

Abstract
Hypoxia occurs in many human solid tumors and activates multiple cellular adaptive-response pathways, including the unfolded protein response (UPR) in the endoplasmic reticulum (ER). Wnt/β-catenin signaling plays a critical role in tumorigenesis, and β-catenin has been shown to enhance hypoxia-inducible factor 1α (HIF1α)-activated gene expression, thereby supporting cell survival during hypoxia. However, the molecular interplay between hypoxic ER stress, Wnt/β-catenin signaling, and HIF1α-mediated gene regulation during hypoxia remains incompletely understood. Here, we report that hypoxic ER stress reduces β-catenin stability, which, in turn, enhances the activity of spliced X-box–binding protein 1 (XBP1s), a transcription factor and signal transducer of the UPR, in HIF1α-mediated hypoxic responses. We observed that in the RKO colon cancer cell line, which possesses a Wnt-stimulated β-catenin signaling cascade, increased ER stress during hypoxia is accompanied by a reduction in low-density lipoprotein receptor-related protein 6 (LRP6), and this reduction in LRP6 decreased β-catenin accumulation and impaired Wnt/β-catenin signaling. Of note, β-catenin interacted with both XBP1s and HIF1α, suppressing XBP1s-mediated augmentation of HIF1α target gene expression. Furthermore, Wnt stimulation or β-catenin overexpression blunted XBP1s-mediated cell survival under hypoxia. Together, these results reveal an unanticipated role for the Wnt/β-catenin pathway in hindering hypoxic UPR-mediated responses that increase cell survival. Our findings suggest that the molecular cross-talks between hypoxic ER stress, LRP6/β-catenin signaling, and the HIF1α pathway may represent an unappreciated mechanism that enables some tumor subtypes to survive and grow in hypoxic conditions.
Keywords: endoplasmic reticulum stress (ER stress), cancer biology, hypoxia-inducible factor (HIF), X-box–binding protein 1 (XBP1), Wnt signaling
Introduction
Hypoxia, a common stress state in most human solid tumors due to the highly chaotic architecture of blood vessels, exerts critical influences upon tumorigenesis and tumor growth (1). Tumor cells must survive in a hostile microenvironment characterized by low levels of oxygen, glucose, and pH value. In response to hypoxia, tumor cells undergo a variety of adaptive alterations for survival and growth, including O2-sensitive pathways that are orchestrated by the hypoxia-inducible factor family of transcription factors (HIFs)3 (2). HIF1 is composed of two subunits, the constitutive nuclear HIF1β subunit and the O2-regulated cytosolic HIF1α subunit. Under normoxic conditions, HIF1α is subjected to hydroxylation catalyzed by prolyl hydroxylase enzymes, leading to its proteasomal degradation. Under hypoxia (typically less than 3–5% O2) states, however, HIF1α is stabilized and translocates to the nucleus where it can dimerize with HIF1β. The HIF1 heterodimer in turn recognizes and binds to hypoxia-response elements (HREs) in the genome to activate the transcription of a series of hypoxia-responsive genes (3). Clinical studies utilizing oxygen electrodes and molecular markers of hypoxia have shown that tumors are poorly oxygenated, whereas such oxygenation is extremely heterogeneous (4). Depending on the intensity and duration of the hypoxic stimuli, HIF1 can trigger activation of the transcription of genes involved in either adaptive responses or detrimental processes. When exposed to hypoxia, cells undergo temporary cell cycle arrest, reduced energy consumption, and secrete various survival and pro-angiogenic factors.
Hypoxia can also cause perturbations in the loading and processing of newly synthesized proteins, thus triggering endoplasmic reticulum (ER) stress in cancer cells (5–7). In order to cope with hypoxic tumor microenvironment-induced ER stress, tumor cells initiate an adaptive response that aims to restore cellular homeostasis, referred to as the unfolded protein responses (UPR) (8). The ER-localized transmembrane proteins, inositol-requiring enzyme 1 (IRE1), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), act to mediate three canonical UPR signaling branches. Activation of the UPR allows cells to alleviate ER stress under hypoxic conditions within tumors. However, chronic activation of the UPR can also lead to cell death under severe and/or persistent ER stress (9). Emerging evidence indicates that activation of the UPR is linked to hypoxia tolerance and tumor growth (5). For instance, activation of PERK and the subsequent phosphorylation of eukaryotic initiation factor 2α (eIF2α) act through two major mechanisms in protein translational control in response to hypoxic ER stress (10). Although globally repressing the initiation of mRNA translation and rapidly down-regulating protein synthesis, activation of the PERK–eIF2α branch can also selectively up-regulate the expression of a subset of genes by the transcription factor ATF4, thereby promoting cell survival (11). The transcription factor X-box–binding protein 1 (XBP1) has also been documented as another important player in handling hypoxic ER stress (12). XBP1 is regulated by the protein Ser/Thr kinase/endoribonuclease IRE1α, which is activated through autophosphorylation and dimerization/oligomerization upon ER stress (13). IRE1α catalyzes the nonconventional splicing of Xbp1 mRNA, thereby generating the spliced form XBP1s that activates a key transcriptional program of the UPR (8). Under hypoxic conditions, XBP1s has been shown to be a critical cell survival factor in vitro and required for optimal tumor growth in vivo (12). Recently, genome-wide mapping of the XBP1 transcriptional regulatory network revealed that XBP1s drives tumorigenesis of triple-negative breast cancer by assembling a transcriptional complex with HIF1α to cooperatively activate the expression of the HIF1α gene-expression program (14).
Many signaling pathways in cell development and growth control are engaged in hypoxia responses, including the evolutionarily conserved Wnt/β-catenin pathway that has essential roles in embryonic development, tissue homeostasis, and tumorigenesis (15–17). In the absence of Wnt stimulation, cytoplasmic β-catenin protein forms a destruction complex with the scaffolding protein Axin, the tumor suppressor adenomatous polyposis coli gene product (APC), casein kinase 1 (CK1), and glycogen synthase kinase (GSK) 3β. Upon phosphorylation by CK1 and GSK3β, β-catenin is targeted by the E3 ubiquitin ligase β-Trcp for proteosomal degradation. Canonical Wnt/β-catenin signaling is initiated by binding of Wnt proteins to the Frizzled family member receptors and subsequent complex formation with the low-density lipoprotein receptor–related protein 5/6 (LRP5/6) co-receptors (18–20). Stimulation by Wnt signals leads to disassembly of the destruction complex and thus inhibition of the β-catenin breakdown, allowing for its accumulation and localization in the nucleus. As a transcriptional co-activator, β-catenin interacts with the T-cell transcription factor (TCF)/lymphoid enhancer–binding factor family of DNA-binding proteins to activate the expression of Wnt target genes such as CyclinD1 and c-MYC proto-oncogenes. It has been well-documented that deregulated Wnt/β-catenin signaling is associated with cancer (21–23). Genetic alterations in the APC and CTNNB1 (β-catenin) genes leading to abnormal accumulation of intracellular β-catenin occur very commonly in human colon cancer as well as other malignancies (24, 25). Moreover, LRP6 expression has also been found to be frequently up-regulated in several types of cancer (26, 27). Notably, canonical Wnt/β-catenin signaling was reported to cross-talk with hypoxia-response pathways in tumor progression and metastasis, and direct interaction has been found between β-catenin and HIF1α, implying potential competition for β-catenin between HIF1α and TCF-4 (28).
