

Abstract
Human frataxin is an iron binding protein involved in the mitochondrial Fe-S clusters assembly, a process fundamental for the functional activity of mitochondrial proteins. Decreased level of frataxin expression is associated with the neurodegenerative disease Friedreich ataxia. Defective function of frataxin may cause defects in mitochondria, leading to increased tumorigenesis. Tumour initiating cells show higher iron uptake, a decrease in iron storage and a reduced Fe-S clusters synthesis and utilization. In this study we selected, from COSMIC database, the somatic human frataxin missense variants found in cancer tissues p.D104G, p.A107V, p.F109L, p.Y123S, p.S161I, p.W173C, p.S181F, and p.S202F to analyze the effect of the single amino acid substitutions on frataxin structure, function and stability. The spectral properties, the thermodynamic and the kinetic stability, as well as the molecular dynamics of the frataxin missense variants found in cancer tissues point to local changes confined to the environment of the mutated residues. The global fold of the variants is not altered by the amino acid substitutions, however some of the variants show a decreased stability and a decreased functional activity in comparison to that of the wild type protein.
Introduction
In cancer cells metabolic activities are altered relative to normal cells. The metabolic reprogramming, and the consequent metabolic alterations, guarantees the acquisition of the malignant properties, necessary to enable the cancer cells to thrive despite the adverse conditions, like for example the oxygen deficiency (Kroemer and Pouyssegur, 2008). In particular, the metabolism and bioenergetics of cancer cells is altered to support proliferation and malignant progression: the glycolytic flux is increased with respect to normal cells, and amino acid and lipid metabolisms are accelerated and upregulated. Overall, macromolecule biosynthesis and mitochondrial biogenesis, as well as cellular redox homeostasis, are perturbed (DeBerardinis et al., 2016; Liu et al., 2019; Orang et al., 2019; Phan et al., 2014). Indeed, the process of tumorigenesis and mitochondrial biology interplays at multiple levels, ranging from direct oncogenic signaling from mitochondria to perturbation of mitochondrial functions, often caused by mutation in mitochondrial proteins (Sreedhar and Zhao, 2018).
In this respect, mutations of proteins involved in the assembly of those mitochondrial proteins, like aconitase and complex I, II and III that depend on iron-sulfur clusters (Fe-S) for their functions, could be at the origin of cancer onset and responsible for its progression (Thierbach et al., 2005). Frataxin (FXN protein, coded by FXN gene: LRG_339), whose primary function is still under debate, is directly involved in the Fe-S cluster assembly inside the mitochondria (Thierbach et al., 2005) and since Fe-S clusters are important for the correct functioning of the mitochondrial proteins, frataxin impaired function may cause defects in mitochondria then leading to increased tumorigenesis (Thierbach et al., 2005). Indeed, it has been reported that tumour initiating cells show higher iron uptake, a decrease in iron storage and a reduced Fe-S clusters synthesis and utilization (Rychtarchikova et al., 2017).
In the general reprogramming of cancer cell metabolism, iron viability is an essential requirement for the invasion. Notably, this is not only important for the cancer cells but also to its particular microenvironment constituted by different type of normal cells ranging from endothelial and stromal cells to macrophages (Torti and Torti, 2011). Iron could simply be regarded as a tumour initiator because of its involvement in the production of DNA damaging oxygen radicals, which might increase the rate of mutagenesis (Torti and Torti, 2013). On the other hand, iron could be regarded as a tumour growth factor when examined in the context of its cellular roles: cellular energy generation, increased rate of cellular cycle, and the increased demand of Fe-S clusters essential for the accelerated DNA metabolism (Torti et al., 2018). In synthesis, the multifaceted role of iron in cancer can be simply considered as beneficial and essential for tumour growth, as well as deleterious at the same time for its potentially toxic effects (Torti and Torti, 2013).
Fe-S clusters are protein cofactors involved in several cellular processes from respiration to DNA replication and repair. The clusters are assembled in the presence of the pyridoxal dependent cysteine desulfurase (NFS1 in human), the iron delivery protein frataxin (FXN), reductant ferredoxin, and a scaffold protein (IscU) (Cai et al., 2018). Decreased level of expression of the iron binding protein human frataxin (FXN) involved in Fe-S cluster assembly is associated with Friedreich ataxia (FRDA; MIM# 229300), a neurodegenerative disease characterized by neuronal death, cardiomyopathy and diabetes (Correia et al., 2008). The FXN deficiency, at the molecular level, is related to the deregulation of iron homeostasis and alteration of the Fe-S protein biogenesis (Correia et al., 2008). Notably, young patients affected by FRDA are also affected by different cancer type, unusual for their age, even though there is no evidence of a direct correlation between this disease and cancer (Schulz et al., 2006).
Somatic mutations of FXN have been identified in various cancer types and reported in COSMIC (Catalogue of Somatic Mutations in Cancer) (http://cancer.sanger.ac.uk/cosmic), a database for the collection of somatic mutations identified in human cancers (Forbes, 2017). Among them, 23 out 32 single nucleotide substitutions are non synonymous single nucleotide variants (nsSNVs) in the FXN coding region leading to a protein sequence with amino acid substitutions.
The effects exerted by somatic mutations on the proteins range from changes in the protein stability, to alterations in the protein functions and in protein-protein interactions (Pasquo et al., 2012; Lori et al., 2013; Lori et al., 2016; Stefl et al., 2013; Petrosino et al., 2017). Computational studies based on protein structural information may provide a rationale for the changes induced by mutation (Casadio et al., 2011, Petukh et al., 2015; Peng et al., 2019), however direct physico-chemical studies of the protein variants may reveal all the local minor structural changes responsible of modifications in protein functions without dramatic alterations of the global protein folding (Lori et al., 2016). In addition, detailed experimental analysis may add new piece of information to computational research and motivate new studies.
In this study eight nsSNVs of FXN present in cancer tissues were selected from those reported in COSMIC database (Fig. 1). The residues carrying the mutations are located in the alpha-1 helix close to the acidic region, presumably involved in iron binding on the protein surface, in the coil regions between alpha-1 and beta-1 and beta-4 and beta-5, respectively, and in the beta-6 and beta-7 strands (Dhe-Paganon et al., 2000) (Fig. 1). We report the effects for the eight FXN nsSNVs on thermodynamic stability by equilibrium and kinetic experiments and we study the impact of each mutation on FXN functional activity.
Figure 1. Amino acid sequence and secondary structure elements of FXN.
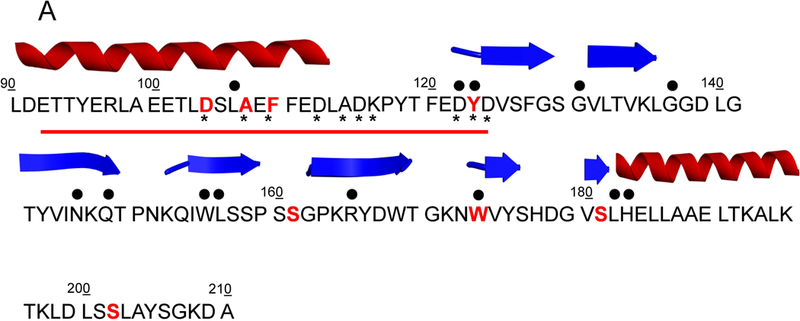
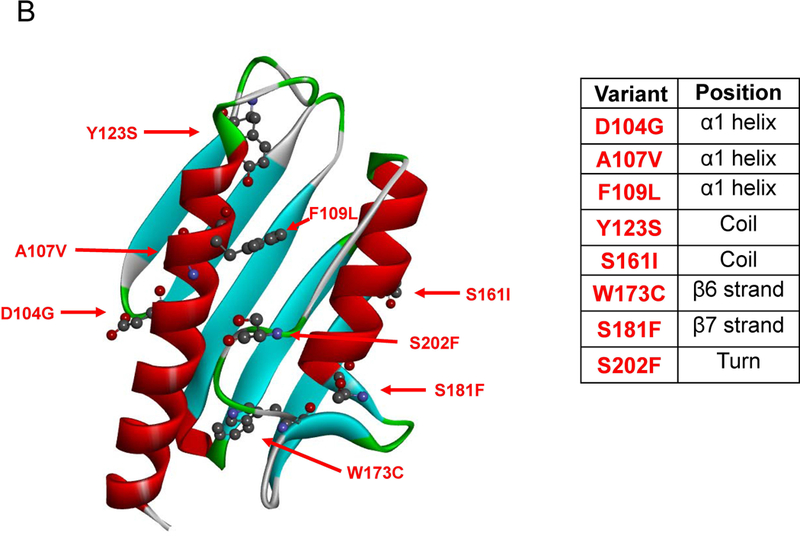
(A) The mutated residues (protein seq. ref. NP_000135.2) object of this study and reported in COSMIC are depicted in bold red, the residues mutated in FRDA are identified by upper black dots. Tyr123 and Trp173 are mutated in FRDA and reported in COSMIC. The lower black stars represent the residues involved in iron binding. The red line highlights the acidic binding region. The secondary structure elements are as reported in 1EKG (Dhe-Paganon et al., 2000). (B) Location of the mutations on FXN structure. Mutated residues are depicted in scaled ball and stick.
Materials and Methods
Site-directed mutagenesis
pET28a plasmid harbouring the FXN wild-type gene was used for E. coli expression. The point mutations on wild type FXN gene were introduced using Quick Change Site-Directed Mutagenesis Kit (Stratagene). The mutagenic oligonucleotides used are listed in Supp. Table S1. The presence of the desired mutations and the absence of unwanted ones were confirmed by sequence analysis.
