

Abstract
Sorafenib is an oral multikinase inhibitor approved for the treatment of differentiated thyroid carcinoma (DTC), renal cell carcinoma, and hepatocellular carcinoma. In the phase III DECISION trial in patients with DTC, sorafenib exposure and the incidence of some adverse events (AEs) were higher than in previous trials; therefore, we analyzed exposure–response relationships, including progression‐free survival (PFS) and selected AEs in patients with DTC. A novel, stratified prediction‐corrected visual predictive check (pc‐VPC) was developed to show robustness of the exposure–response relationships. Time‐to‐event simulations confirmed the benefit of the recommended dosing schedule of 800 mg/day: initial doses of 800 mg/day were associated with the highest PFS, whereas lower doses (600 or 400 mg/day) were associated with improved tolerability but reduced PFS. A simulated dose‐reduction strategy of 800 mg/day for an initial two cycles followed by dose reductions seemed likely to maintain efficacy while possibly mitigating selected AEs (e.g., diarrhea and hand‐foot skin reactions).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The incidence of some adverse events (AEs) was higher in the DECISION trial of sorafenib, an oral multikinase inhibitor, in patients with differentiated thyroid carcinoma (DTC) than in trials in renal cell carcinoma and hepatocellular carcinoma.
WHAT QUESTION DID THIS STUDY ADDRESS?
Because sorafenib exposure is higher in patients with DTC, we explored factors associated with sorafenib exposure and how reduced sorafenib dosing and plasma exposure could affect progression‐free survival (PFS) and the occurrence of selected AEs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A novel, stratified prediction‐corrected visual predictive check was developed to show robustness of the estimated exposure–response relationships for expected efficacy and safety simulations in alternative dosing regimens. No parameters explained the higher sorafenib exposure in patients with DTC. The recommended sorafenib dose (800 mg/day) achieved maximal PFS benefit. Lower doses were associated with improved tolerability but reduced PFS.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The recommended sorafenib dosing schedule was confirmed. Dose reductions after two cycles of 800 mg/day do not affect therapeutic benefit but may mitigate AEs.
Sorafenib, an oral multikinase inhibitor, was approved for the treatment of locally recurrent or metastatic, progressive, radioactive iodine (RAI)–refractory differentiated thyroid carcinoma (DTC)1 based on the results of the randomized, placebo‐controlled, double‐blind, phase III DECISION trial.2 In this trial, sorafenib treatment significantly prolonged progression‐free survival (PFS) in patients with RAI‐refractory DTC (hazard ratio (HR), 0.59; 95% confidence interval (CI), 0.45–0.76; P < 0.0001 vs. placebo). However, there was no statistically significant overall survival benefit in sorafenib‐treated patients, likely because a large number of patients randomized to placebo crossed over to sorafenib after disease progression.2 The sorafenib adverse events (AEs) profile in the DECISION trial was generally consistent with other phase III trials in renal cell carcinoma (RCC) and hepatocellular carcinoma (HCC); however, certain AEs were more common in patients with DTC, possibly due to higher sorafenib exposure.2, 3, 4, 5, 6
To comprehensively investigate the factors associated with sorafenib exposure and the influence of exposure on PFS and selected AEs in DTC, an exposure–response model was developed utilizing a population pharmacokinetic (PK) model that incorporated densely sampled PK data from healthy volunteers and sparsely sampled PK data from patients with HCC, RCC, and DTC enrolled in phase II−IV trials. Simulations investigated whether different starting doses/dose rates (sorafenib 800, 600, or 400 mg/day) or dose reductions (dose reduction from 800 mg/day in the first two cycles to either 600 or 400 mg/day) affected PFS and the frequency of selected AEs in the DTC population.
Methods
DECISION trial design and outcomes
A comprehensive description of the inclusion and exclusion criteria, methods, and primary results of the phase III DECISION trial (NCT00984282; EudraCT 2009‐012007‐25) has been published elsewhere.2 Briefly, patients were ≥ 18 years of age and had locally advanced metastatic RAI‐refractory DTC that had progressed during the previous 14 months (according to Response Evaluation Criteria in Solid Tumors).2 Patients had to have an Eastern Cooperative Oncology Group performance status of 0 or 1 and adequate bone marrow, hepatic and renal function, and serum thyroid‐stimulating hormone level < 0.5 mL U/L. Patients were randomized to either sorafenib 800 mg/day (400 mg twice daily (b.i.d.)) or matching placebo b.i.d. administered without food in a 28‐day cycle. The study continued until disease progression, unacceptable toxicity, noncompliance, or withdrawal of consent.2 It was conducted according to International Conference on Harmonization of good clinical practice guidelines and the Declaration of Helsinki with approval from appropriate ethics committees/institutional review boards. All patients provided written informed consent.7
The protocol permitted treatment interruptions and stepwise dose reductions to manage AEs. According to specific criteria, dose reductions were undertaken in three steps from the initial dose of 800–600 mg/day (400/200 mg am/pm), 400 mg/day (200 mg b.i.d.), and 200 mg once daily. Dose re‐escalation was permitted after AE resolution. In the event of disease progression (investigator assessed), study treatment could be unblinded and open‐label sorafenib could be initiated.2
Outcomes of interest
For each patient, the time from the start of treatment until the first occurrence of specific events was recorded. Events of interest included tumor progression or death due to tumor progression (time to tumor progression was considered equivalent to PFS); any grade ≥ 3 AE, any grade ≥ 4 AE, grade 3 hand‐foot skin reactions (HFSRs), grade ≥ 3 hypertension, and grade ≥ 2 diarrhea. For modeling of tumor progression, the censoring event time (time of exit from the study) was the day of the last radiologic assessment or the day of death. For AEs, the censoring event time was the date of the last recorded dose.
