

Abstract
Prion diseases are a group of incurable neurodegenerative disorders that affect humans and animals via infection with proteinaceous particles called prions. Prions are composed of PrPSc, a misfolded version of the cellular prion protein (PrPC). During disease progression, PrPSc replicates by interacting with PrPC and inducing its conversion to PrPSc. As PrPSc accumulates, cellular stress mechanisms are activated to maintain cellular proteostasis, including increased protein chaperone levels. However, the exact roles of several of these chaperones remain unclear. Here, using various methodologies to monitor prion replication (i.e. protein misfolding cyclic amplification and cellular and animal infectivity bioassays), we studied the potential role of the molecular chaperone heat shock protein 70 (HSP70) in prion replication in vitro and in vivo. Our results indicated that pharmacological induction of the heat shock response in cells chronically infected with prions significantly decreased PrPSc accumulation. We also found that HSP70 alters prion replication in vitro. More importantly, prion infection of mice lacking the genes encoding stress-induced HSP70 exhibited accelerated prion disease progression compared with WT mice. In parallel with HSP70 being known to respond to endogenous and exogenous stressors such as heat, infection, toxicants, and ischemia, our results indicate that HSP70 may also play an important role in suppressing or delaying prion disease progression, opening opportunities for therapeutic intervention.
Keywords: prion, prion disease, protein aggregation, protein folding, endoplasmic reticulum stress (ER stress), heat shock protein (HSP), molecular chaperone, neurodegeneration, bovine spongiform encephalopathy, Creutzfeldt–Jakob disease, scrapie
Introduction
Prions are the causative transmissible agents for a group of neurodegenerative disorders, including scrapie in sheep, bovine spongiform encephalopathy in cattle, chronic wasting disease in cervids, and Creutzfeldt–Jakob disease (CJD)3 in humans. The key component of a prion is PrPSc, which is the misfolded form of the cellular prion protein (PrPC). During disease, PrPSc self-propagates by serving as a template for the conformational conversion of PrPC substrate (1). Through an incompletely characterized and complex mechanism, PrPSc accumulation triggers cellular and endoplasmic reticulum (ER) stress (2). One of the initial cellular responses to PrPSc-induced stress is the up-regulation of specific glucose-regulated proteins (GRPs), protein disulfide isomerases, and heat shock proteins (HSPs) that function as molecular chaperones and folding enzymes (2). Together, these proteins generally assist with proper protein folding, inhibit protein aggregation, and direct the degradation of misfolded proteins.
HSPs belong to a large multigene family and are grouped into subfamilies according to their molecular mass, which can range from 8 to 150 kDa (3, 4). Many HSPs are expressed under normal physiological conditions. These levels can increase dramatically in response to stress, especially stressors that increase production of aberrant proteins. The HSP70 subfamily is highly evolutionarily conserved and has members present in all subcellular compartments (ER, cytoplasm, and nucleoplasm) to mediate protein quality control (3, 4).
The initial possibility of a relationship between HSPs and mammalian prion diseases stems from work in the yeast prion field. Yeast prions have been defined as non-Mendelian genetic elements (e.g. [URE3], [PSI+], [Het-s], and [PIN+]) found to be infectious prion forms of host-encoded proteins (i.e. Ure2p, Sup35p, HET-s, and Rnq1p, respectively) (5–7). Generally speaking, yeast prions are protein aggregates with high β-sheet content that form amyloids and self-propagate this altered conformational state both vertically (as genes composed of protein) and horizontally (as infectious mammalian prions). The first connection between HSPs and yeast prions was the discovery that HSP104 specifically induces their fragmentation, thus enhancing self-propagation (8). Moreover, overproduction or inactivation of HSP104 cured the yeast cells of prions without affecting viability. Therefore, a certain level of disaggregation mediated by HSP104 is necessary to maintain yeast prion propagation, but too much disaggregation completely destroys the replicating prion seeds (8). Although HSP104 is the disaggregase, the primary guide that delivers HSP104 to the yeast prions is HSP70 (9–12), which is a function that can be assisted by HSP40 (13–15). It has been difficult to translate our knowledge about yeast prions to mammalian prions because the mammalian genome does not encode a corresponding HSP104 homolog. Furthermore, despite the ability of yeast HSP104 to induce mammalian PrPSc propagation in vitro (16), prion inoculation of transgenic mice expressing yeast heat shock protein 104 did not modulate PrPSc accumulation or disease incubation time (17). In summary, the current model in yeast states that HSP104 and some members of the HSP70 and HSP40 families are working together in fibril fragmentation and prion propagation in yeast (18).
Among the chaperones discovered to be elevated in CJD patients (19–22) and scrapie-infected rodents (23–25) was HSP70, which is the original prototype of the larger HSP70 subfamily encoded in mammals by the nearly identical Hspa1a and Hspa1b genes. In mice and other mammals, the HSP genes Hspa1a and Hspa1b are induced by both endogenous and exogenous stressors, such as heat and toxicants. They have been shown to be the major and usually only HSP70 members that respond to most physiologically relevant stresses. The Hspa1a/Hspa1b genes differ by a single amino acid and are divergent from all other members of the larger HSP70 gene family, particularly in the C-terminal domain, which contains the substrate binding site. The Hspa1a/Hspa1b genes are not to be confused with the Hsf1 gene that encodes a transcription factor. Knockout of the Hspa1a/Hspa1b genes eliminates their endogenous or induced response to stressors, although other HSP70 gene members are present at endogenous levels. Furthermore, these HSP70 gene family members are the only ones whose cytosolic levels increase in response to stress, further strengthening the potential for a functional role in prion disease.
The physiological role of HSP70 in prion disease has been unclear because it has yet to be specifically explored in a bona fide model for mammalian prion disease. In a previous study, we investigated the role of HSP70 using a model in transgenic Drosophila overexpressing PrPC that naturally accumulated PrP aggregates with aging (26). This insoluble PrP form induced neurotoxicity and locomotor dysfunction, similar to PrPSc during prion disease (26). In these transgenic flies, HSP70 was discovered to reduce the accumulation of PrP aggregates by ∼40% and alleviate the associated neurological deficits. Although HSP70 is a cytosolic/nuclear chaperone and PrPC is a cell surface protein, evidence indicated that the positive effect from HSP70 was mediated by a direct interaction with PrP (26). If such a scenario indeed takes place in a mammalian host, then the lack of HSP70 induction, a major target of heat shock factor (HSF1), may explain why Hsf1−/− knockout mice succumb to prion disease ∼20% faster than WT controls (27). Here we used an in vitro model of PrPC-to-PrPSc conversion, a mammalian cell culture model, and Hsp70 knockout mice (Hspa1a−/− and Hspa1b−/−, hereafter referred to as Hsp70−/−) to better understand the role of HSP70 in prion infection.
