

Abstract
The voltage-gated sodium (Nav) channel is the molecular determinant of action potential in neurons. Protein-protein interactions (PPI) between the intracellular Nav1.6 C-tail and its regulatory protein fibroblast growth factor 14 (FGF14) provide an ideal and largely untapped opportunity for development of neurochemical probes. Based on a previously identified peptide FLPK, mapped to the FGF14:FGF14 PPI interface, we have designed and synthesized a series of peptidomimetics with the intent of increasing clogP values and improving cell perme-ability relative to the parental lead peptide. In-cell screening using the split-luciferase complementation (LCA) assay identified ZL0177 (13) as the most potent inhibitor of the FGF14:Nav1.6 channel complex assembly with an apparent IC50 of 11 μM. Whole-cell patch-clamp recordings demonstrated that ZL0177 significantly reduced Nav1.6-mediated transient current density and induced a depolarizing shift of the channel voltage-dependence of activation. Docking studies revealed strong interactions between ZL0177 and Nav1.6, mediated by hydrogen bonds, cation-π interactions and hydrophobic contacts. All together these results suggest that ZL0177 retains some key features of FGF14-dependent modulation of Nav1.6 currents. Overall, ZL0177 provides a chemical scaffold for developing Nav channel modulators as pharmacological probes with therapeutic potential of interest for a broad range of CNS and PNS disorders.
Keywords: Voltage-gated sodium channels, Fibroblast growth factor, Chemical probes, Protein-protein interactions, Split-luciferase complementation assay
Voltage-gated sodium (Nav) channels consisting of a pore-forming α subunit and accessory regulatory proteins provide the basis for electrical excitability in neurons.1–6 There are nine different α subunits (Nav1.1–1.9) formed by four transmembrane domains (D1–D4) each containing six segments (S1–S6) and extracellular domains.7,8 These portions of the channel share very high (more than 50%) sequence homology across the nine α subunit isoforms, while intracellular loops and N- and C-terminal tails confer structural and functional specificity to each channel complex through protein-protein interaction (PPIs) with specific accessory subunits.9
Nav channels are ubiquitously expressed in the central nervous system (CNS; Nav1.1–1.3 and 1.6), peripheral nervous system (PNS; Nav1.6–1.9), adult skeletal muscle (Nav1.4) as well as cardiac muscle (Nav1.5) and have been linked to a variety of human channelopathies such as epilepsy,10–12 pain, 13–15 schizophrenia,16 cardiac arrhythmias17,18 and Brugada syndromes19. Such translational relevance makes the Nav channel an appealing target for drug development. However, despite strong interest in developing targeted drugs against Nav channels, approved medications such as local anesthetics (i.e. lidocaine) and anti-epileptic drugs (i.e carbamazepine, lamotrigine) target highly conserved sites. There is a need for new drug design strategies that would lead to fine-tune isoform-specific modulation of Nav channels. Hence we propose to use PPI interfaces at the Nav channel complex as targets for new drug development.
We previously identified fibroblast growth factor 14 (FGF14) as an intracellular modulator of Nav channels,20 observed distinct and opposing regulatory roles of FGF14 isoforms on Nav1.2 and Nav 1.6 function,21 and resolved critical amino acid residues at the FGF14:Nav1.6 interface through mutation studies.22 FGF14 modulates amplitude and voltage dependence of Na+ currents through direct interaction with the intracellular Nav channel C-tail.23–26 Thus, probes capable of modulating this highly specific PPI interface might lead to fine-tune regulation of electrical activity in the CNS and PNS and serve as scaffolds for therapeutic development. Here, we present chemical modifications of a short peptide Ac-FLPK-CONH2 (aka. FLPK) that we previously characterized as an FGF14 inhibitor mapped to its PPI surface, and selected one variant, ZL0177,27 as a novel peptidomimetic with improved chemical profiles and demonstrated that it is functionally active against Nav1.6 mediated currents.