Subjects and PK data generation
PK data from 10 phase I to phase IV trials in healthy volunteers and patients were included in the population PK analysis (Table S1 ), which included data from 156 patients with DTC from the DECISION trial. Healthy volunteers in phase I trials received single oral doses of sorafenib with densely sampled PK, whereas patients in phase II to phase IV trials received an oral sorafenib regimen using the recommended starting dose of 400 mg b.i.d. on a continuous schedule with sparsely sampled PK. The lower limit of quantification in plasma was 1–10 μg/L for sorafenib and varied by study because different assays were used during the course of the clinical development, all of which were validated in accordance with the US Food and Drug Administration guidelines. Sorafenib plasma concentrations below the lower limit of quantification were excluded from PK analyses.
PK modeling
The population PK analysis was performed with NONMEM (version 7.2; ICON Development Solutions, Gaithersburg, Maryland, USA) and Perl‐speaks‐NONMEM (PsN; version 3.5.3).8, 9 Population PK modeling was influenced by the earlier work of Prins et al. and Jain et al.6, 10 that used plasma concentration–time profiles, after initial and steady‐state doses, to describe the oral absorption and systematic PK of sorafenib in patients with solid tumors by considering enterohepatic recirculation. Model development was conducted using the first‐order conditional estimation with the η‐ϵ interaction method. The between‐patient variation of the parameter estimates for the volume of distribution (V) and absorption rate constant (k a) could not be estimated reliably with the available data. Covariate models were constructed by considering the impact of body weight, body mass index, race (Asian/non‐Asian), sex, age, serum glutamic oxaloacetic transaminase (aspartate aminotransferase), serum glutamic pyruvic transaminase (alanine aminotransferase), total bilirubin, alkaline phosphatase, lactate dehydrogenase, prothrombin time international normalized ratio, creatinine clearance, and concomitant medication with cytochrome P450 3A4 inducers, cytochrome P450 3A4 inhibitors, uridine 5′‐diphosphate glucuronosyltransferase 1A9 inhibitors, uridine 5′‐diphosphate glucuronosyltransferase 1A9 inducers, or levothyroxine. Covariates were added in the forward inclusion step in a stepwise fashion and retained if the level of significance was P < 0.01. In the backward elimination step, covariates with a level of significance of P < 0.001 were retained in the model.
Prediction‐corrected visual predictive checks (pc‐VPC) stratified on the different covariates were used to qualify the final population PK model.11 A bootstrap procedure, which was stratified by study such that each study was on average equally represented in 2,000 resampled data sets, was carried out to obtain uncertainty estimates around parameter estimates.
Exposure parameters
In the DECISION trial, a PK sample was collected after unsupervised drug administration. This sampling design facilitated estimation of individual clearance, but the individual values of other parameters (e.g., V and k a) could not be estimated. Hence, selection of exposure variables was limited to those directly related to the individual clearance (i.e., area under the concentration‐vs.‐time curve (AUC) in mg h/L). All other possible exposure variables (e.g., maximum concentration or minimum concentration within a dosing interval) were directly correlated to the AUC over a 12‐hour dosing interval (AUC12) and, therefore, did not result in different exposure–response relationships.
The AUC12 was calculated as a time‐varying exposure parameter and updated for each completed 28‐day cycle and any remaining fraction of a cycle as follows:
| (1) |
In addition, the average AUC12 until the time to onset of an event (AUC12AV) was calculated as a constant value of time:
| (2) |
Likewise, another set of time‐varying and constant exposure variables, which were independent of the individual clearance estimate, (average dose, Eq. (3)) and the time‐varying exposure parameter (dose rate, Eq. (4)) were calculated:
| (3) |
| (4) |
Exposure–response modeling
All four covariates (average dose, dose rate, AUC12, and AUC12AV) were tested in hazard models for each outcome of interest. The probability of an event was described through parametric hazard functions.12 The comparison of structurally different base hazard models involved the calculation of the Akaike Information Criterion. The selection of the base hazard model on the basis of Akaike Information Criterion was further supported by pc‐VPCs of the candidate models compared with a Kaplan−Meier plot of the observed event data.
Model qualification through VPCs
A novel pc‐VPC was developed13 to assess the goodness of fit and the ability or robustness of the model to describe the relationship between exposure and response of the selected hazard model (the base model with the exposure–response relationship). In the pc‐VPC simulation, the proportion of patients with an event up to a specified time point was collected into timed bins. Through 1,000 repetitions, a median and 90% prediction interval of the event rate within the given population was simulated with the selected model for each timed bin. In addition, the observed proportion of patients with an event was calculated for each timed bin. A pc‐VPC allows the comparison of simulated and observed event rates and their development over time. To evaluate if the estimated relationship between exposure parameters and response resembles the observations, the patient population was stratified according to categorical exposure parameters. For that purpose, selected exposure parameters were categorized into groups with values above and below the population median. Time‐variant exposure parameters were averaged up to 200 days to facilitate categorization. The pc‐VPCs qualified a hazard model when the observed event rates fell within the 90% prediction interval in most of the timed bins.
Simulations
Simulations of Kaplan−Meier plots using 1,000 repetitions of the given patient population and the selected model were summarized as medians and 95% CIs based on percentiles and were compared with those based on the observed events. For each simulated subject, two events were simulated with their respective hazard models: an adverse or progression event, and the time at which the virtual patient left the study (the censoring event). The event that occurred first was used to derive Kaplan−Meier curves.
The probability of tumor progression and AEs was simulated in patients with DTC treated with sorafenib 800, 600, or 400 mg/day and after dose reductions. Simulated dose reductions included the following four scenarios: 2 cycles of 800 mg/day followed by a reduction to 600 mg/day (group 1), 2 cycles of 800 mg/day followed by a reduction to 400 mg/day (group 2), 2 cycles of 800 mg/day followed by a reduction to 600 mg/day for 2 cycles then re‐escalation to 800 mg/day (group 3), and 2 cycles of 800 mg/day followed by a reduction to 400 mg/day for 2 cycles then re‐escalation to 800 mg/day (group 4). The simulation groups are visualized in Figure S7 .
