐test using Prism version 6·07 for Windows™ (GraphPad Software, San Diego, CA). Differences among three or more groups were evaluated by one‐way analysis of variance (ANOVA) with the Tukey post hoc test or by two‐way ANOVA with the Tukey or Sidak post hoc tests.
Results
The agonistic‐TLR4 mAb facilitates induction of Ag‐specific T‐cells
Pathogen‐derived or synthetic agonists for PRRs exert adjuvant effects on innate and adaptive immunity.20, 21 Previously, we showed that the agonistic‐TLR4 mAb induces the production of Ag‐specific IgG in mice.33 This finding prompted us to investigate the adjuvant activity of the anti‐TLR4 mAb for T‐cell‐mediated antitumour immunity. We assayed the effect of the anti‐TLR4 mAb on the activation of Ag‐specific T‐cells in vivo using OT‐I OVA‐specific T‐cell receptor‐transgenic mice in which CD8 T‐cells recognize the OVA257–264 peptide in the context of H‐2Kb40 (Fig. 1a). According to flow cytometry analysis, immunization with OVA and the anti‐TLR4 mAb in combination, but not individually, significantly induced proliferation of OT‐I CD8 T‐cells, resulting in an increased number of Ag‐specific CD8 T‐cells in the spleen. We performed a similar adoptive transfer study using OT‐II T‐cell transgenic mice in which CD4 T‐cells recognize OVA323–339 in the context of I‐Ab41 (Fig. 1b). Two–three days after immunization, the proliferation and absolute number of OVA‐specific Ly5·1+ OT‐II CD4 T‐cells were induced by OVA, and significantly enhanced by co‐administration of the anti‐TLR4 mAb. In contrast, injection of the anti‐TLR4 mAb alone did not influence the proliferation of OVA‐specific OT‐II CD4 T‐cells. These results indicate that co‐administration of the agonistic‐TLR4 mAb and the Ag results in enhanced induction of Ag‐specific CD4 and CD8 T‐cells.
Figure 1.
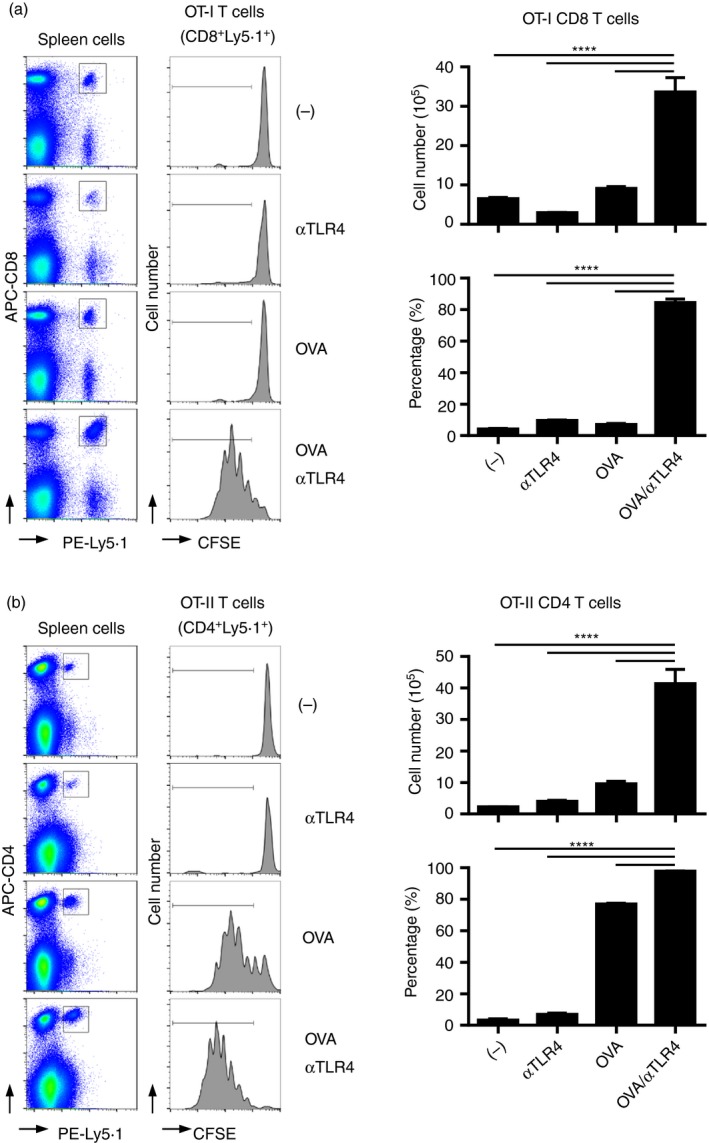
The anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) enhances the proliferation of antigen (Ag)‐specific T‐cells in vivo. 5‐Carboxyfluorescein diacetate succinimidyl ester (CFSE)‐labelled Ly5·1+ (a) OT‐I or (b) OT‐II spleen cells were adoptively transferred into C57BL/6N (Ly5·2+) mice (n = 3 per group). On the following day, transferred mice were immunized with vehicle, (a) 100 μg or (b) 10 μg ovalbumin (OVA), 3 μg of the anti‐TLR4 mAb, or both. (a) Two or (b) 3 days after immunization, spleen cells were subjected to FACS analysis to evaluate the proliferation of OT‐I and OT‐II T‐cells based on the intensity of CFSE fluorescence. Representative histograms and dot plots are shown. The absolute numbers of (a) Ly5·1+ CD8+ OT‐I and (b) Ly5·1+ CD4+ OT‐II T‐cells in the spleen, and the percentages of (a) CFSE low Ly5·1+ CD8+ OT‐I and (b) Ly5·1+ CD4+ OT‐I T‐cells are shown as means ± SEMs. One‐way ANOVA with the Tukey post hoc test; ****P < 0·0001. Data are representative of three independent experiments.