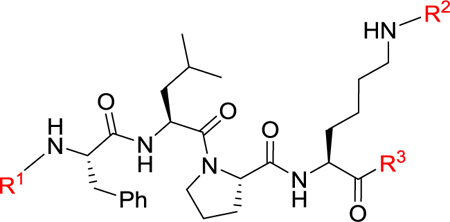
The partition coefficient of the molecule between an aqueous and lipophilic phase (logP), usually water and octanol, determines a molecule’s lipophilicity that is crucial for passive membrane permeability.28–31 The parent tetrapeptide FLPK has a predicted clogP value of 0.9 (calculated by ALOGPS) which is not favorable for crossing the cell membrane. The N- (R1), C-terminal (R3) and free NH2 (R2) in lysine are critical sites to improve clogP of parental peptide. Thus, we designed three series of new peptides through truncation (tripeptides) with diverse functional groups to maintain proper molecule weight, introducing hydrophobic protective groups (tetrapeptide) and incorporating non-peptide small molecules (Table 1). Compared to FLPK, all compounds have improved clogP values that indicate enhanced permeability. The synthetic route exampled by peptides 7–8 and 11–13 is depicted in Scheme 1. (tert-Butoxycarbonyl)-l-leucyl-l-proline (22) and methyl N6-(((9H-fluoren-9-yl) methoxy)carbonyl)-l-lysinate (23) were coupled in the presence of HBTU, HOBt and DIPEA to give tripeptide 7 in a quantitative yield. Boc was removed under TFA leading to 8 which is coupled with (tert-butoxycarbonyl)-l-phenylalanine to produce tetrapeptide 11. Compound 12 was obtained through eliminating the protective group of 11, and subsequent acetylation of 12 generated peptide 13 in a yield of 93%. All the peptides were synthesized in a similar fashion, and their structures were confirmed by 1H NMR, 13C NMR and HR-MS. Compounds 1 and 14 were explored before,32 and are included herein as controls, because the parental peptide does not work in this cellular assay due to its poor cell permeability.
Table 1.
Sequences and clogP values of newly designed peptidomimetics.
 | |||
|---|---|---|---|
| Series | Codes | Sequence | clogPa |
| Parent | – | FLPK | 0.9 |
| Tri-peptide | 1 | Cbz-FLP-NH2 | 2.1 |
| 2 | Cbz-FLK(Boc)-NH2 | 3.3 | |
| 3 | Boc-LPK(Boc)-NH2 | 2.0 | |
| 4 | PLK(Boc)-NH2 | 1.8 | |
| 5 | LPK-NH2 | 1.3 | |
| 6 | Ac-PLK(Boc)-NH2 | 2.1 | |
| 7 | Boc-LPK(Fmoc)-OMe | 4.6 | |
| 8 | LPK(Fmoc)-OMe | 3.2 | |
| 9 | Ac-LPK(Fmoc)-OMe | 3.6 | |
| 10 | Cbz-FLP-OMe | 3.0 | |
| Tetra-peptide | 11 | Boc-FLPK(Fmoc)-OMe | 5.4 |
| 12 | FLPK(Fmoc)-OMe | 4.1 | |
| 13 | Ac-FLPK(Fmoc)-OMe | 4.8 | |
| 14 | Cbz-FLPK(Boc)-NH2 | 3.5 | |
| 15 | Ac-FLPK(Boc)-OH | 2.2 | |
| 16 | FLPK(Boc)-NH2 | 1.9 | |
| 17 | Bz-FLPK(Boc)-NH2 | 3.3 | |
| Incorpo-rationb | 18 | R1 = Ac, R2 = Boc; R3 = Morpholine | 2.5 |
| 19 | R1 = Ac, R2 = Boc; R3 = Ph | 3.6 | |
| 20 | R1 = Ac, R2 = Boc; R3 = 2-thiazol | 2.9 | |
| 21 | R1 = Ac, R2 = H; R3 = Ph | 2.6 | |
Incorporation of non-peptide scaffold into FLPK.
Scheme 1.
