

Abstract
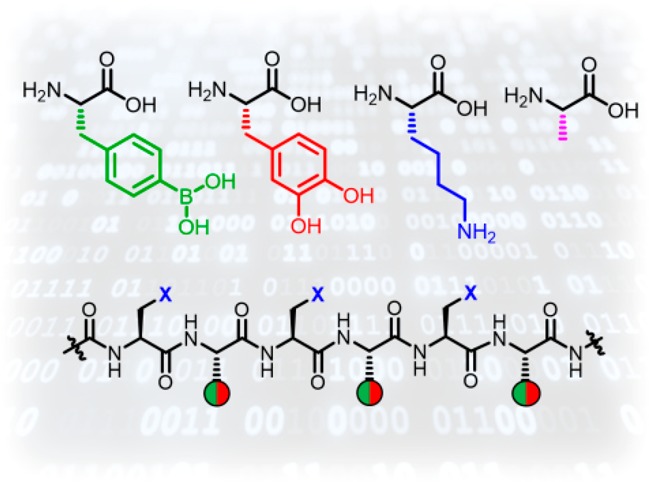
The development of a synthetic code that enables a sequence programmable feature like DNA represents a key aspect toward intelligent molecular systems. We developed herein the well-known dynamic covalent interaction between boronic acids (BAs) and catechols (CAs) into synthetic nucleobase analogs. Along a defined peptide backbone, BA or CA residues are arranged to enable sequence recognition to their complementary strand. Dynamic strand displacement and errors were elucidated thermodynamically to show that sequences are able to specifically select their partners. Unlike DNA, the pH dependency of BA/CA binding enables the dehybridization of complementary strands at pH 5.0. In addition, we demonstrate the sequence recognition at the macromolecular level by conjugating the cytochrome c protein to a complementary polyethylene glycol chain in a site-directed fashion.
Molecular interactions in Nature are often involved in a complex network of energy landscapes where individual components, from small molecules to organelles, form transient systems on demand.1 Proteins have their origin within the genome, where molecular information is encoded and translated based on precise complexation and spatially controlled chemistry. In this respect, there has been much progress in the creation of artificial DNA prototypes to program molecular functions,2,3 adapting both supramolecular4−8 and dynamic covalent interactions.9−12 Despite these successes in pursuit of artificial life, the advent of synthetic chemistry in this field remains far slower than their biochemistry counterparts.13−16
The development of synthetic DNA-type systems is deceptively challenging. On the sequence level, it is necessary that the interacting motifs are small so that both sterical demand and synthesis are not compromised by increasing sequence length. Molecularly, the binding event should be selective for complementary sequences while remaining dynamic and exchangeable in mild aqueous conditions. At first glance, synthetic functional groups in supramolecular chemistry (e.g., cyclodextrin,17 curcubituril,18 ureido-pyrimidinones19) or dynamic covalent chemistry (e.g., spiropyran,20 diarylethenes21) seem to satisfy the above criteria. However, most motifs are either sterically bulky, offer too high binding constants already for the first binding event and/or they are laborious to be incorporated along a sequence.
Unlike most recognition motifs, the interaction between boronic acids (BAs) and electron rich vicinal diols such as catechols (CAs) fulfills these criteria well.22 The binding complex is equipped with (1) small spatial requirement, (2) fast binding kinetics and (3) pH-responsiveness within the physiological range (5.0–7.4).23 Hence, its dynamic covalent binding capabilities have been applied broadly in therapeutics,24 biosensors,25,26 stimulus responsive ligation tools27 and as self-regenerative materials.28,29 Herein, we propose that BA/CA motifs in a binary sequence would encode molecular recognition in a stimulus responsive fashion.
The permutation between binary “1/0” events and the length of the backbone defines the coding space (Figure 1). Inspired by peptide nucleic acids, a peptide backbone was designed to ensure greater hydrolytic stability.30 Moreover, existing peptide synthesis methodologies facilitate the preparation of longer sequences, installation of customizable end groups and flexibility of spacer groups, i.e., lysine or alanine to control solubility, steric demand or surface charges. As a demonstration in a broader context, a protein (cytochrome c) and a polymer (polyethylene glycol) were conjugated via these dynamic covalent tags to demonstrate recognition specificity at the macromolecular level.