Results
Effect of a drug-induced heat shock response on PrPSc in cell culture
The 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG) drug is a water-soluble derivative of geldanamycin known to induce a strong heat shock response in mammalian cells (28). More specifically, 17-DMAG exhibits the broad ability to induce expression of molecular chaperones, including cytosolic HSP40, HSP70, HSP90, and HSP105 as well as GRP94 in the ER. Following our previously established treatment regimen (29, 30), RK13 cells were treated at the beginning and middle of a 6-day period with 60-min exposure to 0, 0.1, 0.5, or 1.5 μm 17-DMAG. We observed a strong induction of HSP70 in RK13 cells treated with 0.5 and 1.5 μm 17-DMAG, which was absent for untreated cells and those treated with 0.1 μm 17-DMAG (Fig. 1A). This is in agreement with our previous results indicating that 17-DMAG induces HSPs and could be detected for 3 to 5 days after transient exposure.4 Furthermore, BCA protein analysis for these treatments demonstrated that 1.5 μm 17-DMAG resulted in a modest reduction in protein content as a result of toxicity, so we chose to utilize 0.5 μm or less for all future treatments to avoid a confounding effect.
Figure 1.

Drug-induced heat shock response induces Hsp70 expression and reduces PrPSc in cell culture. A, Western blot analysis showed HSP70 induction in RK13 cells following transient 6-day treatment consisting of two exposures to 0.1, 0.5, or 1.5 μm 17-DMAG (DMAG). B, PK-resistant PrPSc levels were analyzed in triplicate experiments for chronically infected RKM7-RML cells undergoing a similar 6-day treatment with 0, 0.1, or 0.5 μm DMAG. Non-PK-digested samples from RKM7 and RKM7-RML were used as migration controls. C, densitometry of the Western blot signal in B was used to compare PrPSc levels. ***, p < 0.001. D, PrPC was analyzed by Western blotting in experimental duplicates for uninfected RKM7 lysates derived from cells treated similarly with 0, 0.1, or 0.5 μm DMAG, with GAPDH as the loading control.
RK13 cells are a versatile prion cell culture model that has successfully been genetically engineered to propagate prions derived from sheep (31–33), mice (32, 34, 35), bank voles (34), or cervids (32, 36). Using clonal selection on the previously established RKM7 line expressing mouse PrPC (32), a clone highly sensitive to Rocky Mountain Laboratory (RML) mouse–adapted scrapie prions was isolated and chronically infected, referred to as RKM7-RML. RKM7 and RKM7-RML cells were treated with 0, 0.1, or 0.5 μm 17-DMAG to determine the effect of a heat shock response on prion infection. Interestingly, prion-infected cells treated with 0.5 μm 17-DMAG demonstrated a 50% reduction in PrPSc (p < 0.001; Fig. 1, B and C). A similar 17-DMAG treatment did not alter PrPC levels in RKM7 cells (Fig. 1D), suggesting that the effect is at the level of prion replication. These results indicate that the drug-induced heat shock response was sufficient to significantly reduce PrPSc in RKM7-RML cells. In future studies, we will attempt to analyze in detail the correlation between the magnitude of the changes of HSP70 expression and the reduction in PrPSc replication.
Influence of HSP70 on prion disease onset and progression
We specifically focused on the role of HSP70 in vivo because it is the only HSP induced by 17-DMAG that is also known to be elevated in CJD patients (37) and scrapie-infected mice (24). For this reason, we intracranially inoculated normal brain homogenate (NBH) or RML prions into mice deficient in HSP70 protein (Hsp70−/−) and WT controls expressing HSP70 (Hsp70+/+; Fig. 2A). HSP70-null animals developed terminal prion disease significantly faster compared with WT controls (Fig. 2B), as analyzed by log-rank (Mantel–Cox) test (p < 0.05). The mean incubation period (inoculation to death) for Hsp70−/− mice (145.2 ± 3.06 days) was also significantly shorter than for Hsp70+/+ mice (156.0 ± 3.29 days, p < 0.05; Fig. 2B). These results indicate that HSP70 has a protective role in the onset of prion disease. To further investigate whether HSP70 may also affect the progression of the disease, we blindly analyzed the rate of clinical changes in the two groups of infected animals. Onset and progression of clinical abnormalities were evaluated by a semiquantitative scale commonly employed in the field to analyze clinical signs of prion disease in rodents (38, 39). When normal animals transition from stage 1, the first abnormalities observed in RML prion–infected mice include lack of nesting, rough coat on limbs, and abnormal social behavior (stage 2). This is followed by extensive rough coat, hunchback, and visible motor abnormalities (stage 3). Stage 4 includes more severe progression of previous signs plus urogenital lesions, increased motor abnormalities, and extensive weight loss. At the end stage of the disease (stage 5), animals present with severe ataxia and usually lie in the cage with little movement. For this analysis, stage 6 was scored at termination, after consistently maintaining a score of 5 and as they began to progress further. Clinical signs were first noted for all RML-challenged animals in both groups 123 days post-inoculation (dpi, Fig. 2C). Surprisingly, disease progression began to diverge from this point onward; Hsp70−/− mice exhibited significantly more rapid progression through the clinical stages until becoming terminal (140 dpi, p < 0.01; 144 dpi, p < 0.05; 146 dpi, p < 0.01; 147 dpi, p < 0.001; 148 dpi, p < 0.001; 150 dpi, p < 0.01). In conclusion, our results suggest that the absence of HSP70 reduced the incubation period in prion disease by accelerating clinical progression.
Figure 2.

Comparison of prion incubations in Hsp70−/− and HSP70+/+ mice. A, Western blot analysis was used to confirm HSP70 levels in healthy Hsp70−/− and Hsp70+/+ mice. TH states for B, survival of Hsp70−/− and Hsp70+/+ mice challenged with RML prions or normal brain homogenate (controls) from a healthy animal was determined. Data were analyzed by log-rank (Mantel–Cox) test and found to be significant with p < 0.05. C, prion disease progression was evaluated on a scale of 1 to 6 as described under “Experimental procedures.” A rating of 1 represents healthy animals, and a rating of 6 was used to designate clinically sick animals at the moment when they were sacrificed. Data were analyzed by two-way analysis of variance, using time and type of mice as the variables, and the differences were significant with p < 0.0001. Individual differences were analyzed by Bonferroni post-test. *, p < 0.05; **, p < 0.01; *** p < 0.001.