Inhibition of tumour growth in vivo by immunization with Ag and the anti‐TLR4 mAb
We next investigated whether immunization with Ag and the anti‐TLR4 mAb activates Ag‐specific antitumour immunity and suppresses tumour growth in vivo. To assess the efficacy of the prophylactic regimen, the mice were twice immunized s.c. with OVA and the anti‐TLR4 mAb, and EG7 cells derived from H‐2Kb‐positive EL4 thymoma cells and expressing OVA were inoculated s.c. into their backs (Fig. 2a). Tumour growth was significantly retarded in mice injected with OVA and the anti‐TLR4 mAb compared with the vehicle control. We next investigated the therapeutic efficacy of OVA and the anti‐TLR4 mAb. One day after inoculation of EG7 tumour cells, the mice were injected s.c. around the site of tumour inoculation with OVA, the anti‐TLR4 Ab, or both (Fig. 2b,c). The growth of EG7 tumours was slowed slightly by OVA. Co‐administration of OVA and the anti‐TLR4 mAb resulted in significant suppression of tumour growth compared with administration of OVA alone. In contrast, injection of the anti‐TLR4 mAb did not impact the growth of EG7 tumours compared with the vehicle control. Moreover, immunization with OVA and the anti‐TLR4 mAb did not inhibit the growth of EL4 (OVA‐negative) tumours (Fig. 2d). We also investigated the therapeutic efficacy of OVA and the anti‐TLR4 mAb in mice with H‐2‐negative B16F10 melanoma, which expresses OVA lacking a signal peptide (B16F10‐cOVA; Fig. 2e). Whereas immunization with OVA or the anti‐TLR4 mAb alone did not affect tumour growth, co‐administration of OVA and the anti‐TLR4 mAb significantly suppressed the growth of B16F10‐cOVA tumours. Moreover, this antitumour effect was not observed in mice with parental B16F10 tumours, which are negative for OVA (Fig. 2f). These findings suggest that OVA and the anti‐TLR4 mAb in combination exert an antitumour effect in vivo.
Figure 2.
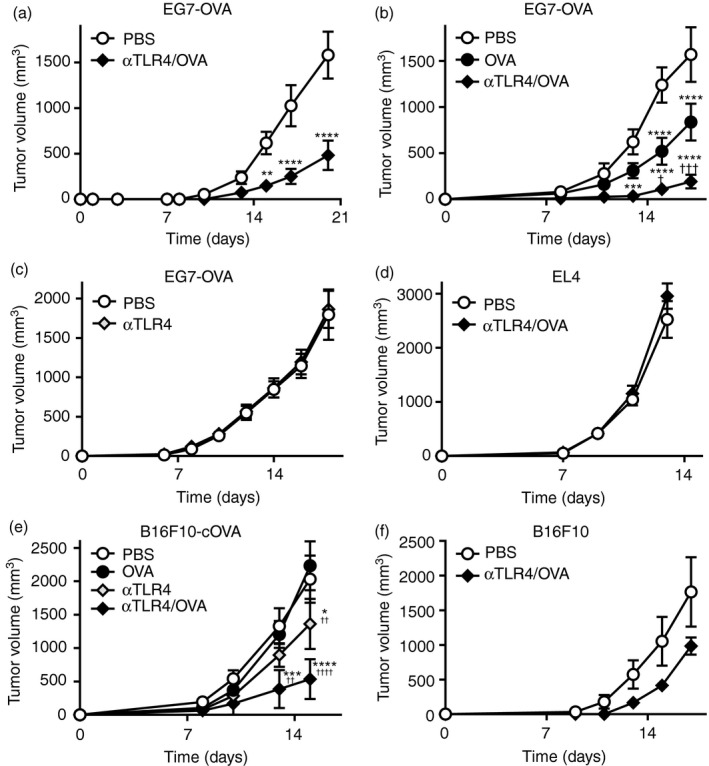
The anti‐Toll‐like receptor 4 (TLR4) agonistic monoclonal antibody (mAb) suppresses tumour growth. C57BL/6N mice (n = 4–6 per group) were inoculated s.c. on the back with (a–c) EG7‐OVA, (d) EL4, (e) B16F10‐cOVA or (f) B16F10 (3 × 105); (a) 21 and 7 days prior to or (b–f) 1 day after inoculation, they were injected s.c. with vehicle (phosphate‐buffered saline; PBS), ovalbumin (OVA; 100 μg), the anti‐TLR4 mAb (3 μg), or the indicated combinations. Tumour volumes are shown as means ± SEMs. Two‐way ANOVA with the Sidak (a, c, d, f) or Tukey (b, e) post hoc test; *P < 0·05, **P < 0·01, ***P < 0·01, ****P < 0·0001 (versus PBS); †† P < 0·01, ††† P < 0·001, †††† P < 0·0001 (versus OVA). Data are representative of two or three independent experiments.
Increased therapeutic efficacy of the anti‐PD‐1 mAb by immunization with Ag and the anti‐TLR4 mAb
As the co‐administration of OVA plus the anti‐TLR4 mAb suppressed tumour growth in single therapy, we next investigated whether it enhances antitumour activity of anti‐PD‐1 mAb when treated in combination with OVA plus the anti‐TLR4 mAb. We used OVA‐expressing MC38 tumour cells, which are responsive to anti‐PD‐1 mAbs. One day after inoculation with MC38‐OVA tumour cells, the mice were injected s.c. with OVA plus the anti‐TLR4 Ab, and then injected twice i.p. with anti‐PD‐1 mAb (Fig. 3). Immunization with OVA plus the anti‐TLR4 mAb significantly suppressed tumour growth, but the efficacy was weaker than that with anti‐PD‐1 mAb single therapy. However, OVA/TLR4 mAb combined with the anti‐PD‐1 mAb enhanced the therapeutic efficacy and suppressed tumour growth significantly compared with the effects with individual single therapies.
Figure 3.
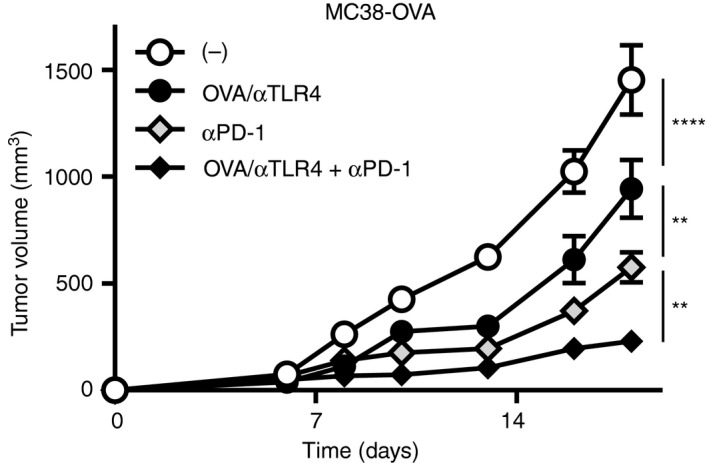
The anti‐Toll‐like receptor 4 (TLR4) agonistic monoclonal antibody (mAb) enhances the therapeutic efficacy of anti‐PD‐1 mAb. C57BL/6N mice (n = 5 per group) were inoculated s.c. on the back with MC38‐OVA (5 × 105). One day after inoculation, the mice were injected s.c. with vehicle (phosphate‐buffered saline; PBS) or the combination of ovalbumin (OVA; 100 μg) and the anti‐TLR4 mAb (3 μg), followed by i.p. injection of the anti‐PD‐1 mAb (100 μg) or vehicle on days 6 and 9. Tumour volumes are shown as means ± SEMs. Two‐way ANOVA with the Tukey post hoc test; **P < 0·01, ****P < 0·0001. Data are representative of three independent experiments.