Figure 1.
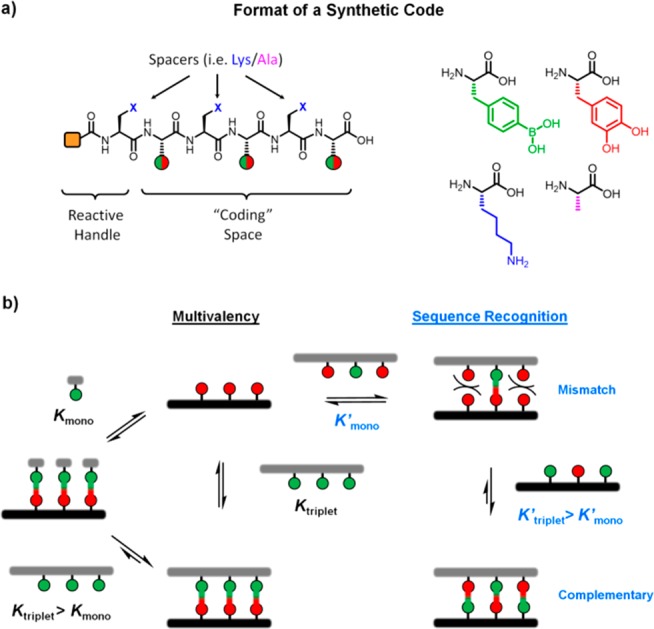
(a) Peptide scaffold to code a binary BA/CA pairing. (b) Thermodynamic processes involved in multivalent effects, complementary and mismatched sequences.
The unnatural amino acid 7 containing the BA was synthesized based on a protocol for iodo-phenylalanine (Scheme 1).31l-Phenylalanine was iodinated to afford 1, with the carboxylic acid and the amine groups protected by the methyl ester (2) and the boc group (3), respectively. Suzuki–Miyaura borylation was conducted to install the BA pinacol ester moiety (4) with a subsequent global deprotection to yield the boronated l-phenylalanine 5. Here, a switch in protecting groups is necessary because the pinacol ester is sensitive to microwave conditions. Additionally, longer sequences containing multiple BA cause aggregation on the solid phase. Thus, after the installation of the fmoc moiety (6), the BA was protected with a highly bulky pinanediol to afford 7. The corresponding CA containing amino acid, dihydroxy-l-phenylalanine, was commercially available.
Scheme 1. Synthesis of BA Containing Amino acid 7 and the Chemical Structures of a Small Library of BA/CA Peptide Codes.

Peptide sequences containing all permutations of BA and CA in a hexa/octa-peptide format were synthesized to elucidate the influence of sequence, length and positional defects. The amino acids that do not participate in the coding segment are filled by lysines (X) to improve water solubility. To demonstrate the chemical versatility, the N-terminus was modified with reactive functionalities, i.e., amine, thiol or maleimide (Scheme 1). In this way, oligopeptides containing one, two and three BA [(AX)1, (AX)2, (AX)3] as well as their complementary CA counterparts [(BX)1, (BX)2, (BX)3] were synthesized. The binding affinities of the dynamic covalent interactions between (AX)1-(BX)1, (AX)2-(BX)2, (AX)3-(BX)3 were evaluated by fluorescence microscale thermophoresis in 300 mM phosphate buffer, pH 7.4 (Figure 2a).
Figure 2.

Characterization of multivalent binding of complementary BA/CA codes. (a) Binding affinity determination of (AX)1-(BX)1 (monovalent, blue), (AX)2-(BX)2 (divalent, red) and (AX)3-(BX)3 (trivalent, black) by fluorescence microscale thermophoresis in 300 mM phosphate buffer, pH 7.4. (b) 1H NMR and (c) 1H NOESY of (AX)3, (BX)3 and (AX)3-(BX)3 complex in 300 mM phosphate buffer, pH 7.4, 9:1 (H2O:D2O), 298 K. (d) DOSY of the (AX)3-(BX)3 complex against (BX)3 in 300 mM phosphate buffer, pH 7.4, 9:1 (H2O:D2O), 298 K. (e) MALDI-TOF of (AX)3-(BX)3 at pH 7.4 (top) and pH 5.0 (bottom). (f) Circular dichroism spectroscopy of (AX)3-(BX)3 and their separate components at phosphate buffer pH 7.4, 298 K.