Absence of HSP70 does not affect pathological hallmarks
The major hallmark of prion disease is proteinase K (PK)–resistant PrPSc deposition, which is accompanied by vacuolation and astrogliosis in the brain. Equal amounts of PrPSc with an identical banding pattern were observed by Western blot analysis for Hsp70−/− and Hsp70+/+ mice terminal with RML prion disease (Fig. 3A). The equal amounts of pathological protein were not unexpected because these lines express indistinguishable PrPC levels (Fig. 3B), and animals were sacrificed at the same stage of the disease in both groups. In parallel, histological analysis for Hsp70−/− and Hsp70+/+ fixed paraffin-embedded samples revealed the typical diffuse PrPSc deposition expected for RML prion disease (Fig. 3C). Moreover, both lines exhibited similar profiles of vacuolation (Fig. 4A) and gliosis (Fig. 4B). Overall, the pathology was most prominent in the thalamus, cerebellum, and hippocampus. These results indicate that the absence of HSP70 does not affect the accumulation or molecular properties of PrPSc or the pathological hallmarks of RML prion disease despite altering clinical progression.
Figure 3.

Evaluation of PrP in Hsp70−/− and Hsp70+/+ mice. A, Western blot analysis was used to detect PK-resistant PrPSc in brain homogenates from five individual Hsp70−/− and HSP70+/+ mice at the end stage of prion disease. B, a similar Western blot analysis was used to detect PrPC in brain homogenate derived from five individual uninfected mice with GAPDH as the loading control. C, histological analysis of PrPSc accumulation in fixed brain tissue from representative RML-infected Hsp70−/− and Hsp70+/+ mice, which were counterstained with hematoxylin to simultaneously observe cell nuclei. Scale bars = 250 μm.
Figure 4.

Histological changes in RML prion–infected Hsp70−/− and Hsp70+/+ mice. A, spongiform degeneration was analyzed in paraffin-embedded brain tissue slices from Hsp70−/− and Hsp70+/+ mice inoculated with RML prions. As controls, we used animals of both genotypes injected with NBH from a healthy animal. Slices were stained with H&E. B, gliosis was evaluated in fixed brain samples from Hsp70−/− and Hsp70+/+ mice inoculated with RML prions or NBH by probing with anti-glial fibrillary acidic protein (GFAP) antibody. Cell nuclei were simultaneously counterstained with hematoxylin. For the studies in this figure, we focused on the thalamus, hippocampus, and cerebellum, areas heavily affected by RML prion infection. Scale bars = 250 μm.
Cell-free PrPSc replication is affected by the presence of HSP70 in the substrate
Infectious prions can be generated in vitro by protein misfolding cyclic amplification (PMCA), where PrPC is converted into PrPSc with high efficiency utilizing cyclic bursts of sonication (40). This assay can be set up to enable the detection of minute quantities of PrPSc for diagnosis or to estimate the concentration of PrPSc present for quantification (41). We utilized this assay to determine whether the absence of HSP70 altered prion conversion by seeding PMCA reactions with RML prions in NBH substrate from three individual Hsp70−/− or Hsp70+/+ mice. Because PrPC levels are equivalent in the two substrates (Fig. 3B), the primary difference in these reactions is the presence or absence of active HSP70 plus any compensatory changes resulting from the removal of this protein (e.g. overexpression of other chaperone proteins). To our surprise, Hsp70+/+ substrate from three individual animals consistently supported the generation of PrPSc following two rounds of PMCA seeded with a 10−8 dilution of RML, whereas no PrPSc was detected in a similar reaction with Hsp70−/− substrate (Fig. 5). However, PrPSc amplification became nearly indistinguishable after a third round. In summary, more PK-resistant PrPSc is produced in Hsp70+/+ substrate than in Hsp70−/− substrate, suggesting that kinetics (rather than eventual yield) were influenced by HSP70.
Figure 5.
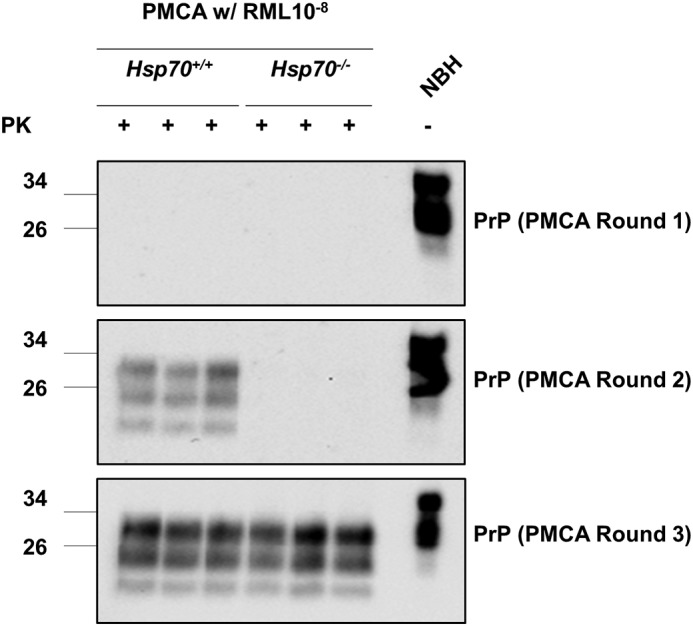
In vitro prion replication by PMCA using Hsp70−/− and Hsp70+/+ brains as substrate. PMCA reactions were seeded with RML prions present in a 10−8 dilution of brain homogenate from terminally ill mice. As substrate for PMCA, we used 10% brain homogenate from three individual Hsp70−/− or Hsp70+/+ mice, and amplification continued for three consecutive rounds (see “Experimental procedures”). PrPSc formation was evaluated by Western blotting after PK digestion. Non-PK-digested samples from NBH derived from a healthy mouse was used as a migration control.
Discussion
In this study, we examined the role of HSP70 in prion replication using an in vitro model of PrPC to PrPSc conversion, a mammalian cell culture model, and a rodent model for prion disease. There is extensive literature supporting the activation of cellular stress mechanisms to regain proteostasis during prion disease (2). One such mechanism is elevated levels of chaperones and foldases triggered to correct the increasing number of misfolded proteins. Indeed, high HSP70 levels have been reported in CJD patients (37) and scrapie-infected mice (24). Moreover, clarifying the influence of HSP70 in mammalian prion disease is particularly intriguing because another member of the HSP family (HSP104) specifically induces fragmentation of yeast prions, enhancing their self-propagation (8). Although the mammalian genome does not encode a corresponding homologue of HSP104, the current model in yeast demonstrates that HSP104 and some members of the HSP70 and HSP40 families are working together in fibril fragmentation and prion propagation (18). Together, our data demonstrate that HSP70 may also play an important role in regulating mammalian prion disease.
