

 Contemporary drug discovery approaches rely on library synthesis coupled with combinatorial methods and high-throughput screening to identify leads.
Contemporary drug discovery approaches rely on library synthesis coupled with combinatorial methods and high-throughput screening to identify leads.
Abstract
Contemporary drug discovery approaches rely on library synthesis coupled with combinatorial methods and high-throughput screening to identify leads. However, due to the multitude of components involved, a majority of optimization techniques face persistent challenges related to the efficiency of synthetic processes and the purity of compound libraries. These methods have recently found an upgradation as fragment-based approaches for target-guided synthesis of lead molecules with active involvement of their biological target. The click chemistry approach serves as a promising tool for tailoring the therapeutically relevant biomolecules of interest, improving their bioavailability and bioactivity and redirecting them as efficacious drugs. 1,2,3-1H-Triazole nucleus, being a planar and biologically acceptable scaffold, plays a crucial role in the design of biomolecular mimetics and tailor-made molecules with therapeutic relevance. This versatile scaffold also forms an integral part of the current fragment-based approaches for drug design, kinetic target guided synthesis and bioorthogonal methodologies.
1. Introduction
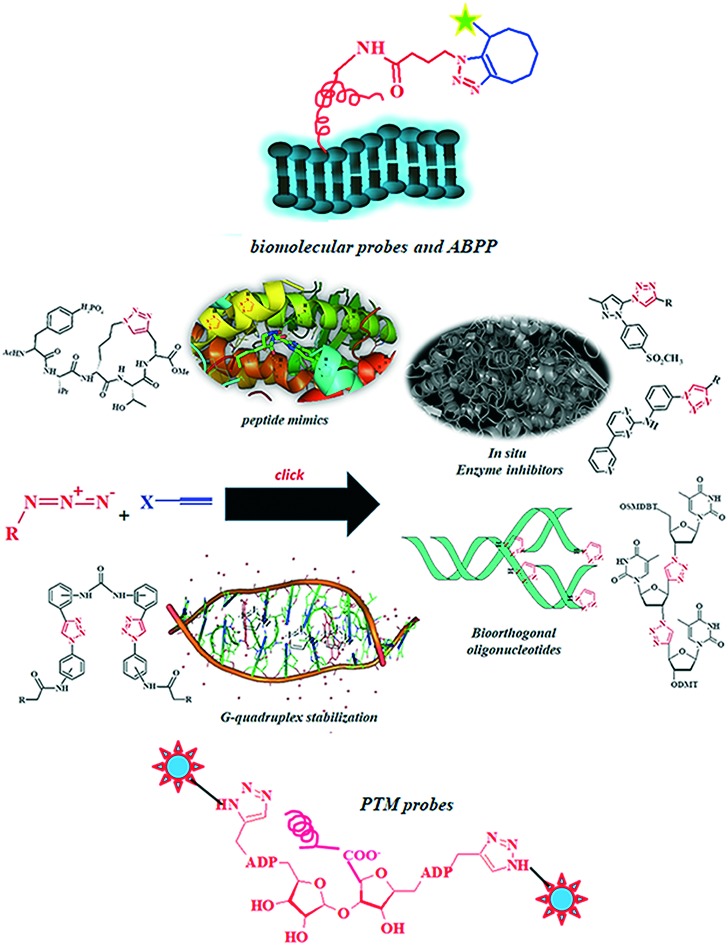
From the burgeoning, conventional virtual molecular screening methodologies based on computer aided drug design (CADD)1 to real world high-throughput organic synthesis2 (HTOS) and high-throughput screening3 (HTS) till the combinatorial bioconjugation-directed bioorthogonal approach4 and fragment-based drug discovery5 by target-templated in situ inhibitor synthesis,6 the mainstream drug discovery paradigm has made a long journey.7 The identification of CADD as a useful tool in pharmaceuticals by Merck8–10 in the 1980s revolutionized the pharma sector;11 however, subsequent developments in HTS soon took precedence over all the contemporary techniques used for drug design and development.12–16 A typical HTS protocol encompasses the evaluation of multiple compound libraries against a purified biological target by technology-driven automated systems for a rapid screening of the ‘hits’ that can be further refined as potential leads.17–19 HTS methodology, when applied on molecules with mass less than 1000 Da,20 produces discrepancies in the form of false positives and false negatives.21–24 Moreover, the HTS methodologies appraise a single dose activity profile for the candidate molecules, thereby giving artifactual hits.25–30 The lead molecules generated from HTS hits are occasionally prone to non-bioavailability, insolubility,31 specificity and toxicity.32 Hence, there are a number of established approaches in the contemporary drug discovery paradigm, but the bioconjugation approach represents the most recent development.33–34 A conventional bioconjugate therapeutic carries an active synthetic molecule appended to a carrier biomolecule by a covalent bond or through a chemical linker.35–37 This strategy holds a promising therapeutic prospective ensuring target specificity, controlled drug release, reduced toxicity and improved drug delivery to the target site.38–41 Moreover, the conjugation between drug and biomolecule functions as a workable probe for an arduous examination of the biochemicals and bioanalytes,42–43 which is critical for the prompt detection and diagnosis of the ensuing diseases.44–46 Methodical developments in the bioconjugation archetype led to the introduction of bioorthogonal chemistry for exploring the molecular information behind various metabolic and essential biochemical processes.47–48 A characteristic bioorthogonal reaction following second-order kinetics49 occurs in an optimum physiological environment without altering the native biochemical machinery.50–51 Initial applications of biochemical modification strategies exclusively defined the amino acid components of proteins,52 but further developments extended their implication to mechanistic enzymology53 and biotechnology.54–55 Presently, kinetic target guided synthesis56 (KTGS) and in situ dynamic combinatorial chemistry57 (DCC) offer the most captivating, unparalleled and robust prototypes for target-templated in situ inhibitor synthesis, thereby facilitating the identification of potential drug candidates.58–63 In the DCC approach, the presence of a biological target shifts the thermodynamic equilibrium between the participating reactants towards the products side that binds to the target via a reversible reaction,64 thus generating the combinatorial libraries.65 In contrast, in KTGC, the target biomolecule stabilizes the reactive relative configurations of the participating reagents,66 thereby accelerating irreversible product formation.67 The click reactions present a viable profile for target templated in situ drug discovery approaches because of their superior selectivity, excellent yield and favorable reaction kinetics.68 Specifically, the in situ click reaction between the bioorthogonal azides and alkynes to generate a cycloaddition product under physiological conditions produces substantial results for the identification of highly potent inhibitors.69–74 However, the cellular performance of KTGS by click reactions meets several challenges for managing an effective concentration of the target macromolecule as well as the reacting biorthogonal building blocks and their interactions with the natural ligands, which usually result in several unwanted background reactions. Still, overcoming these difficulties by the application of metabolomics75 fortifies click chemistry as a superior approach among existing drug discovery methodologies. 1,2,3-1H-Triazoles present a laudable profile for contemporary drug discovery approaches. Their isosteric analogy with the amide bond assists in the fabrication of therapeutically relevant peptidomimetics with improved biological profiles and superior physiological stabilities compared to the parent peptides.76 The triazole modified DNA backbone, customized by replacing the phosphodiester group of the parent nucleotide, reduces its negative charge, thereby improving availability within the target cell and enhancing resistance to nucleases.77 The triazole modified unnatural DNA backbone also assists in nucleotide labelling and can be accurately read by both DNA and RNA polymerases.78 Moreover, the selectivity of click reactions in fragment-based lead discovery has led to in situ development of enzyme inhibitors via kinetic target guided synthesis and biomolecule profiling by dynamic combinatorial chemistry.79 In this review, we present the synthesis and biological profile of tailor-made 1,2,3-1H-triazole based therapeutics vis-à-vis contemporary drug discovery approaches.