In each series, fluorescein labeled BA peptides act as the template strand and are titrated with their complementary CA peptides. For a single BA/CA binding event, (AX)1-(BX)1, a binding affinity of 1300 ± 300 M–1 was observed, which is consistent with published data.32 By increasing the binding event to divalent (AX)2-(BX)2 and trivalent (AX)3-(BX)3, a respective 10-fold (12 500 ± 1100 M–1) and 70-fold (81 400 ± 7300 M–1) increase were observed. Importantly, the absence of binding errors to form unstructured aggregates was supported by the defined fluorescence decay and dynamic light scattering (Figure S4). The binding of (AX)3-(BX)3 was also confirmed independently by Förster resonance energy transfer (FRET) (Figure S5). The lesser increase in binding affinity with each subsequent binding code suggests that energy is required to compensate the backbone structure in the bound state. By bringing the findings into perspective of DNA hybridization, the binding affinity of (AX)3-(BX)3 is comparable to about eight base pairs (with 50% G-C content) on a DNA level.33,34
The binding structure of the (AX)3-(BX)3 complex was characterized by multidimensional 1H NMR spectroscopy. The binding of the boronic acids and catechols leads to the complete transformation of the chemical environment, shifting the 1H aromatic signals of the components (Figure 2b, S6). Resonance peaks of the boronic acids 1HAR in the total correlation spectroscopy (TOCSY) were shifted into high-field confirming that the boron center becomes less electron withdrawing upon binding (Figures S7 and S8). Nuclear Overhauser effect spectroscopy (NOESY) analysis of (AX)3-(BX)3 shows many additional and shifted intramolecular through space 1H-couplings compared to the separate components (Figures 2c, S9 and S10). These new interactions ascertain the formation of chemical environments that increase the intramolecular through space interactions between the aromatic groups and the Hα, Hβ of the boronic acid or catechols. Diffusion ordered NMR (DOSY) confirms the binding event as an increase in the diffusion time of the complex (Figures 2d and S11). Additionally, the monovalent complexes are stable to heat up to 70 °C at the using variable temperature NMR (Figures S15–S19).
The observed BA–CA interaction was supported independently by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS, where the formation of (AX)3-(BX)3 was characterized by an m/z value at 2079.01 at pH 7.4 (Figure 2e, top). Noteworthy, significant fragmentation of the complex occurs due to the acidic nature of the matrix (α-cyano-4-hydroxycinnamic acid, α-CHCA). At pH 5.0, only separate components of (AX)3 and (BX)3 were found (Figure 2e, bottom). In the Fourier-transform infrared (FTIR) of (AX)3-(BX)3, the vibrational mode at 1289 cm–1 was lost while a new peak was observed at 1495 cm–1, suggesting an increase in bond energy and C=C character for the dynamic covalent interaction (Figure S20).35,36 The electron deficient boron withdraws electrons through the vicinal diols, which reduces the electron density of the catechol aromatic system.
Circular dichroism spectroscopy found that, at pH 7.4, the (AX)3-(BX)3 complex shows a strong negative molar ellipticity at 210 nm, corresponding to the n-π* transition of the carbonyl group (Figure 2f, S21). Together with the absence of Cotton effects, these observations imply that the tetrahedral boron center has a strong through-space effect on the C=O bond. Density functional theory (DFT) calculations confirmed that hydrogen bonding interactions between the hydroxyl groups of the tetrahedral boron and the carboxyl groups of lysines contribute to lowering the electronic energy of (AX)3-(BX)3 (Figures 3a, S23 and S24). The combination of these observations seems to corroborate the shifts associated with the TOCSY, NOESY cross-peaks. In contrast, at pH 5.0, there is no interaction between the complementary sequences (Figure S21). Here, DFT calculations using a simplified structure revealed a low activation energy (10 kcal mol–1) for the first boron–oxygen (catechol) bond breaking step, representing the fast hydrolysis of the tetrahedral boron observed at acidic pH (Figure S26).
Figure 3.