Immunization with OVA and the anti‐TLR4 mAb induces activation of Ag‐specific CD8 T‐cells
To determine the mechanism underlying the antitumour effect of OVA plus the anti‐TLR4 mAb, we investigated the activation status of CD4 and CD8 T‐cells by determining their expression of CD44, a marker of activation, by flow cytometry (Fig. 4a). In contrast to OVA, the anti‐TLR4 mAb induced polyclonal activation of CD4 and CD8 T‐cells independently of OVA injection. Next, we investigated production of IFN‐γ, an important mediator of antitumour immunity (Fig. 4b). Ex vivo stimulation with OVA257–264 increased the number of IFN‐γ‐producing CD8 T‐cells following immunization with OVA and the anti‐TLR4 mAb in combination, but not individually. In addition, the number of IFN‐γ‐producing CD4 T‐cells showed an increasing trend following re‐stimulation of spleen cells from mice harbouring OVA323–339 that were administered OVA and the anti‐TLR4 mAb; however, the impact on the number of CD4 T‐cells was milder than that on the number of CD8 T‐cells.
Figure 4.
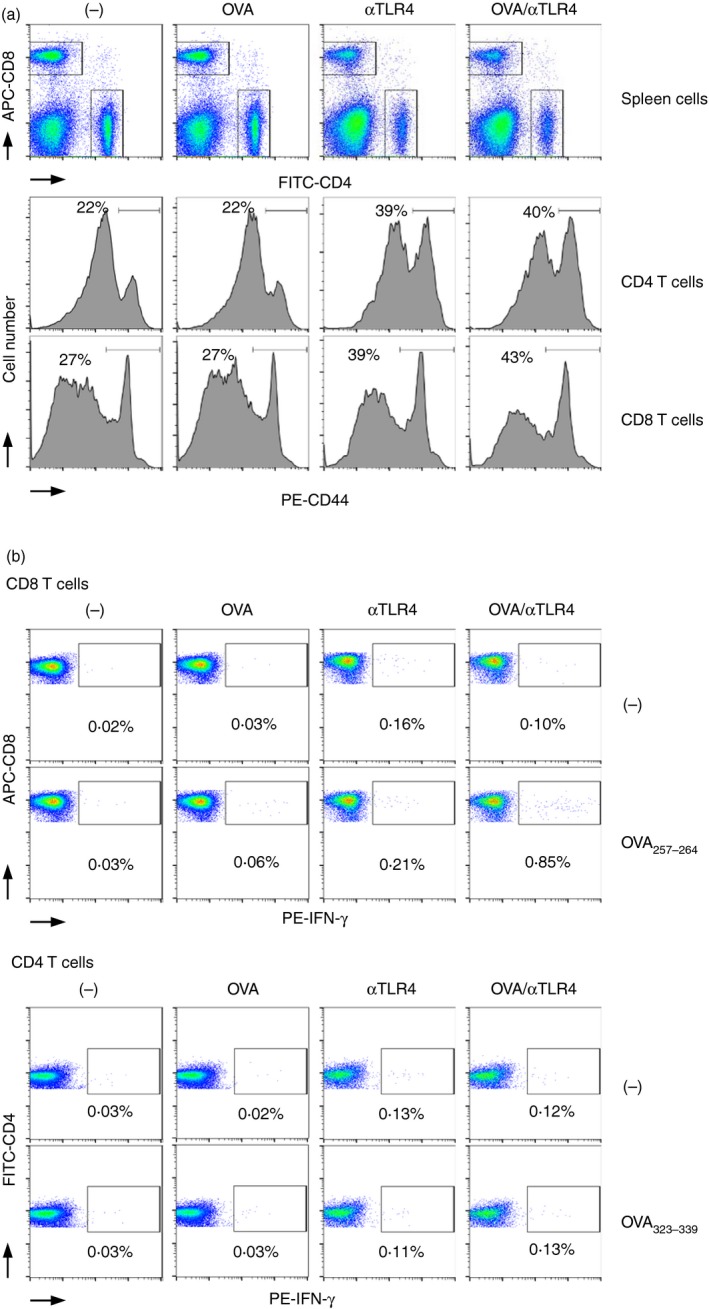
The anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) induces antigen (Ag)‐specific IFN‐γ‐producing CD8 T‐cells. C57BL/6N mice were injected i.p. with vehicle (phosphate‐buffered saline; PBS), ovalbumin (OVA; 100 μg), the anti‐TLR4 mAb (5 μg), or both. (a) Five days later, the CD44hi CD4+ and CD44hi CD8+ T‐cells in the spleen were analysed by FACS. Representative histograms and dot plots of three independent experiments are shown. (b) Five–seven days later, spleen cells were re‐stimulated with the OVA 323–339 (10 μg/ml) or OVA 257–264 (1 μg/ml) peptide for 8 hr in vitro. Intracellular IFN‐γ was stained in CD4 and CD8 T‐cells and analysed by FACS. Representative dot plots are shown. Similar results were obtained from three independent experiments.
Because adoptive transfer activated OVA‐specific CD4 and CD8 T‐cells following co‐administration of OVA and the anti‐TLR4 mAb, we investigated the contributions of CD4 and CD8 T‐cells to OVA/anti‐TLR4 mAb‐induced antitumour immunity in mice with EG7 tumours. An anti‐CD4 or ‐CD8 mAb was administered repeatedly to the mice before and after inoculation of EG7 tumour cells and immunization with OVA and the anti‐TLR4 mAb. This procedure resulted in the near‐total depletion of CD4 and CD8 T‐cells from the peripheral blood (Fig. 5a). The suppression of tumour growth by OVA/anti‐TLR4 mAb was reduced to the level of the control by depletion of CD8, but not CD4, T‐cells (Fig. 5b). Therefore, activation of OVA‐specific CD8 T‐cells is responsible for the antitumour effect of OVA/anti‐TLR4 mAb.
Figure 5.