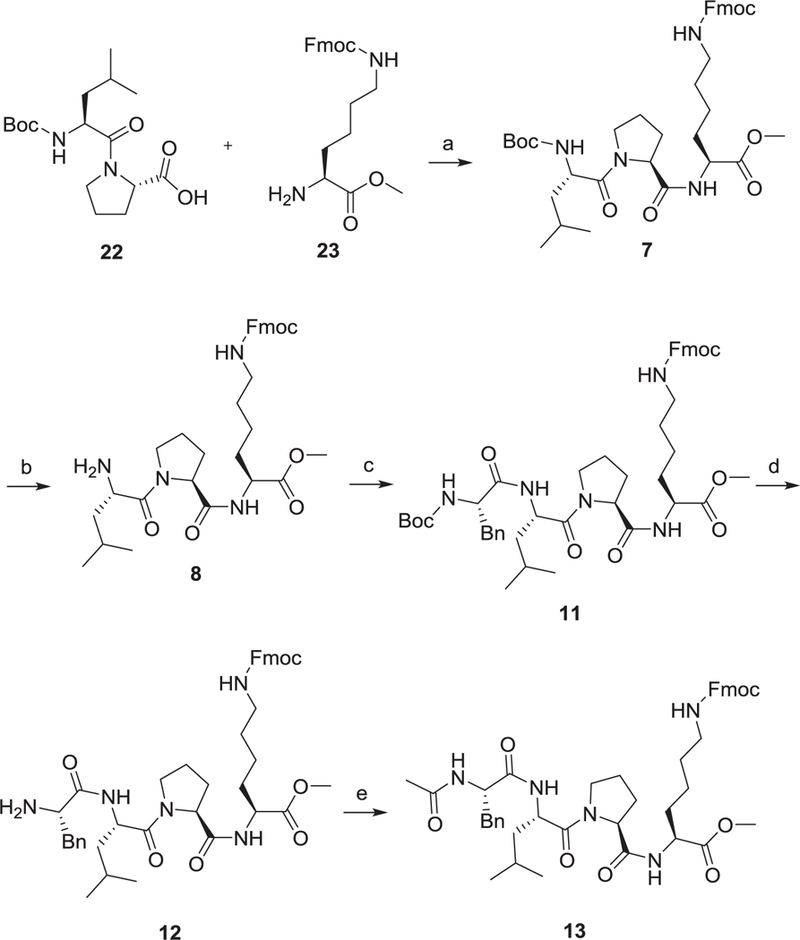
Synthetic route of 7–8 and 11–13. Reagents and conditions: (a) HBTU, HOBt, DIPEA, DCM, rt., quant.; (b) TFA, DCM, rt., 87%; (c) (tert-butoxycarbonyl)-l-phenylalanine, HBTU, HOBt, DIPEA, DCM, rt., 81%; (d) TFA, DCM, rt., quant.; (e) CH3COCl, Et3N, DCM, rt., 91%.
All the newly designed and synthesized peptidomimetics were first evaluated using the split-luciferase complementation assay (LCA) in HEK293 cells, whereby the FGF14:Nav1.6 complex was reconstituted through transient co-transfection of the Cluc-FGF14 and CD4-Nav1.6-Nluc plasmids.22,25,26,33 Peptidomimetics were dissolved in DMSO and delivered to transfected cells via dilution in cell medium to a final concentration of 50 μM in 0.5% DMSO. Interestingly, the luminescent response was slightly enhanced by treatment with several compounds in series I (tripeptides) and series III (incorporation of non-peptide scaffolds), while compounds of series II (tetrapeptides) tended to inhibit complex formation compared to treatment with 0.5% DMSO alone (Fig. 1A). Significant enhancers identified include compounds 4, 5, 6, and 12 (148.7 ± 9.2%, 142.3 ± 11.8%, 126.5 ± 7.8%, and 124.1 ± 7.0%, respectively; p < 0.05), and significant inhibitors identified include compounds 13, 14, and 17 (67.9 ± 6.1%, 71.9 ± 5.6%, and 66.5 ± 5.4%, respectively; p < 0.05). These screening results were validated by testing peptidomimetics against the full-length luciferase (reporter) to ensure that luminescence changes did not arise from modulation of luciferase enzymatic activity alone, and no significant effects were observed (Fig. 1B).
Fig. 1.
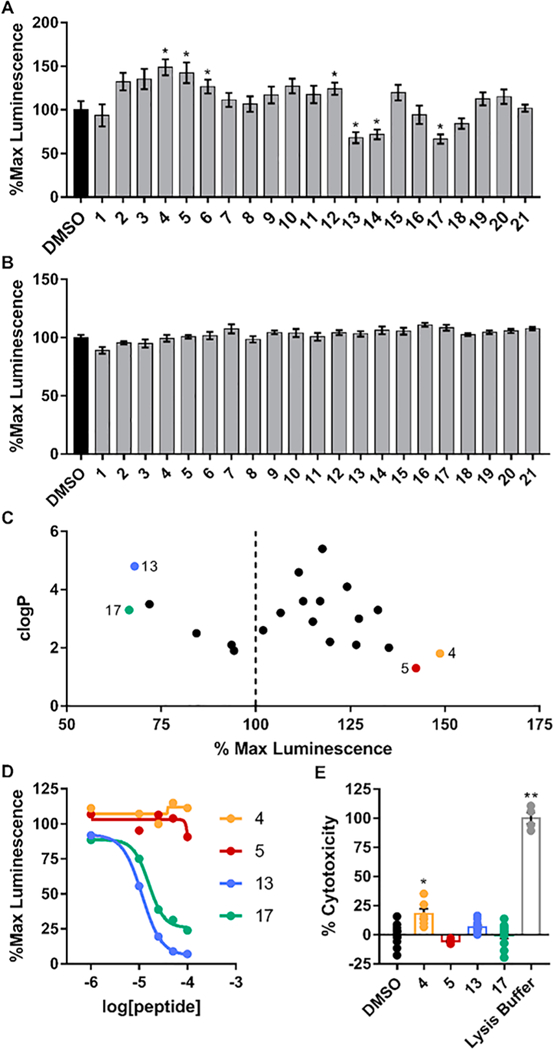
LCA screening of peptidomimetics. A, B) Bar graphs of % maximal luminescence values derived from HEK293 cells stably expressing CLuc-FGF14 and CD4-Nav1.6-NLuc constructs using LCA (A) or transiently transfected with the full-length photinus luciferase (B) treated with either vehicle (0.5% DMSO) or compounds (final concentration=50 μM). C) Comparison of peptidomimetic clogP values and % max luminescence responses. The top two inhibitors and enhancers (Compounds 4, 5, 13, and 17) were selected for further evaluation. D) LCA-based dose-response of compounds 4, 5, 13 and 17 in cells stably expressing CLuc-FGF14 and CD4-Nav1.6-NLuc. E) Bar graph of % cytotoxicity as measured using the LDH cytotoxicity assay of selected compounds at the concentration of 50 μM. Data are mean ± S.E.M. *, p < 0.05; **, p < 0.001.