Our cell culture experiments broadly determined that a heat shock response is beneficial in reducing PrPSc levels. In fact, chemical induction of a heat shock response by incubation with 17-DMAG significantly reduced PrPSc levels in a cell culture model for prion disease by 50%. However, expression of the molecular chaperones HSP40, HSP70, HSP90, GRP94, and HSP105 has been reported to be induced by treatment with 17-DMAG in mammalian cells (28). Although our data confirmed increased levels of HSP70 under our experimental conditions, the complete effect cannot be solely attributed to this chaperone protein. To the best of our knowledge, previous data from in vitro or in vivo studies have yet to link HSP40 and HSP105 to prion disease, but a possible contribution cannot be ruled out. In addition, the induction of the unfolded protein response (UPR) may have impacted this result as well. This finding is also particularly unique because a heat shock response could be induced in normal prion-free neuroblastoma cells but not neuroblastoma cells propagating prions (42). Furthermore, an additional pitfall in experiments using prion-infected cells is the well-established clonal instability of neuroblastoma cells to keep infection (43). This deficiency has been less impactful for prion-infected lines derived from RK13 cells, where each RKM7-RML cell has been shown to contain detectable levels of PrPSc and not just a small fraction of the population (44). Moreover, it is noteworthy that the unaltered amount of PrPC in 17-DMAG–treated RKM7 cells provides additional evidence that the reduction in PrPSc is not an artifact of drug toxicity but the result of the heat shock response.
Although our cell culture data do not allow us to causally link the observed effects specifically to the loss of stress-induced HSP70, this effect enabled us to narrow our investigation to HSP70 because it is the only known HSP induced by 17-DMAG to also be elevated in CJD patients (37) and scrapie-infected mice (24). Impressively, deletion of Hsp70 in vivo significantly shortened the incubation period for RML prion infection. The accelerated disease produced by knocking out HSP70 may be explained by impairment of prion degradation and/or enhancement of PrPSc accumulation. However, animals lacking HSP70 may also exhibit enhanced sensitivity to prion disease because HSP70 is known to be a powerful inhibitor of apoptosis by binding the apoptotic factor apoptotic protease activating factor 1 (APAF-1) (45) as well as apoptosis-inhibitory factor (46). To further investigate the effect of HSP70 in prion disease, we carefully monitored clinical progression of the disease in both groups of infected mice. The results clearly showed that the difference in prion incubation periods was the result of accelerated clinical progression of the disease in Hsp70-null animals. Indeed, Hsp70+/+ or Hsp70−/− mice infected with RML prions began to develop clinical signs of the disease at the same time, but Hsp70−/− mice deteriorated much faster. The accelerated clinical progression observed in the absence of HSP70 suggests that this chaperone is active in preventing prion toxicity. Because prion disease is divided into an infectivity phase and a toxicity phase (47, 48), HSP70 may primarily respond during the toxicity phase. The effect of HSP70 on clinical progression of prion disease observed in our study was comparable with a previous bioassay utilizing HSF1 knockout animals (27). In this study, Hsf1−/− mice also exhibited faster clinical progression, indicating that HSP70 may be at least partially responsible because Hsp70 is known to be a major target of HSF1. Nonetheless, we need to be cautious when interpreting our results because it has been shown that the impact of altered genotypes on prion disease progression requires a larger number of animals than those used in our study to obtain definitive conclusions (49, 50).
An interesting observation in the animal infectivity experiments was that there were no changes in the type of clinical signs, PK-resistant PrPSc, vacuolation, or astrogliosis related to the presence or absence of HSP70. From these results, we can conclude that the absence of HSP70 did not promote generation of a new prion strain. In many ways, this was the expected outcome because strain changes from selection or evolution have only been reported in cases where the PrPC sequence in the host differed from the donor PrPSc sequence, the species barrier was crossed, or resistance to anti-prion drug treatment was observed (51). Although additional experiments would be required, the putative mechanism by which HSP70 altered the clinical course of the disease might be related to changes on PrPSc replication or, more likely, to the cell's ability to withstand the insult of PrPSc toxicity.
Our in vitro studies of prion replication provide evidence that HSP70 may alter PrPC-to-PrPSc conversion. However, these experiments need to consider the impact tissue homogenization and cell lysis have on PrPSc propagation. For example, an interaction between HSP70 and PrPSc could be induced artificially by lysing the cells, whereas under physiological conditions this may not occur. Nonetheless, colocalization was observed between PrP and HSP70 in transgenic flies (26), and interaction analyses provide evidence that other chaperones (i.e. GRP58 and PDIA1) function by directly binding to PrP (21, 23, 52, 53). Also, some redundancy may exist for the molecular chaperones involved in prion disease; thus, other chaperones may play an augmented role upon removal of HSP70. The roles of these chaperones could similarly be exaggerated during in vitro prion replication studies. A simple interpretation of our PMCA results may be that HSP70 alters the kinetics of PrPSc conversion per se. Even with all of the potential technical issues discussed above, it seems that our data may, in fact, be sufficient for favoring an effect (direct or indirect) on prion conversion/replication as the most likely cause of the effect observed during the clinical phase of disease in vivo.
Compared with our mouse model of prion disease, overexpression of HSP70 in Drosophila more effectively prevented PrP accumulation and/or promoted the degradation of aggregated PrP to an extent that largely reversed neurological deficits (26). A major difference in these models is that the mice in this study were infected with bona fide prions, whereas the transgenic flies exhibited age-dependent PrP accumulation as a consequence of overexpression. Like PrPSc in mammalian prion disease, fly PrP aggregates induced spongiform degeneration, resisted high concentrations of denaturing agents, and contained PrPSc-specific conformational epitopes (26). Despite these similarities, the fly PrP aggregates were mostly protease-sensitive. Therefore, HSP70 may effectively target a neurotoxic and protease-sensitive PrPSc isoform, a function potentially compromised by the more heterogeneous PrPSc population in prion-infected mice. Concomitant with the results generated in the transgenic flies, the appearance of disease symptoms in mice has been shown to depend on increasing levels of small protease-sensitive oligomeric PrPSc instead of larger protease-resistant aggregates (47). For this reason, it would be interesting to determine the impact of HSP70 specifically on small protease-sensitive oligomeric PrPSc generated in our mice.