Apparently, complex interplays exist between the cell survival signaling network and multiple adaptive-response pathways in the face of hypoxia. It remains incompletely understood, however, how the Wnt/β-catenin signaling and the UPR branches are integrated with the HIF1α pathway in context-dependent and/or cell type-selective manners to manage hypoxic stress and promote cell survival. In this study, we investigated whether Wnt/β-catenin signaling interconnects with the UPR branch and HIF1α-regulated hypoxia-response program in RKO colon cancer cells possessing normal Wnt/β-catenin signaling. We found that hypoxic ER stress resulted in destabilization of β-catenin, largely because of reduced LRP6 production. Interestingly, we also found that β-catenin could negatively regulate XBP1s-mediated promotion of the HIF1α-activated transcriptional program, suggesting that hypoxic suppression of β-catenin may facilitate a more efficient XBP1s–HIF1α cooperation for cell survival.
Results
Hypoxia leads to activation of the UPR with simultaneously decreased β-catenin signaling
To examine whether hypoxia stress influences both the UPR and Wnt/β-catenin–signaling pathways, we utilized the RKO colon cancer cell line without aberrant β-catenin activation. Consistent with the reported findings that UPR activation constitutes an important component of the hypoxia response (10, 29, 30), we observed that in parallel with a higher accumulation of HIF1α protein (Fig. 1A), phosphorylation of IRE1α (at Ser724 within its kinase domain) as well as eIF2α significantly increased in RKO cells in response to hypoxia (1% O2). Suggesting a hypoxic ER stress state, we also detected increased Xbp1 mRNA splicing along with elevated mRNA abundance of the XBP1 target genes, the ER chaperone BiP, and the ER-localized DnaJ 4 (ERdj4) (Fig. 1B). In contrast to the accumulation of HIF1α protein, we observed significantly decreased β-catenin protein levels (Fig. 1A), but no significant changes in its mRNA levels (Fig. 1C), following exposure to hypoxia. Consistently, gene expression profiling analyses revealed significantly down-regulated mRNA levels of β-catenin target genes, including DKK1, c-MYC, AXIN2, and CyclinD1 (Fig. 1D), along with markedly up-regulated mRNA levels of typical HIF1α targets such as VEGFa, GLUT1, PDK1, DDIT4, ID1, JMJD1a, and MCT4 (Fig. 1E). Moreover, similar hypoxic UPR activation accompanied by decreased β-catenin signaling was also observed in HEK293 cells (Fig. S1). These results showed that hypoxic ER stress accompanied the down-regulation of β-catenin signaling, suggesting that hypoxia may link the Wnt/β-catenin signaling pathway to the UPR-mediated adaptive response.
Figure 1.

Hypoxic ER stress accompanies down-regulation of β-catenin signaling in RKO cells. A, RKO cells were cultured under moderate hypoxia conditions (1% O2) for the indicated time intervals. Protein expression levels of HIF1α and β-catenin, as well as phosphorylation of IRE1α (p-IRE1α) and eIF2α (p-eIF2α), were analyzed by immunoblotting. Quantification of HIF1α and β-catenin levels (relative to α-tubulin) and p-IRE1α/IRE1α and p-elF2α/elF2α ratios is shown after normalization to the value at 0 h. B–D, RKO cells were cultured under hypoxia (H, 1% O2) or normoxia (N, 21% O2) for 12 h. B, quantitative RT-PCR analysis of the expression of the spliced form of XBP1 (XBP1s) mRNA relative to the total amount of XBP1 (XBP1t) mRNA, and the mRNA levels of ERdj4 and Bip (relative to actin). Values were normalized to normoxic levels. C–E, quantitative RT-PCR analysis of the mRNA levels of β-catenin (C), β-catenin target genes (c-MYC, cyclin D1, DKK1, and AXIN2) (D), and hypoxia-responsive HIF1α target genes (VEGFa, GLUT1, PDK1, DDIT4, ID1, JMJD1a, and MCT4) (E). All data are shown as the mean ± S.D. from three independent experiments. ns, not significant; *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by Student's t test.
ER stress results in decreased stability of β-catenin
To affirm that ER stress can affect β-catenin signaling, we treated RKO cells with two chemical ER stressors, tunicamycin (TM, which blocks the first step of N-linked glycosylation) and thapsigargin (TG, which inhibits the ER Ca2+-dependent ATPase to cause depletion of calcium from the ER lumen). Indeed, both TM- and TG-induced ER stress, as indicated by elevated eIF2α phosphorylation, resulted in gradual decreases in the Wnt3a-stimulated accumulation of β-catenin in RKO cells (Fig. 2A). Likewise, similar decreases in Wnt3a-stimulated accumulation of β-catenin were also observed in TM- and TG-treated HEK293 cells (Fig. S2A). Consistently, such ER stress-induced reduction in β-catenin accumulation was associated with decreased luciferase activity from the TOPFlash reporter (31) that can be activated by the β-catenin/TCF complex (Fig. S2B). Next, we tested whether the chemical chaperones, tauroursodeoxycholic acid (TUDCA) and 4-phenylbutyric acid (4-PBA) that are known for their ability to alleviate ER stress (32), could have an effect upon hypoxia-induced decrease of β-catenin protein in RKO cells. Both TUDCA and 4-PBA reduced hypoxia-induced eIF2α phosphorylation and exhibited a partial reversal effect upon the decrease of β-catenin protein following exposure to hypoxia (Fig. S3).
Figure 2.

ER stress results in decreased stability of β-catenin in RKO cells. A, RKO cells were treated with TM (10 μg/ml) (upper panel) or TG (1 μm) (lower panel) in the presence of control cell medium (LCM) or Wnt3a-producing cell medium (WCM) for the indicated time intervals. Immunoblot analysis of β-catenin and phosphorylation of elF2α are shown. Quantification of the expression level of β-catenin relative to α-tubulin is shown. B, RKO cells were preincubated with 5 μm MG132 or 50 μm CQ for 2 h before treatment with DMSO or 1 μm TG for another 12 h. Immunoblot analysis of β-catenin protein levels and quantification are shown in the bar graph after normalization to the value of nontreated LCM control cells. C, RKO cells were preincubated with 10 or 25 μm LiCl for 2 h and then treated by 1 μm TG for another 8 h. Immunoblot analysis of β-catenin protein levels and quantification are shown in the bar graph after normalization to the value of nontreated LCM control cells. All results represent three independent experiments.