2. Ambient conditions for the synthesis of 1,2,3-1H-triazoles by click reactions
Nature-inspired reactions requiring milder conditions for optimum operation fall under ‘click reactions’. Introduced by Sharpless and coworkers80 in 2001, these facile, thermodynamically favorable modular reactions procure high yields of easily purifiable81–83 products from readily available precursor molecules and reagents in environmentally and physiologically benign solvents.84–85 In the 1960s, Huisgen and co-workers introduced 1,3-dipolar cycloaddition reactions for the synthesis of 5-membered heterocycles.86 Unfortunately, a lack of regioselectivity and the requirements of high reaction temperatures and prolonged reaction times limited the synthetic potency of these reactions. The dawn of the 21st century saw developments in Cu(i)-catalyzed azide–alkyne cycloaddition reactions that pioneered the click chemistry archetype, triggering a plethora of applications in both pure and applied chemistry. However, the toxicity of Cu(i) toward biological systems led to development of strained cyclooctynes to sequentially replace metal ion catalysts for the realization of hassle-free dipolar cycloaddition.
2.1. Huisgen cycloaddition reactions
Under Huisgen conditions, two or more polar systems combine by forming sigma bonds between their termini to form a stable cyclic molecule. The synthetic utility of Huisgen cycloaddition reactions has been extended to the production of five membered systems such as 1,2,3-1H-triazole, isoxazoles, isoxazolines, pyrazoles, pyrazolones, and 1,2,4-oxadiazolines.87a,b The suprafacial combination of three 2pz orbitals of a 1,3-dipole (containing 4π electrons) and the 2pz orbitals of a dipolarophile (containing 2π electrons) via a concerted thermochemical pathway88 is considered a probable mechanism, though the concerted mechanism postulated by Huisgen varied considerably from the biradical mechanism postulated by Firestone.89 In addition to the mechanistic ambiguity, stereochemical strains and questionable site specificity hampered the further progress of Huisgen reactions.90
2.2. Cu(i)-Catalyzed azide–alkyne click chemistry (CuAAC) reaction
The CuAAC category of reactions involves a click reaction between an azide-functionalized molecule and a terminal-alkyne functionalized molecule, thereby forming a 1,2,3-1H-triazole conjugate retaining the starting functionalities.91 The presence of a metal catalyst, such as Cu(i), predominantly governs the proficiency of an otherwise less feasible azide–alkyne conjugation under ambient conditions.92 However, the utility of Cu(ii) in the form of CuSO4 salt in combination with an appropriate reducing agent such as ascorbate has been widely substantiated for most biomolecule labeling applications.93,94 The efficacy of this approach is hampered by the toxicity of Cu(i) ions to live cells. Nevertheless, the employment of Cu(i) chelating agents such as tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) allows biomolecular defense by preventing any ensuing oxidative damage95 and accelerates the CuAAC reaction by maintaining the Cu(i) oxidation state.96 There are numerous other CuAAC pathways mediated by Cu nanoparticles and metal organic frameworks which have only recently begun to be explored.97a–d
2.3. Strain-promoted azide–alkyne click chemistry (SPAAC) reaction
Toxicity of the metal ion catalyst in CuAAC reactions restricts their direct employability in biological systems. The development of metal-free click azide–alkyne cycloaddition reactions involving use of strained cyclooctynes, such as (difluoromethylidene) cyclooctane (DIFO), 1-butyl-1-cyclooctene (BCN), dibenzocyclooctyne (DIBO) and azadibenzylcyclooctyne (DIBAC), has overcome this drawback.98 Cyclooctynes possess an extraordinarily lowered activation energy compared to terminal alkynes and therefore do not require an exogenous catalyst for their activation.99 Additionally, the cyclooctyne-based reagents provide extraordinary reactivity100 with azides and ensure adequate hydrophilicity,101 thereby allowing a low background labeling102 of azide-functionalized molecules with superior selectivity103 and efficacy compared to the CuAAC reactions. Additionally, the SPAAC reactions have found extensive utilization for in vivo covalent modification of biomolecules (Fig. 1).104
Fig. 1. Ambient conditions for a dipolar cycloaddition reaction.