Further strengthening the evidence that this pathway is specifically involved in prion disease, we also recently showed that another HSP70 family member localized in the ER, GRP78/Bip, similarly influences prion incubation (54). A comparison of the impact of HSP70 and GRP78 is intriguing because they are both members of the larger HSP70 protein family, with ∼63% amino acid identity. Both are also reportedly activated during prion disease (2). Although the complete absence of HSP70 accelerated the incubation time by ∼7%, reducing GRP78 expression by 50% resulted in 20% shorter incubation times with the same prion strain (54). Substrate specificity may contribute to the shortened incubation time because individual HSP70 family members are likely evolutionally optimized to recognize and bind different peptide sequences or structures. Alternatively, GRP78 may seemingly play a more prominent role in prion disease because of localization in the ER lumen, whereas HSP70 is mainly cytosolic. However, the localization differences may be questioned because the source of toxicity in prion disease and the impact of ER misfolded PrP on PrPSc propagation are controversial topics (2). Another possibility is that, in addition to its activity as a molecular chaperone, GRP78 also acts as a master regulator of the unfolded UPR pathway (39). Thus, the larger effect of GRP78 in prion disease may in part be due to an indirect activity on launching the UPR. Indeed, a substantial amount of evidence exists in favor of the UPR, particularly the Protein Kinase RNA-like Endoplasmic Reticulum Kinase pathway, playing a critical role during prion disease (55–57). Last, GRP78 levels could possibly be affected in HSP70 knockout mice because both proteins are involved in stress response, and some HSP may influence expression of the other family members.
Our study represents the first investigation of a putative influence of HSP70 in a bona fide model of mammalian prion disease. Together, our data support the hypothesis that HSP70 helps reduce toxicity and extend disease incubation. These results complement previous literature utilizing prion bioassays in mice lacking HSF1 and studies with other molecular chaperones. Our findings may open up novel targets for therapeutic intervention in prion diseases.
Experimental procedures
Cell culture conditions and treatment
A rabbit kidney epithelial cell line (RK13, catalog no. CCL-37, ATCC) stably expressing mouse PrPC using pIRESpuro3 (Clontech), called RKM cells, was created previously (32). Here we utilized the RKM7 clone (generated from RKM cells) that was selected for high sensitivity to infection with the RML scrapie prion strain. Following chronic infection with RML, we refer to this line as RKM7-RML. RK13, RKM7, and RKM7-RML were grown in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum and 1:100 antibiotic–antimycotic (Gibco) at 37 °C with 5% CO2. For treatment, 1 × 105 cells were seeded in a 6-well plate (∼10% confluence) and cultured overnight. The cells were treated with 17-DMAG for 60 min twice (first treatment on day 0, second treatment on day 3) over a 6-day period. For these two 60-min exposures, 17-DMAG (dissolved in PBS) or an equal volume of PBS was supplemented into the culture medium. Cells were washed immediately following each 60-min treatment to allow them to recover during the assay. In summary, each exposure was concluded by aspiration, a PBS wash, and complete replacement with fresh medium lacking the supplement. Cells were harvested in 250 μl of lysis buffer (50 mm Tris (pH 7.5), 0.5% Triton X-100, and 0.5% sodium deoxycholate).
Mouse lines, prion bioassays, and tissue harvesting
All mice were housed at the University of Texas Health Science Center at Houston in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care in accordance with animal protocols approved by the Institutional Animal Care and Use Committee. Mice genetically deficient in HSP70 (Hspa1a−/− and Hspa1b−/−, referred to as Hsp70−/−) were maintained as a homozygous colony, with genotyping by PCR using primers and conditions as reported previously (58), whereas WT animals sharing the same C57BL/6J genetic background (Hspa1a+/+ and Hspa1b+/+, referred to as Hsp70+/+) were purchased from The Jackson Laboratories (code no. 664). Because of the high sensitivity of prions to genetic backgrounds, the C57BL/6J substrain was chosen because it is the exact genetic match for the animals in which the Hsp70−/− mice were created. In brief, Hsp70−/− mice were initially backcrossed by speed congenics onto this C57BL/6J strain for six generations and then further backcrossed onto the same strain by standard breeding for an additional four crosses. According to guidelines established by Prusiner et al. (59), groups of five female Hsp70−/− and WT Hsp70+/+ mice (ranging in age from 72 to 78 days) were infected with the RML mouse–adapted scrapie strain via stereotactic intracranial inoculation of 10 μl of 10% (w/v) brain homogenate (BH) from a terminally ill animal into the hippocampus (right hemisphere). Similarly, five age-matched mice for each line received an equal volume of 10% (w/v) NBH derived from a healthy animal. Animals were evaluated five times per week for health conditions and appearance of prion-associated clinical signs. Because RML infection has been carefully characterized in our laboratory, blind evaluation of the mice was conducted to score the progression of prion disease using the following version of a scale published previously (38, 39): 1, normal; 2, no nesting, rough coat on limbs, abnormal social behavior; 3, extensive rough coat, hunchback, and visible motor abnormalities; 4, progression of observed signs plus urogenital lesions, increased motor activity, and extensive weight loss; 5, end stage of disease where the animal presents ataxia and lies in the cage with little movement. Animals were terminated following consecutive scores of 5 as they became terminal. Upon termination, mouse brains were harvested by freezing the right cerebral hemisphere and fixing the left cerebral hemisphere in Carnoy fixative. Frozen samples were homogenized at 10% (w/v) in PBS containing complete protease inhibitors (Roche).
PMCA
PMCA was carried out according to our published protocol (41), using BH from Hsp70−/− or Hsp70+/+ mice (both expressing equal WT levels of endogenous mouse PrPC) as substrate. Briefly, brain substrate was prepared at a concentration of 10% (w/v) in conversion buffer (PBS supplemented with 150 mm NaCl and 1% Triton X-100) with protease inhibitors (Complete, Roche). Debris was removed by low-speed centrifugation (800 × g, 1 min, 4 °C) and stored at −80 °C until use. For PMCA, aliquots of 10 μl of 10% (w/v) BH from terminal RML-infected mice, and 90 μl of brain substrate were added to 0.2-ml PCR tubes (Eppendorf, catalog no. 951010022) containing two Teflon beads (Hoover Precision Products) and subjected to 48 PMCA cycles. Each cycle consisted of 29 min and 40 s incubation at 37/40 °C, followed by a 20-s pulse of sonication set at a potency of 110–120 W using the Qsonica microsonicator (model Q700) equipped with a titanium horn. Subsequent rounds of 48 PMCA cycles were done by taking an aliquot of the amplified material and diluting it 10-fold into fresh brain substrate. After four rounds of PMCA, PK-digested samples were analyzed by Western blots probed with the 6D11 anti-PrP antibody (1:30,000, BioLegend), as described previously (41).