Given that ER stress had no effect upon the levels of β-catenin mRNA in RKO or HEK293 cells (Fig. S4A), we wondered whether the ER stress-induced decrease in β-catenin protein was related to the ubiquitin proteasome system that is considered to be the major mechanism for controlling the stability of β-catenin via its phosphorylation by GSK3β and CK1α. In the absence or presence of Wnt3a stimulation, treatment of RKO cells with the proteasome inhibitor MG132, but not the inhibitor of lysosomal endopeptidases chloroquine (CQ), resulted in dramatic increases in β-catenin protein levels and prominently diminished the suppression by TG-induced ER stress of β-catenin protein accumulation (Fig. 2B). Furthermore, treatment with LiCl, which inhibits GSK-3β activity, led to increased accumulation of β-catenin in a dose-dependent fashion even in the presence of TG-induced ER stress (Fig. 2C), despite that such effects were less pronounced in the presence of Wnt3a stimulation. Together, these data suggest that ER stress could promote the degradation of β-catenin, likely through impairing the Wnt-signaling pathway upstream of GSK-3β.
ER stress and hypoxia cause destabilization of β-catenin via down-regulation of LRP6
To determine how experimental ER stress and hypoxia could promote β-catenin's destabilization via affecting the Wnt signaling cascade, we examined the level of LRP6 protein, the single-span transmembrane co-receptor that is essential for canonical Wnt signaling (19, 20). Notably, chemically-induced ER stress resulted in significant reduction of matured LRP6 protein in RKO or HEK293 cells treated with TM and TG (Fig. 3A) without alterations in its mRNA abundance (Fig. S4B), and an LRP6 protein band of lower molecular weight was detected in TM-treated cells, most likely representing its nonglycosylated form due to TM inhibition of N-glycosylation. Because LRP6 protein contains an extracellular domain with auto-inhibitory activity, we utilized the N-terminally truncated LRP6ΔN, a constitutively activated receptor that lacks most of the extracellular domain and can bind to Axin in the absence of Wnt ligand stimulation (31), in order to test the effect of restored LRP6 signaling upon ER stress-promoted β-catenin degradation. Interestingly, overexpression of LRP6ΔN in HEK293 cells markedly increased β-catenin protein accumulation and prevented its degradation upon TM or TG treatment (Fig. 3B), whereas Wnt3a stimulation that did not affect LRP6 protein levels had no such effect. In addition, gradual decreases in LRP6 protein levels, but not its mRNA abundance (Fig. S4C), were also detected in RKO and HEK293 cells following exposure to hypoxia (Fig. 3C), and restored overexpression of LRP6ΔN or full-length LRP6 was able to blunt hypoxia-induced reduction in β-catenin accumulation in HEK293 cells (Fig. 3D). These results indicate that hypoxia and ER stress resulted in impaired cellular production of LRP6, which in turn led to promotion of β-catenin destabilization and down-regulation of Wnt/β-catenin signaling.
Figure 3.

Hypoxia- and ER stress-responsive destabilization of β-catenin results from impaired production of LRP6. A, RKO or HEK293 cells were treated with 10 μg/ml TM or 1 μm TG for 8 h, and LRP6 protein level was analyzed by immunoblotting. B, HEK293 cells were transfected with vector or plasmid expressing LRP6ΔN and then subjected to TM or TG treatment as indicated. Immunoblot analysis of LRP6, LRP6ΔN, and β-catenin protein levels is shown. C, RKO or HEK293 cells were cultured under 1% O2 for the indicated time intervals, and LRP6 protein was analyzed by immunoblotting. D, HEK293 cells transfected with vector, LRP6ΔN, or LRP6 plasmid were subjected to 1% O2 hypoxia or normoxia for 12 h. Shown are immunoblot analyses of LRP6 and β-catenin proteins. All quantitation data represent the mean ± S.D. from three independent experiments after normalization to the values of nontreated or vector control cells. ns, not significant; *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by Student's t test.
Activated Wnt/β-catenin pathway blunts the HIF1α transcriptional program in RKO cells
To test whether hypoxic ER stress-induced down-regulation of β-catenin is implicated in the regulation of the hypoxia response, we examined the effects of Wnt/β-catenin signaling activation upon the HIF1α-responsive gene expression program. Wnt3a stimulation of RKO cells with WCM under normoxia dramatically increased β-catenin accumulation, whereas exposure to hypoxia significantly reduced it (Fig. 4A) without affecting its mRNA abundance (Fig. 4B). However, Wnt3a treatment had no effect upon HIF1α protein induction following exposure to hypoxia (Fig. 4A) but resulted in significant suppression of hypoxia-evoked up-regulation of HIF1α target genes (Fig. 4B). Next, we utilized cobalt chloride (CoCl2) that can enhance the stability of HIF1α to mimic the inducing effect of hypoxia upon HIF1α without affecting the stability of β-catenin in RKO cells under normoxia (Fig. 4C). Exposure to CoCl2 significantly enhanced the expression of HIF1α targets genes, whereas Wnt3a treatment blunted such induction by CoCl2 (Fig. 4D). Furthermore, overexpression of β-catenin could also markedly diminish CoCl2-induced up-regulation of HIF1α target genes (Fig. 4, E and F). Therefore, activation of the Wnt/β-catenin pathway could exert a negative regulatory action upon the HIF1α-directed gene expression program, and ER stress-associated suppression of Wnt/β-catenin signaling might facilitate the fine-tuning of the adaptive response to hypoxia.
Figure 4.
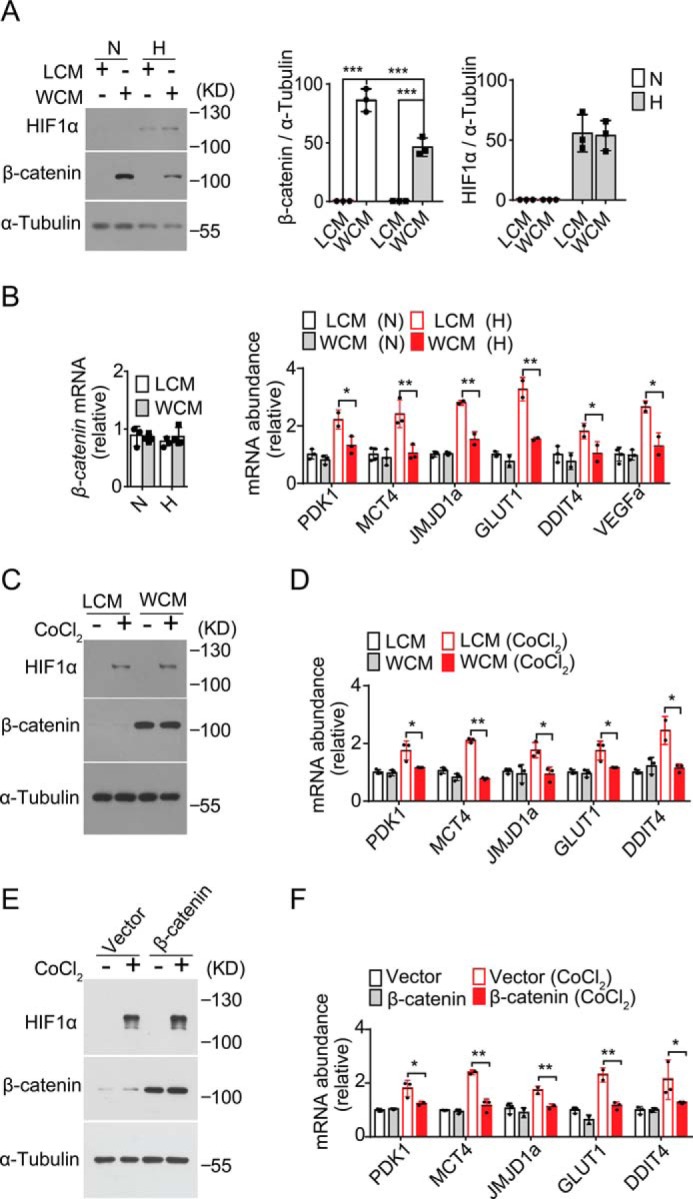
Wnt/β-catenin suppresses HIF1α transcriptional program. A and B, RKO cells cultured with control cell medium (LCM) or Wnt3a-producing cell medium (WCM) were subjected to 1% O2 (H) or 21% O2 (N) for 12 h. A, HIF1α and β-catenin proteins were analyzed by immunoblotting. B, quantitative RT-PCR analysis of the mRNA abundance for β-catenin as well as the indicated hypoxia-responsive HIF1α target genes. Data are shown after normalization to the values of LCM-cultured cells under normoxia. C and D, RKO cells cultured with LCM or WCM were nontreated or treated with 100 μm CoCl2 for 12 h. C, HIF1α and β-catenin protein levels were analyzed by immunoblotting. D, quantitative RT-PCR analysis of the mRNA abundance for the indicated HIF1α target genes. E and F, RKO cells were transfected with vector or β-catenin–expressing plasmid and then nontreated or treated with CoCl2 for 12 h. E, immunoblot analysis of HIF1α and β-catenin protein levels. F, quantitative RT-PCR analysis of HIF1α target genes. All RT-PCR results represent the mean ± S.D. from three independent experiments after normalization to the values of LCM-cultured or vector control cells. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by Student's t test.