One thousand simulations with 1,000 Kaplan−Meier plots were performed with the selected model for each AE of interest and each dosing schedule (800, 600, or 400 mg/day). In cases of tumor progression, the simulated interval censoring was based on scheduled assessment times in the study protocol.
Results
Population PK model
A total of 3,141 sorafenib concentrations obtained from 859 individuals (including 39 healthy subjects; Table S1 ) were included in the population PK analysis. Patient characteristics are listed in Table S2 . Multiple peaks appeared in the concentration‐timecurve around the time of the initial maximum concentration and for ~ 24 hours thereafter. These curves exhibited a relatively monophasic exponential decline indicative of one‐compartment elimination. The sparsely sampled concentration data from patients with cancer, however, did not allow for fitting of multiple peaks with more complex models, such as proposed by Prins et al. and Jain et al.6, 10
When sorafenib concentrations were plotted by study, it was clear that patients with DTC (the DECISION trial) had consistently higher plasma sorafenib concentrations than other patients (Figure S1 ). The data supported a one‐compartment model with linear elimination and three sequential transit absorption compartments. The only covariates that had a significant influence on sorafenib clearance (CL) were sex and enrollment in the sole DTC study, DECISION. CL was estimated to be lower in women than men, with estimates of 7.5 and 9.4 L/hour, respectively (Table 1), resulting in a 26.7% higher exposure to sorafenib in women than men. An approximate twofold increase in sorafenib exposure in patients with DTC in DECISION vs. other studies was observed (Table 1).
Table 1.
Estimates of sorafenib clearance and exposure by sex and study
| Sex | CL/F, L/hour | AUC0–12,ss, mg·h/L | ||||
|---|---|---|---|---|---|---|
| HCC, RCC | DTC | Difference, DTC vs. HCC, RCC, % | HCC, RCC | DTC | Difference, DTC vs. HCC, RCC, % | |
| Male | 9.4 | 4.7 | –50.6 | 42.4 | 85.8 | 103 |
| Female | 7.5 | 3.7 | –50.6 | 53.7 | 109 | 103 |
| Difference, female vs. male, % | –21.1 | –21.1 | — | 26.7 | 26.7 | — |
AUC0–12,ss, area under the concentration vs. time curve from 0–12 hours at steady state; CL/F, oral clearance; DTC, differentiated thyroid cancer; HCC, hepatocellular carcinoma; RCC, renal cell carcinoma.
In the final covariate model (Table S3 and Figure S2 ), the effect of enrollment in the DECISION trial could not be differentiated from the potential effect of the indication (DTC) because only one study in DTC was available for inclusion in the analysis. There were no significant effects of race (Asian vs. non‐Asian patients) or of renal or hepatic function on CL.
The pc‐VPC (Figure S3 ) showed that the final covariate model adequately predicted the model data set over the range of time values after the last dose. In particular, the predicted variability in exposure was in close agreement with the observed data up to 12 hours postdose at steady state. Beyond 12 hours postdose, the curves indicated that the model underpredicts exposure; however, fewer data points were available in this time range.
DTC population for exposure–response analysis
The analysis of efficacy (i.e., PFS) included 401 patients with centrally assessed tumor progression measurements, of whom 197 received sorafenib and 204 received placebo. Among the patients who received placebo, 155 (76%) had crossed over to sorafenib treatment after unblinding. The AE analysis included 413 patients: 206 received sorafenib and 207 received placebo. In the placebo group, 155 patients (75%) had crossed over to sorafenib.
Time to tumor progression
Among the 401 patients included in the analysis of efficacy (PFS), 261 (65.1%) experienced tumor progression before exiting the study (Table 2). A Weibull model was chosen as the base hazard model. Among the four exposure parameters derived from the population PK model and tested in the model (AUC12, AUC12AV, dose rate, and average dose), only AUC12 did not significantly improve the goodness of fit of the base model. The modeling indicated that the hazard of tumor progression decreased with increasing average sorafenib plasma exposure (AUC12AV), with increasing time‐varying sorafenib dose rate and with increases in the average dose. The selected hazard model was qualified through a stratified pc‐VPC, which demonstrated that the progression rate was lower in patients with AUC12AV values greater than the median (Figure 1).
Table 2.
Frequency of events
| Event | Number of patients | Number of events (%) | Hazard model | Exposure covariatea |
|---|---|---|---|---|
| Efficacy | ||||
| Tumor progression, locally assessed | 416 | 321 (77.2) | Not done | Not done |
| Tumor progression, centrally assessed | 401 | 261 (65.1) | Weibull | AUC12AV |
| Safety | ||||
| Grade ≥ 3 AEs | 413 | 259 (62.7) | Constant + Gompertz | Average dose |
| Grade ≥ 4 AEs | 413 | 47 (11.4) | Gompertz | None |
| Grade ≥ 2 diarrhea | 413 | 59 (14.3) | Weibull | Dose rate |
| Grade 3 HFSRs | 413 | 47 (11.4) | Constant + Gompertz | Average dose |
| Grade ≥ 3 hypertension | 413 | 24 (5.8) | Constant | AUC12AV |
AE, adverse event; AUC12AV, average area under the curve during the dosing interval of 12 hours (exposure); HFSRs, hand‐foot skin reactions.
Selected exposure covariate that influences the hazard significantly (P < 0.01).
Figure 1.