Based on the LCA results and taking clogP values into consideration (Fig. 1C), we selected compounds 13 and 17 as potential inhibitors, and 4, 5 as potential enhancers for further validation. Compound potency and efficacy were subsequently assessed using a 5-point dose response (1, 10, 25, 50, and 100 μM), and percent luminescence (normalized to 0.5% DMSO) is plotted in Fig. 1D. Compounds 4 and 5 did not display a reasonable dose-dependent response, while compounds 13 and 17 exhibited a sigmoidal dose-response inhibition curve, with apparent IC50 values of 11 and 16 μM, respectively (Fig. 1_SI). Additionally, these compounds were validated using the lactate dehydrogenase (LDH) cytotoxicity assay to ensure that changes in cell viability were not responsible for changes in luminescence response (Fig. 1E). Only compound 4 demonstrated significant toxicity.
To functionally validate the identified top peptidomimetic (compound 13; ZL0177), we performed whole-cell patch-clamp electro-physiology of HEK293 cells stably expressing Nav1.6 (HEK-Nav1.6) (Fig. 2 and Suppl. Table 1).34 In HEK-Nav1.6 cells pretreated for 1 h with ZL0177 (10 μM), Nav1.6-mediated peak density derived from transient Na+ current (INa) was significantly lower (−26.65 ± 6.3 pA/pF) compared to DMSO treatment (−54.88 ± 7.4 pA/Pf, Fig. 2C–D). Further analysis revealed that ZL0177 slowed the transition of the channel from the open to the inactive state in HEK-Nav1.6 cells (2.05 ± 0.3 ms) compared to DMSO control group in (1.18 ± 0.1ms, Fig. 2B and E). These results suggest that ZL0177 is able to mimic FGF14-induced suppression of Nav1.6 currents and effect on tau of fast inactivation.32 This finding is different from 14 (ZL0181) which requires the presence of FGF14.32
Fig. 2.
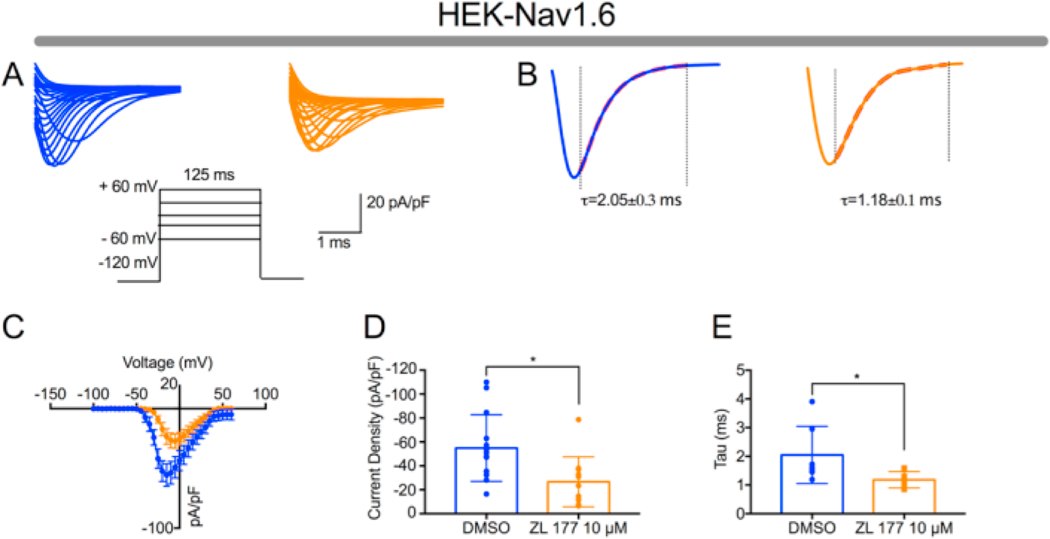
Peptidomimetic 13 (ZL0177) modulates Nav1.6 mediated Na + currents. (A) Representative traces of Nav1.6-mediated transient Na+ currents (INa+) recorded from HEK-Nav1.6 stable cells treated with either DMSO (0.02%; blue) or ZL0177 (10 μM; orange) in response to voltage steps from −120 mV to +60 mV from a holding potential of −70 mV (inset). (B) Representative traces of experimental groups described in A in which tau (τ) of transient INa+ was estimated from a one-term exponential fitting function (red dotted line). (C) Current-voltage relationships of transient INa+ from experimental groups described in A and B. (D) Summary bar graph of peak current densities derived from C. (E) Summary bar graph of tau calculated at −10 mV in the indicated experimental groups. Data are mean ± S.E.M. *, p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Further effects of ZL0177 on V1/2 of activation and steady-state inactivation were investigated. ZL0177 induced a 26.37 ± 1.5(12)mV (DMSO) to 19.2 ± 1.3 (9) depolarizing shift (p < 0.05) of V1/2 of activation (Fig. 3A and B), while it displayed no effects on V1/2 of steady-state of inactivation (Fig. 3C and D). Furthermore, ZL0177 exhibited no effects on long-term inactivation of Nav1.6 channels (Fig. 3E and F).
Fig. 3.