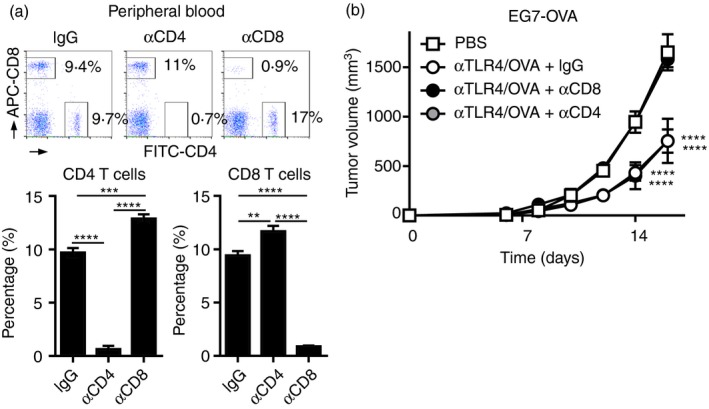
The antitumour effect of the anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) is mediated by CD8 T‐cells in vivo. C57BL/6N mice (n = 5 per group) were inoculated s.c. on the back with EG7‐OVA (3 × 105), and on the following day they were injected s.c. with vehicle (phosphate‐buffered saline; PBS) or the combination of ovalbumin (OVA; 100 μg) and the anti‐TLR4 mAb (3 μg). In mice immunized with OVA and the anti‐TLR4 mAb, an anti‐CD4 (GK1·5), ‐CD8 (YTS169·4·2·1) or rat isotype control mAb (100 μg) was injected i.v. on day −2 and i.p. on days −1 and 6. (a) On day 6, the percentages of CD4 and CD8 T‐cells in the peripheral blood were analysed by FACS and are shown as the means ± SEMs of five mice per group. One‐way ANOVA with the Tukey post hoc test; **P < 0·01, ***P < 0·001, ****P < 0·0001. (b) Tumour volumes are shown as means ± SEMs. Two‐way ANOVA with the Tukey post hoc test; ****P < 0·0001 (versus PBS). Data are representative of two independent experiments.
The anti‐TLR4 mAb induces a more potent immune response and less marked inflammation than does LPS
Lipopolysaccharide and its derivatives have been used as the basis of small‐molecule adjuvants;21, 34, 35 however, their clinical use is hampered by the potent inflammatory response. Therefore, we compared the inflammatory reactions to the anti‐TLR4 mAb and LPS in vivo and in vitro. Activation of NF‐κB was significantly induced by both stimulants in a reporter assay using Ba/F3 cells stably expressing TLR4 and MD‐2 (Fig. 6a). Additionally, TNF‐α and IL‐6 secretion by BMMs was significantly increased by the anti‐TLR4 mAb and LPS (Fig. 6b,c). However, the proinflammatory effect of the anti‐TLR4 mAb at 1–10 μg/ml was lesser than or comparable to that of 10–100 ng/ml LPS. We next determined the TNF‐α and IL‐6 levels in the peripheral blood of mice after i.p. injection of LPS or the anti‐TLR4 mAb (3 μg; Fig. 6d,e). Consistent with the in vitro findings, the TNF‐α and IL‐6 levels in mice administered the anti‐TLR4 mAb were similar to those after i.p. injection of 0·03 and 0·1 μg LPS, respectively. Therefore, the proinflammatory effect of the anti‐TLR4 mAb is of lesser magnitude than that of LPS in vivo.
Figure 6.
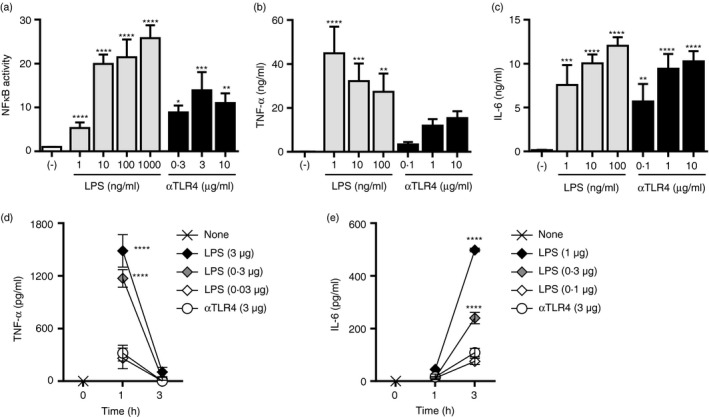
The agonistic anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) induces an inflammatory reaction of lesser magnitude than does lipopolysaccharide (LPS). (a) Mouse TLR4/MD‐2‐expressing Ba/F3‐transfected cells carrying NF‐κB‐responsive luciferase reporter genes were stimulated with the anti‐TLR4 mAb or LPS at the indicated concentrations for 5–7 hr. Luciferase activities are shown as mean ± SD fold increases of triplicate cultures compared with non‐stimulated cells. One‐way ANOVA with the Tukey post hoc test; *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001 (versus the control). Data are representative of three independent experiments. (b, c) Bone marrow‐derived macrophages (BMMs) were stimulated with the anti‐TLR4 mAb or LPS at the indicated concentrations for 24 hr. The concentrations of (b) TNF‐α and (c) IL‐6 in the culture supernatants were evaluated by ELISA. One‐way ANOVA with the Tukey post hoc test; **P < 0·01, ***P < 0·001, ****P < 0·0001 (versus the control). Data are means ± SDs of triplicate cultures and are representative of three independent experiments. (d, e) The mice (n = 3 per group) were administered the anti‐TLR4 mAb or LPS i.p. One or three hours later, plasma was collected, and the concentrations of (d) TNF‐α and (e) IL‐6 were determined by ELISA. Two‐way ANOVA with the Tukey post hoc test; ****P < 0·0001 (versus the anti‐TLR4 mAb). Data are means ± SEMs of two independent experiments.
Next, we compared the ability of the anti‐TLR4 mAb and LPS to induce Ag‐specific immunity. In adoptive OT‐I and ‐II T‐cell transfer experiments (Fig. 7a,b), the anti‐TLR4 mAb (3 μg) significantly induced the proliferation of OT‐I CD8 and OT‐II CD4 T‐cells in vivo. In contrast, injection of 1–3 μg LPS exerted no and a mild effect on the proliferation of OVA‐induced OT‐I CD8 and OT‐II CD4 T‐cells, respectively. In contrast to the anti‐TLR4 mAb (3 μg), LPS (0·1 μg) did not exert an effect on tumour growth, irrespective of the presence of OVA (Fig. 7c). Therefore, the anti‐TLR4 mAb is a potent adjuvant for Ag‐specific tumour immunity, and induces an inflammatory reaction of lesser magnitude compared with LPS.
Figure 7.