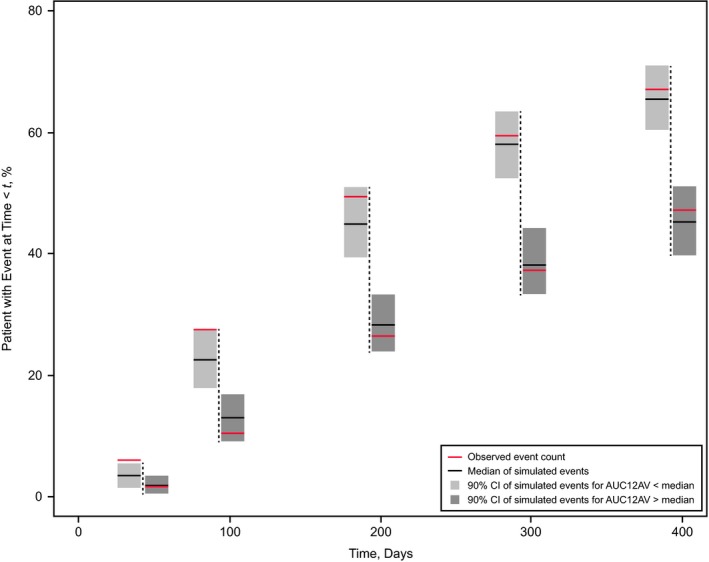
Qualification of the Weibull hazard model for tumor progression by stratified prediction‐corrected visual predictive check (pc‐VPC). The pc‐VPC shows 5 prediction bins at 50, 100, 200, 300, and 400 days (vertical dashed lines) after start of sorafenib treatment. For each time bin, light gray and dark gray fields represent the 90% prediction interval of the 1,000 simulated event rates for tumor progression in patients with average area under the curve (AUC) over the 12‐hour dosing interval during the entire treatment period (AUC12AV) below and above the population median, respectively. Black horizontal bars indicate the respective simulated medians, and red horizontal bars show the observed rates of tumor progression in the two strata. Event rates are calculated cumulatively up to the time of the bin. Each of the 1,000 simulated trials used all 401 patients with centrally assessed tumor progression measurements, of whom 204 received initially placebo. CI, confidence interval.
The Kaplan−Meier curve of the observed tumor progression rate during sorafenib treatment is shown in Figure 2 overlaid with Kaplan−Meier curves from 1,000 simulations of three different continuous dosing regimens. These plots show that the risk of tumor progression increases with each decrease in dose rate from 800 to 600 and 400 mg/day. This is supported by the HRs for the comparison of continuous dosing with 800 vs. 600 mg/day (HR, 0.814; 95% CI, 0.752–0.881) and 400 mg/day (HR, 0.638; 95% CI, 0.537–0.758), and with 600 vs. 400 mg/day (HR, 0.783; 95% CI, 0.713–0.860).
Figure 2.
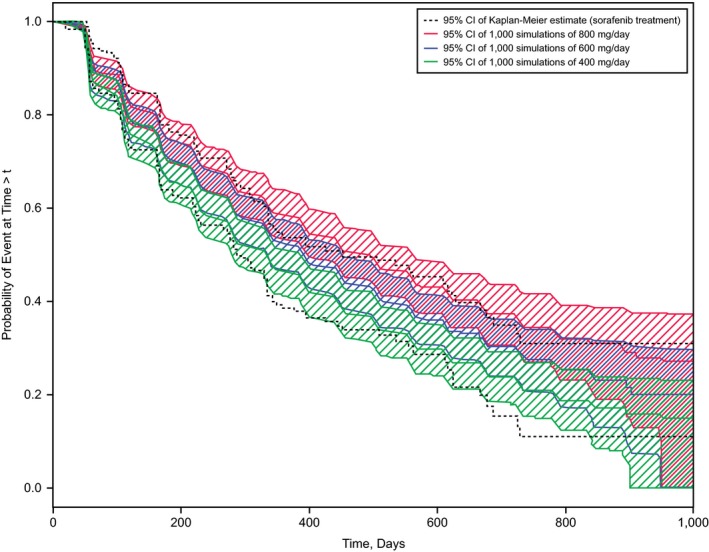
Observed and simulated tumor progression rates. Kaplan−Meier plots of the observed tumor progression events are shown as 95% confidence intervals (CIs; dashed lines). The colored bands represent the 95% CIs of simulated Kaplan−Meier plots for the dose rates described in the key.
The simulated impact of the four protocol‐specified dose‐reduction strategies implemented after two complete cycles of sorafenib 800 mg/day treatment is shown in Figure 3. The broadly overlapping plots for dose‐reduction schemes from 800 to 600 mg/day (group 1) or 400 mg/day (group 2) and for temporary dose reductions with re‐escalation to 800 mg/day (groups 3 and 4) indicate that such dose reductions that begin after two complete cycles of sorafenib 800 mg/day have limited influence on tumor progression.
Figure 3.
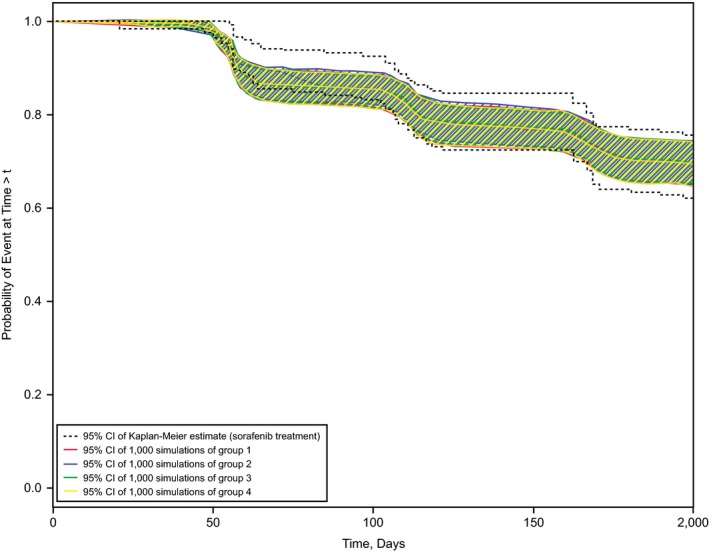
Effect of dose‐reduction schemes on tumor progression rates. Kaplan−Meier plots of the observed tumor progression events are shown as 95% confidence intervals (CIs; dashed lines). The colored lines represent the 95% CIs of simulated Kaplan−Meier plots for the dose‐reduction schemes described in the key.