Brain histology
The left cerebral hemisphere of the mouse brain was fixed in Carnoy fixative (60), dehydrated, and embedded in paraffin. 10-μm slices were mounted on glass slides, dewaxed with xylene, and hydrated in solutions of decreasing concentrations of alcohol (100% to 70%). Serial sections were treated with 6% H2O2 for 20 min, rinsed in water, and stained with H&E or immunostained with monoclonal antibodies for PrP (6H4, 1:1000, Prionics) or astrocytes (anti-glial fibrillary acidic protein (GFAP), 1:2000, Abcam). To visualize PrPSc, the sections were treated with 10 μg/ml PK for 5 min at room temperature and subsequently with 3 m guanidine isothiocyanate for 20 min at room temperature. To prevent nonspecific binding, the Animal Research Kit (Dako, Glostrup, Denmark) was used. Immunostaining was developed using HRP-conjugated streptavidin and visualized with 3–3′-diaminobenzidine (Vector Laboratories) as chromogen. Tissue was later counterstained with hematoxylin for 30 s and rinsed in tap water for 10 min. Sections were examined under a bright-field DMI6000B microscope (Leica).
Western blotting
Protein concentrations were quantified for cell lysate and BH using the BCA assay kit (Pierce). PrPC was analyzed in 40-μg 10% (w/v) BH and 200-μg cell lysate samples precipitated with 5 volumes of MeOH and resuspended in 1× NuPAGE Lithium Dodecyl Sulfate sample loading buffer (Invitrogen). For PrPSc, 40-μg 10% (w/v) BH and 650-μg cell lysate samples were digested with 100 μg/ml PK for 1 h at 37 °C with shaking, which was stopped by adding 1 mm phenylmethylsulfonylfluoride. Digested samples were centrifuged at 4 °C for 1 h at 20,000 × g, and pellets were resuspended in 1× NuPAGE LDS sample loading buffer. All samples were boiled for 10 min before running in 14% Novex Tris/glycine gels (Invitrogen) and transferring onto nitrocellulose membranes (0.45 μm, Amersham Biosciences). Blots were blocked for 1 h at room temperature with 5% milk. Then PrP was probed with the 6H4 mAb (1:10,000). Following incubation with anti-mouse IgG secondary antibody conjugated to horseradish peroxidase (1:3,000), the signal was visualized using the ECL chemicochemical reagent (Amersham Biosciences) and a Chemidoc imaging system (Bio-Rad). As a loading control, some blots were reprobed for GAPDH (anti-GAPDH, 1:2,000, Pierce) after stripping (Restore Western blotting stripping buffer, Thermo Scientific) and blocking. Similarly, HSP70 was detected in 20 μg of BH (anti-HSP70 ADI-SPA-810, 1:2,000, Enzo Life Sciences) with tyrosine hydroxylase as a loading control (anti-tyrosine hydroxylase ab112, 1:2,000, Abcam). For analysis of cultured cells, ∼10 μg of total cell extract was probed with HSP70 antibody, and equivalent gel protein loading was confirmed by simultaneous probing with monoclonal mouse anti-actin (MP Biomedicals, 08691001, 1:100,000).
Statistical analysis
ImageJ was utilized to perform densitometry on the western blots. GraphPad Prism was used to calculate statistical significance via Tukey's multiple comparisons test for Western blots, Mann–Whitney test for in vivo incubation, and two-way analysis of variance for clinical progression. Statistical significance is shown as follows: *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Author contributions
C. E. M., C. R. H., and C. S. conceptualization; C. E. M., E. A., R. M., and C. K. data curation; C. E. M., E. A., and C. S. formal analysis; C. E. M. investigation; C. E. M., E. A., and R. M. methodology; C. E. M. writing-original draft; C. E. M., E. A., R. M., C. K., A. F., A. T., J. B., G. C. T., T. K. P., C. R. H., and C. S. writing-review and editing; A. F., A. T., J. B., G. C. T., T. K. P., and C. R. H. resources; C. S. supervision; C. S. funding acquisition; C. S. project administration.
This work was supported in part by NIAID, National Institutes of Health Grants P01 IA106705 and P01 IA077774 (to C. S.) and NIGMS, National Institutes of Health Grant GM109768 (to T. K. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
C. E. Mays, E. Armijo, R. Morales, C. Kramm, A. Flores, A. Tiwari, J. Bian, G. C. Telling, T. K. Pandita, C. R. Hunt, and C. Soto, unpublished data.
- CJD
- Creutzfeldt–Jakob disease
- ER
- endoplasmic reticulum
- GRP
- glucose-regulated protein
- HSP
- heat shock protein
- HSF
- heat shock factor
- 17-DMAG
- 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin
- RML
- Rocky Mountain Laboratory
- NBH
- normal brain homogenate
- dpi
- day post-inoculation
- PK
- proteinase K
- PMCA
- protein misfolding cyclic amplification
- UPR
- unfolded protein response
- BH
- brain homogenate
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase.