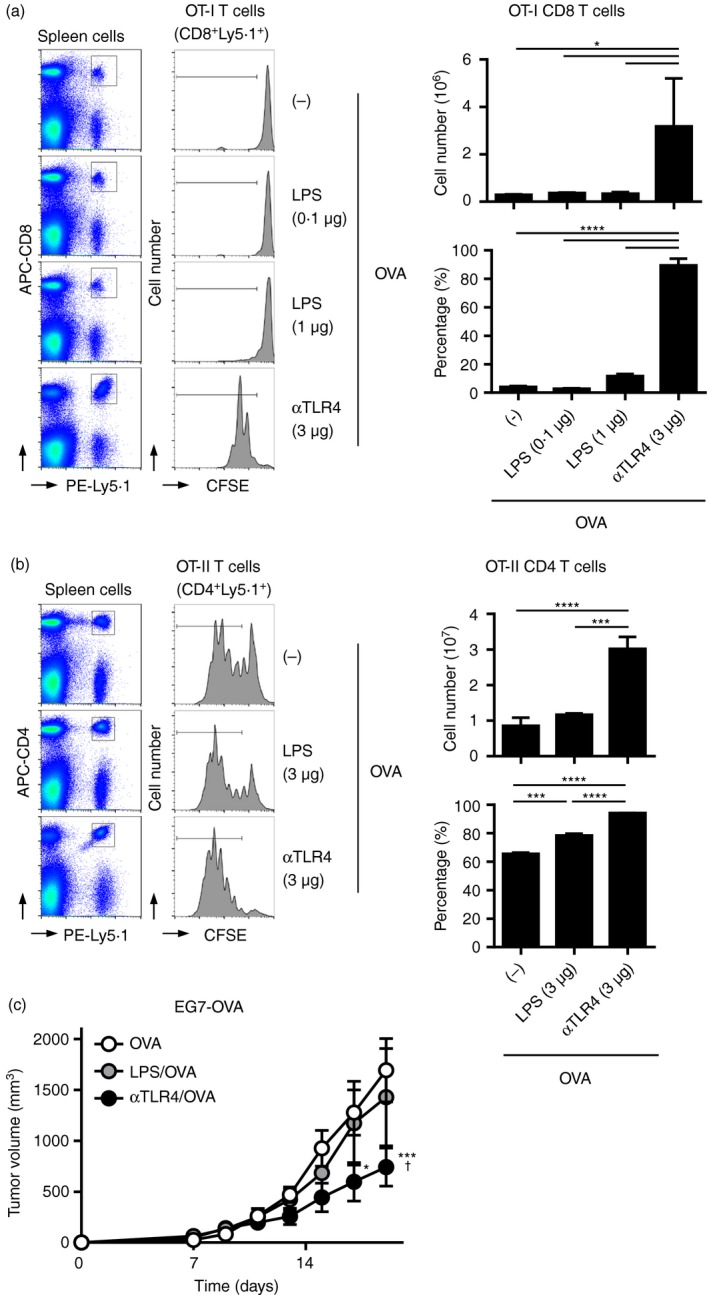
The antitumour effect of the anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) is more potent than that of lipopolysaccharide (LPS). 5‐Carboxyfluorescein diacetate succinimidyl ester (CFSE)‐labelled Ly5·1+ (a) OT‐I or (b) OT‐II spleen cells were adoptively transferred into C57BL/6N (Ly5·2+) mice (n = 3 per group). On the following day, the mice were immunized with the anti‐TLR4 mAb or LPS at the indicated doses with (a) 100 μg or (b) 10 μg ovalbumin (OVA). (a) Two or (b) three days after immunization, spleen cells were subjected to FACS analysis to evaluate the proliferation of transferred OT‐I and OT‐II T‐cells. Representative histograms and dot plots are shown. The absolute numbers of (a) Ly5·1+ CD8+ OT‐I and (B) Ly5·1+ CD4+ OT‐II T‐cells in the spleen, and the percentages of (a) CFSE low Ly5·1+ CD8+ OT‐I and (B) Ly5·1+ CD4+ OT‐I) T‐cells are shown as means ± SEMs. One‐way ANOVA with the Tukey post hoc test; *P < 0·05, ***P < 0·001, ****P < 0·0001. Data are representative of two independent experiments. (c) C57BL/6N mice (n = 5 per group) were inoculated s.c. on the back with EG7‐OVA (3 × 105 cells), and the following day they were injected s.c. with vehicle (phosphate‐buffered saline; PBS), the anti‐TLR4 mAb (3 μg), or LPS (0·1 μg) with OVA (100 μg). Tumour volumes are shown as means ± SEMs. Two‐way ANOVA with the Tukey post hoc test; *P < 0·05, ***P < 0·001 (versus OVA); † P < 0·05 (versus OVA/LPS). Data are representative of two independent experiments.
The anti‐TLR4 mAb stimulates expression, but not cleavage, of pro‐IL‐1β
To investigate further the mechanism underlying the decreased magnitude of inflammatory reaction induced by anti‐TLR4 mAb, we focused on the proinflammatory cytokine IL‐1β, which is activated by non‐canonical inflammasomes consisting of the cytoplasmic LPS sensor, caspase‐11. BMMs were primed with the anti‐TLR4 mAb or LPS, and then transfected with the identical stimulants using polyethyleneimine. IL‐1β was secreted into the culture medium after priming and transfection with LPS, but not with the anti‐TLR4 mAb (Fig. 8a). Next, we engaged the TLR2 ligand, Pam3CSK4. Pam3CSK4‐primed BMMs were transfected with the anti‐TLR4 mAb or LPS to exclude the impact of the anti‐TLR4 mAb and LPS on the priming phase. ELISA and Western blot analyses of the culture media revealed that the secretion of IL‐1β was significantly stimulated by the transfection of LPS, but not by anti‐TLR4 mAb (Fig. 8b,c). We investigated the expression levels of pro‐IL‐1β and caspase‐11 in primed cells to elucidate the impact of the anti‐TLR4 mAb and LPS on the priming phase. Like LPS stimulation, the anti‐TLR4 mAb induced the expression of pro‐IL‐1β and caspase‐11. However, 1 and 10 μg of anti‐TLR4 mAbs induced a lower magnitude than that induced by 0·01 and 0·1 μg LPS, showing a clear contrast to the impact on the activation phase, but a similar impact on the activation of NF‐κB and induction of TNF‐α and IL‐6. Following these in vitro experiments, we determined the plasma levels of IL‐1β in mice administered with 1 μg LPS and 3 μg anti‐TLR4 mAb (Fig. 8e). Whereas LPS increased plasma IL‐1β levels in unprimed mice, such levels were undetectable in mice primed with poly(I:C) and then challenged with the anti‐TLR4‐mAb. These results suggest that the lower magnitude of inflammatory reaction of the anti‐TLR4 mAb could, in part, be accounted for by the differing impact on non‐canonical inflammasomes, as compared with that with LPS.
Figure 8.