(a) Model of the DFT optimized structure of (AX)3-(BX)3 at (B3LYP/6-31G(d) theory level (see Supporting Information). (b) Displacement reaction on a fluorescein-(AX)3 template: Monovalent (BX)1 was titrated to achieve maximum binding (fluorescence, blue) followed by the addition of increasing amounts of Dylight650-(BX)3 to displace (BX)1 (FRET, black). (c) Sequence dependent discrimination between complementary AAYA-BYBB and mismatching (AX)3-BYBB peptide based on titration. (d) Quantification of binding of PEG5000-(AX)3 with CytC-(BX)3 at 100 μM using Alizarin Red S assay. (e) Characterization of the recognition by CytC-(BX)3 (13 886 g mol–1) with PEG5000-(AX)3 (6468 g mol–1) yielding PEG5000-(AX)3(BX)3-CytC (calcd. MW: 20 036 g mol–1, detected MW 20 243 g mol–1, matrix CHCA) using MALDI-TOF MS.
Interestingly, sequences containing consecutive BA/CA residues without the alternating spacers (A3X3, B3X3) do not bind to their complementary partners ≤500 μM (Table 1, Figure S22, S27). Above this concentration, precipitation of A3X3 occurs, indicating the importance of the lysine spacer in the alternating (AX) arrangement providing both solubility and relief of steric constraints for the binding event.
Table 1. Summary of Association Constants of Different Matching/Mismatching Sequences.
| Binding Partner | Ka (M–1) | Type | Binary Code |
|---|---|---|---|
| (AX)1-(BX)1 | 1300 ± 300 | Monovalent | 1 |
| (AX)2-(BX)2 | 12 500 ± 1100 | Divalent | 11 |
| (AX)3-(BX)3 | 81 400 ± 7300 | Trivalent | 111 |
| A3X3-B3X3 | <2000 | No Spacer | 111 |
| ABA-BAB | 79 400 ± 5200 | Mixed Trivalent | 111 |
| (BX)3-BAB | – | – | Mismatch |
| AAYA-BYBB | 21 100 ± 6100 | Divalent (Spaced) | 1101 |
| (AX)3-BYBB | 9400 ± 800 | Divalent | Mismatch |
Next, we encoded a sequence specific binding event by using (1) mixed sequences (ABA and BAB) and (2) inclusion of a nonbinding event Y. Importantly, mixed sequences contain partially complementary parts, whereas the nonbinding event provides an “error” in the sequence. We observed that the ABA-BAB interacts with a binding affinity of 79 400 ± 5200 M–1, which is comparable to the homogeneous (AX)3-(BX)3 pairs (Table 1). However, the interaction took significantly longer (about 8 h) suggesting that error correction requires a certain time frame, similar to DNA.37,38 On the other hand, sequence hybridization can be weakened correspondingly by AAYA-BYBB, where Y is a nonbinding, noninteracting amino acid (alanine). The increase in the chain length also increases the energy needed to compensate, resulting in a lower binding affinity (21 100 ± 6100 M–1) to a divalent level. Mismatched partners such as (AX)3-BYBB further weaken complementarity. Taking another mismatch pair of identical length, BAB-(BX)3, nonbinding residues appear in 1H NMR (Figure S12), indicating very weak interactions.
Since strand displacement dynamics is an important tool in DNA nanotechnology,38−40 we investigate the displacement dynamics of our conjugate by the binding of monovalent (BX)1 and a trivalent (BX)3 against a (AX)3 template. The fluorescein-(AX)3 was first titrated with (BX)1 until binding saturation (Figure 3b, black). Displacement and sorting was then achieved by titrating the (AX)3-(BX)1 against Dylight650-(BX)3, monitored independently by FRET. The intersection between the measurements indicates a stoichiometric ratio where (BX)1 and (BX)3 can competitively displace each other by at least 50% (Figure 3b). For (BX)1 to displace (BX)3 by 50%, a stoichiometric factor of >4000 mol % is required. On the other hand, (BX)3 would only require >2.5 mol % due to its multivalent effect.