Protein Expression and Purification
FXN wild type and variants were expressed as N-terminally His-tagged proteins using a pET28a vector. Wild type and mutant proteins were expressed in E. coli Rosetta cells transformed with the selected plasmid, grown at 37°C in LB medium containing kanamycin as antibiotic at a final concentration of 30 μg/mL until optical density OD600 reached 0.6. The protein expression was induced overnight by adding 0.5 mM isopropyl-β-D-thiogalactoside (Sigma-Aldrich) and grown overnight at 18°C with energic shaking. The culture was harvested by centrifugation and resuspended in 40 mL of Binding buffer (20 mM Tris-HCl, 500mM NaCl, 5 mM Imidazole, pH 8.0) containing 1 mM tris(2-carboxyethyl)phosphine (TCEP) in the presence of a cocktail of EDTA-free protease inhibitors (Sigma). The cells were sonicated in a Vibracell 75115 sonicator with 5 s boots and 9 s pause, on ice and the lysate was cleared by centrifugation. The soluble fraction was applied to a 5mL pre-packed His trap column (GE Healthcare) pre-equilibrated with Binding buffer. The column was washed with Binding buffer to elute weakly bound contaminants and the recombinant protein was eluted by passing over the column Binding buffer solutions containing 250 mM Imidazole. The eluted protein was concentrated to a final volume of 2.5 mL on an Amicon concentrator Ultra-15 (Millipore) and then applied to a PD-10 pre-packed column (GE Healthcare) to remove imidazole. The pure fraction in Binding buffer was incubated overnight at 4°C with tobacco etch virus (TEV) protease to cleave the hexahistidine tag. After digestion, the mixture containing TEV protease, the His-tag and the cleaved protein was applied to a 5 mL pre-packed His Trap column (GE Healthcare) previously equilibrated in Binding buffer. The flow through containing the protein without His-tag was collected, and purity and size were checked by SDS–PAGE on a pre-casted NuPage 4–12% bis-Tris polyacrylamide gel (Invitrogen). Protein concentration was determined spectrophotometrically using a molar absorptivity of 26930 M−1 cm−1 at 280 nm based on a molecular mass of 13.886 kDa.
Spectroscopic measurements
Intrinsic fluorescence emission spectra were recorded from 300–450 nm (at 295 nm excitation wavelength, 1 nm sampling interval), at 50.0 μg/mL protein concentration in 20 mM Tris/HCl, pH 8.0 containing 0.1 M NaCl, 1mM EDTA and 200 μM DTT with a LS50B spectrofluorimeter (Perkin-Elmer). Fluorescence measurements were carried out using a 1.0 cm path length quartz cuvette at 20°C. Far-UV (190–250 nm) CD spectra were recorded either at a protein concentration ranging over 100–140 μg/mL (0.4 mM DTT, 0.1 cm path length quartz cuvette), corresponding to an absorbance of the protein solutions at 280 nm of 0.24 AU, either at a protein concentration ranging over 50–60 μg/mL (0.2 mM DTT, 0.2 cm path length quartz cuvette), corresponding to an absorbance of the protein solutions at 280 nm of 0.10 AU. For near-UV (250–320 nm) CD spectra, the absorbance of the protein solutions at 280 nm was 2.2 AU, corresponding to a protein concentration ranging over 1.0–1.3 mg/mL (1.0 mM DTT, 1.0 cm path length quartz cuvette). CD measurements were performed in a Jasco-815 spectropolarimeter (Jasco, Easton, MD, Usa) and the results obtained were expressed as the mean residue ellipticity [Θ], assuming a mean residue molecular mass of 110 per amino acid residue. All spectroscopic measurements were carried out at 20°C in 20 mM Tris/HCl, pH 8.0 containing 0.1 M NaCl, 1mM EDTA and 200 μM or 1 mM DTT.
Urea-induced equilibrium unfolding.
For equilibrium transition studies, FXN wild type and variants (final concentration 50.0 μg/mL) were incubated at 20 °C at increasing concentrations of urea (0−9 M) in 20 mM Tris/HCl, pH 8.0, in the presence of 0.1 M NaCl, 1mM EDTA and 200 μM DTT. After 10 min, equilibrium was reached and intrinsic fluorescence emission and far-UV CD spectra (0.2-cm cuvette) were recorded in parallel at 20 °C. To test the reversibility of the unfolding, FXN wild-type and variants were unfolded at 20 °C in 9.0 M urea at 0.5 mg/mL protein concentration in 20 mM Tris-HCl, pH 8.0, in the presence of 2 mM DTT and 0.1 M NaCl. After 10 min, refolding was started by 10-fold dilution of the unfolding mixture at 20 °C into solutions of the same buffer used for unfolding containing decreasing urea concentrations. The final protein concentration was 50 μg/mL. After 24 h, intrinsic fluorescence emission and far-UV CD spectra were recorded at 20 °C. All denaturation experiments were performed in triplicate.
Stopped-flow folding kinetics experiments
Kinetic folding experiments were performed on a single-mixing SX-18 stopped-flow instrument (Applied Photophysics); the excitation wavelength was 280 nm and the fluorescence emission was collected using a 320 nm cut-off glass filter for unfolding experiments and a 360 nm cut-off glass filter for refolding experiments. Experiments were conducted at 37°C, and the buffer used was Tris-HCl 50mM, NaCl 100mM, DTT 1mM, pH 8.0, and different concentrations of urea, ranging from 0 to 8 M. At least 5 individual traces were acquired and then averaged for each experiments. Protein concentration was typically 2 μM.
Thermal denaturation experiments.
FXN wild type and variants (protein concentration ranging over 100–140 μg/mL) were heated from 20°C to 95°C and then cooled from 95°C to 20°C in a 0.1 cm quartz cuvette with a heating rate of 1.0 degree x min−1 controlled by a Jasco programmable Peltier element. The dichroic activity at 222 nm and the PMTV were continuously monitored in parallel every 0.5°C (Benjwal et al., 2006). All the thermal scans were corrected for the solvent contribution at the different temperatures. Melting temperature (Tm) values were calculated by taking the first derivative of the ellipticity at 222 nm with respect to temperature. All denaturation experiments were performed in triplicate.
Molecular Dynamics
Molecular Dynamics (MD) simulations were performed with the GROMACS package (Berendsen et al., 1995; Lindahl et al., 2001; Van der Spoel et al., 2005; Hess et al., 2008). The initial coordinates of the wild type protein were taken from the crystal structure of the FXN (Dhe-Paganon et al., 2000) (PDB ID: 1EKG). The coordinates of the seven variants were obtained from the wild type coordinates by performing a single amino acid mutation by using PyMOL (The PyMOL Molecular Graphics System). Each system was placed in a cubic box of dimensions such that nearby images lay more than 10 Å away. The box was filled with water molecules and an appropriate number of counterions to make the whole system neutral. The AMBER03 force field (Duan et al., 1999) was used to simulate wild type FXN and all its variants. The equilibration strategy adopted for the eight systems is quite standard and is explained in detail in (Petrosino et al., 2017; Di Carlo et al., 2015). The temperature, for the production simulations, was held fixed at 300 K and 355 K using the v-rescale thermostat (Bussi et al., 2007) with a coupling time of 0.1 ps. The simple point charge (SPC) model was employed for water molecules. Periodic boundary conditions were used throughout the simulation. The Particle Mesh Ewald algorithm (Darden et al., 1993) was employed to treat Coulomb interaction. A time step of 2 fs and a non-bond pair list cutoff of 1.0 nm were used. The list was updated every 10 steps. Each one of the eight systems was simulated for 150 ns (at 300 K) and 200 ns (at 355 K) in the NVT ensemble. The analysis of the numerical data obtained in the simulation was carried out by GROMACS and VMD (Humphrey et al., 1996) tools according to needs.
Reconstitution experiments
Enzymatic cluster formation was achieved under strict anaerobic conditions in a Belle chamber kept under nitrogen atmosphere. The reaction was followed by absorbance spectroscopy using a Cary 50 Bio Spectrophotometer (Varian). Absorbance variations at 456 nm was measured as a function of time. A solution of 3 mM DTT, 1μM IscS, 50 μM IscU, of 50 μM of FXN wild type or mutated and 25 μM Fe(NH4)2(SO4)2 was incubated in sealed cuvette for 30 minutes in 20 mM Tris-HCl pH 8, 150 mM NaCl. The reaction was initiated by adding 250 μM of the substrate L-cysteine.
Crosslinking
A mixture of IscS 40 μM and 40 μM FXN wild type or mutated was prepared in PBS buffer. Dimethyl adipimidate or Bis[sulfosuccinimidyl]suberate was added to the protein sample to a final concentration of 2 mM. The reaction mixture was incubated at room temperature for 30 minutes and quenched by adding Tris-HCl at pH 8 to a final concentration of 20 mM. The quenching reaction was incubated at room temperature for 15 minutes. SDS PAGE was run to determine crosslinked species.
Data analysis
The changes in intrinsic fluorescence emission spectra at increasing urea concentrations were quantified as the intensity-averaged emission wavelength,, (Royer et al., 1993) calculated according to
| (1) |
where λi and Ii are the emission wavelength and its corresponding fluorescence intensity at that wavelength, respectively. This quantity is an integral measurement, negligibly influenced by the noise, which reflects changes in the shape and position of the emission spectrum. Urea-induced equilibrium unfolding transitions monitored by far-UV CD ellipticities changes was analysed by fitting baseline and transition region data to a two-state linear extrapolation model (Santoro and Bolen, 1988) according to:
| (2) |
where ΔGunfolding is the free energy change for unfolding for a given denaturant concentration, ΔGH2O the free energy change for unfolding in the absence of denaturant and m a slope term which quantifies the change in ΔGunfolding per unit concentration of denaturant, R the gas constant, T the temperature and Kunfolding the equilibrium constant for unfolding. The model expresses the signal as a function of denaturant concentration:
| (3) |
where yi is the observed signal, yU and yN are the baseline intercepts for unfolded and native protein, sU and sN are the baseline slopes for the unfolded and native protein, [X]i the denaturant concentration after the ith addition, ΔGH2O the extrapolated free energy of unfolding in the absence of denaturant, m the slope of a ΔGunfolding versus [X] plot. The denaturant concentration at the midpoint of the transition, [Urea]0.5, according to equation 2, is calculated as:
| (4) |
All unfolding transition data were fitted by using Graphpad Prism 5.04.