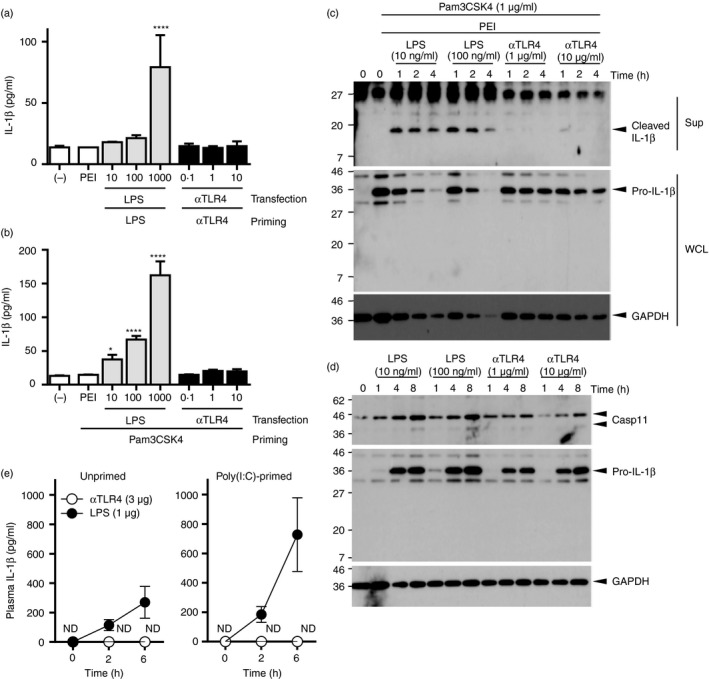
The agonistic anti‐Toll‐like receptor 4 (TLR4) monoclonal antibody (mAb) primes the expression of IL‐1β but does not stimulate its secretion. (a) Bone marrow‐derived macrophages (BMMs) were primed with the anti‐TLR4 mAb (0·1, 1, 10 μg/ml) or lipopolysaccharide (LPS; 10, 100, 1000 ng/ml) for 4 hr and then transfected with identical stimulants using polyethylenimine for 20 hr. (b) Pam3CSK4‐primed BMMs were transfected with the anti‐TLR4 mAb (0·1, 1, 10 μg/ml) or LPS (10, 100, 1000 ng/ml) as in (a). (a, b) The concentrations of IL‐1β in the culture supernatants were evaluated by ELISA. One‐way ANOVA with the Tukey post hoc test; *P < 0·05, ****P < 0·0001 (versus primed and mock‐transfected control). Data are means ± SDs of triplicate cultures and are representative of two independent experiments. (c) BMMs were stimulated with the anti‐TLR4 mAb or LPS for the indicated periods. (d) Pam3CSK4‐primed BMMs were transfected with the anti‐TLR4 mAb or LPS for the indicated periods. (c, d) Following stimulation, culture supernatants and whole cell lysates were analysed by Western blot. Data are representative of two independent experiments. (e) The mice (n = 3 per group) were primed with i.v. injection of poly(I:C) (200 μg) for 21 hr and then challenged i.p. with the anti‐TLR4 mAb (3 μg) or LPS (1 μg) i.p. Plasma samples were collected at 2 or 6 hr, and the concentration of IL‐1β was determined by ELISA. Data are means ± SEMs of two independent experiments.
Discussion
Immune‐checkpoint blockade is a novel therapy for cancer,5, 6, 7 but its response rate and/or therapeutic efficacy need to be improved.6, 15, 16 In this study, we showed that an agonistic anti‐TLR4 mAb induces tumour‐specific immune responses in mice immunized with a tumour‐specific Ag, which enhanced the therapeutic efficacy of the anti‐PD‐1 mAb. Moreover, the anti‐TLR4 mAb induced more potent antitumour immunity and an inflammatory response of lesser magnitude compared with LPS. In contrast to LPS, the anti‐TLR4 mAb did not induce Il‐1β activation. These findings will facilitate the development of novel Ab‐based immune adjuvants.
In EG7 tumour‐bearing mice, the anti‐TLR4 mAb in combination with OVA, but not the mAb alone, inhibited tumour growth in vivo. This result is consistent with the induction of OVA‐specific T‐cell activation in vivo following adoptive transfer. In addition, depletion of CD8 T‐cells eliminated the antitumour immune response in EG7‐bearing mice. The induction of IFN‐γ‐producing CD8 T‐cells by co‐administration of OVA and anti‐TLR4 mAb suggests that the effect is mediated by CD8 T‐cells. In addition, CD4 T‐cells were activated by co‐administration of OVA and the anti‐TLR4 mAb; however, the activation of Ag‐specific CD4 T‐cells was not required for the antitumour effect of the anti‐TLR4 mAb because depletion of CD4 T‐cells did not impact the suppression of tumour growth in EG7‐bearing mice. Previously, we showed that immunization with an anti‐TLR4 mAb enhances Ag‐specific IgG production.33 Therefore, activation of Ag‐specific CD4 T‐cells may contribute to the induction of humoral, rather than cellular, immunity, which is mediated primarily by Ag‐specific CD8 T‐cells.
The OVA/TLR4 mAb combination suppressed the growth of B16F10‐cOVA tumours in mice. B16 melanoma expressed H‐2Kb in the presence, but not the absence, of IFN‐γ stimulation (Fig. S1).46 The IFN‐γ produced by Ag‐specific CD8 T‐cells in OVA/anti‐TLR4 mAb‐immunized mice may have induced H‐2Kb in B16F10 tumour cells in vivo. This possibility suggests that OVA/anti‐TLR4 mAb therapy may be effective against some MHC I‐negative tumours. Indeed, B16F10 tumours can be eradicated by cytotoxic CD8 T‐cells in vivo.46
Whereas EG7 and MC38 cells secrete OVA extracellularly, B16F10 cells retain OVA intracellularly because the transfected OVA lacks a signal peptide. The subcellular distribution of the tumour Ag targeted by the mAb seems not to be critical if it contains MHC‐I‐restricted peptides, such as OVA257–264. We used OVA as a model tumour Ag in mice; however, this approach is unlikely to be feasible in the clinic. Rather, peptide immunogens are commonly used to activate tumour‐specific immune responses.17 However, peptide‐vaccine therapy has failed to show clinical efficacy, despite its safety and tolerability in patients with cancer.15, 16, 17, 35 Thus, most tumour Ags expressed exclusively or preferentially by tumours, but not normal tissues, are likely not suitable targets for tumour vaccine therapy.15, 16, 17 The application of next‐generation sequencing and bioinformatics has shown that neoantigens produced by mutations in individual tumours are targets of antitumour immunity.15, 47 Neoantigen‐derived peptides show promise for adjuvant therapy. In clinical trials, neoantigen‐based vaccines elicited robust and polyfunctional T‐cell responses in patients with melanoma.48, 49, 50 Adjuvants such as our agonistic anti‐TLR4 mAb could be used to enhance the tumour‐specific immune response.