Interactions between β-catenin, HIF1α, and XBP1s
Given that hypoxia could induce the activation of the IRE1α–XBP1 pathway (Fig. 1, A and B) and XBP1s has been documented to enhance the HIF1α-activated gene expression program in hypoxia (14), we wondered if there exist mutual interactions between β-catenin, HIF1α, and XBP1s that may constitute a dynamic regulatory network to govern the transcriptional program in response to hypoxia. Co-immunoprecipitation analyses showed that in HEK293 cells under hypoxia, HIF1α could associate with both β-catenin and XBP1s, and decreased HIF1α-XBP1s interaction was detectable in the presence of overexpressed β-catenin (Fig. 5A). Moreover, XBP1s was able to interact with both β-catenin and HIF1α, and the presence of overexpressed HIF1α apparently reduced β-catenin–XBP1s association (Fig. 5B), whereas overexpression of XBP1s resulted in decreased β-catenin–HIF1α association under hypoxia (Fig. 5C). Curiously, neither β-catenin nor XBP1s overexpression influenced HIF1α–HIF1β interaction in HEK293 cells under hypoxia (Fig. 5, A and C). Although the precise interactions and their regulatory modes, or possible competitions between these transcriptional regulators on the target chromatins are currently unclear, these data suggest that there might be a favorable HIF1α–XBP1s interaction relative to their association with β-catenin under hypoxic stress.
Figure 5.

β-Catenin interacts with XBP1s and HIF1α. A, HEK293 cells were co-transfected with XBP1s overexpression plasmid together with empty vector (−) or β-catenin–FLAG plasmid for 24 h and were then cultured under hypoxia (1% O2) for 12 h. Cell lysates were immunoprecipitated (IP) with anti-HIF1α antibody and analyzed by immunoblotting using the indicated antibodies. B, HEK293 cells were co-transfected with XBP1s-FLAG plasmid along with empty vector (−) or HIF1α–HA plasmid for 24 h and cultured under normoxia conditions. Cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by immunoblotting using the indicated antibodies. C, HEK293 cells were co-transfected with β-catenin–FLAG plasmid along with empty vector (−) or XBP1s plasmid for 24 h and were subsequently cultured under hypoxia (1% O2) for 12 h. Cell lysates were immunoprecipitated with anti-HIF1α antibody and analyzed by immunoblotting using the indicated antibodies. All results represent three independent experiments.
Wnt/β-catenin blunts XBP1s augmentation of HIF1α-activated transcription
We then asked whether Wnt/β-catenin signaling was able to affect the transcriptional cooperation between XBP1s and HIF1α. Remarkably, using HIF1α-responsive 5xHRE luciferase reporter, we observed that overexpression of XBP1s not only markedly activated its transcription in HEK293 cells under normoxia, but also enhanced its induction in response to hypoxia (Fig. 6A). Interestingly, Wnt3a treatment had no effect upon XBP1s-activated transcription, but it prominently abolished XBP1s' augmentative effect in response to hypoxia (Fig. 6A). Overexpressed β-catenin exhibited similar blunting effects upon XBP1s-promoted HIF1α activation of the 5xHRE reporter (Fig. 6B). In addition, Wnt3a also suppressed XBP1s-mediated up-regulation of HIF1α target genes in CoCl2-treated RKO cells (Fig. 6C). These results indicate that Wnt/β-catenin signaling could act to disrupt the cooperative activation by XBP1s–HIF1α of the hypoxic gene expression program.
Figure 6.
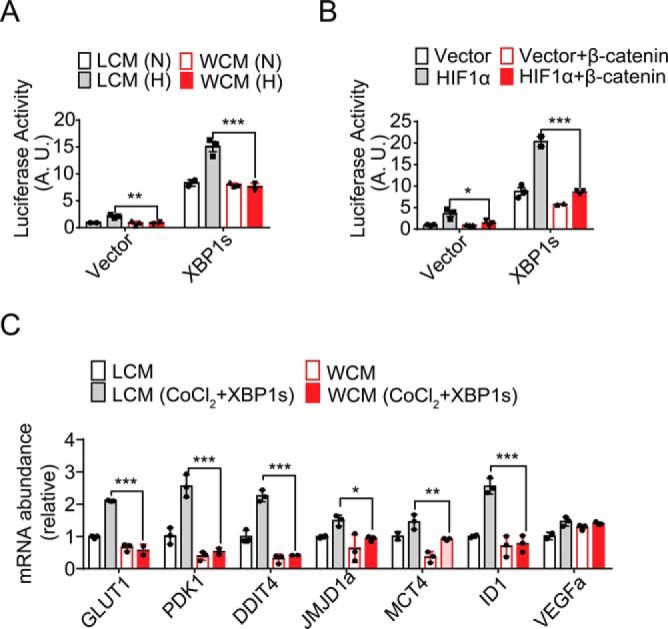
Suppression by Wnt/β-catenin of XBP1s promotion of HIF1α transcriptional activity. A and B, HIF1α-activated luciferase reporter assays. A, HEK293 cells were transiently co-transfected with 5xHRE-luciferase reporter plasmid together with vector or XBP1s-expressing plasmid for 24 h. Cells were cultured in LCM or WCM and exposed to normoxia (N) or 1% O2 hypoxia (H) for 12 h. Cell lysates were used for luciferase activity measurement. B, HEK293 cells were co-transfected with 5xHRE-luciferase reporter together with the indicated plasmids for 24 h, followed by measurement of luciferase activity. C, RKO cells were transfected with vector or XBP1s plasmid for 24 h and cultured in LCM or WCM in the absence or presence of 100 μm CoCl2 for 12 h. Quantitative RT-PCR analysis of the mRNA abundance for the indicated HIF1α target genes is shown. All data are shown as the mean ± S.D. from three independent experiments after normalization to the values of vector control or LCM-cultured cells. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by Student's t test.