3. Tailor-made biomolecular mimics based on disubstituted 1,2,3-1H-triazoles
3.1. Backbone tailored therapeutic peptidomimetics based on 1,2,3-1H-triazole
Disubstituted 1,2,3-1H-triazole ring displays a substantial functional overlap with the amide bond.105 1,5-Disubstituted 1,2,3-1H-triazole nucleus mimics E-amide, where the negatively polarized N atom in the triazole nucleus replaces the electrophilic carbonyl carbon, with the relative distance between the two substituents remaining almost the same (Fig. 2). In contrast, 1,4-disubstituted 1,2,3-1H-triazole nucleus displays an analogy with the Z-amide, where the lone pair on the third nitrogen of the triazole ring mimics the carbonyl oxygen on an amide with an overall increase in the relative distance between the two substituents of 1.1 Å in the triazole ring (Fig. 2).106
Fig. 2. 1,2,3-1H-Triazole nucleus as an amide bond bioisostere.

Additionally, the 1,2,3-1H-triazole moiety displays commendable stability against acidic and basic hydrolysis and adverse oxidative or reductive environments, coupled with a notable resistance to metabolic degradation and extraordinary aromatic stabilization.107 Furthermore, the triazole nucleus possesses a superior dipole moment (around 5 Debye)108 compared to the amide bond, affirming substantial hydrogen bonding donor and acceptor characteristics and dipole–dipole interactions and justifying its use in enhanced peptide mimicry109 and in designing bioactive molecules and prospective drugs.110 The evidence for the 1,2,3-1H-triazoles as bioisosteres of an amide bond was provided by Mohammed et al. (2012, 2016)111,112 by developing novel RN-18 analogues (6a–h, Chart 1, Scheme 1). The validation of disubstituted-1,2,3-1H-triazoles (6b, Chart 1) as a plausible amide bond replacement in RN-18 (8, Chart 1) was also done by comparison studies with 1,3,4-oxadiazole (6a, Chart 1), 1,2,4-oxadiazole (6c, Chart 1) and 1,5-disubstituted-1,2,3-1H-triazoles (6d, Chart 1).111 The functional analogues of RN-18 (4 and 7, Chart 1, Scheme 1) were prepared by replacing the amide functionality with 1,4-disubstituted-1,2,3-1H-triazoles, thereby generating a greater HIV-1 Vif antagonism propensity compared to the parent RN-18 molecule. RN-18, an HIV-1 Vif-APOBEC3G axis inhibitor, was further modified at rings A, B and C, bridge A–B, and bridge B–C to optimize the critical structural features responsible for its activity. The synthesis of molecule 5 required the synthons 2-ethynylaniline (1, Chart 1, Scheme 1) and 1-azido-2-methoxybenzene (2, Chart 1, Scheme 1) to undergo regioselective CuAAC reaction in t-BuOH/H2O to produce 1,4-disubstituted triazolamine I (3, Chart 1, Scheme 1). The same synthons undergo ruthenium-catalyzed click chemistry in the presence of Cp*RuCl(PPh3)2 catalyst in benzene at 80 °C to generate 1,5-disubstituted triazolamine II (5, Chart 1, Scheme 1). A two-step conversion of the triazolamines involving diazotization followed by iodination generated the active molecules (4 and 7, Chart 1, Scheme 1). SAR analysis on the test compounds (6a–h, Chart 1) indicated a remarkable sensitivity of the Z-bridge and R1 and R2 substituents towards HIV-1 Vif activity. Sulfide (–S–) as the bridge ‘Z’ displayed superior activity (6e–h, Chart 1) compared with the sulfone (–SO2–) bridge (6a–d). Conversely, the sulfones (–SO2–) exhibited enhanced activities when the R2 substituent was an amino group. Compound 6h, with an IC50 of 10 nM, presented the most significant: a 1000-fold higher potency than the original lead molecule RN-18.112 These investigations established a bioisosteric analogy between the amide bond and 1,2,3-1H-triazoles, where the replacement of the former with the latter in the B–C bridge of RN-18 led to retained bioactivity and enhanced bioavailability. These investigations also validated the significance of 1,2,3-1H-triazole as a favorable scaffold for tailoring the amide backbone of biologically relevant therapeutics.
Chart 1. Syntheses and biological activities of disubstituted 1,2,3-1H-triazoles as bioisosteres of the amide bond.

Bioactive natural peptides endowed with superior specificity and low toxicity present an excellent archetype for therapeutics design.113 Similarly, bioactive oligomers with macrocyclic constraints ease the conformational flexibility and pre-organize the side-chain pharmacophores into ideal orientations for binding.114 The cyclic peptides with favorable conformational stability, superior proteolytic resistance and capacity to exhibit protein-like epitopes present potential as therapeutic agents.115 However, the therapeutically relevant peptides often suffer a rapid in vivo degradation of the amide-backbone by proteases116 and display low bioavailability. A tailor-made peptide must therefore incorporate a biologically stable nucleus that also displays compatibility with the amino acid side chains and improved stability under physiological conditions. 1,4-Disubstituted-1,2,3-1H-triazole presents a favorable profile for modeling the peptide backbone by acting as a surrogate for biologically important amide bonds.117 Bock et al. (2007)118 developed triazole-containing peptidomimetics (12, Scheme 2 and 16, Scheme 3, Chart 2) of naturally occurring tyrosinase inhibitor cyclo-[Pro-Val-Pro-Tyr] (17, Chart 2). The macrocyclic peptide presented synthetic complications during head to tail ring closure, resolved by introducing a 1,4-disubstituted-1,2,3-1H-triazole nucleus as a peptide bond surrogate connecting proline and valine residues. Synthesis of 1,2,3-1H-triazole analogue 12 began from the azido and alkyne synthons (9, 10), which produced the linear precursor to cyclotetrapeptide analogue N3-Tyr-(OBn)-Pro-Val-Pro-C CH (11) by base-assisted amide bond formation in the presence of N,N-diisopropylethylamine (DIPEA) using dichloromethane as the solvent. A subsequent intramolecular SPAAC reaction in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) yielded the cyclic peptide with a triazole nucleus. A similar synthetic route produced the cyclic-peptide decorated with two triazole nuclei (16, Chart 2, Scheme 2) by an intramolecular SPAAC reaction on linear peptide 15 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
Chart 2. Syntheses and biological activities of disubstituted 1,2,3-1H-triazoles as cyclotetrapeptide analogues.