Immune‐checkpoint therapy using anti‐PD‐1 and CTLA‐4 mAbs is effective in patients with highly immunogenic tumours.4, 9, 10 The mutation burden of a tumour may be predictive of its response to immune‐checkpoint blockade therapy.4, 10 In addition, microsatellite‐instable and mismatch repair‐deficient tumours are responsive to immune‐checkpoint blockade, likely because they frequently express neoantigens.9, 11 However, poorly immunogenic tumours may be resistant to immune‐checkpoint blockade because of the small number of tumour‐infiltrating T‐cells in the tumour microenvironment.5, 12, 13, 51 Such poorly immunogenic tumours could be targets of tumour vaccine therapy using an anti‐TLR4 mAb. The induction of tumour‐specific CD8 T‐cells may enhance the response rate and therapeutic efficacy of immune‐checkpoint inhibitors. Recently, we showed that administration of an anti‐TLR4 mAb to mice in the absence of Ag induces immunosuppressive myeloid‐derived suppressor cells with high PD‐L1 expression.32 Therefore, the efficacy of combinations of an anti‐TLR4 mAb and checkpoint inhibitors, particularly an anti‐PD‐1 or ‐PD‐L1 mAb, is of interest. We showed that the OVA/TLR4 mAb combination enhances the efficacy of single therapy anti‐PD‐1 mAb in MC38‐OVA‐bearing mice. In this study, we found that the suppression of tumour growth by the OVA/TLR4 mAb single therapy appears to be weaker in MC38‐OVA‐bearing mice, as compared with that observed in EG7‐ and B16F10‐cOVA‐bearing mice. This observation may be explained by the higher immunogenicity of MC38 than that of EL4 and B16F10 tumours. Without boosting the immune responses by the anti‐TLR4 mAb, inherent tumour‐specific cytotoxic lymphocytes might be stimulated to a certain extent in the MC38‐bearing mice. Peptide vaccines could be used not only for therapeutic purposes but also to prevent cancer in high‐risk patients and those who have undergone surgery to prevent tumour progression and/or recurrence.16 Use of an anti‐TLR4 mAb may enhance the preventive effect of such vaccines; indeed, monophosphoryl lipid A reportedly prevents human papilloma virus‐induced cervical carcinoma.35
Some inflammation is necessary for the induction of Ag‐specific adaptive immunity.18, 19, 20, 21 However, an excessive inflammatory response is detrimental to the patient and should be avoided. The inflammatory response to LPS hampers the development of LPS‐based adjuvants. LPS reportedly induces inflammation in a TLR4‐independent manner.36, 37 Similar to TLR4 and its associated factor MD‐2, lipid A is recognized by the intracellular LPS sensors caspase‐4 and ‐11 in humans and mice, respectively, resulting in inflammasome activation.52, 53 In contrast, the anti‐TLR4 agonistic mAb activates TLR4, but not caspases, because of its high specificity and inability to penetrate the cell membrane. The secretion of IL‐1β is controlled by TLR‐dependent priming followed by ‐independent cleavage.36, 37 The latter step, which is stimulated by non‐canonical inflammasomes,52, 53 represents an obvious difference in the point of action between LPS and the anti‐TLR4 mAb. In clear contrast to the action of LPS, the anti‐TLR4 mAb did not stimulate cleavage of pro‐IL‐1β in macrophages. Furthermore, the priming effects on pro‐IL‐1β and caspase‐11 were slightly lower in magnitude than the effects of LPS as shown in production of TNF‐α and IL‐6. In line with these in vitro findings, IL‐1β production was undetectable in mice injected with the anti‐TLR4 mAb, whereas LPS stimulated production. Injection of low‐dose LPS induced levels of inflammatory cytokines (TNF‐α, IL‐6 and IL‐1β), comparable to or higher than those induced by the anti‐TLR4 mAb, which failed to activate OVA‐specific CD8 T‐cells after adoptive transfer in vivo. In addition, LPS at a dosage comparable to that of the anti‐TLR4 mAb did not suppress tumour growth in EG7‐bearing mice. The activation of TLR4, but not inflammatory caspases, by the anti‐TLR4 mAb may explain the less intense inflammatory reaction. These findings suggest that activation of TLR4 alone is sufficient to activate Ag‐specific CD8 T‐cells. Theoretically, higher dosages of LPS or its derivatives could induce tumour‐specific T‐cells and eradicate the tumours by strengthening TLR4 stimulation. However, in these situations, host toxicity would be likely to impede clinical development.
In conclusion, we showed that an agonistic anti‐TLR4 mAb induces more potent CD8 T‐cell‐dependent antitumour immunity, and an inflammatory reaction of lesser magnitude, than does LPS. These findings will facilitate the development of novel antitumour Ab adjuvants to enhance the response rate and therapeutic efficacy of immune‐checkpoint blockade and/or to prevent tumour progression and recurrence in high‐risk patients and those who have undergone surgery.
Disclosures
The authors declare no conflict of interest.
Supporting information
Figure S1. B16F10 cells (5 × 104) were cultured on 48‐well plates overnight in 500 μl culture medium and stimulated with the anti‐TLR4 mAb or IFN‐γ for 24 hr.
Acknowledgements
HT conceived and supervised the study; HT, KK and AS performed experiments; HT wrote the manuscript; MM, NM, HY, SO and YT reviewed the manuscript. All authors approved the final version of the manuscript. This work was supported in part by JSPS KAKENHI grant numbers 18890140 (HT), 70423605 (HT) and 16H04704 (YT), grants provided by the SGH foundation (HT) and the Osaka Cancer Research Foundation (HT). The authors thank Dr N. Ishii for providing them with OT‐I and ‐II mice. The authors also thank Ms Sao Kozakai, Ms Misaki Okubo and Mr Yohei Kobayashi in their laboratory for the assistance of experiments.