Wnt/β-catenin hinders XBP1s-promoted cell survival under hypoxia
To determine the functional impact of Wnt/β-catenin–mediated suppression of the XBP1s–HIF1α transcriptional cooperation upon cell survival in the face of hypoxia stress, we first examined XBP1s' ability to affect cell survival under hypoxia. Overexpression of XBP1s significantly improved the survival of RKO cells following exposure to hypoxia (Fig. 7C), which might largely result from its alleviating effects upon apoptosis induced by such prolonged exposure to hypoxia (Fig. 7, A and B). Notably, Wnt3a treatment significantly blunted the promoting effect of XBP1s upon the survival of RKO cells (Fig. 7D), and overexpression of β-catenin also diminished XBP1s-mediated improvement of survival in HEK293 cells (Fig. 7E). Thus, these data suggest that hypoxic ER stress suppression of Wnt/β-catenin signaling may facilitate the efficiency of XBP1s–HIF1α cooperation in promoting the ability of cells to survive the hostile hypoxia environment.
Figure 7.

Effects of Wnt/β-catenin upon XBP1s promotion of cell survival under hypoxia. A, RKO cells were infected with adenoviruses expressing EGFP alone (Ad-EGFP) or EGFP and XBP1s (Ad-XBP1s) for 48 h and subsequently exposed to hypoxia for 48 h. Cells were then fixed and subjected to TUNEL analysis. EGFP- and Cy3.0-positive cells were quantified by flow cytometry, and the percentage of Cy3.0-positive apoptotic cells is shown as the mean ± S.D. from three independent experiments. B, RKO cells were transfected with vector or XBP1s overexpression plasmid for 24 h before exposure to hypoxia for 48 h. Cells were then fixed and analyzed by TUNEL. Representative images are shown for Cy3.0-positive signals among 4′,6-diamidino-2-phenylindole-stained cells, and the percentage of Cy3.0-positive apoptotic cells is shown as the mean ± S.D. from three independent experiments. C–E, cell survival assays. C, RKO cells were transfected with vector or XBP1s-expressing plasmid for 24 h and then exposed to 1% O2 hypoxia for the indicated time intervals. Representative images of cells stained with crystal violet are shown, and the number of cells alive was counted. D, RKO cells transfected with vector or Xbp1s plasmid for 24 h were exposed to 1% O2 hypoxia in the presence of vehicle (Con) or recombinant Wnt3a (10 ng/ml) for 48 h. Cells alive were counted, and cell viability is shown. E, HEK293 cells transfected with the indicated plasmids for 24 h were exposed to 1% O2 hypoxia for 48 h. Relative cell viability is determined and shown. All data are shown as the mean ± S.D. from three independent experiments. Normalization was done to the values of vector control in D and E. *, p < 0.05; **, p < 0.01, and ***, p < 0.001 by Student's t test. F, schematic model. Hypoxic ER stress results in suppression of β-catenin, thereby promoting XBP1s–HIF1α cooperation for enhanced cell survival under hypoxia.
Discussion
Hypoxia is a hallmark of many solid tumors, eliciting multiple cellular adaptive-response pathways to promote angiogenesis, metastasis, and therapy resistance (5). The hypoxic tumor microenvironment disturbs cellular proteostasis to trigger ER stress (33), which is thought to affect tumor progression. It remains to be better understood how tumor cells employ their hypoxic stress-response pathways in connection with developmental control signaling, such as the Wnt/β-catenin cascade to survive the hostile microenvironment during tumor progression. As depicted by the schematic model (Fig. 7F), this study shows that hypoxic ER stress may exert dual actions in promoting the survival of RKO cells during deprivation of oxygen: activation of the IRE1α–XBP1 branch of the UPR to elicit XBP1s-mediated enhancement of the HIF1α-regulated gene expression program, and suppression of Wnt/β-catenin signaling to alleviate its negative effect upon XBP1s–HIF1α cooperation in regulating the hypoxia stress response. Our results indicate that to gain adaptive advantage for cell survival, dynamic interplays are at work between these signaling regulators of the hypoxic stress response and developmental control mechanism.
Wnt/β-catenin signaling is a key cascade in developmental regulation, which is tightly associated with many types of cancer (34, 35). Aberrant Wnt/β-catenin cascade in tumorigenesis has been most commonly documented in colorectal cancer. A previously reported study (28) showed that in SW480 and HCT116 colon cancer cells with a constitutively high level of β-catenin, hypoxia could inhibit β-catenin/TCF-4 complex formation and transcriptional activity, and β-catenin was found to bind directly to HIF1α and enhance its transcriptional activity, thereby promoting cell survival under hypoxia. In contrast, here we found that in RKO colon cancer cells with normal Wnt/β-catenin signaling capacity, hypoxia could inhibit β-catenin accumulation by reducing the LRP6 protein level, leading to impairment of Wnt signaling transduction. Furthermore, Wnt stimulation or β-catenin overexpression exhibited an attenuating effect upon HIF1α-regulated gene expression program in hypoxia. More notably, β-catenin was able to diminish the cooperative cross-talk between the HIF1α and XBP1s pathways that could be provoked by hypoxic ER stress. Thus, our results from RKO cells suggest that hypoxia may influence the Wnt/β-catenin cascade through distinct mechanisms with different functional consequences in different colon cancer subtypes. The net effect of hypoxia upon the interplays between β-catenin, HIF1α, and XBP1s is most likely context-dependent and can be influenced by tumor cell origins or its subtypes. It is also noteworthy that hypoxia has been shown to block the processing and secretion of Wnt proteins through ER stress induction, leading to suppression of the Wnt/β-catenin pathway and growth of RKO cells (36). Our results revealed hypoxic ER stress-associated β-catenin destabilization resulted from decreases in matured protein levels of the LRP6 co-receptor, mimicking its alterations under TG-induced experimental ER stress. It remains to be defined, however, whether this hypoxia-induced reduction of LRP6 involves suppression of global cellular protein translation, membrane protein processing and maturation within the ER, or ER-associated degradation processes. Another open question that has yet to be deciphered is the exact mechanism(s) implicated in regulating the modes of the mutual interactions between β-catenin, HIF1α, and XBP1s, as well as their functional output(s) as the key nodes in reprogramming metabolism, protein synthesis, and cell cycle progression to fine-tune the adaptive hypoxia responses.