Cycloaddition reaction between alkyne 13 and triazolyl-dipeptide 14 procured 15 with clickable azide and alkyne functionalities, which was further cyclized to a triazole ring in 16. The investigations revealed that 1,4-disubstituted 1,2,3-1H-triazoles mimicked trans peptide bonds and also demonstrated retained inhibitory activity for the tyrosinase enzyme.118 Compound 16, where 1,2,3-1H-triazole ring replaced both the transoid amide groups, displayed a similar inhibitory profile as 17; compound 12 (Chart 2, Scheme 3), where a single amide bond was replaced, displayed superior tyrosine inhibition potency compared to 17. The rational design of synthetic ligands for binding a protein of interest also involves utilization of a known ligand as a structural model for postulating accurate conformational and pharmacophoric necessities for binding. However, a noteworthy difficulty in this approach is that, in the absence of their associated receptors, the free ligands frequently adopt multiple conformations in solution or in the solid state, which can render the models, designed based on free-ligand structure, misleading and challenging to construct. Horne et al. (2009) validated a cis–trans–trans–trans (c–t–t–t) conformation in apicidin119 (22, Chart 3, Scheme 4), a representative member of a family of naturally occurring cyclic tetrapeptide inhibitors of histone deacetylases (HDACs), to be more predominant in contrast to the all trans (t–t–t–t) structure that predominates in solution.120 A linear azido–alkyne tetrapeptide 19, obtained by solution-phase peptide synthesis on the precursor propargylic-amine derivative 18, served as synthon for the synthesis of active molecules. A copper(i)-catalyzed intramolecular azide–alkyne cycloaddition of compound 19 yielded the heterocyclic peptide 21 containing the trans-amide surrogate. Peptide 20, with a 1,5-substituted 1,2,3-1H-triazole, was prepared by a thermal Huisgen [3 + 2] dipolar cycloaddition of 19, (N,N-dimethylformamide (DMF), microwave, 220 °C). The cyclic pseudo-tetrapeptides containing 1,5- and 1,4-disubstituted 1,2,3-1H-triazole amino acids (20 and 21, Chart 3, Scheme 4) served as surrogates for trans and cis amide bonds, respectively.121 These outcomes confirmed the efficacy of 1,2,3-1H-triazole-modified cyclic peptides for the rational design of pragmatic bioactive probes and three-dimensional pharmacophore models for selective HDAC inhibitors serving as potent molecules in anticancer chemotherapy.
Chart 3. Syntheses and biological activities of disubstituted 1,2,3-1H-triazoles as trans and cis-amide bond surrogates.

β-Turn mimetics based on small-molecules function as vital probes to assess conformations and establish the pharmacophoric requirements of the biologically relevant ligands. The utilization of cyclic tetrapeptides serves as a customary approach for constraining the peptides into turn conformations. Owing to their appropriate size, shape, and synthetic modularity, the cyclic tetrapeptides reportedly mimic the protein turn regions. However, due to a low synthetic efficiency while constructing the strained 12-membered ring and an inability to control the cis–trans geometry of backbone amides, these structures remain mostly unexplored.122a Additionally, the requirement for incorporating a proline or other turn-forming residue at the expense of one of four amino acid residues poses challenges for developing β-turn mimetics. Beierle et al. (2009)122b synthesized 2 sets of dipeptides, each comprised of four diastereomers (from compounds 23 and 24, Chart 4, Scheme 5) from which all 16 required diastereomeric tetramers could be synthesized. The precursor compounds 23 and 24 underwent copper(i)-mediated [3 + 2] Huisgen dipolar cycloaddition to give the product 25 in 31–90% yield. Inclusion of the copper-chelating ligand tris(benzyltriazolylmethyl)amine (TBTA) during the [3 + 2] macrocyclization step preferred the formation of the desired cyclic tetramer over the cyclic octamer resulting from head-to-tail dimerization of two tetrapeptide substrates. The 1,4-disubstituted 1,2,3-1H-triazole-based peptidomimetic (25, Chart 4, Scheme 5) served as a functional ligand for the human somatostatin receptor (hSSTR). The conformational analysis of the tailored ligand within the hSSTR indicated the four Cα positions and the Cα–Cβ vectors of Lys and Trp to be the key elements of hSSTR1 affinity, whereas Phe and Thr side chains guided the receptor selectivity. Additionally, the 1,4-disubstituted-1,2,3-1H-triazole moiety mimicked a trans-peptide bond within tetrapeptide 25 that displayed a diverse selectivity against the five human SSTR subtypes.122b Among contemporary anticancer therapeutics, STAT3 serves as a chief molecular target. The dimerization of STAT3 is required for its activation; obstructing this event using small-molecule antagonists is an attractive approach for treating human cancers by targeting the pathways contributing to constitutive activation of STAT3. Ren et al. (2003) identified that the phosphotyrosine group in peptide 29 acts as a key ingredient in the inhibition of STAT3 dimerisation.123 Contemporary approaches for designing small-molecule antagonists to block STAT3 dimerization involve peptide-based antagonists and non-peptidic small-molecules. The peptidomimetic ligands attain substantial binding affinities to STAT3; however, their utility faces persistent challenges related to poor cell-permeability due to negatively charged phosphotyrosine groups in the ligands. The non-peptide small-molecule inhibitors, on the other hand, exhibit relatively poor binding affinities to STAT3. Chen et al. (2007)124 reported the structure-based design, synthesis, and biochemical evaluation of a conformationally constrained macrocyclic peptidomimetic as STAT3 inhibitor to block its dimerization. Side chain modified macrocyclic peptidomimetic 28 has been identified as a potent mimic of 29 (Chart 4, Scheme 6),124 an inhibitor of STAT3. CuAAC ring closure of azido norleucine and propargylglycine in 26 (Chart 4, Scheme 6) yielded precursor compound 27 that then gave the constrained peptidomimetic 28 in 80% yield. A crystal structure of STAT3 bound with peptide 29 revealed the side chains of two key lysine residues exposed to the solvent to be dormant to any crucial interactions. The fluorescence-polarisation-based competitive binding assays performed by Chen and coworkers revealed the mimic 28 (Chart 4, Scheme 6) to have three-fold superior potency (Ki = 7.3 μM) against STAT3 compared to the linear peptide 29 (Chart 4, Scheme 6) (Ki = 21.9 μM).