References
- 1. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy–revisited. Nat Rev Drug Discov 2011; 10:591–600. [DOI] [PubMed] [Google Scholar]
- 2. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L et al Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–30. [DOI] [PubMed] [Google Scholar]
- 3. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J et al Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet 2017; 389:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014; 371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L et al PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018; 359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gong J, Chehrazi‐Raffle A, Reddi S, Salgia R. Development of PD‐1 and PD‐L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 2018; 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD‐L1 checkpoint. Immunity 2018; 48:434–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK et al Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017; 357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA et al Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): an open‐label, multicentre, phase 2 study. Lancet Oncol. 2017; 18:1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C et al International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018; 391:2128–39. [DOI] [PubMed] [Google Scholar]
- 13. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69–74. [DOI] [PubMed] [Google Scholar]
- 14. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour‐specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018; 18:168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finn OJ. The dawn of vaccines for cancer prevention. Nat Rev Immunol 2018; 18:183–94. [DOI] [PubMed] [Google Scholar]
- 17. Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R et al Trial watch: peptide‐based vaccines in anticancer therapy. Oncoimmunology 2018; 7:e1511506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M et al Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 2001; 194:769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol 2017; 198:2223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gutjahr A, Tiraby G, Perouzel E, Verrier B, Paul S. Triggering intracellular receptors for vaccine adjuvantation. Trends Immunol 2016; 37:573–87. [DOI] [PubMed] [Google Scholar]
- 21. Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol 2016; 28:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol 2015; 16:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Medzhitov R, Preston‐Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388:394–7. [DOI] [PubMed] [Google Scholar]
- 24. Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Ohta S, Nagai Y et al Reduced surface expression of TLR4 by a V254I point mutation accounts for the low lipopolysaccharide responder phenotype of BALB/c B cells. J Immunol 2013; 190:195–204. [DOI] [PubMed] [Google Scholar]
- 25. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y et al Cutting edge: Toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162:3749–52. [PubMed] [Google Scholar]
- 26. Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Ihara H, Ikeda Y et al Multiple potential regulatory sites of TLR4 activation induced by LPS as revealed by novel inhibitory human TLR4 mAbs. Int Immunol 2012; 24:495–506. [DOI] [PubMed] [Google Scholar]
- 27. Tsukamoto H, Takeuchi S, Kubota K, Kobayashi Y, Kozakai S, Ukai I et al Lipopolysaccharide (LPS)‐binding protein stimulates CD14‐dependent Toll‐like receptor 4 internalization and LPS‐induced TBK1‐IKK‐IRF3 axis activation. J Biol Chem 2018; 293:10 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsukamoto H, Ihara H, Ito R, Ukai I, Suzuki N, Kimoto M et al MD‐2‐dependent human Toll‐like receptor 4 monoclonal antibodies detect extracellular association of Toll‐like receptor 4 with extrinsic soluble MD‐2 on the cell surface. Biochem Biophys Res Commun 2013; 440:31–6. [DOI] [PubMed] [Google Scholar]
- 29. Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Kimoto M. Lipopolysaccharide‐binding protein‐mediated Toll‐like receptor 4 dimerization enables rapid signal transduction against lipopolysaccharide stimulation on membrane‐associated CD14‐expressing cells. Int Immunol 2010; 22:271–80. [DOI] [PubMed] [Google Scholar]
- 30. Tsukamoto H, Hishinuma T, Tayama R, Narahara K, Suzuki N, Tomioka Y et al The induction of prostaglandin E synthase and upregulation of cyclooxygenase‐2 by 9‐cis retinoic acid. Prostaglandins Other Lipid Mediat 2004; 74:61–74. [DOI] [PubMed] [Google Scholar]
- 31. Tsukamoto H, Hishinuma T, Suzuki N, Tayama R, Hiratsuka M, Yoshihisa T et al Thiazolidinediones increase arachidonic acid release and subsequent prostanoid production in a peroxisome proliferator‐activated receptor gamma‐independent manner. Prostaglandins Other Lipid Mediat 2004; 73:191–213. [DOI] [PubMed] [Google Scholar]
- 32. Tsukamoto H, Kozakai S, Kobayashi Y, Takanashi R, Aoyagi T, Numasaki M et al Impaired antigen‐specific lymphocyte priming in mice after Toll‐like receptor 4 activation via induction of monocytic myeloid‐derived suppressor cells. Eur J Immunol 2019; 49:546–63. [DOI] [PubMed] [Google Scholar]
- 33. Rachmawati NM, Fukudome K, Tsuneyoshi N, Bahrun U, Tsukamoto H, Yanagibashi T et al Inhibition of antibody production in vivo by pre‐stimulation of Toll‐like receptor 4 before antigen priming is caused by defective B‐cell priming and not impairment in antigen presentation. Int Immunol 2013; 25:117–28. [DOI] [PubMed] [Google Scholar]
- 34. Reed SG, Carter D, Casper C, Duthie MS, Fox CB. Correlates of GLA family adjuvants’ activities. Semin Immunol 2018; 39:22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lehtinen M, Paavonen J, Wheeler CM, Jaisamrarn U, Garland SM, Castellsague X et al Overall efficacy of HPV‐16/18 AS04‐adjuvanted vaccine against grade 3 or greater cervical intraepithelial neoplasia: 4‐year end‐of‐study analysis of the randomised, double‐blind PATRICIA trial. Lancet Oncol 2012; 13:89–99. [DOI] [PubMed] [Google Scholar]
- 36. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S et al Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341:1246–9. [DOI] [PubMed] [Google Scholar]
- 37. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 2013; 341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bahrun U, Kimoto M, Tsukamoto H, Tsuneyoshi N, Kohara J, Fukudome K. Preparation and characterization of agonistic monoclonal antibodies against Toll‐like receptor 4‐MD‐2 complex. Hybridoma (Larchmt) 2007; 26:393–9. [DOI] [PubMed] [Google Scholar]
- 39. Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. Induction of long‐term lipopolysaccharide tolerance by an agonistic monoclonal antibody to the toll‐like receptor 4/MD‐2 complex. Clin Vaccine Immunol 2006; 13:1131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin‐specific TCR transgenic line OT‐I: MHC elements for positive and negative selection. Immunol Cell Biol 2000; 78:110–7. [DOI] [PubMed] [Google Scholar]
- 41. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA‐based alpha‐ and beta‐chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 1998; 76:34–40. [DOI] [PubMed] [Google Scholar]
- 42. Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988; 54:777–85. [DOI] [PubMed] [Google Scholar]
- 43. Tsukamoto H, Yamagata Y, Ukai I, Takeuchi S, Okubo M, Kobayashi Y et al An inhibitory epitope of human Toll‐like receptor 4 resides on leucine‐rich repeat 13 and is recognized by a monoclonal antibody. FEBS Lett 2017; 591:2406–16. [DOI] [PubMed] [Google Scholar]
- 44. Tsukamoto H, Chernogorova P, Ayata K, Gerlach UV, Rughani A, Ritchey JW et al Deficiency of CD73/ecto‐5′‐nucleotidase in mice enhances acute graft‐versus‐host disease. Blood 2012; 119:4554–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsukamoto H, Ukai I, Yamagata Y, Takeuchi S, Kubota K, Kozakai S et al Leucine‐rich repeat 2 of human Toll‐like receptor 4 contains the binding site for inhibitory monoclonal antibodies. FEBS Lett 2015; 589:3893–8. [DOI] [PubMed] [Google Scholar]
- 46. Nagato T, Lee YR, Harabuchi Y, Celis E. Combinatorial immunotherapy of polyinosinic‐polycytidylic acid and blockade of programmed death‐ligand 1 induce effective CD8 T‐cell responses against established tumors. Clin Cancer Res 2014; 20:1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hackl H, Charoentong P, Finotello F, Trajanoski Z. Computational genomics tools for dissecting tumour‐immune cell interactions. Nat Rev Genet 2016; 17:441–58. [DOI] [PubMed] [Google Scholar]
- 48. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M et al Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature 2017; 547:222–6. [DOI] [PubMed] [Google Scholar]
- 49. Carreno BM, Magrini V, Becker‐Hapak M, Kaabinejadian S, Hundal J, Petti AA et al Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen‐specific T cells. Science 2015; 348:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ et al An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017; 547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK et al Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187–92. [DOI] [PubMed] [Google Scholar]
- 53. Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F et al Human caspase‐4 detects tetra‐acylated LPS and cytosolic Francisella and functions differently from murine caspase‐11. Nat Commun 2018; 9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. B16F10 cells (5 × 104) were cultured on 48‐well plates overnight in 500 μl culture medium and stimulated with the anti‐TLR4 mAb or IFN‐γ for 24 hr.