Sustained ER stress is thought to endow malignant cells with a higher capacity for tumorigenesis, metastasis, and drug resistance (6, 37). Hypoxic activation of the UPR-signaling pathways may be exploited by tumor cells to cope with the hostile microenvironment, and such adaptation to hypoxia is a critical determinant of tumor progression. Increasing lines of evidence have indicated that the IRE1α–XBP1 branch of the UPR is implicated in promoting the progression of various types of tumor, including triple-negative breast cancer (14, 38), hepatocellular carcinoma (39), glioblastoma (40), ovarian cancer (41, 42), prostate cancer (43), as well as colon cancer (44). Preclinical studies have also shown promising anti-tumor potential of targeted inhibition of the IRE1α–XBP1 pathway (45), particularly when employed in combination with standard chemotherapy drugs (46). Although therapeutic agents targeting the IRE1α–XBP1 and HIF1α pathways have been actively pursued (47, 48), chemical modulators of the Wnt/β-catenin cascade are also being developed as anti-tumor entities (34, 35, 49). Therefore, it is of great significance for precision targeting to dissect the molecular hallmarks of hypoxia and ER stress responses together with the activation state of Wnt/β-catenin signaling in different tumors or subtypes. Given the presumably distinct inter-relationships between XBP1s, HIF1α, and β-catenin in different tumors, it requires more in-depth characterization of the molecular signatures of the cross-talk between these signaling cascades in order to develop more effective combination anti-tumor therapies.
In summary, our findings revealed the occurrence of tumor cell subtype-selective or context-dependent cross-talks between the Wnt/β-catenin signaling cascade and the UPR and HIF1α stress-response pathways, which is likely to allow tumor cells for more efficient adaptation to the unfavorable microenvironment of hypoxia. Further elucidation of the molecular dialogues in these signaling networks will advance our understanding of the molecular basis of malignant progression.
Experimental procedures
Chemicals, plasmids, and antibodies
TM, TG, MG132, LiCl, CQ, and G418 were purchased from Sigma. The TOPFlash reporter plasmid, the LRP6-vsvg, and LRP6ΔN expression plasmids were kindly provided by Dr. Xi He from Harvard University. The β-catenin–FLAG expression plasmid was purchased from Addgene. The XBP1s and XBP1s–FLAG expression plasmids and adenoviruses were used as described previously (50). HIF1α–HA and 5xHRE reporter plasmids were gifts from Dr. Mingliang Zhang at Shanghai Jiao-Tong University.
Antibodies for the detection of IRE1α (no. 3294), p-eIF2α (no. 9721), eIF2α (no. 9722), XBP1s (no. 12782), and HIF1β (no. 5537) were purchased from Cell Signaling Technology (Danvers, MA). HIF1α (no. 610958) and β-catenin (no. 610153) antibodies were from BD Biosciences. Antibody (no. NB100-2323) against the phosphorylated IRE1α at Ser724 (p-IRE1α) was from Novus Biologicals (Littleton, CO). Antibodies against LRP6 (no. ab134146) was from Abcam, and α-tubulin (no. T6199), FLAG (no. F3165), and HA (no. H6908) antibodies were from Sigma. Antibodies were used at a dilution of 1:1000, except for α-tubulin that was used with a dilution of 1:5000.
Cell culture and transfection
HEK293T cells, mouse L cells, Wnt3a-producing mouse L cells (stably transfected with the Wnt3a expression plasmid and maintained in the presence of G418, kindly provided by Dr. Xi He), and RKO cells were maintained in DMEM supplemented with 10% fetal bovine serum and 100 μg/ml penicillin and 100 μg/ml streptomycin. HEK293T cells were transfected in 12-well or 24-well plates by Lip2000 (Invitrogen) at 0.5 μg/well (12-well plates) or 0.2 μg/well (24-well plates). Cells were used for subsequent analysis after incubation for 24 h at 37 °C with 5% CO2.
Hypoxia
For exposure to hypoxia, cells in fresh medium were maintained for varying periods of time in a hypoxia incubator chamber (STEMCELL Technologies Inc.) containing 1% O2, 5% CO2-balanced N2. For normoxia, cells were cultured in an incubator containing 21% O2, 5% CO2. For CoCl2 treatment, cells were grown to 50–80% confluency and cultured with 100 μm CoCl2 for 4 h.
Wnt stimulation
To analyze Wnt/β-catenin signaling activation, cells were treated with Wnt3a-conditioned medium (Wnt3a CM) or control medium (LCM) that was prepared as described previously (31). Briefly, Wnt3a-producing mouse L cells or control L cells were cultured to full confluency in DMEM and then replaced with serum-free DMEM. The culture medium was collected after 3 and 7 days, and the two collections were mixed and filtered using 0.22-μm filters. After a 2-fold dilution, medium was used for cell treatment after the concentration of Wnt3a was evaluated by its ability to stabilize β-catenin as compared with recombinant Wnt3a protein (R&D Systems, Minneapolis, MN), usually in the range of 150–200 ng/ml.
Cell survival assay
RKO or HEK293 cells at 2.5 × 105 cells per well (24-well plate) or 5 × 105 cells per well (12-well plate) were exposed to hypoxia and counted after 1, 2, or 3 days. Cells were stained with crystal violet dye after fixation with methanol, followed by microscopic analysis.
TUNEL assay
RKO cells were transfected with the vector or XBP1s expression plasmid, or they were infected with Ad-EGFP or Ad-XBP1s adenovirus (50) for 24 h. Cells were then incubated under hypoxia conditions for 48 h. After fixation with 4% paraformaldehyde for 10 min, cells were subjected to TUNEL analysis according to the manufacturer's instructions (one-step TUNEL apoptosis assay kit, Beyotime). Apoptotic cells labeled with Cy3.0-dUTP were quantified from micrograph images or by flow cytometry.
Immunoblot analysis and co-immunoprecipitation
Cell lysates were prepared using RIPA buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris-HCl, pH 7.4). Immunoprecipitation was conducted using the desired antibody by incubation overnight on the shaker to allow formation of protein/antibody complexes. Thirty μl of protein G–agarose beads (GE Healthcare) were added for another 2 h, and the beads were collected by centrifugation at 2000 × g for 1 min and washed four times with lysis buffer. For immunoblotting, SDS-PAGE was performed, and separated proteins from cell lysates or the beads were transferred onto a polyvinylidene difluoride membrane filter and incubated with the desired antibodies. The blots were then developed with Thermo Fisher Scientific SuperSignal West Pico Chemiluminescent substrate or Millipore Immobilon Western Chemiluminescent horseradish peroxidase substrate.