Based on these findings, the established binary codes could be used to enable recognition of macromolecules, i.e., proteins and polymers. As PEGylation of proteins remain an important aspect in protein therapeutics,41,42 we functionalized PEG5000 and yeast cytochrome c (CytC) with (AX)3 and (BX)3, respectively. The conversion to PEG5000-(AX)3(BX)3-CytC was quantified by a fluorogenic sensor, Alizarin Red S in a titration assay (Figure 3d). The construct was characterized additionally with MALDI-TOF MS albeit with partial dissociation of the construct due to the acidic matrix (Figure 3e). Additionally, topological height increases due to complexation was visualized by atomic force microscopy (Figure S19).43
In summary, we have demonstrated, to the best of our knowledge, the first application of boronic acid chemistry in molecular sequence programming under physiological conditions. By combining the recognition and binding customization of boronic acid/catechol chemistry with a peptide scaffold, synthetic binary codes were created, in which binding and dynamic displacement was directed in a predictable and logical manner. In addition, the pH responsiveness of the chemistry adds an extra trigger to control the hybridization process. Assisted by the burgeoning field of site-selective protein chemistry,44 the application of complementary sequences to the protein cytochrome c and PEG5000 further underlines the potential to create defined and dynamic macromolecular assemblies. We envision that the technology provides a synthetic platform for programming macromolecular architectures to possess complex dynamic features.
Acknowledgments
The project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 316249678 – SFB 1279 (C01) and Project number: 318290668 – SPP 1923 as well as by an ERC Synergy Grant under grant agreement No. 319130 (BioQ). M. M. Zegota thanks the Marie Curie International Training Network Protein Conjugates for a research scholarship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b03107.
Materials and methods, characterization techniques and instrumentation as well as supporting figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Grzybowski B. A.; Huck W. T. S. The nanotechnology of life-inspired systems. Nat. Nanotechnol. 2016, 11, 585–592. 10.1038/nnano.2016.116. [DOI] [PubMed] [Google Scholar]
- Hirao I.; Kimoto M.; Yamashige R. Natural versus Artificial Creation of Base Pairs in DNA: Origin of Nucleobases from the Perspectives of Unnatural Base Pair Studies. Acc. Chem. Res. 2012, 45, 2055–2065. 10.1021/ar200257x. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Lehn J.-M.; Hirsch A. K. H. Molecular Biodynamers: Dynamic Covalent Analogues of Biopolymers. Acc. Chem. Res. 2017, 50, 376–386. 10.1021/acs.accounts.6b00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J. A.; Iadevaia G.; Hunter C. A. H-Bonded Duplexes based on a Phenylacetylene Backbone. J. Am. Chem. Soc. 2018, 140, 11526–11536. 10.1021/jacs.8b08087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stross A. E.; Iadevaia G.; Núñez-Villanueva D.; Hunter C. A. Sequence-Selective Formation of Synthetic H-Bonded Duplexes. J. Am. Chem. Soc. 2017, 139, 12655–12663. 10.1021/jacs.7b06619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B. Molecular Duplexes with Encoded Sequences and Stabilities. Acc. Chem. Res. 2012, 45, 2077–2087. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cao R.; Shen J.; Detchou C. S. F.; Zhong Y.; Wang H.; Zou S.; Huang Q.; Lian C.; Wang Q.; Zhu J.; Gong B. Hydrogen-Bonded Duplexes with Lengthened Linkers. Org. Lett. 2018, 20, 1555–1558. 10.1021/acs.orglett.8b00283. [DOI] [PubMed] [Google Scholar]
