

Abstract

KRAS regulates many cellular processes including proliferation, survival, and differentiation. Point mutants of KRAS have long been known to be molecular drivers of cancer. KRAS p.G12C, which occurs in approximately 14% of lung adenocarcinomas, 3–5% of colorectal cancers, and low levels in other solid tumors, represents an attractive therapeutic target for covalent inhibitors. Herein, we disclose the discovery of a class of novel, potent, and selective covalent inhibitors of KRASG12C identified through a custom library synthesis and screening platform called Chemotype Evolution and structure-based design. Identification of a hidden surface groove bordered by H95/Y96/Q99 side chains was key to the optimization of this class of molecules. Best-in-series exemplars exhibit a rapid covalent reaction with cysteine 12 of GDP-KRASG12C with submicromolar inhibition of downstream signaling in a KRASG12C-specific manner.
Keywords: Cancer, KRAS, G12C, irreversible inhibitor, nonsmall cell lung cancer, FBLD
RAS proteins are GTPases with essential roles in controlling the activity of several critical signaling pathways that regulate cell differentiation, proliferation, and survival.1−3 The three members of the RAS family of proteins, HRAS, NRAS, and KRAS, function as binary switches with an active GTP-bound state and an inactive GDP-bound state.1,3,4 KRAS is the most frequently mutated family member, with mutations observed in 90% of pancreatic adenocarcinomas, 45% of colorectal cancers, and 35% of lung adenocarcinomas.1,2 The KRAS p.G12C mutation results in a glycine-to-cysteine substitution at residue 12 and is found in approximately 14% of lung adenocarcinomas, 3–5% of colorectal cancers, and low levels in other solid tumor types.5,6 The presence of cysteine at position 12 in KRASG12C protects bound GTP from the rapid regulated hydrolysis catalyzed by GTPase activating protein (GAP) family proteins, resulting in overall pathway activation. While GTP-bound mutant KRAS has been termed “undruggable” due to a lack of accessible pockets for small molecule binding, several features of KRASG12C suggest its tractability as a drug discovery target. These include the cysteine residue as a target for covalent binding, relatively rapid cycling between inactive and active states, enhanced flexibility of the inactive state leading to a shallow small molecule binding site adjacent to the cysteine, and a favorable microenvironment for activation of acrylamides toward nucleophilic addition by cysteine 12.
While the KRASG12C target is compelling, it has been difficult to identify chemical starting points for covalent inhibitor programs. Shokat and co-workers were the first to report the use of tethering7 to identify reversible covalent binders of the inactive GDP-bound form of KRASG12C and successfully advanced the hits to a series of irreversible covalent inhibitors.8 These molecules form an irreversible covalent attachment to the mutant cysteine near the switch II region of the protein, binding to an allosteric site referred to as the P2 pocket.9 Compound binding to the P2 pocket impairs RAS function by trapping the protein in the inactive state.10 Building on this foundational work, Wellspring Biosciences reported optimized P2-pocket KRASG12C inhibitors ARS-85311 and ARS-1620.12 ARS-1620 was the first molecule reported to demonstrate in vivo efficacy in a mouse xenograft model.12
This Letter details a successful electrophile screen conducted by Amgen and Carmot Therapeutics, which led to a series of cell active, irreversible covalent small-molecule KRASG12C inhibitors that are chemically unique from previously reported structures.
Carmot Therapeutics has developed a technology called Chemotype Evolution,13 which provides rapid access to novel chemical diversity. The process begins with design of an anchor molecule or “bait”. The bait can be derived from known inhibitors, substrates, cofactors, peptides, hits from a fragment screen, or covalent warheads. In addition to its target-interacting components, the bait contains a reactive functionality that can be individually linked to molecules from Carmot’s collection of fragments, which has grown from the time this work was conducted to >20,000 members. The linked, two-component molecules constitute a biased, custom library that is screened against the target in a plate-based assay with one compound per well. Importantly, the linked molecules are made on a nanogram scale and screened without purification, which greatly accelerates the process. Hit compounds are resynthesized and purified on a milligram scale for confirmation. The process can be repeated iteratively, with new baits designed based on hits from prior screens, as illustrated below.