Chart 4. Syntheses and biological activities of disubstituted-1,2,3-1H-triazole-based peptidomimetics.

3.2. 1,4-Disubstituted-1,2,3-1H-triazole based bioactive triazolamers
Due to their participation in crucial biological pathways, protein–protein interactions have emerged as potential therapeutic targets. However, these interactions are difficult to target with small molecules because the domains in which the inhibitor binds, predominantly by hydrophobic interaction, are often large, shallow and not well defined.125 These challenges created a mandate for the development of novel methodologies to tailor small molecule inhibitors of protein–protein interactions. Fragment-based drug discovery presents a workable approach in the identification of useful fragments, which may offer lower activity and affinity for binding sites when used independently but considerably improved activity when appropriately linked.126 Triazolamers represent a class of backbone-modified distinctly folded nonpeptidic oligomers with the amide bond substituted by rigid and physiologically stable hetero-aromatic rings, while maintaining the chiral main chain and amino acid side chains.127 These molecules adopt definite conformations, presenting different sets of side chains without the limitations imposed by the secondary amide bond.128 Triazolamers obtained by swapping the amide bond with an appropriate hetero-aromatic nucleus and projecting the attached main chains at different angles from a given ring offer numerous distinct backbone conformations, likely from α- and β-amino acids, that potentially add a “drug-like” characteristic to these oligomers.129 Arora et al. reported the synthesis of backbone-modified peptidomimetics, termed triazolamers (32, Chart 5, Scheme 7),130–132 by carefully optimizing the CuAAC conditions on synthon 30 (carboxyl-protected phenylalanine) followed by deprotection and subsequent diazo transfer reaction on compound 31 (Chart 5, Scheme 7). Reports on using proteases to bind their substrates in a β-strand conformation133 inspired a library of triazolamers tested as HIV-1 protease inhibitors, with the best compound having an IC50 of 257 μM. Montagnat and coworkers reported CuAAC synthesis of racemic triazolamers (35 and 36; Chart 5, Scheme 8) using building blocks134 containing β3-substituted homopropargyl alcohol (33, Chart 5, Scheme 8) and chiral β3-substituted trimethyl-silylhomopropargyl azides from l-amino acids (34, Chart 5, Scheme 8).135 Sola et al. (2015) synthesized 1,4-substituted triazole oligomers made from quaternary amino acid derivatives that demonstrated similar conformation and behaviour to natural peptides (40, 41 and 42; Chart 5, Scheme 9).136 Unprotected benzylic azide 37 underwent Cu-mediated azide–alkyne cycloaddition with N-Boc-protected 2-methyl-3-butyn-2-amine coupled to yield benzylic azide (38, Chart 5, Scheme 9), which further underwent Cu(ii)-mediated conversion of deprotected amine to azide in the presence of imidazole-1-sulfonyl azide hydrochloride and K2CO3 to yield 39 (Chart 5, Scheme 9). 2-D NMR analysis and ab initio calculations confirmed the conformational preference of triazolamer 42 and that the anti-conformation is responsible for the turn-backbone structure. The solution NMR analysis validated the co-existence of several conformations together with aggregation, whereas the X-ray diffraction investigations offered convincing evidence for the existence of oligomers as twisted strands or zigzag structures, depending on the appended substituents. The residues with bulky N-groups and rotationally restricted benzylic protons exhibited folded structures in solvents such as chloroform and acetonitrile. Additionally, the dipole–dipole interactions between neighboring triazole rings in a triazolamer appear to play a critical role in stabilizing the conformations observed in polar solvents, such as DMSO and acetone. One surface of the zigzag triazolamer efficiently mimics a β-strand and hence offers admirable utility to target protein pockets and surfaces involved in β-strand identification. The backbone of the triazolamer does not afford the hydrogen bond functionality of a β-strand; however, the N-2 and N-3 electron pairs serve as hydrogen bond acceptors.
Chart 5. Syntheses and biological activities of backbone-modified acyclic peptidomimetic triazolamers.