AEs
Among the 413 patients included in the safety analysis, the incidences of grade ≥ 3 and grade ≥ 4 AEs were 62.7% and 11.4%, respectively, and the incidences of grade ≥ 2 diarrhea, grade 3 HFSRs, and grade ≥ 3 hypertension were 14.3%, 11.4%, and 5.8%, respectively (Table 2). The incidences of grade ≥ 3 AEs, grade ≥ 2 diarrhea, and grade 3 HFSRs were significantly associated with covariates related to sorafenib dosing (average dose or dose rate) but not with sorafenib exposure (AUC12 and AUC12AV) in the hazard models (Table 2).
Qualification of the models was achieved through stratified pc‐VPCs. The selected covariate hazard model for grade ≥ 3 AEs predicted the AEs reasonably well in all timed bins for average doses above the median but underpredicted the event rate for average doses below the median (Figure 4 a). Therefore, the model was not considered to be qualified for simulations of alternative dose schedules for grade ≥ 3 AEs.
Figure 4.
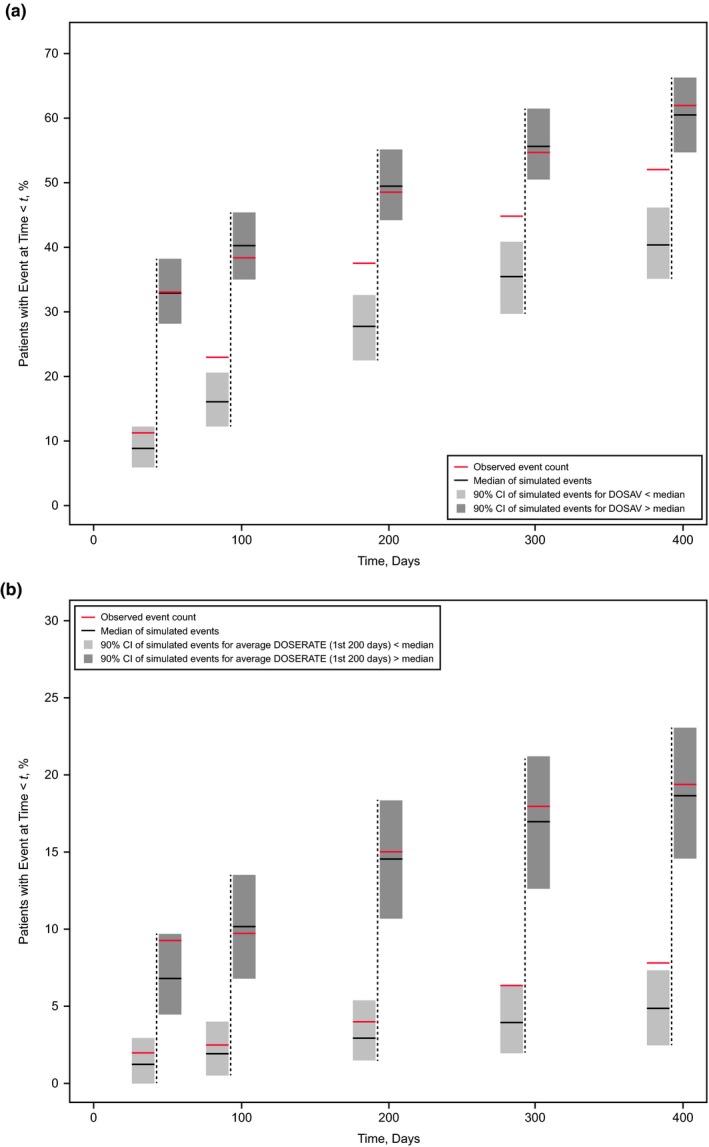
Qualification of the (a) Constant + Gompertz hazard model for adverse events (AEs) grade ≥ 3 and (b) Weibull hazard model for grade ≥ 2 diarrhea stratified by prediction‐corrected visual predictive check (pc‐VPC). The pc‐VPC shows 5 prediction bins at 50, 100, 200, 300, and 400 days (vertical dashed lines) after start of sorafenib treatment. For each time bin, light gray and dark gray fields represent the 90% prediction interval of the 1,000 simulated event rates for AEs in patients with average dose below and above the population median, respectively. Black horizontal bars indicate the respective simulated medians, and red horizontal bars show the observed rates of (a) grade ≥ 3 AEs and (b) grade ≥ 2 diarrhea events in the 2 strata. Event rates are calculated cumulatively up to the time of the bin. Each of the 1,000 simulated trials used all 413 patients, of whom 207 initially received placebo; 155 of these 207 patients (75%) were crossed over to sorafenib after disease progression. DOSAV, distribution of the average sorafenib dose.
The hazard for a grade ≥ 2 diarrhea event was influenced by dose rate. The stratified pc‐VPC (Figure 4 b) demonstrated that for up to 200 days of treatment, a dose rate above the population median is associated with a higher event rate than a time‐averaged dose rate below the population median. As the pc‐VPC showed that the agreement between predicted and observed event rates was good (i.e., the observed percentage of patients with an event falls into the 90% CI of the model predictions for 4 of 5 time bins), simulations for lower constant dosing rates were performed (Figure 5 a).
Figure 5.