Using Chemotype Evolution, multiple electrophilic baits were derivatized and screened against GDP-KRASG12C in a series of assays including a thiol reactive probe assay,14 a RAF-coupled nucleotide exchange assay, and intact protein mass spectrometry. Aiming for diversity, we selected a number of structurally distinct bifunctional heterocycles for screening that were derivatized with an acryloyl group as well as a fragment library attachment site (Figure 1). The intrinsic reactivity of representatives of each class was determined in a glutathione (GSH) assay,15,16 and all were within the range of clinical covalent kinase inhibitors (t1/2 from 30 to >512 min). The relative reactivity of each heterocycle type follows an order that agrees with previously published data.17 We did not observe significant changes in intrinsic reactivity between hits and model acrylamides, likely due to the distance between the fragment and acrylamide in these systems. Azetidine bait A produced hits that were the most attractive and amenable to optimization and are the subject of this Letter. The success of azetidine bait A relative to the other baits may be due to relatively higher intrinsic reactivity or shape complementarity/interactions with KRAS when modified with appropriate fragments, or a combination of both.
Figure 1.
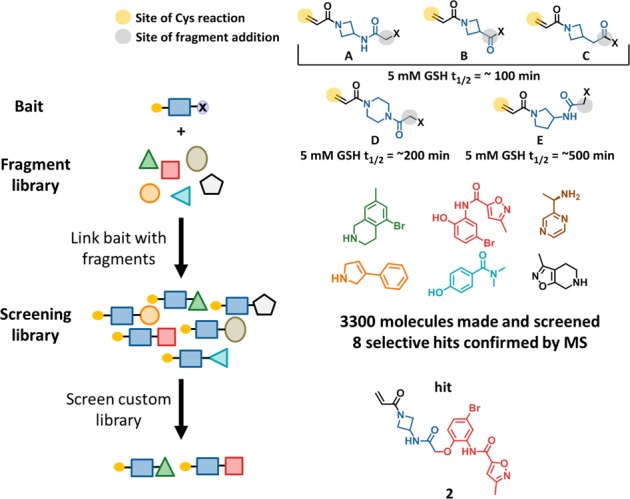
Chemotype Evolution leading to the discovery of 2. Yellow circle in top right illustrates reactive site of the acrylamide, blue illustrates the spacing element, and black indicates the reactive handle that can be coupled to individual fragments, examples of which are shown above. GSH half-life values shown are representative of bait-fragment conjugates.
A library was built from N-(1-acryloylazetidin-3-yl)-2-bromoacetamide (Figure 1), and each of the 3300 members was screened individually at 20 μM for 20 h against 2 μM protein. Compound 2 was one of the eight molecules that had confirmed binding by mass spectrometry (MS) (Figure 1). The mass spectrometry-based assay14 directly and quantitatively measures covalent adduct formation at cysteine-12 (C12) of KRAS, and reports the extent of conversion at a given time point under pseudo-first order kinetics. For convenience in comparing data collected at different time points and inhibitor concentrations, we convert the MS data between 10% and 90% conversion to an estimated rate constant kobs/[I] with units of M–1 s–1, using eq 1.
| 1 |
For 2, 2-h and 20-h incubations of 10 μM inhibitor and 2 μM KRASG12C exhibited 11% and 63% conversion, respectively. The estimated average rate (kobs/[I] M–1 s–1) for 2 by eq 1 was therefore 2 M–1 s–1.
Figure 2 depicts the X-ray cocrystal structure of 2 bound in the P2 pocket of KRASG12C. As expected, the acrylamide is covalently bound to C12. The acrylamide carbonyl forms a hydrogen bond to the K16 side chain (2.7 Å), while the secondary amide linker hydrogen bonds with the A59 backbone (2.9 Å) and the Y96 side chain hydroxyl group (3.1 Å). Switch II is in a closed conformation. The bromophenyl group projects deep into the pocket, with the bromine atom surrounded by relatively hydrophobic side chains. The pendant isoxazole-amide lies between the Q99 side chain and switch II. One unusual feature of this structure at the time it was obtained is the position of H95. Most often, the imidazole side chain is observed to pack closely between Y96 and Q99, yet in this structure H95 adopts an alternative position, leaving a gap between residues Y96 and Q99. The gap in this structure is occupied by resolved water molecules (not shown).