Far-UV CD spectra recorded as a function of urea concentration were analyzed by a singular value decomposition algorithm (SVD) using the software MATLAB (Math-Works, South Natick, MA) to remove the high frequency noise and the low frequency random errors and to determine the number of independent components in any given set of spectra. CD spectra in the 213–250 nm or in the 250–320 nm region were placed in a rectangular matrix A of n columns, one column for each spectrum collected at each time. The A matrix is decomposed by SVD into the product of three matrices: A = U*S*VT, where U and V are orthogonal matrices and S is a diagonal matrix. The U matrix columns contain the basis spectra and the V matrix columns contain the urea dependence of each basis spectrum. Both U and V columns are arranged in terms of decreasing order of the relative weight of information, as indicated by the magnitude of the singular values in S. The diagonal S matrix contains the singular values that quantify the relative importance of each vector in U and V. The signal-to-noise ratio is very high in the earliest columns of U and V while the random noise is mainly accumulated in the latest U and V columns. The wavelength averaged spectral changes induced by increasing denaturant concentrations are represented by the columns of matrix V; hence, the plot of the columns of V versus the denaturant concentrations provides information about the observed transition.
Results
The eight human frataxin (FXN) variants, associated to human carcinoma and reported in this study (Fig. 1), were mined from the COSMIC database (http://cancer.sanger.ac.uk/cosmic) (Forbes, 2017). The variant p.D104G is reported to be present in liver carcinoma, p.A107V, p.Y123S and p.W173C are reported in carcinoma from diverse locations of the digestive tract, p.F109L and p.S161I from endometrium carcinoma and p.S181F and p.S202F from skin malignant melanoma and carcinoma, respectively. In Fig. 1 is reported the location of the selected mutants mapped onto the FXN structure. It is noteworthy that most of the mutated residues are solvent exposed. Residues Asp104, Ala107 and Phe109 are located in the alpha1 helix, which belongs to the FXN acidic region (Gomes and Santos, 2013; Foury et al., 2007) formed at the interface with strand beta1, a region placed at the protein surface and presumably involved in iron binding (Foury et al., 2007). The residues Tyr123 and Ser161, located in the coils between helix alpha1 and strand beta1, and beta5 and beta6, respectively, are both the last coil residues prior to the next strand. Residues Trp173 and Ser181 belong to strand beta6 and beta7, respectively, and Ser202 is located in the turn at the C-terminus. Notably, the residues Tyr123 and Trp173 (Galea et al., 2016 for review) are mutated in Freidreich ataxia (FRDA; MIM# 229300), and reported in COSMIC. For each of the identified mutant we generated recombinant protein using site directed mutagenesis. With the exception of p.W173C, all mutations resulted in soluble recombinant proteins and allowed us to investigate the results of the single amino acid substitution on the FXN thermal and thermodynamic stability and the biological activity.
Spectroscopic characterization of FXN wild-type and variants
To evaluate the effect of the FXN variants found in cancer tissues on the protein conformation in solution, spectroscopic analysis was performed using CD and fluorescence spectroscopy. The spectral contributions of all aromatic residues present in FXN wild type are evidenced in the near-UV CD spectrum that is characterized by an intense negative peak between 260 and 270 nm and by two other negative contributions at around 285 and 295 nm (Fig. 2A). Most of the variants display near-UV CD spectra almost identical to that of the wild type with slight differences in the intensity of the peaks suggesting that their tertiary structure arrangements are similar respect to that of the corresponding wild type protein. The most significant alterations in the intensity of the negative contributions are observed in the near-UV CD spectrum of p.Y123S variant that shows a significant decrease of the dichroic activity specially in the region between 265–285 nm, a region where the typical spectral contribution of tyrosine is expected. In line with the near-UV CD results, the fluorescence emission spectra of variants are similar to that of wild-type protein. They show the same maximum emission wavelength around 343 nm, with a decreased emission fluorescence intensity (Fig. 2B) with respect to that of the wild type.
Figure 2. Spectral properties of FXN wild type and variants.
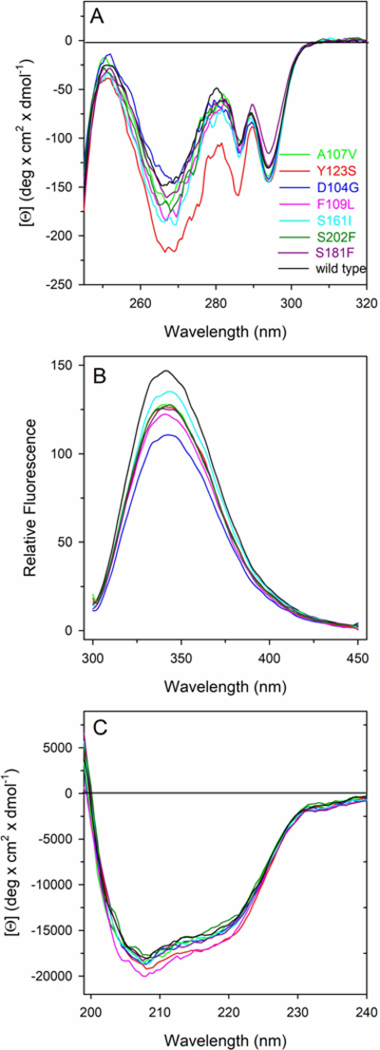
(A) Near-UV CD spectra were recorded in a 1.0-cm quartz cuvette at 1.1–1.2 mg/mL protein concentration. (B) Intrinsic fluorescence emission spectra were recorded at 60–80 μg/mL variants (0.09 AU280nm, 295 nm excitation wavelength). (C) Far-UV CD spectra were recorded in a 0.1-cm quartz cuvette at 103–130 μg/mL. All spectroscopic measurements were carried out at 20°C in 20 mM Tris-HCl, pH 8.0 containing 0.1 M NaCl and 0.2 mM DTT.
Far-UV CD spectra of FXN wild type and variants show local minima at around 208 and 220 nm and a zero intercept at around 200 nm, indicating the major contribution of alpha helical secondary structural elements, slightly influenced by the contribution of β sheet (Fig. 2C). As judged by the shape and the intensities of the signals, the secondary structure of the FXN variants is similar to that of the wild type (Fig. 2C) suggesting that the effect of the point mutations are mainly directed and localized to the mutated residue with minor modification of tertiary and secondary arrangements. The 222/208 ellipticity ratio gives information about interhelical contacts and is generally used to distinguish between coiled coil helices and non-interacting helices (<0.9) (Choy et al., 2003; Kiss et al., 2003). The ratio of the molar ellipticity at 222 and at 208 nm ([Θ]222/[Θ]208) observed for the wild type is 0.70 and is quite similar for the variants ranging from 0.69 for p.D104G, to 0.75 for p.Y123S, suggesting that the single amino acid substitutions do not alter interhelical interactions in solution.
Thermal and thermodynamic stability studies
The thermal stability of FXN wild-type and variants was investigated by continuously monitoring the ellipticity changes at 222 nm between 20 and 95°C (Supp. Figure S1) and between 95 and 20°C (data not shown). By monitoring the temperature-induced ellipticity changes at 222 nm, where the main amplitude was observed, an apparent cooperative transition for FXN wild type and all the variants was observed, with apparent Tm values ranging from 56.0 to 73.0°C. The thermal unfolding transitions were reversible as judged by the signals upon cooling protein solutions to the starting temperature. As reported in Table 1, a modest increase in Tm value was observed for p.D104G and p.S202F variants, whereas all the other variants show a significant reduction of the Tm values ranging from 3 degrees below that of the wild type, as in the case of p.A107V, to 14 degrees below that of the wild type, as in the case of p.Y123S.
Table 1.
Melting temperatures and thermodynamic parameters for urea-induced unfolding equilibrium of FXN wild type and variants measured by far-UV CD and fluorescence spectroscopy
| Protein Variant |
aTm
(°C) |
b ΔGH2O
(kcal/mol) |
b
m
(kcal/mol•M) |
c [Urea]0.5
(M) |
|||
|---|---|---|---|---|---|---|---|
| CD ([Θ]222) | Fluorescence ( ) | CD ([Θ]222) | Fluorescence ( ) | CD ([Θ]222) | Fluorescence ( ) | ||
| Wild type | 71.0 | 9.23 ± 0.47 | 9.77 ± 0.40 | 1.88 ± 0.10 | 2.00 ± 0.08 | 4.91 | 4.87 |
| p.D104G | 74.0 | 9.44 ± 0.46 | 10.39 ± 0.31 | 1.82 ± 0.09 | 1.99 ± 0.06 | 5.20 | 5.22 |
| p.A107V | 68.0 | 10.03 ± 0.91 | 9.06 ± 0.22 | 2.13 ± 0.19 | 1.95 ± 0.05 | 4.71 | 4.64 |
| p.F109L | 59.6 | 7.14 ± 0.34 | 6.18 ± 0.16 | 2.09 ± 0.10 | 1.85 ± 0.05 | 3.42 | 3.34 |
| p.Y123S | 56.6 | 4.31 ± 0.18 | 4.48 ± 0.18 | 1.60 ± 0.06 | 1.59 ± 0.06 | 2.69 | 2.84 |
| p.S161I | 60.0 | 5.88 ± 0.58 | 6.95 ± 0.24 | 1.88 ± 0.19 | 2.11 ± 0.07 | 3.12 | 3.30 |
| p.S181F | 59.9 | 6.12 ± 0.27 | 6.86 ± 0.33 | 1.84 ± 0.08 | 1.99 ± 0.09 | 3.32 | 3.45 |
| p.S202F | 70.7 | 9.07 ±0.32 | 9.60 ±0.33 | 1.89 ± 0.07 | 2.18 ± 0.07 | 4.78 | 4.40 |
Tm values were calculated by taking the first derivative of the ellipticity at 222 nm with respect to temperature, as described in the text. Urea-induced unfolding equilibrium data were obtained by monitoring the ellipticity at 222 nm ([Θ222]) and fluorescence intensity averaged emission wavelength (), as described in Materials and Methods.
ΔGH2O and m values were obtained from Eq (2);
[Urea]0.5 was calculated from Eq (4). Data are reported as the mean ± SE of the fit.