Quantitative RT-PCR analysis
Total cellular RNA was isolated using the TRIzol reagent according to the manufacturer's protocol (Invitrogen). For cDNA synthesis, Moloney murine leukemia virus reverse transcriptase and random hexamer primers (Invitrogen) were used. Real-time quantitative PCR was conducted using SYBR Green (Roche Applied Science) and the StepOne plus Real-Time PCR System (Applied Biosystems). For normalization, ACTB mRNA was utilized as the internal control. The oligonucleotide primer pairs used were as follows: ACTB, 5′-catgtacgttgctatccaggc-3′ and 5′-ctccttaatgtcacgcacgat-3′; XBP1s, 5′-ctgagtccgcagcaggtgcag-3′ and 5′-ctccaggctggcaggctctg-3′; XBP1t, 5′-gtggccgggtctgctgagtc-3′ and 5′-ctccaggctggcaggctctg-3′; ERdj4, 5′-tcttaggtgtgccaaaatcgg-3′ and 5′-tgtcagggtggtacttcatgg-3′; BiP, 5′-catcacgccgtcctatgtcg-3′ and 5′-cgtcaaagaccgtgttctcg-3′; c-MYC, 5′-ttcgggtagtggaaaaccag-3′ and 5′-cagcagctcgaatttcttcc-3′; CyclinD1, 5′-aactacctggaccgcttcct-3′ and 5′-ccacttgagcttgttcacca-3′; DKK1, 5′-ccttgaactcggttctcaattcc-3′ and 5′-caatggtctggtacttattcccg-3′; AXIN2, 5′-ttatgctttgcactacgtccctcca-3′ and 5′-cgcaacatggtcaaccctcagac-3′; VEGFa, 5′-agggcagaatcatcacgaagt-3′ and 5′-agggtctcgattggatggca-3′; GLUT1, 5′-tggacccatgtctggttgta-3′ and 5′-atggagcccagcagcaa-3′; PDK1, 5′-ggaggtctcaacacgaggtc-3′ and 5′-gttcatgtcacgctgggtaa-3′; DDIT4, 5′-catcaggttggcacacaagt-3′ and 5′-cctggagagctcggactg-3′; ID1, 5′-ctgctctacgacatgaacgg-3′ and 5′-gaaggtccctgatgtagtcgat-3′; JMJD1a, 5′-tcaggtgactttcgttcagc-3′ and 5′-caccgacgttaccaagaagg-3′; MCT4, 5′-tacatgtagacgtgggtcgc-3′ and 5′-ctgcagttcgaggtgctcat-3′; LRP6, 5′-ttgttgctttatgcaaacagacg-3′ and 5′-cgtttaatggcttcttcgctgac-3′; and CTNNB1, 5′-aaagcggctgttagtcactgg-3′ and 5′-cgagtcattgcatactgtccat-3′.
Luciferase reporter assay
HEK293T cells were transfected with TOPFlash or 5xHRE reporter plasmid and Renilla (10:1) for 24 h (51). Cell lysates were used for luciferase activity measurement with the Dual-Luciferase reporter system (Promega).
Statistical analysis
All data represent results from at least three independent experiments. Statistical significance was determined by Student's t test, and p < 0.05 was considered significant.
Author contributions
Z. X., Y. Liu, Jianmiao Liu, and Jianfeng Liu conceptualization; Z. X. and X. W. data curation; Z. X. and Jianmiao Liu formal analysis; Z. X., X. W., Y. Liao, Y. Liu, Jianmiao Liu, and Jianfeng Liu investigation; Z. X., S. W., and P. Y. methodology; Z. X. and Jianmiao Liu writing original draft; Z. X. and Jianmiao Liu project administration; Y. Liu and Jianfeng Liu writing review and editing; Jianmiao Liu and Jianfeng Liu resources; Jianmiao Liu supervision; Jianmiao Liu and Jianfeng Liu funding acquisition; Jianmiao Liu and Jianfeng Liu validation.
Supplementary Material
Acknowledgment
We thank Dr. Xi He from Harvard University for advice on this work and for providing valuable reagents.
This work was supported by National Natural Science Foundation of China Grants 91539107 and 91739303 (to Jianmiao Liu) and Grants 31420103909, 81720108031, 31721002, and 81872945 (to Jianfeng Liu), Ministry of Science and Technology of China Grant 2018YFA0507003, and Program for Introducing Talents of Discipline to the Universities of the Ministry of Education Grant B08029, and the Mérieux Research Grants Program of the Institut Mérieux. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- HIF
- hypoxia-induced factor
- UPR
- unfolded protein response
- PERK
- protein kinase R (PKR)-like endoplasmic reticulum kinase
- APC
- adenomatous polyposis coli gene product
- eIF2α
- eukaryotic initiation factor 2α
- ER
- endoplasmic reticulum
- HRE
- hypoxia response element
- CQ
- chloroquine
- TM
- tunicamycin
- TG
- thapsigargin
- TUDCA
- tauroursodeoxycholic acid
- 4-PBA
- 4-phenylbutyric acid
- DMEM
- Dulbecco's modified Eagle's medium
- EGFP
- enhanced GFP
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling
- WCM
- Wnt3a-producing cell medium
- TCF
- T-cell transcription factor.
References
- 1. Harris A. L. (2002) Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- 2. Majmundar A. J., Wong W. J., and Simon M. C. (2010) Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 10.1016/j.molcel.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weidemann A., and Johnson R. S. (2008) Biology of HIF-1α. Cell Death Differ. 15, 621–627 10.1038/cdd.2008.12 [DOI] [PubMed] [Google Scholar]
- 4. Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 10.1038/nrc1187 [DOI] [PubMed] [Google Scholar]
- 5. Wouters B. G., and Koritzinsky M. (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 8, 851–864 10.1038/nrc2501 [DOI] [PubMed] [Google Scholar]
- 6. Cubillos-Ruiz J. R., Bettigole S. E., and Glimcher L. H. (2017) Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168, 692–706 10.1016/j.cell.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mohamed E., Cao Y., and Rodriguez P. C. (2017) Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: a promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 66, 1069–1078 10.1007/s00262-017-2019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 9. Koumenis C., and Wouters B. G. (2006) “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Cancer Res. 4, 423–436 10.1158/1541-7786.MCR-06-0150 [DOI] [PubMed] [Google Scholar]
- 10. Koumenis C., Naczki C., Koritzinsky M., Rastani S., Diehl A., Sonenberg N., Koromilas A., and Wouters B. G. (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 22, 7405–7416 10.1128/MCB.22.21.7405-7416.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blais J. D., Filipenko V., Bi M., Harding H. P., Ron D., Koumenis C., Wouters B. G., and Bell J. C. (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 24, 7469–7482 10.1128/MCB.24.17.7469-7482.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Romero-Ramirez L., Cao H., Nelson D., Hammond E., Lee A. H., Yoshida H., Mori K., Glimcher L. H., Denko N. C., Giaccia A. J., Le Q. T., and Koong A. C. (2004) XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 64, 5943–5947 10.1158/0008-5472.CAN-04-1606 [DOI] [PubMed] [Google Scholar]
- 13. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 10.1038/nrm3270 [DOI] [PubMed] [Google Scholar]
- 14. Chen X., Iliopoulos D., Zhang Q., Tang Q., Greenblatt M. B., Hatziapostolou M., Lim E., Tam W. L., Ni M., Chen Y., Mai J., Shen H., Hu D. Z., Adoro S., Hu B., et al. (2014) XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 508, 103–107 10.1038/nature13119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Logan C. Y., and Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- 16. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- 17. MacDonald B. T., Tamai K., and He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He X. (2003) A Wnt-Wnt situation. Dev. Cell 4, 791–797 10.1016/S1534-5807(03)00165-5 [DOI] [PubMed] [Google Scholar]
- 19. Cong F., Schweizer L., and Varmus H. (2004) Wnt signals across the plasma membrane to activate the beta-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development 131, 5103–5115 10.1242/dev.01318 [DOI] [PubMed] [Google Scholar]