Figure 2.
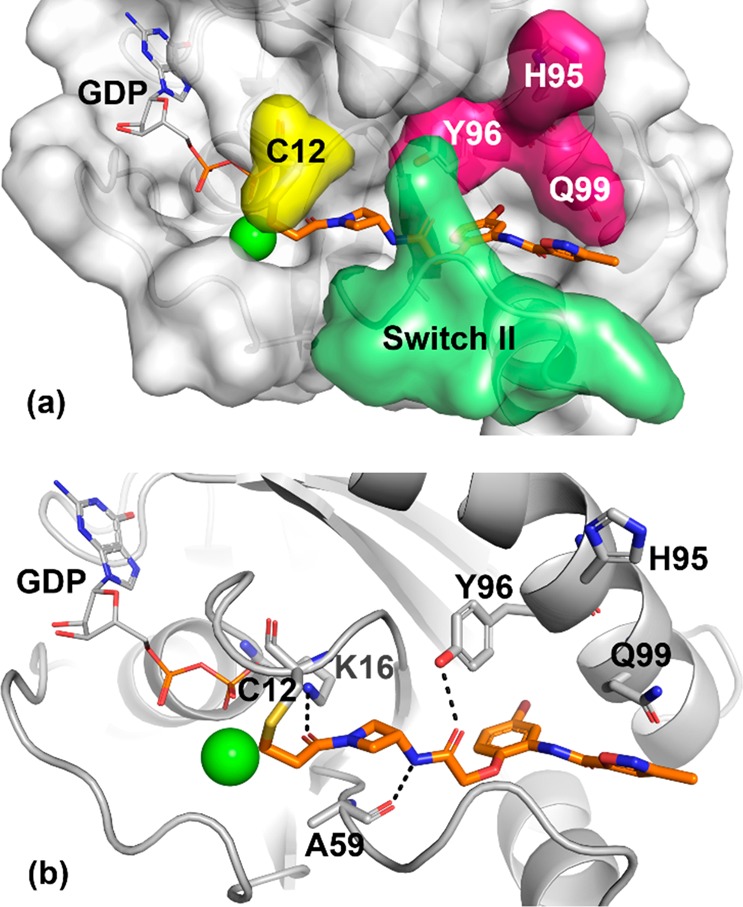
X-ray cocrystal structure of 2 (orange, PDB code: 6P8W) in complex with KRASG12C at 2.1 Å resolution. (a) Surface rendering. (b) Ribbon rendering.
Chemotype Evolution was used to improve the activity of the initial hit molecule. Second-generation baits were designed to provide substitution vectors that would further probe the binding site (Figure 3). This involved simplification of the initial hit followed by installation of a reactive moiety to enable library construction.
Figure 3.
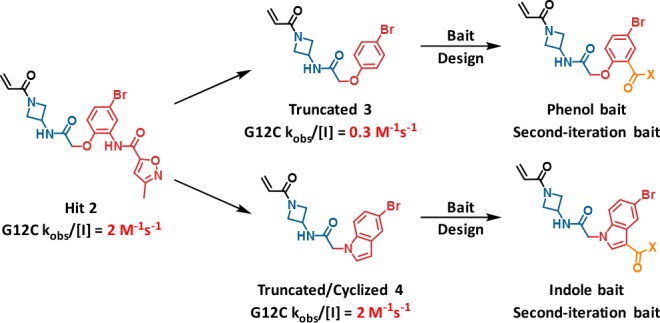
Design of second-generation baits.