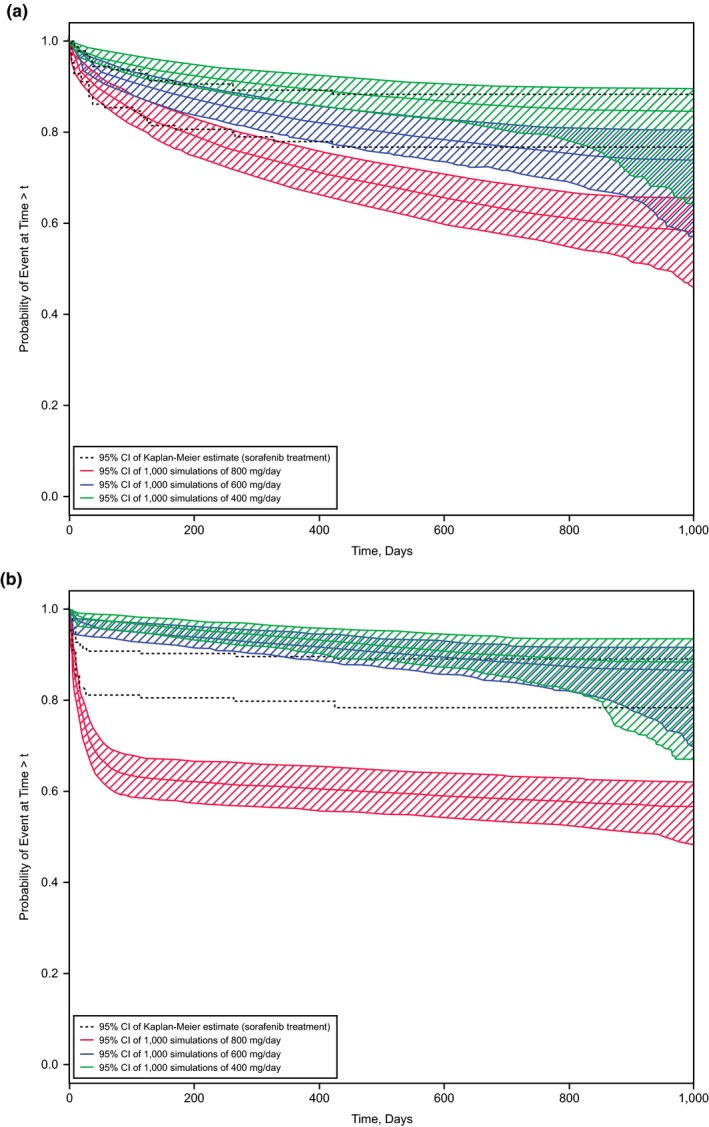
Observed and simulated risk of adverse events. Kaplan−Meier plots of the observed risk are shown by 95% confidence intervals (CIs; dashed lines). The colored bands represent the 95% CIs of simulated Kaplan−Meier plots for the doses described in the keys. (a) Grade ≥ 2 diarrhea. (b) Grade 3 hand‐foot skin reactions.
The hazard model for a grade 3 HFSR event was influenced by the average dose. The stratified pc‐VPC (Figure S4 a) demonstrated that an average dose above the population median is associated with a higher event rate than an average dose below the population median. The agreement between predicted and observed event rates was good; therefore, simulations for lower constant average doses were performed (Figure 5 b). The hazard for grade ≥ 4 AEs was not influenced by any exposure covariate. The pc‐VPC (Figure S4 b) demonstrated good agreement between predicted and observed event rates.
The hazard for grade ≥ 3 hypertension AEs was influenced by AUC12AV. The stratified pc‐VPC (Figure S4 c) demonstrated that AUC12AV above the population median was associated with a lower event rate than AUC12AV below the population median. No simulations for lower constant dosing rates were performed for this counterintuitive effect of exposure on event rate of hypertension.
None of the four protocol‐specified dose‐reduction strategies that were implemented after complete cycles of treatment with sorafenib 800 mg/day had an influence on the incidence of grade ≥ 2 diarrhea or grade 3 HFSR indicating that any dose reduction that occurs after two complete cycles of treatment with sorafenib 800 mg/day is likely to have no or only a minimal influence on the incidence of these AEs.
Discussion
PK analysis
It is tempting to speculate about the underlying cause of the higher sorafenib exposure in the DECISION trial compared with other studies. Obvious external reasons like different assays or different formulations have been investigated and can be ruled out. The comparison of the available PK data from all eight studies in patients with cancer (Figure S1 ) illustrates that there were also patients with HCC and RCC who had unusually high plasma concentrations. Furthermore, as the single PK sample was obtained in the DECISION trial after unsupervised drug intake, it cannot be ruled out that patients may have taken their drugs shortly before visiting the clinic but recorded intake to be earlier. This could result in higher drug exposure than expected on the basis of the recorded time after dose.
The simplification of the current model compared with the population PK model by Jain et al.10 was necessary because the current model focuses on sparse PK data from patients with cancer treated with sorafenib. A protracted absorption process here accounts for the enterohepatic recycling represented in the Jain model. We show through pc‐VPC displays that our simplified model represents the time course over the entire 12‐hour dosing interval correctly and without bias (Figure S3 ). In addition, the model correctly predicts the higher plasma exposure in the DECISION study (Table 1).
Exposure–response analysis
The exposure–response analysis of sorafenib in patients with DTC from the DECISION trial used a parametric modeling approach rather than Cox proportional hazard modeling typically used in oncology.14 Parametric modeling was chosen based on the objective of this analysis: to offer guidance to physicians when their patient with thyroid cancer experiences AEs thought to be caused by sorafenib treatment. To simulate the outcome of various changes to the standard sorafenib dosing schedule, parametric hazard models are required. The models presented here passed qualification through stratified pc‐VPCs before they were used in outcome simulations from alternative schedules.
Outcome modeling was performed in two stages. First, a baseline hazard model was selected from a range of candidate models. Then, the influence of exposure on the hazard was tested by including the selected exposure covariates in the hazard model as continuous variables. The stratified pc‐VPCs, however, were only stratified by exposure above and below the population median. Although it would have been more informative if pc‐VPCs allowed the direct comparison between observed and model‐simulated events under the influence of a continuous variable, such detailed qualification would have required the subdivision of exposure into more than two categories with fewer subjects and wider CIs, and the resulting display would have been less informative.