The impact of point mutations on the thermodynamic stability of FXN was investigated at 20°C in the presence of urea as a chemical denaturing agent. FXN wild type and variants reversibly unfold in urea in 20 mM Tris-HCl, pH 8.0, containing 0.1 M NaCl, 1mM EDTA and 200 μM DTT with an apparent two-state cooperative sigmoidal transitions (Fig. 3). The effect of increasing urea concentrations (0–9 M) on the structure of FXN variants was analyzed by far-UV CD and intrinsic fluorescence spectroscopy. Incubation of FXN wild type and variants at increasing urea concentrations at 20°C resulted in a progressive change of the intrinsic fluorescence emission intensity and a red-shift of the maximal emission wavelength. At the end of the transition, above 9 M urea, the intrinsic fluorescence emission intensity is increased about 1.5 fold and the maximal fluorescence emission wavelength shifts from around 343 nm to around 357 nm either for the wild type and all the variants (Fig. 3A). The intrinsic fluorescence emission changes were expressed by calculating the intensity averaged emission wavelength,, at increasing urea concentration (Fig. 3A). This parameter is an integral measurement, negligibly influenced by the noise, and reflects changes in both the shape and the position of the emission spectrum.
Figure 3. Urea-induced equilibrium unfolding of FXN wild type and variants.
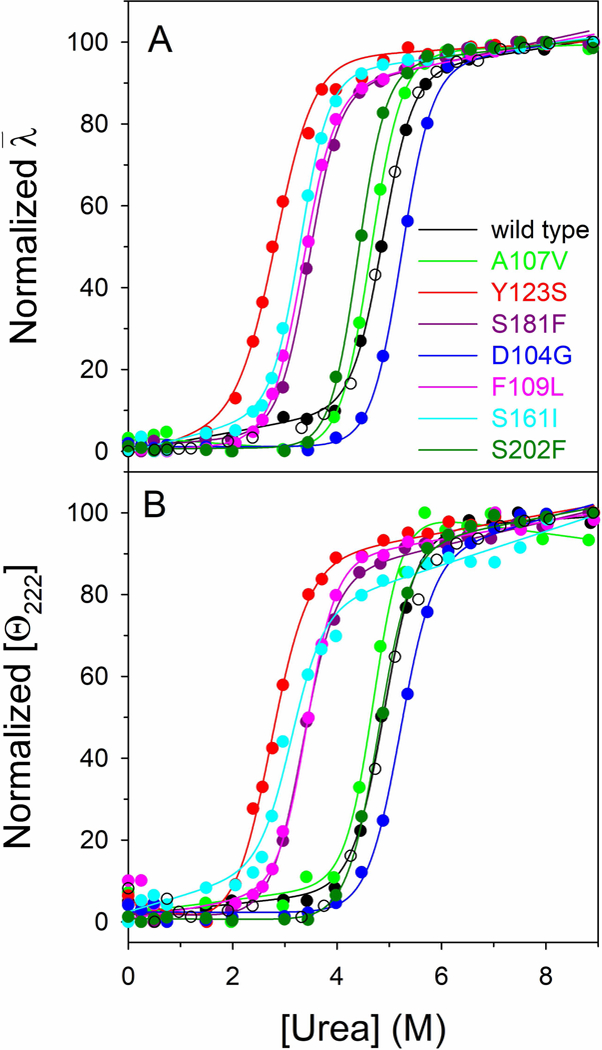
(A) Normalized intensity-averaged emission wavelength . (B) Normalized molar ellipticity at 222 nm ([Θ222]) reported after removal of the high-frequency noise and the low-frequency random error by SVD. The continuous lines represent the nonlinear fitting of the normalized and [Θ222] data to Eq (3). The reversibility points (empty circles) are shown, for clarity, only for the wild type and were not included in the nonlinear regression analysis. All spectra were recorded at 20 °C, as described in Materials ad Methods.
The urea induced unfolding transitions monitored by far-UV CD (Fig. 3B) are coincident with those obtained by monitoring fluorescence changes. The thermodynamic parameters obtained from the analysis of the far-UV CD and fluorescence changes transitions are reported in Table 1. p.D104G and p.S202F show conformational stability closely similar to that of the wild type, whereas the free energy change for unfolding in the absence of denaturant, ΔGH2O, indicates a significant decrease in thermodynamic stability of 2–4 kcal/mol for of p.F109L, p.Y123S, p.S161I and p.S181F and an increase of about 1 kcal/mol for p.A107V variant. The changes in m values may indicate differences in the solvent exposed surface area upon unfolding between the variants and the wild type: decrease in m values is usually referred to a decrease in the solvent-exposed surface area upon unfolding. This is frequently ascribed to an increase in the compactness of the residual structure in the non-native state ensemble, rather than to an increase of the accessible surface area of the native state (Pradeep and Udgaonkar, 2004; Wrabl and Shortle, 1999).
Thermodynamic stability data obtained by monitoring far-UV CD and fluorescence changes as a function of urea concentration have been compared with those obtained by computing the changes in folding free energy for each variant utilizing the servers PoPMuSiC (Dehouck et al., 2011) and I-Mutant 2.0 (Capriotti et al., 2005) (Supp. Table S2). The data reported in Supp. Table S2 are expressed as the difference in the free energy change upon unfolding of the variant and that of the wild type, ΔΔG; thus, a negative ΔΔG value < −1.0 kcal/mol indicates that the mutation is destabilizing. Notably, for the variants p.F109L, p.Y123S, p.S161I and p.S181F there is a general agreement between the experimental data and the predicted ΔΔG values, with the exception of p.S161I (Supp. Table S2).
Folding Kinetics
In order to better understand the folding of FXN wild type and variants we resorted to perform kinetic experiments. In an effort to fully characterize the folding properties of FXN and its variants we performed stopped-flow kinetic unfolding and refolding experiments, at different concentrations of urea ranging from 0 to 8 M, in 50 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, pH 8.5, at 37°C. In all the experiments, the kinetics of folding and unfolding were perfectly described by a single exponential behaviour, an aspect that is usually interpreted with the absence of stable intermediates accumulating in the ms to s time range. The dependences of the logarithm of the observed rate constants versus the concentration of urea (chevron plot) for wild type is reported in Fig. 4. In a simple two-state folding mechanism, the resulting chevron plot presents a typical V-shape with the logarithm of the observed rate constants of the unfolding and refolding arms increasing and decreasing linearly as the concentration of urea changes. However, as it might be noted from inspection of Fig. 4, the arms of the chevron plot of wild type FXN deviate from linearity, which suggests the presence of a more complex scenario. In a previous work (Bonetti et al., 2014) the folding kinetics of yeast frataxin were extensively characterized. The chevron plots obtained for yeast frataxin presented a curvature in both the unfolding and refolding arms and were fitted with an equation that describes a folding pathway characterized by the presence of a broad energy barrier in the transition state (Oliveberg et al., 1998).
Figure 4. Chevron plots.
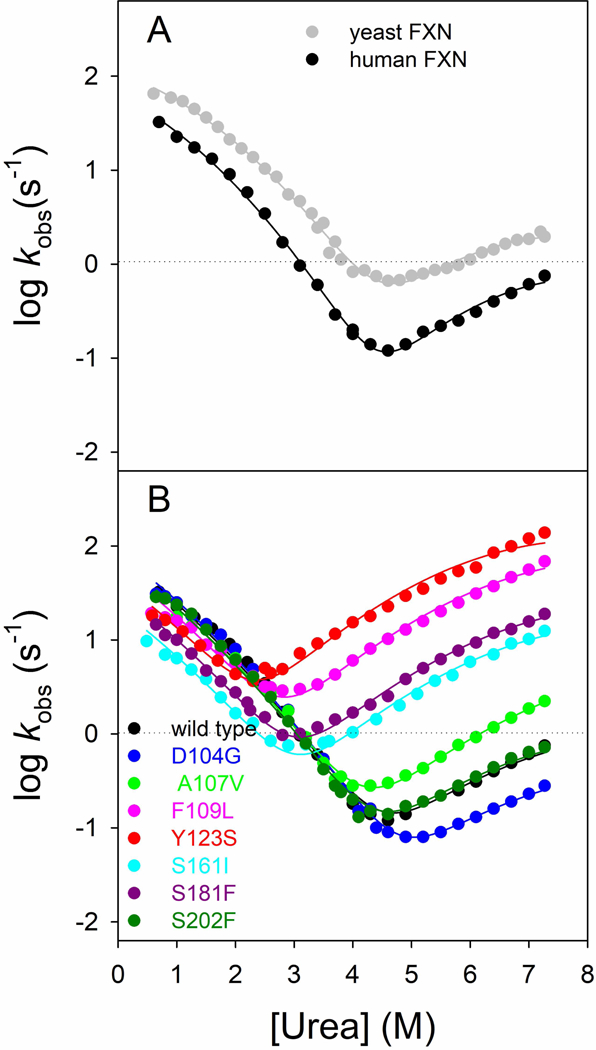
(A) Chevron plots of human (black squares) and yeast frataxin (data from Bonetti et al., 2014, gray circles). Data are fitted with an equation that takes into account a broad energy barrier in the transition state. (B) Chevron plots of FXN wild type and variants p.D104G, p.A107V, p.F109L, p.Y123S, p.S161I, p.S181F, p.S202F in 50 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, pH 8.5, at 37°C. A global fit analysis was performed, sharing the mf and mu values for all the data sets (see details in the text).
| (5) |
Thus, we resorted to compare folding kinetics data of the wild type FXN with the ones obtained from the yeast FXN, which returned an excellent fit (Fig. 4A). This result suggests that the folding of the human variant of FXN can be described with a comparable mechanism.
Then we performed kinetic folding experiments on the variants of FXN. The resulting chevron plots are reported in Fig. 4B. A global fit was performed to analyse data, sharing the mu and mf values for all the data sets, which, as Fig. 4B reports, returned an excellent fit for every variant analysed, suggesting that mutations inferred to the protein do not have any effect on the folding mechanism of FXN. Kinetic data obtained from the fit are shown in Table 2. Analysis of kinetic data allowed us to calculate the difference in free energy between the native and denatured state in absence of denaturant as
| (6) |
Table 2.