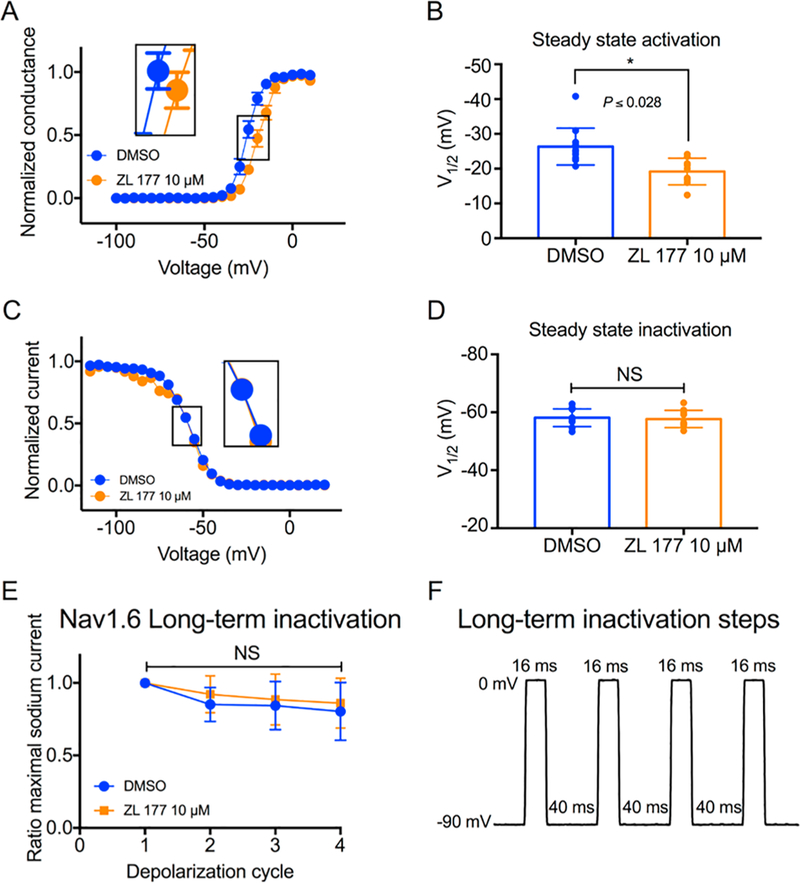
ZL0177 leads to a depolarizing shift in voltage-dependence of Nav1.6 channel activation. A) Voltage-dependence of Nav1.6 activation is measured as normalized conductance vs. membrane potential (mV) from cells treated vehicle with (DMSO 0.02%; blue) or ZL0177 (10μM); data were fitted with the Boltzmann function. (B) Bar graph represents summary of V1/2 for voltage-dependence of activation in the indicated experimental groups. (C) Steady-state inactivation is measured as normalized current vs the membrane potential (mV) from cells treated vehicle with (DMSO 0.02%; blue) or ZL0177 (10 μM); data were fitted with the Boltzmann function. (D) Bar graph represents summary of V1/2 for voltage-dependence of steady-state in the indicated experimental groups. (E) Accumulating long-term inactivation of Nav1.6 channels (channels available as a function of depolarization cycle) treated with DMSO or ZL0177. (F) Representing long-term in-activation steps: cells were subjected to four 0mV 16 ms depolarizations separated by −90mV 40 ms recovery intervals. Data are mean S.E.M.*, p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We then performed a docking study based on FGF14:Nav1.6 homology model (Fig. 4) to characterize the specific structural features that may enable ZL0177 binding.35 ZL0177 docked into the interface of the Nav1.6 C-tail (Fig. 4A), and it occupies a large and flat surface of Nav1.6 and forms critical hydrogen bonds with residues Arg1866, Asp1833 and Arg1891. Tri-peptides may be not big enough to occupy the whole surface. The hydrogen bond between O atom of acetyl group explains why compounds 12 and 16 are less active. Additionally, the phenylalanine moiety on the N-terminal of ZL0177 has a cation-π interaction, and the Fmoc group on the C-terminal is surrounded by hydrophobic and aromatic interactions with Phe1873, Tyr1883 and His1843. R3 group (For ZL0177, it is OMe) surrounded by Phe 1873 and Arg1866. The limited space appears to be not large enough to accommodate phenyl, thiazole or morpholine rings (compounds 18–21). The overlay of ZL0177 with FGF14:Nav1.6 homology model clearly illustrates that ZL0177 mimics the critical regions (two loops highlighted in red) at the interface of FGF14. In previous studies, we have reported that mutation of two FGF14 residues, Tyr158 and Val160, impaired FGF14:Nav1.6 complex formation and prevented FGF14-dependent modulation of Nav1.6 currents.22 ZL0177 plays the role of Tyr158 and Val160, interacting with Nav1.6 C-terminal.
Fig. 4.
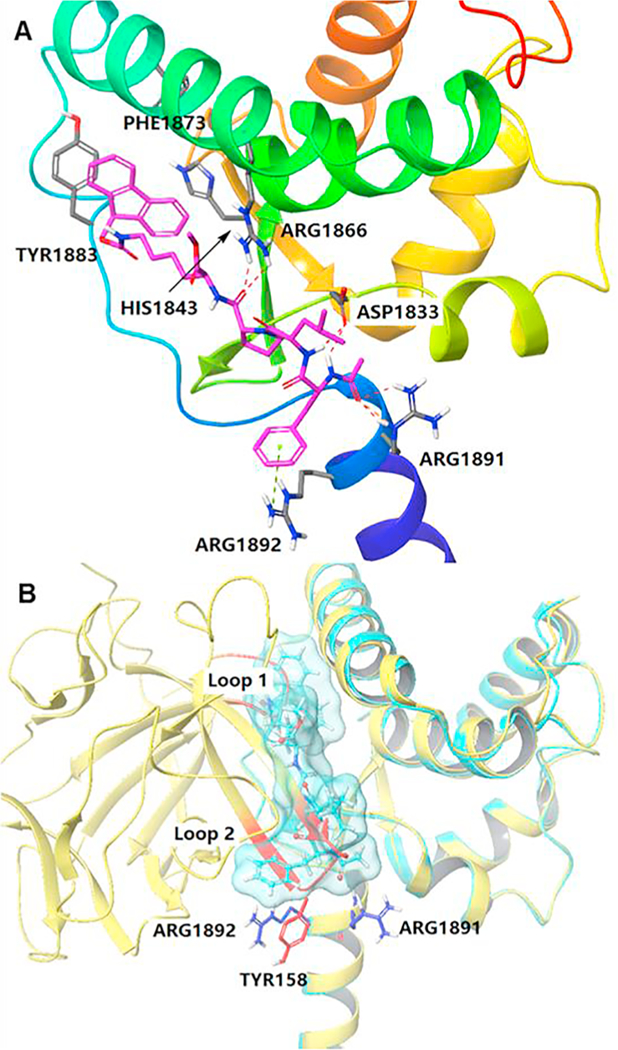
Docking of compound 13 with the Nav1.6 homology model. A) Ribbon representation of docking studies on peptidomimetic 13 (magenta) with Nav1.6 C-tail homology model. Residues PHE1873, TYR1883, HIS1843, ARG1866, ASP1833, ARG1891 and ARG1892 were highlighted in grey. Hydrogen bonds are highlighted by red dash line and cation-π interaction is indicated in green dash line. B) Overlay of FGF14/Nav1.6 homology model (yellow) with compound 13 docked into Nav1.6 (light blue). Two important loops containing TYR158 of FGF14 were highlighted in red. ARG1891 and ARG1892 were colored in blue. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In summary, we have designed, synthesized, and investigated a batch of cell permeable peptidomimetics based on the previously identified parental lead peptide FLPK. Among those, ZL0177 displayed potent in vitro activity in disrupting PPIs between FGF14:Nav1.6 with an in cell IC50 value of 11 μM. Importantly, ZL0177 is capable of modulating the biophysical properties of Nav1.6 currents, mimicking previously reported modulatory effects of FGF14 on Nav1.6 currents.22,32 Docking studies revealed multiple interactions between ZL0177 and the Nav1.6 C-tail including hydrogen bonds, cation-π interactions and hydrophobic contacts. Overlay study indicated that ZL0177 mimics the critical loop of FGF14 that encompass Tyr158 and Val160, two previously identified hot-spots at the FGF14:Nav1.6 channel interface. Further investigations on ZL0177 are underway to determine target specificity and usefulness of this compound as an in vivo probe and therapeutic potential in the CNS and PNS.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) Grants R01 MH095995 (F.L.), R01 MH111107 (F.L. and J.Z.), P30 DA028821 (J.Z.), John Sealy Memorial Endowments Fund (F.L.), Jeane B. Kempner Postdoctoral Fellowship (O.F.), NIA T32 Fellowship Grant No. T32 AG051131 (P.A.W.), and Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia NIGMS Grant T32 GM089657 (S.R.A.).
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2018.12.031.
References
- 1.Cantrell AR, Tibbs VC, Yu FH, et al. Mol Cell Neurosci. 2002;21(1):63–80. [DOI] [PubMed] [Google Scholar]
- 2.de Lera Ruiz M, Kraus RL. J Med Chem. 2015;58(18):7093–7118. [DOI] [PubMed] [Google Scholar]
- 3.Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. Proc Natl Acad Sci USA. 1998;95(15):8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Neuron. 2005;45(2):233–244. [DOI] [PubMed] [Google Scholar]
- 5.Qu Y, Rogers JC, Chen SF, McCormick KA, Scheuer T, Catterall WA. J Biol Chem 1999;274(46):32647–32654. [DOI] [PubMed] [Google Scholar]
- 6.Yu FH, Westenbroek RE, Silos-Santiago I, et al. J Neurosci. 2003;23(20):7577–7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catterall WA. Exp Physiol. 2014;99(1):35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marban E, Yamagishi T, Tomaselli GF. J Physiol. 1998;508(Pt 3):647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z, Chen H, Wold EA, Zhou J. Chapter 2.13 - Small-Molecule Inhibitors of Protein-Protein Interactions In: Chackalamannil S, Rotella D, Ward SE, eds. Comprehensive Medicinal Chemistry III. Oxford: Elsevier; 2017:329–353. [Google Scholar]
- 10.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. Am J Hum Genet 2001;68(6):1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mantegazza M, Gambardella A, Rusconi R, et al. Proc Natl Acad Sci USA. 2005;102(50):18177–18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullen SA, Scheffer IE. Arch Neurol. 2009;66(1):21–26. [DOI] [PubMed] [Google Scholar]