3.3. Amino acid mimics based on 1,2,3-1H-triazole
Phosphatases and protein kinases are useful drug targets, as the post-translational phosphorylation of proteins defines their diverse functions. O-Phosphorylation of serine, tyrosine, and threonine is well studied; however, instability of phosphorylated histidine and its existence as two isomers restricts understanding of histidine phosphorylation. Kee et al. (2010) reported a facile synthesis of phosphorylated histidine (pHis) mimics (44 and 46, Chart 6, Scheme 10) using CuAAC and RuAAC chemistry, respectively137 on synthons 43 and 45 (Chart 6, Scheme 10). Compound 49 interacted with phosphorylated His-containing proteins, which reportedly produced antibodies towards the synthetic peptidomimetics. Upon isolation, these antibodies selectively recognized the native phosphorylated protein instead of its non-phosphorylated equivalent. The triazole containing pHis analogues also considerably upregulated histone H4 histidine kinase (HHK) activity by 200-fold in human hepatocellular carcinomas compared to normal liver tissues. The peptides incorporated with stable pHis analogues serve as useful tools for studying the histone histidine phosphorylation. Additionally, the most significant application of pHis analogues is the generation of antibodies that can selectively recognize pHis residues in phosphoproteins. These analogues also find useful applications in solid-phase peptide synthesis and protein semi-synthesis. Gajewski et al. (2007) studied the application of triazole-modified amino acids in ligand–protein binding interactions of the neutral amino acid transporter SN1.138 This neutral amino acid transport protein facilitates the movement of l-glutamine, the most abundant free amino acid, found profusely in mammalian blood plasma and cerebral spinal fluid and involved in numerous essential biochemical processes. An accurate binding-model pharmacophore for the quantization of the ligand–protein binding interactions involving SN1 transporter develops useful pharmacological tools for the SN1 transporter protein. Studies validated that the binding of the ligand to neutral amino acid transport protein SN1 involves interactions other than the ligand acting simply as a hydrogen-bond acceptor on the amino acid side chain functionality within the ligand-binding site. These interactions include hydrogen bonding with a protein or protein-bound water near an endogenous ligand binding site. Further investigations suggested that an amino acid appended with 1,4-disubstituted triazole in the side chain affords hydrogen-bonding interactions with the target protein (49, Chart 6, Scheme 11). However, the fully protected triazole amino acids 48 (Chart 6, Scheme 11), prepared by CuAAC of β-azido alanine 47 (Chart 6, Scheme 11) with a range of acetylenes, and the amino acids mimic 49 (Chart 6, Scheme 11), though proved to be inactive at the SN1 transporter protein. Still, these analogues displayed significant interactions with a number of other glutamine-utilizing proteins. Targeting the central nervous system glutamate receptors led Stanley et al. (2010) to explore the potential of 1,4- and 1,5-disubstituted-1,2,3-triazolyl amino acid glutamate homologues as selective AMPA receptor ligands139 (54 and 55, Chart 6, Scheme 12). The synthons 50 and 51 (Chart 6, Scheme 12) underwent ruthenium-catalyzed cycloaddition to yield 1,5-disubstituted triazolyl amino acid (53, Chart 6, Scheme 12) and Cu(ii)-catalyzed cycloaddition to yield 1,4-disubstituted triazolyl amino acid (52, Chart 6, Scheme 12). Subsequent deprotection procured the amino acid mimics 54 and 55 (Chart 6). The triazole nucleus in the test compounds creates a similar association with key amino acid residues within the binding pocket of the corresponding receptor as the π-excessive heterocycles of HIBO, AMPA and TDPA. Moreover, a structural similarity supported by in silico molecular docking analysis validates a similar affinity profile for the proposed triazole compounds and the famously competitive ionotropic glutamate-receptor ligands. The in vitro analysis on analogues 54 and 55 at native ionotropic glutamate receptors (NMDA, AMPA) and in functional assays of recombinant metabotropic glutamate-receptors revealed that these compounds acted as competitive binders with IC50 values of 63 and 49 μM, respectively.
Chart 6. Syntheses of triazole-based amino acid mimics.

3.4. G-quadruplex stabilizers based on 1,2,3-1H-triazole
The self-association of guanine residues140 by hydrogen-bonding141,142 to form four-stranded double helical conserved DNA sequence repeats of telomeres143 that regulate multiple biological processes represents the quadruplex nucleic acids. Physiological buffer conditions maintained by monovalent Na+ and K+ ions favor the formation of a G-quadruplex.144 Their structures display topological polymorphism arising from intra- or inter-molecular folding of guanosine-rich strands.145 The primary functions of G-quadruplex structures are transcriptional regulation of proto-oncogenes146–149 cMYC, c-KIT, κRAS, and BCL2 and maintenance of telomere integrity, which makes them an attractive clinical target for developing anticancer therapeutics. G-quadruplex DNA offers a wide array of structures owing to dissimilarities in the topology, strand orientation, molecularity, and glycosidic conformation. Telomeres are structures on the ends of chromosomes that play a critical role in chromosome stabilization. Their shortening, reportedly, is implicated in cellular senescence.150 However, the presence of a single-stranded G-rich DNA in telomere may adopt a G-quadruplex structure. Telomerase is an enzyme that synthesizes the G-rich strand of telomere DNA and its activity is associated with cancer.151 However, targeting telomerase offers difficulties, including the existence of telomerase-independent mechanisms for conserving the telomere and an extended lag time between telomerase inhibition and effects on cancer cell proliferation.152 The ligands that specifically bind G-quadruplex DNA may interact directly with telomeres, in addition to inhibiting telomerase, and produce more immediate anti-proliferative effects. The development of quadruplex binding ligand fluoroquinolone CX-3543, or quarfloxin,153 and AS1411, the quadruplex-forming DNA aptamer,154 and studies validating their ability to interact with the nucleolin protein establish an encouraging clinical profile of G-quadruplex ligands.155 Nevertheless, the design of G-quadruplex target specific leads poses numerous challenges because the drug molecule must specifically target the quadruplexes over the abundant double stranded DNA present in human chromosomes. Additionally, the candidate molecule must display an ability to differentially identify the individual target quadruplex, as entire G-quadruplexes comprise common structural units, known as G-quartets. Therefore, owing to its intrinsic regioselectivity and characteristic specificity, the click chemistry tool could profitably influence the design and development of novel ligands that specifically target G-quadruplex structures.156 Moorhouse et al. (2006)157 developed the first generation of ligands to stabilize a G-quadruplex by incorporating the “clicked” triazole linkers into a pharmacophore proficient in π-stacking interactions with G-tetrads. Treatment of precursor compound 56 (Chart 7, Scheme 13) with cyclic amine followed by a subsequent conversion of nitro-group to triazide procured synthon 57 (Chart 7, Scheme 13) in 74% yield. The active products 1,4-disubstituted triazoles 58a–c (Chart 7, Scheme 13) were eventually obtained by Cu(i)-mediated Huisgen-type azide–alkyne cycloaddition reaction between triazide 57 (Chart 7, Scheme 13) and 1,3-diethynylbenzene and displayed substantial affinity and selectivity for the G-quadruplex. Molecule 58a (Chart 