The inactivation rate of the truncated phenol analog 3 was 0.3 M–1 s–1, while the truncated and cyclized indole analog 4 possessed a similar rate constant to 2 and was of considerably reduced molecular weight. The “phenol bait” and “indole bait” with activated esters at the indicated positions were prepared and used in Chemotype Evolution to generate 2600 and 2900 amide-containing molecules, respectively. Screening as before against GDP-KRASG12C yielded a higher hit rate, with 71 compounds confirming in the MS-based assay. Compound 5 was the most active hit from the phenol bait, with a KRASG12C modification rate of 26 M–1 s–1, approximately 10-fold greater than the initial hit 2. X-ray crystallography revealed that 5 binds in a similar orientation to the hit 2, with the key difference being that the phenyl dihydropyrrole substituent occupies the space between Y96 and Q99 (Figure 4a,b). In addition, the phenyl group engages H95 in an edge to π-face stacking interaction. We believe the successful occupancy of the Y96/H95/Q99 groove by the phenyl dihydropyrrole is responsible for the improved activity of the second-iteration hit molecule 5.
Figure 4.

Chemotype Evolution to generate phenol hit 5 and indole hit 6. X-ray cocrystal structure of 5 (blue, PDB code: 6P8X) in complex with KRASG12C at 2.1 Å resolution, (a) surface rendering and (b) ribbon rendering. (c–f) X-ray cocrystal structure of 6 (green, PDB code: 6P8Y) in complex with KRASG12C at 2.3 Å resolution: (c, d) surface and ribbon rendering of chain A, respectively and (e, f) surface and ribbon rendering of chain B, respectively.
In the screen with the indole bait, compound 6 was the most active hit, with a KRASG12C modification rate of 230 M–1 s–1, approximately 100-fold greater than the initial hit 2. An X-ray cocrystal structure of 6 bound to KRASG12C was obtained, with clear electron density for the ligand in two different conformations in chain A (Figure 4c,d) and chain B (Figure 4e,f) of the asymmetric unit cell. In chain A, 6 is covalently bound to C12 and extends into the switch II region, with switch II residing in an open conformation. The middle amide carbonyl forms a hydrogen bond with a conserved water between the G10 and T58 residues (3.0 Å), in addition to the Y96 side chain hydroxyl group (2.6 Å). The indole core is positioned over the R68 side chain and engages in van der Waals interactions. The amide at the indole 3-position exhibits a dihedral angle (CCCO dihedral = 155°) nearly coplanar with the indole. The 5-methoxy-tetrahydroisoquinoline (THIQ) motif is positioned in the space between Y96 and Q99, where it engages H95 in π–π stacking interactions. In chain B, the middle amide and indole are rotated relative to chain A. The amide at the indole 3-position exhibits a more twisted conformation (CCCO dihedral = 69°), allowing the THIQ to engage the space between Y96 and Q99 differently, with an edge to π-face stacking interaction with Y96 (3.8 Å) in addition to π–π stacking with H95. Nine residues of switch II are unresolved in chain B; therefore, it is difficult to understand protein–ligand interactions in this region. Overall, the X-ray cocrystal structure of 6 supports the hypothesis that, like 5, successful occupancy of the Y96/H95/Q99 groove is likely responsible for the improved activity of this second-iteration hit molecule 6.
Compounds 5 and 6 were further characterized in additional assays. A RAF-coupled nucleotide exchange assay was used to assess functional inactivation as a result of covalent binding to GDP-KRASG12C. Following preincubation of inhibitor and GDP-KRASG12C, guanine exchange factor son of sevenless (SOS) and GTP were added to exchange GTP into the nucleotide binding site. The extent of GTP loading could be read by determining the RAS-RAF Ras binding domain (RBD) proximity, as the RBD of RAF binds with high affinity to only the active state of KRAS. With a 2 h preincubation, 5 and 6 exhibited IC50 values of 2.78 and 0.638 μM, respectively, which was qualitatively consistent with the improved reaction rate of 6 with KRASG12C. A cellular assay was employed to determine inhibitory activity downstream of KRASG12C in the pancreatic adenocarcinoma cell line MIA PaCa-2. Cells were incubated with inhibitor for 4 h, and then the RAS pathway was stimulated by EGF addition, and ERK phosphorylation was determined. Compounds 5 and 6 exhibited IC50 values in the low double digit micromolar range in this assay.