Folding kinetics data for FXN wild type and variants calculated from chevron plot fitting.
|
akf (s−1) |
amf (kcal/ mol•M) |
aku (s−1) |
amu (kcal/ mol•M) |
am’ (kcal/ mol•M) |
bΔGN-D (kcal/mol) |
|
|---|---|---|---|---|---|---|
| Wild type | 78.5 ± 5.8 | 0.64 ± 0.02 | 7.8·10−5 ± 0.8·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 8.0 ± 0.8 |
| p.D104G | 84.5 ± 5.9 | 0.64 ± 0.02 | 3.2·10−5 ± 0.3·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 8.6 ± 0.9 |
| p.A107V | 70.0 ± 5.4 | 0.64 ± 0.02 | 1.6·10−5 ± 0.2·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 7.5 ± 0.8 |
| p.F109L | 57.7 ± 3.6 | 0.64 ± 0.02 | 1.1·10−5 ± 0.1·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 5.0 ± 0.5 |
| p.Y123S | 43.8 ± 2.9 | 0.64 ± 0.02 | 3.4·10−5 ± 0.3·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 4.1 ± 0.4 |
| p.S161I | 21.7 ± 1.4 | 0.64 ± 0.02 | 1.9·10−5 ± 0.2·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 5.4 ± 0.5 |
| p.S181F | 32.7 ± 2.1 | 0.64 ± 0.02 | 3.0·10−5 ± 0.3·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 5.4 ± 0.5 |
| p.S202F | 71.4 ± 4.9 | 0.64 ± 0.02 | 9.4·10−5 ± 0.9·10−5 | 1.22 ± 0.06 | 0.06± 0.01 | 7.9 ± 0.8 |
Results reveal that some mutations clearly affect the stability of FXN, with the ΔGN-D values obtained from kinetics being in good agreement with the ΔGH2O values obtained from equilibrium fluorescence experiments.
Functional activity
It is well reported in literature that frataxin effects on the Fe-S cluster enzymatic constitution are caused by its binding to the desulfurase (IscS in this case). The functional ability of the FXN variants was compared to that of the wild type protein and to this purpose we used bacterial desulfurase IscS, using high sequence conservation between human desulfurase NFS1 and bacterial IscS. It was previously demonstrated that the two systems are functionally interchangeable, except that frataxin works as an inhibitor in bacteria and as an activator in eukaryotes. This functional inversion resides in the desulfurase (Bridwell-Rabb et al., 2012) which has a distinct assembly in the two families of organisms (Cory et al., 2017, Boniecki et al., 2017). Hence, we expected to see an effect that could be compared among the mutants. In order to prove the interaction, first a chemical cross-linking assay was performed to test the binding ability of the mutants. Bis[sulfosuccinimidyl]suberate, BS3, a cross-linking agent that reacts with primary amino groups, was added to a mixture containing IscS and each FXN mutant in PBS. SDS-PAGE revealed that a new species at around 58KDa that corresponds to FXN-IscS covalently bound is present, even if in small amount (Fig. 5).
Figure 5. Cross-linking.
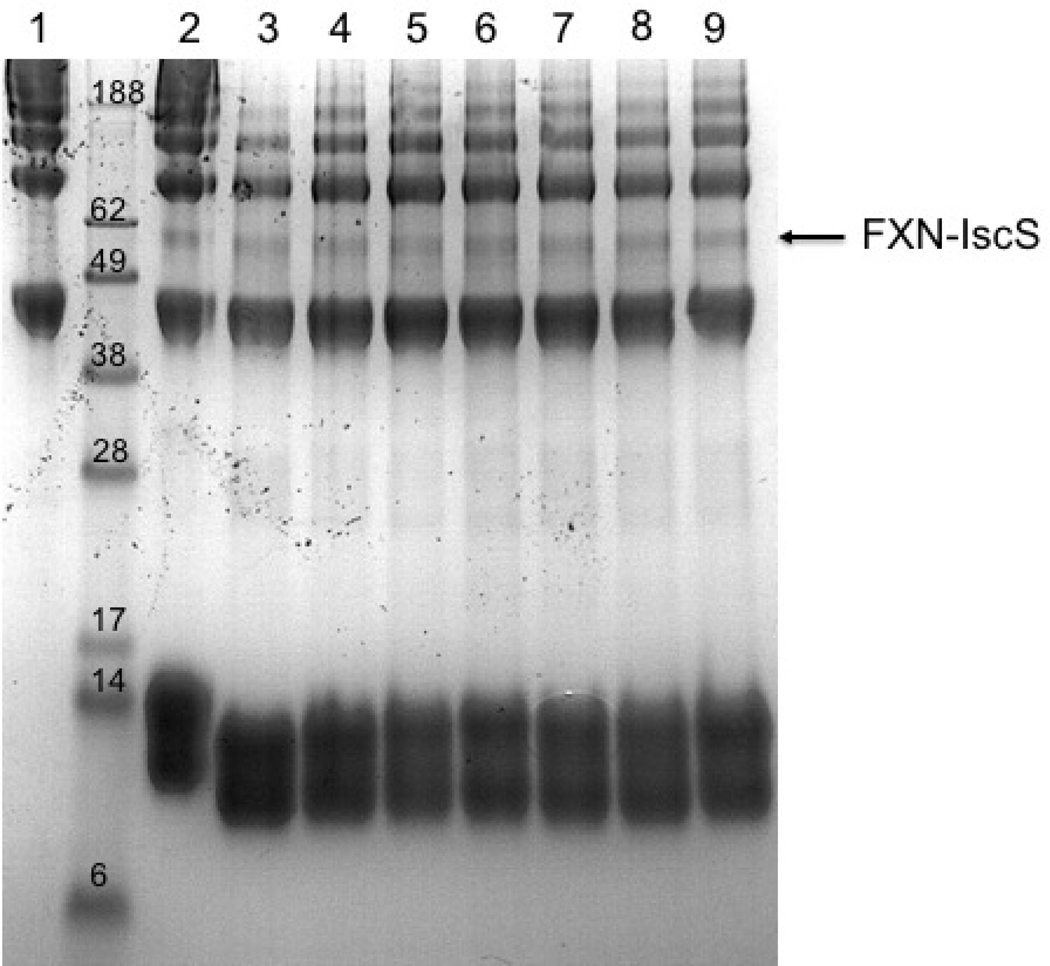
Cross-linking experiments with BS3 to identify the interaction between 40 μM IscS and 40 μM FXN wild-type (2) or mutated: p.D104G (3), p.A107V (4), p.F109L (5), p.Y123S (6), p.S161I (7), p.S181F (8), p.S202F (9). The first lane on the left is the control carried out using isolated IscS (1). The new species at ca. 58 kDa corresponds to the complex FXN-IscS.
We then checked the ability of the mutants to modulate the activity of IscS. Enzyme activity was measured using E. coli Isc proteins IscS and IscU. This in vitro assay is a sensitive method to follow the enzymatic rates by detecting the absorbance as function of time. As shown by Bridwell-Rabb, addition of FXN to the IscS/IscU system inhibited Fe-S assembly less efficiently than bacterial frataxin, CyaY (Bridwell-Rabb et al., 2012). Proteins were mixed with 25 μM Fe2+ and 3 mM DTT in 20 mM Tris-HCl, 150mM NaCl under inert atmosphere of nitrogen to avoid oxidation. After 30 minutes, when the system was equilibrated, 250 μM cysteine was added to start the reaction. Fe-S cluster formation was then easily followed by measuring the change in absorbance at the specific wavelength at 456 nm because this is the characteristic wavelength for the absorbance of Fe-S cluster on IscU. The concentration of IscU was set to 50 μM which is a value that allows easy measurement of the absorbance signal from the cluster. IscS was set to catalytic concentration of 1μM and FXN at 50 μM because at this concentration it did not significantly change the desulfurase activity (Bridwell-Rabb et al., 2012).
Introduction of FXN wild type does not produce appreciable effects on the enzymatic rates (Fig. 6). On the contrary, its variants p.D104G, p.A107V, p.S161I, p.S181F and p.S202F remarkably inhibits the kinetic at the point that no Fe-S cluster is detected (Fig. 6). On the other side p.F109L and p.Y123S have only a partial inhibitor effect on the Fe-S cluster formation (Fig. 6).
Figure 6. Cluster formation.
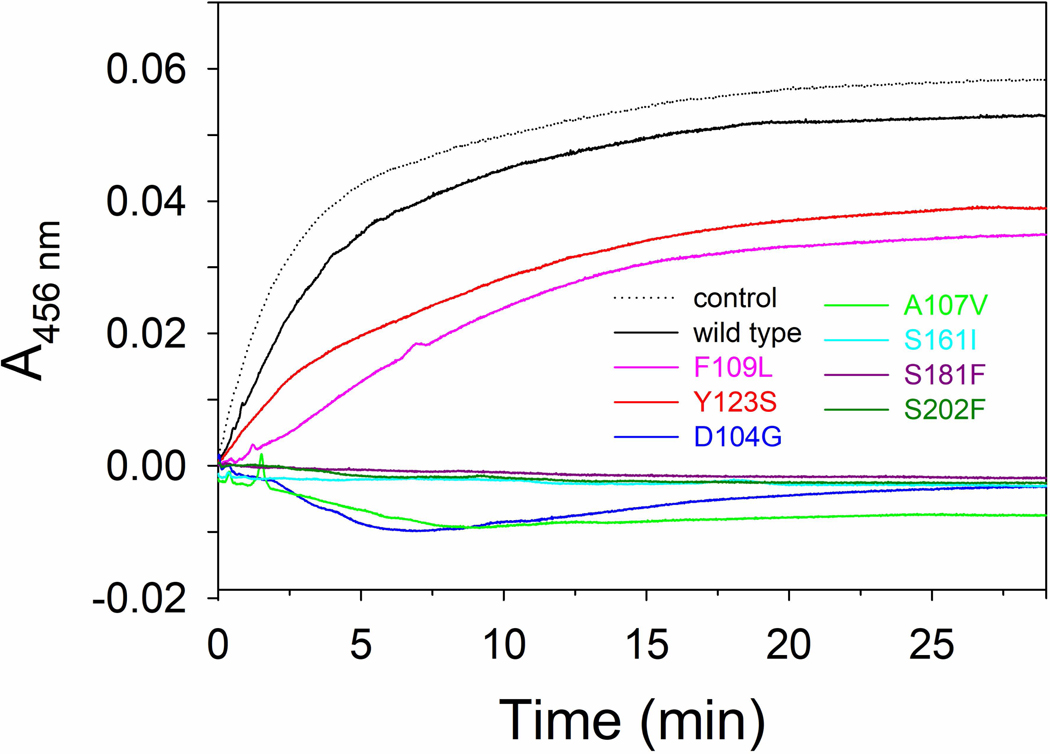
Cluster formation rates on IscU (50μM) in the presence of 1μM IscS, 3 mM DTT with 250 μM Cys, and 25 μM Fe2+. The curves were recorded in the absence (dotted line) or in the presence of 50 μM of FXN wild type and variants. Negative values are due to imperfect subtraction from the baseline and should be considered as zero (full inhibition).