Although dosing‐related exposure parameters could be obtained directly from the clinical database, the AUC12 measures for 156 patients were obtained from a single PK measurement, the patient's sex, the individual dosing history, and the individual post hoc estimates of the PK parameters derived from the population PK model. There were no PK data for 41 patients randomized to active treatment and for 155 of 204 patients initially randomized to placebo and later switched to active treatment after disease progression, and for these patients we predicted AUC12 solely on the basis of the population PK model with sex as a covariate.
We modeled the hazard of tumor progression, which is equivalent to PFS. In the DECISION trial, there is an incident rate of 65% for tumor progression and 63% for AE grade ≥ 3. Incidence rates > 30% favor, in general, parametric hazard modeling. Conversely, we also modeled AE grade ≥ 4, diarrhea grade ≥ 2, HFSR grade 3, and hypertension grade ≥ 3 with incidence rates of 11%, 14%, 11%, and 6%, respectively. The lower rates of more severe AEs were deliberately accepted to approach the situation when a physician considers a dose reduction. Consequently, we found no exposure influence on AE grade ≥ 4 and a counterintuitive influence of AUC12AV on hypertension grade ≥ 3. The latter may have been caused by the first hypertension grade ≥ 3 event occurring in some patients receiving placebo after disease progression (i.e., after their switch to active treatment). In such patients, the AUC12AV would be particularly small because of the initial placebo period with zero sorafenib exposure. There was also a general trend (Figure S5 ) for lower sorafenib doses and exposures the longer a patient stayed on treatment without disease progression. This trend may be the underlying reason for the counterintuitive relationship with hypertension. On the other hand, AE grade ≥ 3 is least expected to be affected by this general trend because no clinical decision of lowering the dose was made before the first occurrence of AE grade ≥ 3.
A high level of scrutiny was applied before a model was used in simulations. For example, in the hazard model for AE grade ≥ 3, the influence of average dose leading up to the event was statistically significant but the stratified pc‐VPC (Figure 4 a) showed the simulated event rate for average dose below the median to be consistently lower than the observed event rate in that stratum of patients. On the basis of this plot, the model with its covariate effect was not qualified to give guidance through simulations of alternative dosing schedules. Conversely, the close resemblance among stratified, observed, and estimated event rates for grade ≥ 2 diarrhea (Figure 4 b) led to the conclusion that this model was qualified for simulations of alternative dosing schedules.
In clinical studies, patients are typically randomized based on treatment and other potentially confounding factors that might also have an effect on the outcome but not according to exposure. Hence, there could be differences in exposure among these confounding factors, which could potentially bias the exposure–response analysis if confounding factors are not considered in a multivariate analysis. To investigate the potential limitation of our monovariate analysis, we investigated the association between exposure variables and the prognostic factors, histology and age, which were found to be significantly associated in the multivariate analysis of PFS in the DECISION trial2; exposure was equally distributed among these factors (Figure S6 ). The relationships we found are, therefore, unlikely to be confounded by underlying associations with influential clinical baseline covariates. As a comparison, we refer to the exposure–response analysis for ado‐trastuzumab emtansine15 in which the distribution of some baseline covariates was clearly different among ado‐trastuzumab emtansine exposure quartiles, which necessitated a multivariate analysis.
There is an additional question behind the current simulations: do patients who experience AEs later benefit from longer PFS? This question cannot be addressed by the methodology presented here, as this would require an assessment of competing risk in which also right‐censoring is opened up into relevant categories (e.g., death, dropout due to AE, or end of observation period). Although methodologies are available to consider competing risk in the analysis of survival data16 and progress is being made in the methodological development of its use in parametric exposure‐response analyses,16 further development is needed. This was beyond the scope of this paper.
In conclusion, we present two models for AEs (diarrhea grade ≥ 2 in Figure 5 a and HFSR grade 3 in Figure 5 b) where incidence rates could be reduced with sorafenib b.i.d. schedules that started with 400 or 600 mg/day instead of the standard 800 mg/day. However, a change to lower starting doses would not be in the best interest of the patient, as demonstrated in Figure 2, which shows shorter expected PFS for sorafenib doses lower than 800 mg/day. As a compromise, we simulated PFS for various dose‐reduction schedules, all starting with 2 cycles of 800 mg/day. The simulation in Figure 3 suggests that the therapeutic benefit of sorafenib in patients with DTC would be preserved, provided treatment started with 2 cycles of 800 mg/day before any dose reduction.
Funding
The study was supported by Bayer HealthCare Pharmaceuticals and Onyx (a wholly owned subsidiary of Amgen). Scientific writing was funded by Bayer HealthCare Pharmaceuticals.
Conflict of Interest
J.L., F.H., G.M., C.P., and B.P. are employees of Bayer. F.H., C.P., and B.P. own stock in Bayer. M.B. and M.S. were (principle) investigators of the DECISION study. Both have received consultancy fees and research support from Bayer. J.G., G.J., and R.A. are employed by BAST Inc. Ltd., a Contract Research Organization under contract with Bayer. N.H.P. is employed by qPharmetra, LLC, a Contract Research Organization under contract with Bayer. D.M. is employed by Mitchell Pharmaceutical Consulting, a CRO under contract with Bayer. Employees of BAST qPharmetra and Mitchell Pharmaceutical Consulting are barred from owning stock in client companies.
Author Contributions
All authors wrote the manuscript. J.G., G.J., R.A., J.L., G.M., C.P., B.P., N.P., D.M., and F.H. designed the research. J.G., G.J., R.A., J.L., G.M., C.P., B.P., N.P., D.M., and F.H. performed the research. J.G., G.J., R.A., J.L., G.M., C.P., B.P., N.P., D.M., and F.H. analyzed the data. M.B. and M.S. contributed new reagents/analytical tools.