- Makiguchi W.; Tanabe J.; Yamada H.; Iida H.; Taura D.; Ousaka N.; Yashima E. Chirality- and sequence-selective successive self-sorting via specific homo- and complementary-duplex formations. Nat. Commun. 2015, 6, 7236. 10.1038/ncomms8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida H.; Ohmura K.; Noda R.; Iwahana S.; Katagiri H.; Ousaka N.; Hayashi T.; Hijikata Y.; Irle S.; Yashima E. Double-Stranded Helical Oligomers Covalently Bridged by Rotary Cyclic Boronate Esters. Chem. - Asian J. 2017, 12, 927–935. 10.1002/asia.201700162. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Sisco S. W.; Moore J. S. Homochiral Self-Sorting of BINOL Macrocycles. Chem. Sci. 2014, 5, 81–85. 10.1039/C3SC52018H. [DOI] [Google Scholar]
- Epstein E. S.; Martinetti L.; Kollarigowda R. H.; Carey-De La Torre O.; Moore J. S.; Ewoldt R. H.; Braun P. V. Modulating Noncovalent Cross-links with Molecular Switches. J. Am. Chem. Soc. 2019, 141, 3597–3604. 10.1021/jacs.8b12762. [DOI] [PubMed] [Google Scholar]
- Powell K. How biologists are creating life-like cells from scratch. Nature 2018, 563, 172–175. 10.1038/d41586-018-07289-x. [DOI] [PubMed] [Google Scholar]
- Göpfrich K.; Platzman I.; Spatz J. P. Mastering Complexity: Towards Bottom-up Construction of Multifunctional Eukaryotic Synthetic Cells. Trends Biotechnol. 2018, 36, 938–951. 10.1016/j.tibtech.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Hu S.; Chen X. Artificial cells: from basic science to applications. Mater. Today 2016, 19, 516–532. 10.1016/j.mattod.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman N. C.; Sleiman H. F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. 10.1038/natrevmats.2017.68. [DOI] [Google Scholar]
- Crini G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. 10.1021/cr500081p. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Kasera S.; Rowland M. J.; del Barrio J.; Scherman O. A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- Webber M. J.; Appel E. A.; Meijer E. W.; Langer R. Supramolecular biomaterials. Nat. Mater. 2016, 15, 13–26. 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- Klajn R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. 10.1039/C3CS60181A. [DOI] [PubMed] [Google Scholar]
- Kathan M.; Eisenreich F.; Jurissek C.; Dallmann A.; Gurke J.; Hecht S. Light-driven molecular trap enables bidirectional manipulation of dynamic covalent systems. Nat. Chem. 2018, 10, 1031–1036. 10.1038/s41557-018-0106-8. [DOI] [PubMed] [Google Scholar]
- Brooks W. L. A.; Sumerlin B. S. Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine. Chem. Rev. 2016, 116, 1375–1397. 10.1021/acs.chemrev.5b00300. [DOI] [PubMed] [Google Scholar]
- Akgun B.; Hall D. G. Boronic Acids as Bioorthogonal Probes for Site-Selective Labeling of Proteins. Angew. Chem., Int. Ed. 2018, 57, 13028–13044. 10.1002/anie.201712611. [DOI] [PubMed] [Google Scholar]
- Santos F. M. F.; Matos A. I.; Ventura A. E.; Gonçalves J.; Veiros L. F.; Florindo H. F.; Gois P. M. P. Modular Assembly of Reversible Multivalent Cancer-Cell-Targeting Drug Conjugates. Angew. Chem., Int. Ed. 2017, 56, 9346–9350. 10.1002/anie.201703492. [DOI] [PubMed] [Google Scholar]
- Wu X.; Li Z.; Chen X.-X.; Fossey J. S.; James T. D.; Jiang Y.-B. Selective sensing of saccharides using simple boronic acids and their aggregates. Chem. Soc. Rev. 2013, 42, 8032–8048. 10.1039/c3cs60148j. [DOI] [PubMed] [Google Scholar]
- Sun X.; James T. D.; Anslyn E. V. Arresting “Loose Bolt” Internal Conversion from – B(OH)2 Groups is the Mechanism for Emission Turn-On in ortho-Aminomethylphenylboronic Acid-Based Saccharide Sensors. J. Am. Chem. Soc. 2018, 140, 2348–2354. 10.1021/jacs.7b12877. [DOI] [PubMed] [Google Scholar]
- Seidler C.; Zegota M. M.; Raabe M.; Kuan S. L.; Ng D. Y. W.; Weil T. Dynamic Core–Shell Bioconjugates for Targeted Protein Delivery and Release. Chem. - Asian J. 2018, 13, 3474–3479. 10.1002/asia.201800843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler C.; Ng D. Y. W.; Weil T. Native protein hydrogels by dynamic boronic acid chemistry. Tetrahedron 2017, 73, 4979–4987. 10.1016/j.tet.2017.06.066. [DOI] [Google Scholar]