Overall these results confirm that FXN is able to affect the Fe-S cluster formation by interacting with IscS. We investigated FXN carrying different missense mutations, as identified in disease, with an inversion of polarity or change of steric effect. When a non-polar or a charged aminoacid is changed to one with polar characteristics (Ser with Ile or Phe or Asp with Gly) the effect on the kinetic is more pronounced as well as with the replacement of a smaller residue with one of different steric characteristics (Ala with Val). A minor, but still remarkable, effect is observed when an aromatic residue is mutated to one with an aliphatic side chain (Phe with Leu or Tyr with Ser).
Molecular dynamics
MD simulations give us atomistic (microscopic) information on what is observed at the macroscopic level, thus they may be very helpful in the interpretation of experimental data.
The stability of the eight models, both in the simulation at 300 K and in that at 355 K, is assured by the stability of the root mean square displacement (r.m.s.d.) and the radius of gyration, shown in Supp. Figures S2 and S3, respectively. In Table 3 we report the values of r.m.s.d. and the radius of gyration of the simulated systems averaged respectively over the last 50 ns/100 ns of the 300 K/355 K simulation. The radius of gyration is identical (within errors) for all the systems, while the r.m.s.d. values of p.D104G and p.S161I at 300 K and of p.S202F at 355 K are smaller than that of the wild type thus meaning that the three mutants are more similar to the starting structure than the wild type.
Table 3 –
Average r.m.s.d. values computed in the last 50 ns (100 ns) of the simulations at 300 K (355 K).
| System | ar.m.s.d. 300K | ar.m.s.d. 355K | bRgyr 300K | bRgyr 355K |
|---|---|---|---|---|
| Wild type | 0.16(1) nm | 0.21(4) nm | 1.35(1) nm | 1.39(1) nm |
| p.D104G | 0.13(1) nm | 0.18(2) nm | 1.36(1) nm | 1.39(1) nm |
| p.A107V | 0.14(2) nm | 0.23(3) nm | 1.36(1) nm | 1.40(1) nm |
| p.F109L | 0.17(2) nm | 0.15(3) nm | 1.36(1) nm | 1.40(1) nm |
| p.Y123S | 0.18(1) nm | 0.18(2) nm | 1.35(1) nm | 1.39(1) nm |
| p.S161I | 0.13(1) nm | 0.21(2) nm | 1.36(1) nm | 1.40(1) nm |
| p.S181F | 0.19(2) nm | 0.19(2) nm | 1.35(1) nm | 1.39(1) nm |
| p.S202F | 0.15(2) nm | 0.13(2) nm | 1.36(1) nm | 1.39(1) nm |
The r.m.s.d. is calculated on the backbone of the protein and taking as reference structure the one at the end of the equilibration procedure of the wild type simulation.
The average values of the radius of gyration (Rgyr) are computed in the last 50 ns (100 ns) of the simulations at 300 K (355 K) considering the backbone of the protein.
In Supp. Figure S4 the root mean square fluctuations (r.m.s.f.) for the side chains of the simulated proteins are shown. At 300K (left side of Supp. Figure S4), we observe the highest mobility nearby residue 140 (for most of the simulated systems) and in the segment 176–179 for p.S181F and p.S202F variants. At 355 K, one of the most fluctuating systems is the wild type one, around residue 140 and at the C-terminal.
Experimental data show that variants p.D104G, p.A107V, p.S161I, p.S181F and p.S202F remarkably inhibit Fe-S cluster formation while p.F109L and p.Y123S inhibit the process only partially. Since FXN binding to the desulfurase (IscS) is known to have effects on the Fe-S cluster enzymatic constitution, to obtain information on the propensity of the variants to interact with IscS, we computed (for wild type and the seven variants) the solvent accessible surface area (SASA) for the side chains of the FXN residues potentially involved in the interaction (Prischi et al., 2010).
As shown in Supp. Figure S5, the main differences between wild type and its mutants are in the side chains SASA of the residues 136 and 140, for all the mutants except p.Y123S, for which only residue 136 is less accessible and p.S181F for which there are not differences with respect to wild type.
Since higher SASA values correspond to greater exposure of the amino acids and to larger distances between amino acids within the protein, we evaluated the distances between Leu136-Ala193 (d1), Leu140-Ala93 (d2) and Leu140-Leu194 (d3). In Table 4 we report the average values of the three distances computed along the last 50 ns of the simulations at 300 K. As expected, we observe that the values of three distances are significantly different from the values they assume in the wild type samples for all variants except p.Y123S and p.S181F.
Table 4.
Average distances between Leu136-Ala193 (d1), Leu140-Ala193 (d2) and Leu140-Leu194 (d3).
| System | d1 (Å) | d2 (Å) | d3 (Å) |
|---|---|---|---|
| Wild type | 7 ± 1 | 11 ± 2 | 16 ± 2 |
| p.D104G | 4 ± 1 | 5 ± 1 | 7 ± 3 |
| p.A107V | 4 ± 1 | 5 ± 1 | 8 ± 2 |
| p.F109L | 5 ± 1 | 6 ± 1 | 10 ± 2 |
| p.Y123S | 6 ± 2 | 8 ± 2 | 15 ± 3 |
| p.S161I | 4 ± 1 | 5 ± 1 | 7 ± 2 |
| p.S181F | 6 ± 1 | 12 ± 2 | 17 ± 2 |
| p.S202F | 5 ± 1 | 5 ± 1 | 8 ± 2 |
Distances and errors were computed along the last 50 ns of the simulations at 300 K.
The present results indicate that the inhibition of Fe-S cluster formation may depend on the weaker interaction of the mutants, and in particular of Leu136 and Leu140, with respect to wild type FXN with IscS. From MD data analysis it turns out that the only two mutants that have the same behaviour as wild type FXN (with respect to the interaction with IscS) are p.Y123S and p.S181F. On the contrary, considering the SASA of the residues involved in the interaction with IscU (Cai et al., 2018) it turns out that Trp155 is less exposed to the solvent (has a smaller SASA value than the wild type) only in p.Y123S and p.S181F thus possibly meaning that the two mutants have a weaker interaction than all the other mutants (and the wild type) with IscU.
MD data analysis point out that despite there are no significant differences in secondary structure (data not shown), r.m.s.d. and gyration radius between wild type FXN and its mutants, Leu136 and Leu140 may be important in the interaction with IscS and hence in FXN involvement in Fe-S cluster formation.
DISCUSSION
The mitochondrial protein frataxin (FXN) is expressed in several tissues, in particular liver, adrenal, bone marrow, heart and skin (Campuzano et al., 1996). The function of this protein is still under debate and, amongst various hypotheses, the involvement in iron binding as a component of the Fe-S cluster assembly complex is the most supported by experimental evidence (Pastore and Puccio, 2013).
The biosynthesis of Fe-S clusters is necessary for normal functions of human mitochondria and defects in their complex assembly mechanism, which involves at least 18 proteins, have been associated with a number of different diseases (Cai et al., 2018). As a matter of fact, Fe-S clusters are required for the correct functioning of proteins involved in several cellular activities ranging from electron transport in respiratory chain complexes to DNA repair (Rouault, 2012). The defects of Fe-S clusters assembly in mitochondria influence a great number of extra-mitochondrial pathways, as seen for example in the nucleus for DNA synthesis and repair, or in the cytosol for the synthesis of ribosomal proteins and, in general, in cellular iron regulation and homeostasis (Stehling et al., 2014).
FXN gene does not seem to possess cancer driver properties (Davoli et al., 2013), and an increased risk of tumour has been apparently excluded in Friedreich ataxia (FRDA) patients (Bürk, 2017; Martelli et al., 2012). However, alteration of FXN levels has been found in different types of cancer, as reported in the Cancer Genome Atlas (Hutter and Zenklusen, 2018), and several cases of FRDA are associated with the occurrence of neoplastic diseases and alteration of DNA in peripheral blood cells (Ackroyd at al., 1996; Ramos et al., 1997, Kidd et al., 2001, Deutsch et al., 2012, Misiakos et al., 2011, Barr et al, 1984; Haugen et al., 2010). Notably, alteration of iron homeostasis (Rychtarchikova et al., 2017) and perturbation of the redox equilibrium, as well as of the macromolecule biosynthesis and mitochondrial biogenesis (DeBerardinis et al., 2016; Phan et al., 2014), are concomitant to the general reprogramming of cancer cell metabolism (DeBerardinis et al., 2016; Phan et al., 2014). Indeed, it has been reported that FXN impaired function may cause defects in mitochondria then leading to increased tumorigenesis (Thierbach et al., 2005). However, the role of frataxin in cancer onset and progression could only be indirect, e.g. related to the genomic instability caused by defective biosynthesis of Fe-S cluster specific for DNA-repair enzyme (Thierbach et al., 2010; Tsai and Barondeau, 2010). On the other hand, hypoxia mediated increase of FXN expression may protect against oxidative stress and apoptosis, and suggest a tumour suppressor function for this protein (Guccini et al., 2011). The interconnection between alteration in iron metabolism and cancer is further evident from the fact that several variants of human mitochondrial ferritin, a protein that may rescue the defects caused by FXN silencing (Zanella et al., 2007; Campanella et al., 2004), are present in the same cancer tissues and cell lines where also FXN variants are reported to be present (Hutter and Zenklusen, 2018).
For this study, we selected from COSMIC database (http://cancer.sanger.ac.uk/cosmic) (Forbes et al., 2017) some somatic FXN non synonymous single nucleotide variants (nsSNVs) found in cancer tissues. In these variants, the mutations mainly involve surface residues, exposed to the solvent (Fig. 1). Three out of them, are located at the alpha-1 and beta-1 interface and presumably involved in iron binding in the acidic binding region of FXN (Gomes and Santos, 2006). None of the residues mutated in the cancer associated variants is placed in a conserved position (Dhe-Paganon et al., 2000). Notably, two residues mutated in the variants reported in COSMIC database, Tyr123 and Trp173, are also mutated in FRDA (Galea et al., 2016). However, the mutations observed in FRDA, p.Y123D and p.W173G, are different from those reported in COSMIC and object of this study (Fig. 1).