7, Scheme 13) displayed the most significant telomerase inhibition, with telEC50 = 13.3 μM comparable to naturally occurring telomerase inhibitor telomestatin, in the two-step TRAP assay.157 The protonable amine side-chains in series 58 (Chart 7, Scheme 13) reportedly interacted with the groove regions of quadruplex DNA. The compounds in this series exhibited superior selectivity for quadruplexes over duplex DNA because the large aromatic surfaces of the bis-triazoles hinder intercalative binding with duplex DNA. Moses et al. (2010)158 designed non-planar and non-fused heteroaromatic tris-triazoles containing three symmetric triazolyl substituents tethered to the central benzene core through tetrahedral carbon atoms: compounds 63a–e (Chart 7, Scheme 14). The precursors triester 59 and 4-ethynylaniline 61 yielded synthons 60 and 62 (Chart 7, Scheme 14) which underwent Cu(i)-mediated cycloaddition followed by Michael addition to procure compound series 63 (Chart 7, Scheme 14). The triazole moieties seem to expedite the differential groove-specific interactions of the molecule with different quadruplexes. Nevertheless, due to minimized conformational flexibility and their planarity, the compounds exhibited a weak stabilization potential for DNA quadruplexes.158 Lombardo et al. (2010)159 further designed non-polycyclic tris-triazoles with the central benzene ring directly appended to 1,4-disubstituted triazoles without any methylene bridges (66a–c, Chart 7, Scheme 15) from the precursor synthon 65 (Chart 7, Scheme 15) by click reaction with 1,3,5-triethynylbenzene. The precursor aromatic amine 64 (Chart 7, Scheme 13) produced synthon 65 by conversion of amine functionality to triazide. The ligands in series 66 (Chart 7, Scheme 15) mimicked h-TELO G-quadruplex selective ligand 3,6,9-trisubstituted acridine derivative BRACO-19 in relation to the three distended alkylamino side chains. Interestingly, the C3-symmetric structure of 66 (Chart 7, Scheme 15) displays significant stacking interactions with the nearby G-quartets, whereas the alkylamino side-chains show substantial interactions with the groove regions of quadruplex DNA via electrostatic interactions. Overall, the metasubstituted tris-triazoles display 41 000-fold selectivity for quadruplex over duplex in KD values owing to shorter alkyl side chains containing diethylamine end groups.159
Chart 7. First and second-generation triazole bridged ligands for G-quadruplex stabilization.

Ritson et al. (2012) developed third generation triazole compounds 69a–c (Chart 8, Scheme 16), containing a central triazole ring appended by phenyloxazoles.160 A Cu-mediated click cycloaddition between synthons 67 and 68 (Chart 8, Scheme 16) yielded the desired ligands, which contained oxazole motifs similar to telomestatin. These ligands, however, presented a reserved yet selective affinity for the intramolecular telomeric G-quadruplex, meagre conformational flexibility and superior selectivity for quadruplexes over duplex DNA.
Chart 8. Third generation triazole bridged ligands for G-quadruplex stabilization.

Drewe et al. (2008)161 reported a series of positional isomers of diarylurea-based ligands (74a–d, Chart 8, Scheme 17) procured by Cu-catalyzed click-cycloaddition reaction between synthons 71 and 73 obtained from precursor molecules 70 and 72 (Chart 8, Scheme 17), respectively. The 1,3-diphenyl urea scaffold in the ligands reportedly displays significant pharmacophoric activities with curtailed short-term acute cytotoxicity, suitable for telomerase inhibition. The analysis by molecular docking indicated the ligand urea oxygen atom to be located over the G-quartet ion cavity, while the 1,3-diphenyl urea moiety lies along the planar heteroaromatic triazole rings, effectively stacking over the guanine quartets and maximizing the p–p stacking interactions.161 Further investigation by Förster resonance energy transfer (FRET) melting studies revealed that meta-substituted analogues exhibited enhanced G-quadruplex stabilizing activity (dTm = 16–20 °C) compared to the ortho-substituted (dTm = 12–16 °C) and para-substituted analogues (dTm = 0–4 °C).
3.5. Nucleobase analogues based on 1,2,3-1H-triazole
Tailored oligonucleotides are noteworthy principally in molecular biology as potential materials for oligonucleotide-based therapy and as diagnostic probes.162 Rationally modified nucleobases display superior base-discrimination potency and improved stability of duplex or triplex nucleic acids that caters to their application in several oligonucleotide-based technologies.163–167 Nakahara et al. (2009) developed oligonucleotides and C-nucleotides containing 1-substitued 1H-1,2,3-triazoles as artificial nucleobases (76a–e, Chart 9, Scheme 18) by post-elongation modification method with utilization of Cu(i)-catalyzed alkyne–azide 1,3-dipolar cycloaddition (CuAAC) reaction168 between the 1-ethynyl-2-deoxy-β-d-ribofuranose moiety in oligonucleotide 75 (Chart 9, Scheme 18) and several azide compounds RN3 (R = a–e, Chart 9). Thermal shift (Tm) experiments appraised the base-pairing properties of these triazole nucleobase analogs in forming duplexes with oligonucleotides. The Tm values indicated that the nucleobase analogue 1-(phenylthio) methyl-1H-1,2,3-triazole (76b, Chart 9, Scheme 18) could act as a universal base and be used as a diagnostic probe in the formation of ambiguous sites in primers for PCR and sequencing. Chittepu et al. (2008)169 synthesized alkynyl nucleobases of pyrimidines and purines (R1, Chart 9) as starting materials in the Cu(i)-catalyzed Huisgen–Sharpless–Meldal alkyne–azide cycloaddition ‘click’ reaction to attach an azido residue (77, Chart 9, Scheme 19), procuring the strongly fluorescent final products 78a–d (Chart 9, Scheme 19). Upon their incorporation into DNA using solid phase oligonucleotide synthesis, these tailored nucleosides reportedly behaved as abasic sites, displaying a destabilizing effect on the DNA duplexes.169 The melting experiments validated the negative effect of these conjugates on the stability of a duplex upon their placement opposite to the canonical bases as well as abasic sites. Additionally, no base pair formation with the opposite bases occurred for the triazole-tethered nucleobases due to the structural restraints prompted by the triazole nucleus. Nuzzi et al. (2007)170 developed oligothymidines with triazole internucleosidic linkages replacing the native phosphodiester groups. The compounds 81, 82 and 83 (Chart 9, Scheme 20) were designed as non-natural oligodeoxyribonucleotide antisense agents from synthons 79 and 80 (Chart 9, Scheme 20) by a repetition of copper(i)-catalyzed azide–alkyne cycloaddition as the principal ligation process of suitably functionalized thymidine building blocks.170 Later, Shivalingam et al. (2017)171 developed oligonucleotides containing several structurally and electronically variable artificial linkages (84, Chart 9, Scheme 20) and investigated their kinetics and fidelity of replication through these artificial linkages by primer extension, PCR, and deep sequencing. The examinations revealed an elusive relationship between the backbone flexibility, steric factors, and ability to hydrogen bond to the polymerase which modulates activation of rapid and accurate information decoding. The studies also deposed the essentiality of the phosphate group, as some radical triazole and amide DNA backbones performed significantly well.171 El-Sagheer et al. (2011)172 presented the first ever report of transcription through tailored DNA backbone linkage (85, Chart 9), validating the applicability of click-ligated DNA for the direct synthesis of biologically active RNA and proteins. The tailored DNA strands containing a triazole phosphodiester mimic served as effective templates for in vitro transcription.172
Chart 9. Triazole based tailored oligonucleotides I.