Due to the improved activity of 6 relative to 5 and a lower number of rotatable bonds, the indole series was investigated further. A number of analogs were made to better understand the effect of the THIQ substituent in the indole hit 6, as summarized in Table 1. Aliphatic ring-opened variants (N-benzyl-carboxamide (7), N-(2-phenylethyl)-carboxamide (8), and N-benzyl-N-methyl-carboxamide (9)) improved activity relative to the unsubstituted indole 4. However, all three compounds demonstrated weaker activity than the THIQ. Substitution around the THIQ moiety itself was generally tolerated, with unsubstituted 10 showing a high rate of modification (308 M–1 s–1). This translated into improved biochemical potency (IC50 = 1.27 μM), but the cellular activity remained weak (IC50 = 36.5 μM). Overall, further modification of the THIQ portion of the molecule was not productive.
Table 1. Impact of 3-Indole Substitution on Potencya.
Relative % bound at 10 μM of compound at 0.02, 0.2, 2, and 20 h.
Observed rate (kobs/[I] M–1 s–1) derived from 0.02, 0.2, 2, and 20 h of relative % bound at 10 μM compound.
Dose response GDP-KRASG12C/C118A (1–169) SOS1-catalyzed GTP exchange assay.
Quantification of phospho-ERK1/2 following compound treatment and EGF stimulation by MSD assay in MIA PaCa-2 cells. For number of replicates, mean, and standard deviations, see the Supporting Information.
To further improve the activity of 6, the substitution pattern on the indole scaffold was explored (Table 2). Methyl group scanning was employed to quickly identify fruitful positions of substitution. Addition of a methyl group to the 2-position of the indole improved biochemical potency of 15 in the GDP-KRASG12C/C118A SOS1-catalyzed GTP 2 h exchange assay 2-fold (IC50 = 0.299 μM), while the cellular potency improved 7-fold (IC50 = 1.68 μM) compared to 6. Introduction of a 2-cyclopropyl motif further improved the cellular potency of 16 (IC50 = 0.604 μM, 19-fold improvement) over 6. 7-Methylindole 17 did not show any improvement in cellular potency (IC50 = 12.8 μM); however, 2,7-dimethyl substitution on the indole improved the cellular potency (IC50 = 0.299 μM) of 18 38-fold over 6. The corresponding 5-chloro analogs also showed similar improvements in activity. 5-Chloro-2-cyclopropyl-7-methylindole analog 1 showed the best overall activity profile, with p-ERK inhibition IC50 of 0.219 μM, and was therefore selected for further profiling.
Table 2. Lead Optimization by Substitution on the Indole Scaffolda.

Relative % bound at 10 μM of compound at 0.02, 0.2, 2, and 20 h.
Observed rate (kobs/[I] M–1 s–1) derived from 0.02, 0.2, 2, and 20 h of relative % bound at 10 μM of compound.
Dose response GDP-KRASG12C/C118A (1–169) SOS1-catalyzed GTP exchange assay.
Quantification of phospho-ERK1/2 following compound treatment and EGF stimulation by MSD assay in MIA PaCa-2 cells. For number of replicates, mean, and standard deviations, see the Supporting Information.
An X-ray cocrystal structure of 1 covalently bound to KRASG12C is shown in Figure 5. The binding mode of 1 is very similar to that observed for 6 in chain B (Figure 4e,f and Figure 5c, overlay). From comparison of these cocrystal structures, we identify two possible sources for improved activity of compound 1 relative to compound 6. First, the cyclopropyl group at the 2-position of the indole likely reduces the energetic cost of the twisted CCCO dihedral (64 degrees) between indole and C3-carbonyl through steric destabilization of the coplanar conformation, as well as through intramolecular van der Waals interactions of the cyclopropane and THIQ ring. Second, the methyl group at the 7-position of the indole is within van der Waals distance of a bound water molecule, T58, Y71, and G60. It may also be that switch II plays a role in the differences between 1 and 6, but the unresolved residues for this loop near the ligand binding site preclude further analysis. Finally, we note that throughout this optimization program, we have observed a large variety of switch II positions, including closed, open, and unresolved, in a manner that does not appear to be correlated to the activity of the covalent ligand.