The thermodynamic stability of the missense variants p.F109L (Supp. Figure S8), p.Y123S (Supp. Figure S9), p.S161I (Supp. Figure S10), and p.S181F (Supp. Figure S11), is decreased in comparison to that of the wild type and is unchanged for p.D104G, where a charged polar residue is mutated into a hydrophobic one (Supp. Figure S6), for p.S202F (Supp. Figure S12), where a polar residue is substituted by a hydrophobic one and for p.A107V (Supp. Figure S7), where there is a steric difference between the two hydrophobic residues involved. To rationalise the effect of the mutations on the thermodynamic parameters, we analysed the interactions in the protein structure of the residues mutated in cancer tissues. In the case of Phe109, the aromatic ring is involved in a network of hydrophobic interactions with Phe110, Leu113 and Leu200 that might be hampered by the substitution of the phenylalanine benzene ring with the aliphatic side chain of leucine, in particular for the lack of the pi interaction with Phe110 (Supp. Figure S8). Indeed, MD simulations show that the distance between residue Leu109 (in p.F109L) and Leu200 is significantly larger than in the wild type. Tyr123 residue is involved in a pi-alkyl hydrophobic interaction, that requires the presence of an aromatic ring, with the methyl group of Ala114, the first residue at the end of the alpha1 helix (Supp. Figure S9). In addition, the hydroxyl group of Tyr123 is hydrogen bonded to the peptide carbonyl group of Phe110 on alpha1 helix (Supp. Figure S9). p.Y123S is the only variant that shows a modest decrease in m value that may suggest a modest decrease of the surface area exposed to the solvent upon unfolding. The hydroxyl group of p.S161I (Supp. Figure S10) is involved in a hydrogen bond with the carboxyl moiety of Glu189 that is lost upon substitution with the hydrophobic isoleucine (Supp. Figure S10), as indicated by MD simulation. In addition, the nitrogen of the Ser161 peptide bond is hydrogen bonded to the hydroxyl group of Ser158. Notably, from MD simulations comes out that the residue 161, that was near to Tyr143 and Ser158 in the wild type, in p.S161I is quite far from the two residues. The Ser181 residue (Supp. Figure S11), in the wild type crystal structure is involved in a network of hydrogen bonds with Glu184, that may be lost upon the variation from a polar serine into phenylalanine, hence the effect of the amino acid substitution on the FXN structural stability might be related to the hydrophobic nature of the phenylalanine side chain (Supp. Figure S11). The mutation of residues Asp104, Ala107 and Ser202 does not affect significantly thermodynamic stability of FXN and, in the wild type crystal structure, these residues do not engage in any specific interactions with other amino acid side chains (Supp. Figure S6, S7 and S12). Notably, the variants p.D104G, p.A107V, p.S161I, p.S181F and p.S202F that show a remarkable inhibition of Fe-S clusters formation involve residues that do not form a network of interactions with other side chains (Supp. Figure S6, S7, S10, S11 and S12). On the other hand, the variants p.F109L and p.Y123S that affect residues involved in a network of interaction with other side chains (Supp. Figure S8 and S9), are thermodynamically destabilized and show only a partial inhibitor effect on Fe-S cluster formation. Notably, the failure to obtain p.W173C as a soluble recombinant protein may be ascribed to the disruption of the network of hydrophobic interactions caused by the substitution of Trp173 with a cysteine residue (Supp. Figure S13).
The similarities in the spectral properties of the FXN variants with those of the wild type suggest that the effect of the mutations is localised to the neighbourhoods of the mutated residues, that involve non conserved surface residues, without affecting the global protein fold. Indeed, mutations of surface residues are usually well tolerated in proteins, they do not significantly affect the global folding and may promote new protein functions, though may be destabilizing (Tokuriki and Tawfik, 2009). In the case of FXN, the substitution of non conserved and solvent exposed residues, that do not alter the structural global fold, may give rise to a protein with alternative functions or promote new protein-protein interactions. The presence of the nsSNVs in some particular cancer types may suggest a possible advantage for their expression over the wild type in cancer cells. The main question that arise from our results concerns the importance of these variants. How a less stable, and apparently less efficient, FXN may be an advantage for the metabolic reprogramming of particular cancer cells and, more generally, for tumour onset and progression, is still an open question and will be a matter of further investigations.
Supplementary Material
Acknowledgments
The CAGI experiment coordination is supported by NIH U41 HG007446 and the CAGI conference by NIH R13 HG006650
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
REFERENCES
- Ackroyd R, Shorthouse AJ., & Stephenson TJ (1996). Gastric carcinoma in siblings with Friedreich’s ataxia. European Journal of Surgical Oncology, 22, 301–303. PMID:. [DOI] [PubMed] [Google Scholar]
- Barr H, Page R, & Taylor W (1986). Primary small bowel ganglioneuroblastoma and Friedreich’s ataxia. Journal of the Royal Society of Medicine, 79, 612–613. doi: 10.1177/014107688607901018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjwal S, Verma S, Rohm KH., Gursky O (2006). Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Science, 15, 635–639. doi: 10.1110/ps.051917406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, van der Spoel D, & van Drunen R (1995). GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications, 91, 43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Bonetti D, Toto A, Giri R, Morrone A, Sanfelice D, Pastore A, Temussi P, Gianni S, & Brunori M (2014). The kinetics of folding of frataxin. Physical Chemistry Chemical Physics,16, 6391–6397. doi: 10.1039/c3cp54055c. [DOI] [PubMed] [Google Scholar]
- Boniecki MT., Freibert SA, Mühlenhoff U, Lill R, & Cygler M Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. (2017). Nature Communications, 8, 1287. doi: 10.1038/s41467-017-01497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridwell-Rabb J, Iannuzzi C, Pastore A, & Barondeau DP (2012). Effector role reversal during evolution: the case of frataxin in Fe-S cluster biosynthesis. Biochemistry, 51–2506-2514. doi: 10.1021/bi201628j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürk K (2017). Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias, eCollection 2017, 4, 4. doi: 10.1186/s40673-017-0062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D, & Parrinello M (2007). Canonical sampling through velocity rescaling. Journal of Chemical Physics, 126, 014101. doi: 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- Cai K, Frederick RO, Tonelli M, & Markley JL (2018). Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. Journal of Inorganic Biochemistry, 183, 107–116. doi: 10.1016/j.jinorgbio.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Frederick RO , Tonelli M, & Markley JL (2018). ISCU(M108I) and ISCU(D39V) differ from wild-type ISCU in their failure to form cysteine desulfurase complexes containing both frataxin and ferredoxin. Biochemistry, 57, 1491–1500. doi: 10.1021/acs.biochem.7b01234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella A, Isaya G, O’Neill HA, Santambrogio P, Cozzi A, Arosio P, & Levi S (2004). The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Human Molecular Genetics,13, 2279–2288. doi: 10.1093/hmg/ddh232. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI., Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, & Pandolfo M Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. (1996). Science, 271, 1423–1427. PMID:. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, & Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research, 33(Web Server issue), W306–W10. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadio R, Vassura M, Tiwari S, Fariselli P, & Martelli LP (2011). Correlating disease-related mutations to their effect on protein stability: a large-scale analysis of the human proteome. Human Mutation, 32, 1161–1170. doi: 10.1002/humu.21555. [DOI] [PubMed] [Google Scholar]
- Choy N, Raussens V, & Narayanaswami V (2003). Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. Journal of Molecular Biology, 334, 527–539. PMID: . [DOI] [PubMed] [Google Scholar]
- Correia AR, Pastore C, Adinolfi S, Pastore A, & Gomes CM Dynamics., stability and iron-binding activity of frataxin clinical mutants. (2008). FEBS Journal, 275, 3680–3690. doi: 10.1111/j.1742-4658.2008.06512.x. [DOI] [PubMed] [Google Scholar]
- Cory SA., Van Vranken JG, Brignole EJ, Patra S, Winge DR, Drennan CL, Rutter J, & Barondeau DP (2017). Structure of human cysteine desulfurase complex. Proceedings of the National Academy of Sciences, 114, E5325–E5334; doi: 10.1073/pnas.1702849114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T York D & Pedersen L (1993). Particle mesh Ewald: An N·log (N) method for Ewald sums in large systems. Journal of Chemical Physics, 98, 10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM., Yoon JC, Park PJ, & Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell, 155, 948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, & Chandel NS. (2016). Fundamentals of cancer metabolism. Science Advances, 2: e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck Y, Kwasigroch JM, Gilis D, & Rooman M (2011). PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics, 12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EC, Seyer LA, Perlman SL, Yu J, & Lynch DR (2012). Clinical monitoring in a patient with Friedreich ataxia and osteogenic sarcoma. Journal of Child Neurology, 27:1159–1163. doi: 10.1177/0883073812448460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, & Shoelson SE (2000). Crystal structure of human frataxin. Journal of Biological Chemistry, 275, 30753–30756. doi: 10.1074/jbc.C000407200. [DOI] [PubMed] [Google Scholar]
- Di Carlo MG; Minicozzi V, Foderà V, Militello V, Vetri V, Morante S, & Leone M (2015). Thioflavin-T templates amyloid-β (1–40) conformation and aggregation pathway. Biophysical Chemistry, 206, 1–11. doi: 10.1016/j.bpc.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, & Kollman P (2003). A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. Journal of Computational Chemistry, 24, 1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research, 45(D1): D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foury F, Pastore A, & Trincal M (2007). Acidic residues of yeast frataxin have an essential role in Fe-S cluster assembly. EMBO Reports, 8, 194–199. doi: 10.1038/sj.embor.7400881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea CA, Huq A, Lockhart PJ., Tai G, Corben LA, Yiu EM, Gurrin LC, Lynch DR, Gelbard S, Durr A, Pousset F, Parkinson M, Labrum R, Giunti P, Perlman SL, Delatycki MB, & Evans-Galea MV (2016). Compound heterozygous FXN mutations and clinical outcome in Friedreich ataxia. Annals of Neurology, 79, 485–495. doi: 10.1002/ana.24595. [DOI] [PubMed] [Google Scholar]