- 13.Woods CG, Babiker MO, Horrocks I, Tolmie J, Kurth I. Eur J Hum Genet. 2015;23(10):1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Brain Res. Rev. 2009;60(1):65–83. [DOI] [PubMed] [Google Scholar]
- 15.Tang Z, Chen Z, Tang B, Jiang H. Orphanet J Rare Dis. 2015;10:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Large CH, Bison S, Sartori I, et al. J Pharmacol Exp Ther. 2011;338(1):100–113. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Shen J, Splawski I, et al. Cell. 1995;80(5):805–811. [DOI] [PubMed] [Google Scholar]
- 18.Musa H, Kline CF, Sturm AC, et al. Proc Natl Acad Sci USA. 2015;112(40):12528–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Probst V, Kyndt F, Potet F, et al. J Am Coll Cardiol. 2003;41(4):643–652. [DOI] [PubMed] [Google Scholar]
- 20.Lou JY, Laezza F, Gerber BR, et al. J Physiol. 2005;569(Pt 1):179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laezza F, Lampert A, Kozel MA, et al. Mol Cell Neurosci. 2009;42(2):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali SR, Singh AK, Laezza F. J Biol Chem 2016;291(21):11268–11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laezza F, Gerber BR, Lou JY, et al. J Neurosci. 2007;27(44):12033–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goetz R, Dover K, Laezza F, et al. J Biol Chem 2009;284(26):17883–17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shavkunov AS, Wildburger NC, Nenov MN, et al. J Biol Chem. 2013;288(27):19370–19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu WC, Nenov MN, Shavkunov A, Panova N, Zhan M, Laezza F. PLoS One. 2015;10(2):e0117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali S, Shavkunov A, Panova N, Stoilova-McPhie S, Laezza F. CNS Neurol Disord Drug Targets. 2014;13(9):1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Testa B, Fahr A. Pharm Res. 2011;28(5):962–977. [DOI] [PubMed] [Google Scholar]
- 29.Refsgaard HH, Jensen BF, Brockhoff PB, Padkjaer SB, Guldbrandt M, Christensen MS. J Med Chem. 2005;48(3):805–811. [DOI] [PubMed] [Google Scholar]
- 30.Leung SS, Mijalkovic J, Borrelli K, Jacobson MP. J Chem Inf Model. 2012;52(6):1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennion BJ, Be NA, McNerney MW, et al. J Phys Chem B. 2017;121(20):5228–5237. [DOI] [PubMed] [Google Scholar]
- 32.Ali SR, Liu Z, Nenov MN, et al. ACS Chem Neurosci. 2018;9(5):976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shavkunov A, Panova N, Prasai A, et al. Assay Drug Dev Technol. 2012;10(2):148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stable HEK-Nav1.6 cells were plated at low density on glass cover slips for 3–4 hours and subsequently transferred to the recording chamber. Recordings were performed at room temperature (20–22 °C) 24 h post-transfection using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA). The recording solutions were consisted of the following salts: extracellular (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.3; intracellular (mM): 130 CH3O3SCs, 1 EGTA, 10 NaCl, 10 HEPES, pH 7.3. Membrane capacitance and series resistance were estimated by the dial settings on the amplifier and compensated