Figure 5.
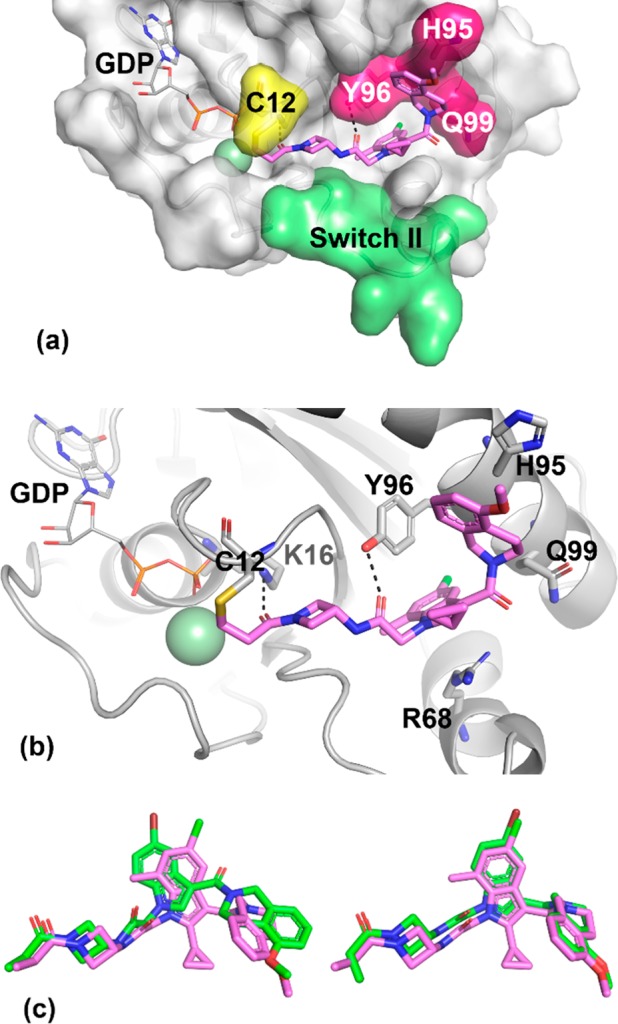
X-ray cocrystal structure of advanced lead 1 (pink, PDB code: 6P8Z) in complex with KRASG12C at 1.65 Å resolution. (a) Surface rendering. (b) Ribbon rendering. (c) Overlay of indole hit 6 (green, chain A on left and chain B on right) and advanced lead 1 (pink).
Further in vitro and in vivo profiling of 1 was conducted (Table 3). As previously noted, 1 reacted rapidly with KRASG12C and was capable of inhibiting SOS-catalyzed nucleotide exchange. The noncovalent binding of 1 to KRASG12 (containing wild-type glycine at amino acid 12) could be measured by proton direct deconvolution NMR (1H-ddNMR), and the KD was estimated to be 20 ± 5 μM (Figure S1). With respect to cellular activity, 1 exhibited IC50 = 0.219 μM for inhibition of MAPK signaling (p-ERK) in MIA PaCa-2, and this translated to a 0.067 μM IC50 for inhibition of cellular viability in a 72 h CellTiter-Glo experiment. Importantly, 1 exhibited only marginal activity when tested against the nontarget (KRAS p.G12S) cell line A549. Compound 1 exhibited reasonable stability to 5 mM glutathione (GSH) and showed acceptable solubility in fasted-state simulated intestinal fluid (FaSIF), but high turnover in both rodent microsomes and hepatocytes was correlated with rapid clearance in both mouse and rat. Oral bioavailability was low likely due to high first-pass clearance. Therefore, 1 was not advanced into further studies. However, the efforts leading to 1 identified previously unobserved, actionable subsites in the P2 pocket. These structural learnings formed the basis of a scaffold-hopping approach that led to the identification of a highly successful hybrid series of molecules, exemplified by AMG 510,18 which has advanced into human clinical trials for the treatment of advanced cancers with the KRAS p.G12C mutation.19 The SAR leading up to the discovery of AMG 510 will be reported in due course.