- Gomes CM, & Santos R (2013). Neurodegeneration in Friedreich’s Ataxia: From Defective Frataxin to Oxidative Stress. Oxidative Medicine and Cellular Longevity, 2013, 487534. doi: 10.1155/2013/487534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccini I, Serio D, Condò I, Rufini A, Tomassini B, Mangiola A, Maira G, Anile C, Fina D, Pallone F, Mongiardi MP., Levi A, Ventura N, Testi R, & Malisan F (2011). Frataxin participates to the hypoxia-induced response in tumors. Cell Death and Disease, 2: e123. doi: 10.1038/cddis.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, Halweg C, Collins JB, Durr A, Fischbeck K, & Van Houten B (2010). Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLOS Genetics, 6, e1000812. doi: 10.1371/journal.pgen.1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, & Lindahl E (2008). Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, 4, 435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, & Schulten K (1996). VMD-Visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38. PMID: . [DOI] [PubMed] [Google Scholar]
- Hutter C, & Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell, 173, 283–285. doi: 10.1016/j.cell.2018.03.042. [DOI] [PubMed] [Google Scholar]
- Karki R, Pandya D, Elston RC, & Ferlini C (2015). Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Medical Genomics, 8, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd A, Coleman R, Whiteford M, Barron LH, Simpson SA, & Haites NE (2001). Breast cancer in two sisters with Friedreich’s ataxia. European Journal of Surgical Oncology, 27, 512–514. doi: 10.1053/ejso.2000.1093. [DOI] [PubMed] [Google Scholar]
- Kiss RS, Weers PM, Narayanaswami V, Cohen J, Kay CM, & Ryan RO (2003). Structure-guided protein engineering modulates helix bundle exchangeable apolipoprotein properties. Journal of Biological Chemistry, 278, 21952–21959. doi: 10.1074/jbc.M302676200. [DOI] [PubMed] [Google Scholar]
- Kroemer G, & Pouyssegur J (2008). Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell, 13, 472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Hess B, & van der Spoel D (2001). Gromacs 3.0: A package for molecular simulation and trajectory analysis. Journal of Molecular Modeling, 7, 306–317. [Google Scholar]
- Liu Y, Zhang Z, Wang J, Chen C, Tang X, Zhu J, & Liu J (2019). Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. OncoTargets and Therapy, 12, 1195–1204. doi: 10.2147/OTT.S189687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori C, Lantella A, Pasquo A, Alexander LT, Knapp S, Chiaraluce R, & Consalvi V (2013). Effect of single amino acid substitution observed in cancer on Pim-1 kinase thermodynamic stability and structure. PLoS One, 8(6):e64824. doi: 10.1371/journal.pone.0064824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori L, Pasquo A, Lori C, Petrosino M, Chiaraluce R, Tallant C, Knapp S, & Consalvi V (2016). Effect of BET missense mutations on bromodomain function., inhibitor binding and stability. PLoS One,11(7): e0159180. doi: 10.1371/journal.pone.0159180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Friedman LS, Reutenauer L, Messaddeq N, Perlman SL., Lynch DR, Fedosov K, Schulz JB, Pandolfo M, & Puccio H (2012). Clinical data and characterization of the liver conditional mouse model exclude neoplasia as a non-neurological manifestation associated with Friedreich’s ataxia. Disease Models & Mechanisms, 5, 860–869. doi: 10.1242/dmm.009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiakos EP., Siama E, Schizas D, Petropoulos C, Zavras N, Economopoulos N, Charalabopoulos A, & Macheras A (2011). Massive Uterine Leiomyoma in a Patient with Friedreich’s Ataxia: Is There a Possible Association? Case Reports in Medicine, 2011, 648217. doi: 10.1155/2011/648217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveberg M, Tan YJ, Silow M, & Fersht AR (1998). The changing nature of the protein folding transition state: implications for the shape of the free-energy profile for folding. Journal of Molecular Biology, 277, 933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]
- Orang AV, Petersen J, McKinnon RA, & Michael MZ (2019). Micromanaging aerobic respiration and glycolysis in cancer cells. Molecular Metabolism, pii: S2212–8778(18)31112–8. doi: 10.1016/j.molmet.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquo A, Consalvi V, Knapp S, Alfano I, Ardini M, Stefanini S, & Chiaraluce R (2012). Structural stability of human protein tyrosine phosphatase rho catalytic domain: effect of point mutations. PLoS One, 7(2), e32555. doi: 10.1371/journal.pone.0032555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore A, & Puccio H (2013). Frataxin: a protein in search for a function. Journal of Neurochemistry, 126 Suppl 1, 43–52. doi: 10.1111/jnc.12220. [DOI] [PubMed] [Google Scholar]
- Peng Y, Alexov E, & Basu S (2019). Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. International Journal of Molecular Sciences, 20(3). pii: E548. doi: 10.3390/ijms20030548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino M. Lori, ., Pasquo A, Lori C, Consalvi V, Minicozzi, Morante S, Laghezza A, Giorgi A, Capelli D, & Chiaraluce R (2017). Single-Nucleotide Polymorphism of PPARγ, a Protein at the Crossroads of Physiological and Pathological Processes. International Journal of Molecular Sciences, 18(2). pii: E361 pmid:28208577. doi: 10.3390/ijms18020361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukh M, Kucukkal TG, & Alexov E (2015). On human disease-causing amino acid variants: statistical study of sequence and structural patterns. Human Mutation, 36, 524–534. doi: 10.1002/humu.22770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan LM., Yeung SC, & Lee MH (2014). Cancer metabolic reprogramming: importance., main features., and potentials for precise targeted anti-cancer therapies. Cancer Biology and Medicine, 11, 1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeep L, & Udgaonkar JB (2004). Effect of salt on the urea-unfolded form of barstar probed by m value measurements. Biochemistry, 43, 11393–11402. doi: 10.1021/bi049320b. [DOI] [PubMed] [Google Scholar]
- Prischi F, Konarev PV, Iannuzzi C, Pastore C, Adinolfi S, Martin SR, Svergun DI, & Pastore A (2010). Structural bases for the interaction of frataxin with the central components of iron–sulphur cluster assembly. Nature Communications, 95, 1. doi: 10.1038/ncomms1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos E, Latash MP., Hurvitz EA, & Brown SH (1997). Quantification of upper extremity function using kinematic analysis. Archives of Physical Medicine and Rehabilitation, 78, 491–496. PMID:. [DOI] [PubMed] [Google Scholar]
- Rouault TA (2012). Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Disease Models & Mechanisms, 5, 155–164. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer CA, Mann CJ, & Matthews CR (1993). Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Science, 2, 1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychtarcikova Z, Lettlova S, Tomkova V, Korenkova V, Langerova L, Simonova E, Zjablovskaja P, Alberich-Jorda M, Neuzil J, & Truksa J (2017). Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget, 8, 6376–6398. doi: 10.18632/oncotarget.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MM, & Bolen DW (1988). Unfolding free energy changes determined by the linear extrapolation method.1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry, 27, 8063–8068. PMID: . [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Thierbach R, Voigt A, Drewes G, Mietzner B, Steinberg P, Pfeiffer AF, & Ristow M (2006). Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. Journal of Biological Chemistry, 281, 977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- Sreedhar A, & Zhao Y (2018). Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomedical Reports, 8, 3–10. doi: 10.3892/br.2017.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl S, Nishi H, Petukh M, Panchenko AR, & Alexov E (2013). Molecular mechanisms of disease-causing missense mutations. Journal of Molecular Biology, 425, 3919–3936. doi: 10.1016/j.jmb.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehling O, Wilbrecht C, & Lill R (2014). Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie, 100, 61–77. doi: 10.1016/j.biochi.2014.01.010. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- Tokuriki N, & Tawfik DS (2009). Stability effects of mutations and protein evolvability. Current Opinion in Structural Biology, 19, 596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Thierbach R, Schulz TJ, Isken F, Voigt A, Mietzner B. Drewes, ., von Kleist-Retzow JC, Wiesner RJ, Magnuson A, Puccio H, Pfeiffer AF, Steinberg P, & Ristow M (2005). Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function., decreased life span and tumor growth in mice. Human Molecular Genetics, 14, 3857–3864. doi: 10.1093/hmg/ddi410. [DOI] [PubMed] [Google Scholar]
- Thierbach R, Drewes G, Fusser M, Voigt A, Kuhlow D, Blume U, Schulz TJ, Reiche C, Glatt H, Epe B, Steinberg P, & Ristow M (2010). The Friedreich’s ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals. Biochemical Journal, 432, 165–172. doi: 10.1042/BJ20101116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CL, & Barondeau DP (2010). Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry, 49, 9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- Torti SV, & Torti FM (2011). Ironing out cancer. Cancer Research, 71, 1511–4. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV, & Torti FM (2013). Cellular iron metabolism in prognosis and therapy of breast cancer. Critical reviews in oncogenesis, 18, 435–448. doi: 10.1615/CritRevOncog.2013007784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV, & Torti FM (2013). Iron and cancer: more ore to be mined. Nature Reviews Cancer, 13, 342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV., Manz DH, Paul BT, Blanchette-Farra N, & Torti FM (2018). Iron and Cancer. Annual Review of Nutrition, 38, 97–125. doi: 10.1146/annurev-nutr-082117-051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Spoel D, Lindahl E, Hess B, Groenhor G, Mark AE, & Berendsen HJC (2005). Gromacs: Fast, flexible, and free. Journal of Computational Chemistry, 26, 1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Wrabl J, & Shortle D (1999). A model of the changes in denatured state structure underlying m value effects in staphylococcal nuclease. Nature Structural Biology, 6, 876–83. doi: 10.1038/12338. [DOI] [PubMed] [Google Scholar]
- Zanella I, Derosas M, Corrado M, Cocco E, Cavadini P, Biasiotto G, Poli M, Verardi R, & Arosio P (2008). The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochimica et Biophysica Acta, 1782, 90–98. doi: 10.1016/j.bbadis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Zjablovskaja P, Alberich-Jorda M, Neuzil J, & Truksa J (2017). Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget, 8, 6376–6398. doi: 10.18632/oncotarget.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.