- 20. He X., Semenov M., Tamai K., and Zeng X. (2004) LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: arrows point the way. Development 131, 1663–1677 10.1242/dev.01117 [DOI] [PubMed] [Google Scholar]
- 21. Clevers H., and Nusse R. (2012) Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 22. Polakis P. (2012) Drugging Wnt signalling in cancer. EMBO J. 31, 2737–2746 10.1038/emboj.2012.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Polakis P. (2007) The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45–51 10.1016/j.gde.2006.12.007 [DOI] [PubMed] [Google Scholar]
- 24. Polakis P. (2000) Wnt signaling and cancer. Genes Dev. 14, 1837–1851 [PubMed] [Google Scholar]
- 25. Giles R. H., van Es J. H., and Clevers H. (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 1653, 1–24 10.1016/s0304-419x(03)00005-2 [DOI] [PubMed] [Google Scholar]
- 26. Li Y., Lu W., He X., Schwartz A. L., and Bu G. (2004) LRP6 expression promotes cancer cell proliferation and tumorigenesis by altering β-catenin subcellular distribution. Oncogene 23, 9129–9135 10.1038/sj.onc.1208123 [DOI] [PubMed] [Google Scholar]
- 27. Liu C. C., Prior J., Piwnica-Worms D., and Bu G. (2010) LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. U.S.A. 107, 5136–5141 10.1073/pnas.0911220107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaidi A., Williams A. C., and Paraskeva C. (2007) Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217 10.1038/ncb1534 [DOI] [PubMed] [Google Scholar]
- 29. Feldman D. E., Chauhan V., and Koong A. C. (2005) The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 3, 597–605 10.1158/1541-7786.MCR-05-0221 [DOI] [PubMed] [Google Scholar]
- 30. Koritzinsky M., Magagnin M. G., van den Beucken T., Seigneuric R., Savelkouls K., Dostie J., Pyronnet S., Kaufman R. J., Weppler S. A., Voncken J. W., Lambin P., Koumenis C., Sonenberg N., and Wouters B. G. (2006) Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25, 1114–1125 10.1038/sj.emboj.7600998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamai K., Zeng X., Liu C., Zhang X., Harada Y., Chang Z., and He X. (2004) A mechanism for Wnt coreceptor activation. Mol. Cell 13, 149–156 10.1016/S1097-2765(03)00484-2 [DOI] [PubMed] [Google Scholar]
- 32. Uppala J. K., Gani A. R., and Ramaiah K. V. A. (2017) Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation efficiently and resists ER and non-ER stress induced HepG2 cell death. Sci. Rep. 7, 3831 10.1038/s41598-017-03940-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hetz C., Chevet E., and Oakes S. A. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838 10.1038/ncb3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nusse R., and Clevers H. (2017) Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 35. Zhan T., Rindtorff N., and Boutros M. (2017) Wnt signaling in cancer. Oncogene 36, 1461–1473 10.1038/onc.2016.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verras M., Papandreou I., Lim A. L., and Denko N. C. (2008) Tumor hypoxia blocks Wnt processing and secretion through the induction of endoplasmic reticulum stress. Mol. Cell. Biol. 28, 7212–7224 10.1128/MCB.00947-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sim J., and Johnson R. S. (2015) Through a clear cell, darkly: HIF2α/PLIN2-maintained fat droplets protect ccRCCs from ER stress. Cancer Discov. 5, 584–585 10.1158/2159-8290.CD-15-0480 [DOI] [PubMed] [Google Scholar]
- 38. Bernardi R., and Gianni L. (2014) Hallmarks of triple negative breast cancer emerging at last? Cell Res. 24, 904–905 10.1038/cr.2014.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu Y., Shan B., Dai J., Xia Z., Cai J., Chen T., Lv S., Feng Y., Zheng L., Wang Y., Liu J., Fang J., Xie D., Rui L., Liu J., and Liu Y. (2018) Dual role for inositol-requiring enzyme 1α in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology 68, 533–546 10.1002/hep.29871 [DOI] [PubMed] [Google Scholar]
- 40. Lhomond S., Avril T., Dejeans N., Voutetakis K., Doultsinos D., McMahon M., Pineau R., Obacz J., Papadodima O., Jouan F., Bourien H., Logotheti M., Jégou G., Pallares-Lupon N., Schmit K., et al. (2018) Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 10, e7929 10.15252/emmm.201707929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cubillos-Ruiz J. R., Silberman P. C., Rutkowski M. R., Chopra S., Perales-Puchalt A., Song M., Zhang S., Bettigole S. E., Gupta D., Holcomb K., Ellenson L. H., Caputo T., Lee A. H., Conejo-Garcia J. R., and Glimcher L. H. (2015) ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538 10.1016/j.cell.2015.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song M., Sandoval T. A., Chae C. S., Chopra S., Tan C., Rutkowski M. R., Raundhal M., Chaurio R. A., Payne K. K., Konrad C., Bettigole S. E., Shin H. R., Crowley M. J. P., Cerliani J. P., Kossenkov A. V., et al. (2018) IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428 10.1038/s41586-018-0597-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheng X., Nenseth H. Z., Qu S., Kuzu O. F., Frahnow T., Simon L., Greene S., Zeng Q., Fazli L., Rennie P. S., Mills I. G., Danielsen H., Theis F., Patterson J. B., Jin Y., and Saatcioglu F. (2019) IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 10, 323 10.1038/s41467-018-08152-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li X. X., Zhang H. S., Xu Y. M., Zhang R. J., Chen Y., Fan L., Qin Y. Q., Liu Y., Li M., and Fang J. (2017) Knockdown of IRE1α inhibits colonic tumorigenesis through decreasing β-catenin and IRE1α targeting suppresses colon cancer cells. Oncogene 36, 6738–6746 10.1038/onc.2017.284 [DOI] [PubMed] [Google Scholar]
- 45. Zhao N., Cao J., Xu L., Tang Q., Dobrolecki L. E., Lv X., Talukdar M., Lu Y., Wang X., Hu D. Z., Shi Q., Xiang Y., Wang Y., Liu X., Bu W., et al. (2018) Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Invest. 128, 1283–1299 10.1172/JCI95873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie H., Tang C. H., Song J. H., Mancuso A., Del Valle J. R., Cao J., Xiang Y., Dang C. V., Lan R., Sanchez D. J., Keith B., Hu C. C., and Simon M. C. (2018) IRE1α RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Invest. 128, 1300–1316 10.1172/JCI95864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hetz C., Chevet E., and Harding H. P. (2013) Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719 10.1038/nrd3976 [DOI] [PubMed] [Google Scholar]
- 48. Semenza G. L. (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 33, 207–214 10.1016/j.tips.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., and Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 10.1038/nchembio.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shao M., Shan B., Liu Y., Deng Y., Yan C., Wu Y., Mao T., Qiu Y., Zhou Y., Jiang S., Jia W., Li J., Rui L., Yang L., and Liu Y. (2014) Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s-PPARα axis signalling. Nat. Commun. 5, 3528 10.1038/ncomms4528 [DOI] [PubMed] [Google Scholar]
- 51. Cañive J. M., Lewine J. D., Edgar J. C., Davis J. T., Torres F., Roberts B., Graeber D., Orrison W. W. Jr., and Tuason V. B. (1996) Magnetoencephalographic assessment of spontaneous brain activity in schizophrenia. Psychopharmacol. Bull. 32, 741–750 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.