for electronically by 70–80%. Data were acquired at 20 kHz and filtered at 5 kHz prior to digitization and storage. All experimental parameters were controlled by Clampex 9.2 software (Molecular Devices) and interfaced to the electrophysiological equipment using a Digidata 1200 analog-digital interface (Molecular Devices). Voltage-dependent inward currents for HEK-Nav1.6 cells were evoked by depolarizations to test potentials between −100 mV and +60 mV from a holding potential of −70 mV followed by a voltage pre-step pulse of −120 mV (Nav1.6). Steady-state (fast) inactivation of Nav channels was measured with a paired-pulse protocol. From the holding potential, cells were stepped to varying test potentials between −120 mv (Nav1.6) and +20 mV (pre-pulse) prior to a test pulse to −20 mV. Current densities for channels were obtained by dividing Na+ current (INa) amplitude by membrane capacitance. Current–voltage relationships were generated by plotting current density as a function of the holding potential. Conductance (GNa) was calculated by the following equation: GNa = INa/(Vm – Erev) where INa is the current amplitude at voltage Vm, and Erev is the Na+ reversal potential. Steady-state activation curves were derived by plotting normalized GNa as a function of test potential and fitted using the Boltzmann equation: GNa/GNa,Max = 1 + e[(Va–Em)/k] where GNa,Max is the maximum conductance, Va is the membrane potential of half-maximal activation, Em is the membrane voltage and k is the slope factor. For steady-state inactivation, normalized current amplitude (INa/INa,Max) at the test potential was plotted as a function of prepulse potential (Vm) and fitted using the Boltzmann equation: INa/INa,Max = 1/[1 + e[(Vh-Em)/k]] where Vh is the potential of half-maximal inactivation, Em is the membrane voltage, and k is the slope factor. Data analysis was performed using Clampfit 9 software (Molecular Devices) and Origin 8.6 software (OriginLab Corporation, Northampton, MA). Results were expressed as mean ± SEM. The statistical significance of observed differences among groups was determined by unpaired t-test, p < 0.05 was regarded as statistically significant.
- 35.The docking study was performed with the Schrödinger Small-Molecule Drug Discovery Suite using the FGF14:Nav1.6 homology model. The Nav1.6 was kept for docking and prepared with Protein Prepared Wizard. ZL0177 was prepared with LigPrep and the initial lowest energy conformations were calculated. The grid center was chosen on selected hotspot residues at the PPI of FGF14:Nav1.6 with the co-ordination of x = 12.14, y= −13.71, z=−11.09. Grid box size was set to 26 × 26 × 26 Å, and a finer scaling factor of 0.5 was used. Grid generating, and docking were both employed with Glide using SP-Peptide protocol. Docking poses were in-corporated into Schrödinger Maestro for a visualization of ligand–receptor inter-actions. The homology structure of FGF14:Nav1.6 was incorporated into Maestro with the top ranked docking pose of ZL0177 for an overlay analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.