Table 3. Profile of KRASG12C Inhibitor 1.
| calcd and measured properties | |
| MW | ACD_LogP | ACD_LogD (7.4) | PSA | 561 | 2.5 | 2.5 | 84 |
| 5 mM GSH t1/2 (min)a | 80 |
| solubility (μM, PBS | FaSIF | HCl)b | 0 | 252 | 0 |
| biochemical | |
| KRASG12 binding KD(μM)c | 20 ± 5 |
| exchange 2 h IC50 (μM)d | 0.115 |
| cellular | |
| pERK 4 h IC50 MIA PaCa-2 | A549 (μM)e | 0.219 | > 10 |
| viability 72 h IC50 MIA PaCa-2 | A549 (μM)f | 0.067 | 9.33 |
| in vitro ADME | |
| MuLM | RLM | HLM Clint (μL/min/mg)g | 708 | 197 | 161 |
| Mu | R | H hep Clint (μL/min/106 cells)h | 228 | 293 | 53 |
| PPB Mu | R | Hu (5 μM, UC, fu)i | 0.02 | 0.02 | 0.04 |
| in vivo PKDM | |
| mouse Cl (L/h/kg) | Vss(L/kg) | t1/2(h) | %F (10 mpk)j | 6.4 | 2.9 | 0.9 | 8 |
| rat Cl (L/h/kg) | Vss (L/kg) | t1/2 (h) | %F (10 mpk)j | 3.9 | 2.6 | 0.7 | <1 |
Determined by the rate of disappearance of parent.15
Determined by an HPLC method after equilibration at rt for 72 h.
Noncovalent binding to KRASG12 by 1H-ddNMR.
GDP-KRASG12C/C118A (1–169) SOS1-catalyzed GTP exchange assay.
Quantification of phospho-ERK1/2 following compound treatment and EGF stimulation in MIA PaCa-2 (KRAS p.G12C) or A549 (KRAS p.G12S) cells.
Assessed by CellTiter-Glo.
Intrinsic clearance determined by LC/MS after incubation of parent compound (1 μM) in liver microsomes (0.25 mg/mL) in potassium phosphate buffer supplemented with NADPH (1 mM) at 37 °C for 30 min.
Hepatocyte intrinsic clearance assay (mouse, rat, human, 0.5 μM, 0.5 million cells/mL, 60 min).
Plasma protein binding determined by ultracentrifugation at 5 μM.
1 mg/kg iv dose in 100% DMSO, 10 mg/kg po dose in 1% Tween 80, 2% HPMC, 97% water.
Acknowledgments
We thank Trace Tsuruda for protein expression, Yuping Chen for GSH assay, Leszek Poppe for 1H-ddNMR, and Tatiana Didenko for NMR characterization.
Glossary
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, and excretion
- Cl
clearance
- Clint
intrinsic clearance
- EGF
epidermal growth factor
- F
bioavailability
- GSH
glutathione
- HLM
human liver microsome
- HPMC
hydroxypropyl methylcellulose
- LC/MS
liquid chromatography/mass spectrometry
- MS
mass spectrometry
- MW
molecular weight
- MuLM
mouse liver microsome
- NADPH
nicotinamide adenine dinucleotide phosphate
- ND
not determined
- PPB
plasma protein binding
- PSA
polar surface area
- RLM
rat liver microsome
- THIQ
tetrahydroisoquinoline
- Vss
volume of distribution
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00258.