References
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mays C. E., and Soto C. (2016) The stress of prion disease. Brain Res. 1648, 553–560 10.1016/j.brainres.2016.04.009 [DOI] [PubMed] [Google Scholar]
- 3. Hunt C., and Morimoto R. I. (1985) Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc. Natl. Acad. Sci. U.S.A. 82, 6455–6459 10.1073/pnas.82.19.6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benbrook D. M., and Long A. (2012) Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol 34, 286–297 [PubMed] [Google Scholar]
- 5. Wickner R. B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- 6. Coustou V., Deleu C., Saupe S., and Begueret J. (1997) The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. U.S.A. 94, 9773–9778 10.1073/pnas.94.18.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sondheimer N., and Lindquist S. (2000) Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell 5, 163–172 10.1016/S1097-2765(00)80412-8 [DOI] [PubMed] [Google Scholar]
- 8. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., Liebman S. W. (1995) Role of the chaperone protein HSP104 in propagation of the Yeast prion-like Factor [psi+]. Science 268, 880–884 10.1126/science.7754373 [DOI] [PubMed] [Google Scholar]
- 9. Winkler J., Tyedmers J., Bukau B., and Mogk A. (2012) Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 198, 387–404 10.1083/jcb.201201074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jung G., Jones G., Wegrzyn R. D., and Masison D. C. (2000) A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 156, 559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roberts B. T., Moriyama H., and Wickner R. B. (2004) [URE3] prion propagation is abolished by a mutation of the primary cytosolic Hsp70 of budding yeast. Yeast 21, 107–117 10.1002/yea.1062 [DOI] [PubMed] [Google Scholar]
- 12. Sharma D., and Masison D. C. (2008) Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179, 1301–1311 10.1534/genetics.108.089458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma D., Stanley R. F., and Masison D. C. (2009) Curing of yeast [URE3] prion by the Hsp40 cochaperone Ydj1p is mediated by Hsp70. Genetics 181, 129–137 10.1534/genetics.108.098699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reidy M., Miot M., and Masison D. C. (2012) Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 192, 185–193 10.1534/genetics.112.142307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higurashi T., Hines J. K., Sahi C., Aron R., and Craig E. A. (2008) Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. U.S.A. 105, 16596–16601 10.1073/pnas.0808934105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DebBurman S. K., Raymond G. J., Caughey B., and Lindquist S. (1997) Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl. Acad. Sci. U.S.A. 94, 13938–13943 10.1073/pnas.94.25.13938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dandoy-Dron F., Bogdanova A., Beringue V., Bailly Y., Tovey M. G., Laude H., and Dron M. (2006) Infection by ME7 prion is not modified in transgenic mice expressing the yeast chaperone Hsp104 in neurons. Neurosci. Lett. 405, 181–185 10.1016/j.neulet.2006.05.066 [DOI] [PubMed] [Google Scholar]
- 18. Chernova T. A., Wilkinson K. D., and Chernoff Y. O. (2017) Prions, chaperones, and proteostasis in yeast. Cold Spring Harb. Perspect. Biol. 9, a023663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hetz C., Russelakis-Carneiro M., Maundrell K., Castilla J., and Soto C. (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 22, 5435–5445 10.1093/emboj/cdg537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shyu W.-C., Kao M.-C., Chou W.-Y., Hsu Y.-D., and Soong B.-W. (2000) Creutzfeldt-Jakob disease: heat shock protein 70 mRNA levels in mononuclear blood cells and clinical study. J. Neurol 247, 929–934 10.1007/s004150070048 [DOI] [PubMed] [Google Scholar]
- 21. Torres M., Medinas D. B., Matamala J. M., Woehlbier U., Cornejo V. H., Solda T., Andreu C., Rozas P., Matus S., Muñoz N., Vergara C., Cartier L., Soto C., Molinari M., and Hetz C. (2015) The protein-disulfide isomerase ERp57 regulates the steady-state levels of the prion protein. J. Biol. Chem. 290, 23631–23645 10.1074/jbc.M114.635565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoo B. C., Krapfenbauer K., Cairns N., Belay G., Bajo M., and Lubec G. (2002) Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci. Lett. 334, 196–200 10.1016/S0304-3940(02)01071-6 [DOI] [PubMed] [Google Scholar]
- 23. Hetz C., Russelakis-Carneiro M., Wälchli S., Carboni S., Vial-Knecht E., Maundrell K., Castilla J., and Soto C. (2005) The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 25, 2793–2802 10.1523/JNEUROSCI.4090-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenward N., Hope J., Landon M., and Mayer R. J. (1994) Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brain. J. Neurochem. 62, 1870–1877 [DOI] [PubMed] [Google Scholar]
- 25. Wang S.-B., Shi Q., Xu Y., Xie W.-L., Zhang J., Tian C., Guo Y., Wang K., Zhang B.-Y., Chen C., Gao C., and Dong X.-P. (2012) Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 7, e38221 10.1371/journal.pone.0038221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fernandez-Funez P., Casas-Tinto S., Zhang Y., Gómez-Velazquez M., Morales-Garza M. A., Cepeda-Nieto A. C., Castilla J., Soto C., and Rincon-Limas D. E. (2009) In vivo generation of neurotoxic prion protein: role for Hsp70 in accumulation of misfolded isoforms. PLoS Genet. 5, e1000507 10.1371/journal.pgen.1000507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steele A. D., Hutter G., Jackson W. S., Heppner F. L., Borkowski A. W., King O. D., Raymond G. J., Aguzzi A., and Lindquist S. (2008) Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc. Natl. Acad. Sci. U.S.A. 105, 13626–13631 10.1073/pnas.0806319105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herbst M., and Wanker E. E. (2007) Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. Neurodegener. Dis. 4, 254–260 10.1159/000101849 [DOI] [PubMed] [Google Scholar]
- 29. Mays C. E., Joy S., Li L., Yu L., Genovesi S., West F. G., and Westaway D. (2012) Prion inhibition with multivalent PrPSc binding compounds. Biomaterials 33, 6808–6822 10.1016/j.biomaterials.2012.06.004 [DOI] [PubMed] [Google Scholar]
- 30. Bian J., Kang H.-E., and Telling G. C. (2014) Quinacrine promotes replication and conformational mutation of chronic wasting disease prions. Proc. Natl. Acad. Sci. U.S.A. 111, 6028–6033 10.1073/pnas.1322377111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vilette D., Andreoletti O., Archer F., Madelaine M. F., Vilotte J. L., Lehmann S., and Laude H. (2001) Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. U.S.A. 98, 4055–4059 10.1073/pnas.061337998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bian J., Khaychuk V., Angers R. C., Fernández-Borges N., Vidal E., Meyerett-Reid C., Kim S., Calvi C. L., Bartz J. C., Hoover E. A., Agrimi U., Richt J. A., Castilla J., and Telling G. C. (2017) Prion replication without host adaptation during interspecies transmissions. Proc. Natl. Acad. Sci. U.S.A. 114, 1141–1146 10.1073/pnas.1611891114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neale M. H., Mountjoy S. J., Edwards J. C., Vilette D., Laude H., Windl O., and Saunders G. C. (2010) Infection of cell lines with experimental and natural ovine scrapie agents. J. Virol. 84, 2444–2452 10.1128/JVI.01855-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Courageot M.-P., Daude N., Nonno R., Paquet S., Di Bari M. A., Le Dur A., Chapuis J., Hill A. F., Agrimi U., Laude H., and Vilette D. (2008) A cell line infectible by prion strains from different species. J. Gen. Virol. 89, 341–347 10.1099/vir.0.83344-0 [DOI] [PubMed] [Google Scholar]