Supporting information
Table S1. Studies included in the population pharmacokinetic analysis.
Table S2. Patient demographics and baseline clinical characteristics* by type of study participant.
Table S3. Final covariate model parameter estimates.
Figure S1. Sorafenib plasma concentration vs. time at steady state in patients with hepatocellular carcinoma.
Figure S2. Sorafenib final covariate pharmacokinetic model equation.
Figure S3. Visual predictive check of the final covariate pharmacokinetic model.
Figure S4. Qualification of the (a) Constant + Gompertz hazard model for grade 3 hand‐foot skin reactions (HFSRs), (b) Gompertz hazard model for grade ≥ 4 adverse events (AEs), and (c) constant hazard model for grade ≥ 3 hypertension stratified by prediction‐corrected visual predictive check (pc‐VPC).
Figure S5. (a) Correlation between the average sorafenib exposure over the 12‐hour dosing interval during the entire treatment period (AUC12AV) and the estimated individual clearance normalized to a unity dose. (b) General trend of the average AUC12 in each cycle (mean_AUC_12) over time.
Figure S6. Distribution of the average sorafenib dose (DOSAV), the average sorafenib exposure over the 12‐hour dosing interval during the entire treatment period (AUC12AV), and the average sorafenib exposure in cycle 1 (AV_AUC_12) among (a) differentiated thyroid cancer histology categories (papillary, follicular‐Hürthle cell, follicular—other and poorly differentiated) and (b) age quartiles.
Figure S7. Schematic representation of the four simulation scenarios.
Acknowledgments
Scientific writing support was provided by Lisa M. Klumpp Callan, PhD, and Alan J. Klopp, PhD, CMPP, of C4 MedSolutions, LLC (Yardley, PA), a CHC Group company.
References
- 1. Nexavar (sorafenib) . Full Prescribing Information. (Bayer HealthCare Pharmaceuticals, Inc., Whippany, NJ, 2015). [Google Scholar]
- 2. Brose, M.S. et al Sorafenib in radioactive iodine‐refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double‐blind, phase 3 trial. Lancet 384, 319–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Escudier, B. et al Sorafenib in advanced clear‐cell renal‐cell carcinoma. N. Engl. J. Med. 356, 125–134 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Llovet, J.M. et al Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 5. Bastholt, L. et al Population PK modeling and exposure‐response analyses of sorafenib in patients with radioactive iodine‐refractory differentiated thyroid cancer (RAI‐rDTC) in the phase III DECISION trial [abstract 6061]. J. Clin. Oncol. 32, 5s (2014). [Google Scholar]
- 6. Prins, N.H. , Grevel, J. , Mitchell, D. , Lettieri, J. , Ploeger, B.A. & Pena, C. Population pharmacokinetics of sorafenib: a meta‐analysis of patients with hepatocellular carcinoma, renal cell carcinoma, and differentiated thyroid carcinoma [abstract]. J. Pharmacokinet. Pharmacodyn. 41, s23 (2014). [Google Scholar]
- 7. Brose, M.S. et al Rationale and design of decision: a double‐blind, randomized, placebo‐controlled phase III trial evaluating the efficacy and safety of sorafenib in patients with locally advanced or metastatic radioactive iodine (RAI)‐refractory, differentiated thyroid cancer. BMC Cancer 11, 349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 9. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 10. Jain, L. et al Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharmacol. 72, 294–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holford, N. A time to event tutorial for pharmacometricians. CPT Pharmacometrics Syst. Pharmacol. 2, e43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grevel, J. Time to event modelling of competing risks‐ Rcode for VPC (2018) repository.ddmore.eu/model/DDMODEL00000243.
- 14. Lu, D. et al A survey of new oncology drug approvals in the USA from 2010 to 2015: a focus on optimal dose and related postmarketing activities. Cancer Chemother. Pharmacol. 77, 459–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang, J. et al Exposure‐response relationship of T‐DM1: insight into dose optimization for patients with HER2‐positive metastatic breast cancer. Clin. Pharmacol. Ther. 95, 558–564 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Austin, P.C. , Lee, D.S. & Fine, J.P. Introduction to the analysis of survival data in the presence of competing risks. Circulation 133, 601–609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Studies included in the population pharmacokinetic analysis.
Table S2. Patient demographics and baseline clinical characteristics* by type of study participant.
Table S3. Final covariate model parameter estimates.
Figure S1. Sorafenib plasma concentration vs. time at steady state in patients with hepatocellular carcinoma.
Figure S2. Sorafenib final covariate pharmacokinetic model equation.
Figure S3. Visual predictive check of the final covariate pharmacokinetic model.
Figure S4. Qualification of the (a) Constant + Gompertz hazard model for grade 3 hand‐foot skin reactions (HFSRs), (b) Gompertz hazard model for grade ≥ 4 adverse events (AEs), and (c) constant hazard model for grade ≥ 3 hypertension stratified by prediction‐corrected visual predictive check (pc‐VPC).
Figure S5. (a) Correlation between the average sorafenib exposure over the 12‐hour dosing interval during the entire treatment period (AUC12AV) and the estimated individual clearance normalized to a unity dose. (b) General trend of the average AUC12 in each cycle (mean_AUC_12) over time.
Figure S6. Distribution of the average sorafenib dose (DOSAV), the average sorafenib exposure over the 12‐hour dosing interval during the entire treatment period (AUC12AV), and the average sorafenib exposure in cycle 1 (AV_AUC_12) among (a) differentiated thyroid cancer histology categories (papillary, follicular‐Hürthle cell, follicular—other and poorly differentiated) and (b) age quartiles.
Figure S7. Schematic representation of the four simulation scenarios.