Experimental details and synthetic scheme. Crystallography table of statistics. Mean, standard deviation, and n for biological replicates (PDF)
Accession Codes
The X-ray cocrystal structures have been deposited with the Protein Data Bank under Accession ID codes 6P8W, 6P8X, 6P8Y, and 6P8Z.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Adjei A. A. Blocking oncogenic ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001, 93 (14), 1062–1074. 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3 (1), 11–22. 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Wennerberg K.; Rossman K. L.; Der C. J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118 (5), 843–846. 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- Karnoub A. E.; Weinberg R. A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9 (7), 517–531. 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discovery 2017, 7 (8), 818–831. 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A. D.; Fesik S. W.; Kimmelman A. C.; Luo J.; Der C. J. Drugging the undruggable RAS: Mission Possible?. Nat. Rev. Drug Discovery 2014, 13 (11), 828–851. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson D. A.; Braisted A. C.; Raphael D. R.; Randal M.; Stroud R. M.; Gordon E. M.; Wells J. A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (17), 9367–9372. 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (London, U. K.) 2013, 503 (7477), 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; Lukman S.; Hocker H. J.; Sayyah J.; Brown J. H.; McCammon J. A.; Gorfe A. A. Novel allosteric sites on Ras for lead generation. PLoS One 2011, 6 (10), e25711. 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M. L.; Shokat K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 2016, 15 (11), 771–785. 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Janes M. R.; Li L.-S.; Hansen R.; Peters U.; Kessler L. V.; Chen Y.; Kucharski J. M.; Feng J.; Ely T.; Chen J. H.; Firdaus S. J.; Babbar A.; Ren P.; Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery 2016, 6 (3), 316–329. 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L.-S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; Feng J.; Chen J. H.; Li S.; Li S.; Long Y. O.; Thach C.; Liu Y.; Zarieh A.; Ely T.; Kucharski J. M.; Kessler L. V.; Wu T.; Yu K.; Wang Y.; Yao Y.; Deng X.; Zarrinkar P. P.; Brehmer D.; Dhanak D.; Lorenzi M. V.; Hu-Lowe D.; Patricelli M. P.; Ren P.; Liu Y. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell (Cambridge, MA, U. S.) 2018, 172 (3), 578–589.e17. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Hansen S.; Erlanson D. Methods of chemotype evolution. US10107798B2, 2018.
- Campuzano I. D. G.; San Miguel T.; Rowe T.; Onea D.; Cee V. J.; Arvedson T.; McCarter J. D. High-throughput mass spectrometric analysis of covalent protein-inhibitor adducts for the discovery of irreversible inhibitors: a complete workflow. J. Biomol. Screening 2016, 21 (2), 136–144. 10.1177/1087057115621288. [DOI] [PubMed] [Google Scholar]
- Cee V. J.; Volak L. P.; Chen Y.; Bartberger M. D.; Tegley C.; Arvedson T.; McCarter J.; Tasker A. S.; Fotsch C. Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015, 58 (23), 9171–9178. 10.1021/acs.jmedchem.5b01018. [DOI] [PubMed] [Google Scholar]
- Flanagan M. E.; Abramite J. A.; Anderson D. P.; Aulabaugh A.; Dahal U. P.; Gilbert A. M.; Li C.; Montgomery J.; Oppenheimer S. R.; Ryder T.; Schuff B. P.; Uccello D. P.; Walker G. S.; Wu Y.; Brown M. F.; Chen J. M.; Hayward M. M.; Noe M. C.; Obach R. S.; Philippe L.; Shanmugasundaram V.; Shapiro M. J.; Starr J.; Stroh J.; Che Y. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014, 57 (23), 10072–10079. 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- Palkowitz M. D.; Tan B.; Hu H.; Roth K.; Bauer R. A. Synthesis of Diverse N-Acryloyl Azetidines and Evaluation of Their Enhanced Thiol Reactivities. Org. Lett. 2017, 19 (9), 2270–2273. 10.1021/acs.orglett.7b00788. [DOI] [PubMed] [Google Scholar]
- Cee V.Discovery of AMG 510, a first-in-human covalent inhibitor of KRASG12C for the treatment of solid tumors; American Chemical Society: 2019; pp MEDI-0271. [Google Scholar]
- A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation. https://clinicaltrials.gov/ct2/show/NCT03600883.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.