- Smithmyer M. E.; Deng C. C.; Cassel S. E.; LeValley P. J.; Sumerlin B. S.; Kloxin A. M. Self-Healing Boronic Acid-Based Hydrogels for 3D Co-cultures. ACS Macro Lett. 2018, 7, 1105–1110. 10.1021/acsmacrolett.8b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann E. Peptide nucleic acids (PNA) and PNA-DNA chimeras: from high binding affinity towards biological function. Biol. Chem. 1998, 379, 1045–1052. [PubMed] [Google Scholar]
- Lei H.; Stoakes M. S.; Schwabacher A. W.; Herath K. P. B.; Lee J. Efficient Synthesis of a Phosphinate Bis-Amino Acid and Its Use in the Construction of Amphiphilic Peptides. J. Org. Chem. 1994, 59, 4206–4210. 10.1021/jo00094a036. [DOI] [Google Scholar]
- Martínez-Aguirre M. A.; Villamil-Ramos R.; Guerrero-Alvarez J. A.; Yatsimirsky A. K. Substituent Effects and pH Profiles for Stability Constants of Arylboronic Acid Diol Esters. J. Org. Chem. 2013, 78, 4674–4684. 10.1021/jo400617j. [DOI] [PubMed] [Google Scholar]
- Kibbe W. A. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratilainen T.; Holmén A.; Tuite E.; Haaima G.; Christensen L.; Nielsen P. E.; Nordén B. Hybridization of Peptide Nucleic Acid. Biochemistry 1998, 37, 12331–12342. 10.1021/bi9808722. [DOI] [PubMed] [Google Scholar]
- Zhang H.-Y.; Sun Y.-M.; Wang X.-L. Substituent Effects on O-H Bond Dissociation Enthalpies and Ionization Potentials of Catechols: A DFT Study and Its Implications in the Rational Design of Phenolic Antioxidants and Elucidation of Structure–Activity Relationships for Flavonoid Antioxidants. Chem. - Eur. J. 2003, 9, 502–508. 10.1002/chem.200390052. [DOI] [PubMed] [Google Scholar]
- Gulley-Stahl H.; Hogan P. A.; Schmidt W. L.; Wall S. J.; Buhrlage A.; Bullen H. A. Surface Complexation of Catechol to Metal Oxides: An ATR-FTIR, Adsorption, and Dissolution Study. Environ. Sci. Technol. 2010, 44, 4116–4121. 10.1021/es902040u. [DOI] [PubMed] [Google Scholar]
- Zhang J. X.; Fang J. Z.; Duan W.; Wu L. R.; Zhang A. W.; Dalchau N.; Yordanov B.; Petersen R.; Phillips A.; Zhang D. Y. Predicting DNA hybridization kinetics from sequence. Nat. Chem. 2018, 10, 91–98. 10.1038/nchem.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnich C. M.; Hariri A. A.; Rahbani J. F.; Gordon J. B.; Sleiman H. F.; Cosa G. Kinetics of Strand Displacement and Hybridization on Wireframe DNA Nanostructures: Dissecting the Roles of Size, Morphology, and Rigidity. ACS Nano 2018, 12, 12836–12846. 10.1021/acsnano.8b08016. [DOI] [PubMed] [Google Scholar]
- Fern J.; Schulman R. Modular DNA strand-displacement controllers for directing material expansion. Nat. Commun. 2018, 9, 3766. 10.1038/s41467-018-06218-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machinek R. R. F.; Ouldridge T. E.; Haley N. E. C.; Bath J.; Turberfield A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nat. Commun. 2014, 5, 5324. 10.1038/ncomms6324. [DOI] [PubMed] [Google Scholar]
- Mummidivarapu V. V. S.; Rennie M. L.; Doolan A. M.; Crowley P. B. Noncovalent PEGylation via Sulfonatocalix[4]arene–A Crystallographic Proof. Bioconjugate Chem. 2018, 29, 3999–4003. 10.1021/acs.bioconjchem.8b00769. [DOI] [PubMed] [Google Scholar]
- Nischan N.; Hackenberger C. P. R. Site-specific PEGylation of Proteins: Recent Developments. J. Org. Chem. 2014, 79, 10727–10733. 10.1021/jo502136n. [DOI] [PubMed] [Google Scholar]
- Takeda K.; Uchihashi T.; Watanabe H.; Ishida T.; Igarashi K.; Nakamura N.; Ohno H. Real-time dynamic adsorption processes of cytochrome c on an electrode observed through electrochemical high-speed atomic force microscopy. PLoS One 2015, 10, e0116685–e0116685a. 10.1371/journal.pone.0116685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.