- 35. Lawson V. A., Vella L. J., Stewart J. D., Sharples R. A., Klemm H., Machalek D. M., Masters C. L., Cappai R., Collins S. J., and Hill A. F. (2008) Mouse-adapted sporadic human Creutzfeldt-Jakob disease prions propagate in cell culture. Int. J. Biochem. Cell Biol. 40, 2793–2801 10.1016/j.biocel.2008.05.024 [DOI] [PubMed] [Google Scholar]
- 36. Bian J., Napier D., Khaychuck V., Angers R., Graham C., and Telling G. (2010) Cell-based quantification of chronic wasting disease prions. J. Virol. 84, 8322–8326 10.1128/JVI.00633-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kovács G. G., Kurucz I., Budka H., Adori C., Müller F., Acs P., Klöppel S., Schätzl H. M., Mayer R. J., and László L. (2001) Prominent stress response of Purkinje cells in Creutzfeldt-Jakob disease. Neurobiol. Dis. 8, 881–889 10.1006/nbdi.2001.0418 [DOI] [PubMed] [Google Scholar]
- 38. Castilla J., Morales R., Saá P., Barria M., Gambetti P., and Soto C. (2008) Cell-free propagation of prion strains. EMBO J. 27, 2557–2566 10.1038/emboj.2008.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morales R., Estrada L. D., Diaz-Espinoza R., Morales-Scheihing D., Jara M. C., Castilla J., and Soto C. (2010) Molecular cross-talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J. Neurosci. 30, 4528–4535 10.1523/JNEUROSCI.5924-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saborio G. P., Permanne B., and Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 10.1038/35081095 [DOI] [PubMed] [Google Scholar]
- 41. Morales R., Duran-Aniotz C., Diaz-Espinoza R., Camacho M. V., and Soto C. (2012) Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 7, 1397–1409 10.1038/nprot.2012.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tatzelt J., Zuo J., Voellmy R., Scott M., Hartl U., and Prusiner S. B., and Welch W. J. (1995) Scrapie prions selectively modify the stress response in neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 92, 2944–2948 10.1073/pnas.92.7.2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klöhn P.-C., Stoltze L., Flechsig E., Enari M., and Weissmann C. (2003) A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. U.S.A. 100, 11666–11671 10.1073/pnas.1834432100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lyon A., Mays C. E., Borriello F., Telling G. C., Soto C., and Pritzkow S. (2019) Application of PMCA to screen for prion infection in a human cell line used to produce biological therapeutics. Sci. Rep. 9, 4847 10.1038/s41598-019-41055-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sabirzhanov B., Stoica B. A., Hanscom M., Piao C.-S., and Faden A. I. (2012) Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J. Neurochem. 123, 542–554 10.1111/j.1471-4159.2012.07927.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ravagnan L., Gurbuxani S., Susin S. A., Maisse C., Daugas E., Zamzami N., Mak T., Jäättelä M., Penninger J. M., Garrido C., and Kroemer G. (2001) Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 3, 839–843 10.1038/ncb0901-839 [DOI] [PubMed] [Google Scholar]
- 47. Mays C. E., van der Merwe J., Kim C., Haldiman T., McKenzie D., Safar J. G., and Westaway D. (2015) Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased oligomeric PrPSc species. J. Virol. 89, 12418–12426 10.1128/JVI.02142-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandberg M. K., Al-Doujaily H., Sharps B., Clarke A. R., and Collinge J. (2011) Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470, 540–542 10.1038/nature09768 [DOI] [PubMed] [Google Scholar]
- 49. Zhu C., Li B., Frontzek K., Liu Y., and Aguzzi A. (2019) SARM1 deficiency up-regulates XAF1, promotes neuronal apoptosis, and accelerates prion disease. J. Exp. Med. 216, 743–756 10.1084/jem.20171885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Puig B., Altmeppen H. C., Linsenmeier L., Chakroun K., Wegwitz F., Piontek U. K., Tatzelt J., Bate C., Magnus T., and Glatzel M. (2019) GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLOS Pathog. 15, e1007520 10.1371/journal.ppat.1007520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morales R. (2017) Prion strains in mammals: Different conformations leading to disease. PLOS Pathog. 13, e1006323 10.1371/journal.ppat.1006323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin T., Gu Y., Zanusso G., Sy M., Kumar A., Cohen M., Gambetti P., and Singh N. (2000) The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J. Biol. Chem. 275, 38699–38704 10.1074/jbc.M005543200 [DOI] [PubMed] [Google Scholar]
- 53. Watts J. C., Huo H., Bai Y., Ehsani S., Jeon A. H., Won A. H., Shi T., Daude N., Lau A., Young R., Xu L., Carlson G. A., Williams D., Westaway D., and Schmitt-Ulms G. (2009) Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 5, e1000608 10.1371/journal.ppat.1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park K.-W., Eun Kim G., Morales R., Moda F., Moreno-Gonzalez I., Concha-Marambio L., Lee A. S., Hetz C., and Soto C. (2017) The endoplasmic reticulum chaperone GRP78/BiP modulates prion propagation in vitro and in vivo. Sci. Rep. 7, 44723 10.1038/srep44723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., Martin M. G., Halliday M., Morgan J., Dinsdale D., Ortori C. A., Barrett D. A., Tsaytler P., Bertolotti A., Willis A. E., Bushell M., and Mallucci G. R. (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 10.1038/nature11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moreno J. A., Halliday M., Molloy C., Radford H., Verity N., Axten J. M., Ortori C. A., Willis A. E., Fischer P. M., Barrett D. A., and Mallucci G. R. (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138 [DOI] [PubMed] [Google Scholar]
- 57. Halliday M., Radford H., Sekine Y., Moreno J., Verity N., le Quesne J., Ortori C. A., Barrett D. A., Fromont C., Fischer P. M., Harding H. P., Ron D., and Mallucci G. R. (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672 10.1038/cddis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hunt C. R., Dix D. J., Sharma G. G., Pandita R. K., Gupta A., Funk M., and Pandita T. K. (2004) Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol. Cell. Biol. 24, 899–911 10.1128/MCB.24.2.899-911.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prusiner S. B., Safar J., and DeArmond S. J. (2004) in Prion Biology and Diseases (Prusiner S. B., ed.) pp. 143–186, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 60. Giaccone G., Canciani B., Puoti G., Rossi G., Goffredo D., Iussich S., Fociani P., Tagliavini F., and Bugiani O. (2000) Creutzfeldt-Jakob disease: Carnoy's fixative improves the immunohistochemistry of the proteinase K-resistant prion protein. Brain Pathol. 10, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]