Walczak et al. (2017)173 developed mRNA cap analogues (88, Chart 10, Scheme 21) with differently located triazole rings, a polyphosphate chain and different types of linkers joining the phosphate and the triazole moieties by Cu-mediated click cycloaddition between the synthons 86 and 87 (Chart 10, Scheme 21). This work created a novel paradigm for the development of chemically modified, therapeutically valid mRNAs. The biochemical investigations revealed that upon incorporation into mRNA, the produced transcripts translated with a comparable proficiency as the original compounds containing the oligophosphate-bridge attained by customary synthesis.173 Liu et al. (2018)174 utilized copper(i)-mediated intramolecular “click” cyclization of 89 (Chart 10, Scheme 22) in the presence of TBTA to yield nucleoside macrocycles 90 (Chart 10, Scheme 22) without using protecting groups. The macrocycle could freely approach Watson–Crick recognition sites valued for base pairing with nucleic acids or proteins. The macrocycle also exhibited superior lipophilicity, making it a potential lead for transmembrane delivery of nucleotides and oligonucleotides.174 Hemamalini et al. (2014) developed sugar-chalcone based bis-triazole derivatives 92a–c (Chart 10, Scheme 23) by Cu-mediated click reaction of R1N3 (Chart 10) with synthon 91 (Chart 10, Scheme 23) using THF : H2O (1 : 1) as solvent. The NMR investigations established the existence of the β-anomeric form and 1,4-regioisomer of the synthesized sugar-triazole derivatives. These derivatives also displayed commendable interactions with bovine serum albumin (BSA); in particular, the xylose-coupled triazole 6b displayed the strongest interactions compared to 6a and 6c. The investigations by UV-titration discovered the groove binding nature of the chalcone-based bistriazole derivatives with CT-DNA.175 Biological methylation plays a crucial role in directing the mechanisms of gene transcription. The methylation of promoter sequences within both the genome and protein substrates also regulates this process. Weller et al. (2005) developed adenosine-derived N-mustards 94a–c (Chart 10, Scheme 24) which serve as potent synthetic cofactors allowing for subsequent “click” chemistry involving the improved nucleic acid substrate176 by Cu-assisted Huisgen click cycloaddition reaction using alkyne modified DNA as synthon 93 (Chart 10, Scheme 24).
Chart 10. Triazole based tailored oligonucleotides II.

These strategies for tailoring molecules to modulate the activity of the target biomacromolecule have witnessed rapid developments over the past decades. The routine strategies involving parallel and combinatorial synthesis, automated systems and high-throughput screening suffered from the necessity to synthesize extended libraries and the limited success rate of screened bioactive hits.177 Therefore, contemporary research focuses on the validation of rational procedures that could facilitate a prompt exploration of hit molecules from a pool of potential leads. In vivo screening and structure- or fragment-based drug design represent improved drug development approaches that complement each other to afford identification of superior potential drugs.178 These strategies rely on prior knowledge of the targeted biomacromolecule due to the large availability of biochemical data. Kinetic target-guided syntheses and in situ click chemistry represent recent advancements in the development of alternative approaches to finding ligands for therapeutically relevant proteins.179 Specially, the target-guided synthesis (TGS) strategy, owing to its originality of concept, gained a wide acceptance. However, isolation of in situ generated products is practically unmanageable due to their trace amounts, mainly from target poisoning.180 This strict criterion has led to the limited development of reactions since the unanticipated discovery of the first example in 1960s. Predominantly, the use of 1,3-dipolar cycloaddition click reactions proved to be highly compatible for the discovery of ligands of various biomolecules such as enzymes, RNA and DNA, protein–protein interactions, protein-targeted drug delivery and probing the macromolecular complexity and dynamics of biological targets.181
4. Kinetic target guided synthesis of enzyme inhibitors based on 1,2,3-1H-triazoles
Mock and coworkers reported significant acceleration in the rate of azide–alkyne cycloaddition reaction by impounding the participating components inside the macromolecular host,182 promptly leading to the development of a novel drug-discovery archetype reliant on irreversible, target-guided synthesis of high-affinity inhibitors from reagents that are inert under physiological conditions. Using a target enzyme to screen the building blocks and synthesize its own inhibitor is emerging as a novel drug-development prototype183 also referred to as ‘kinetic target guided synthesis’. The concurrent binding of two parent ligands appended with corresponding reactive groups within the active site of the target enzyme expedites the reaction that connects them. Interestingly, when bond formation is blocked by product inhibition, the higher affinity products serve as lead compounds.184
4.1. In situ inhibitors of acetylcholine esterase (AchE)
Lewis et al. (2002) developed in situ inhibitors of the enzyme acetylcholine esterase using click chemistry.185 Krasiński et al. (2005) improved the approach186 with liquid chromatography-mass spectrometry in selected ion mode (LC/MS-SIM). Subsequently, Bourne et al. (2010) validated tacrine-azide hybrid as an anchor molecule187 at concentrations sufficient to saturate the enzyme's active site. This enzyme-inhibitor complex recruited the most favored outlying binding group from a mixture of alkynes. Compound 95 (Chart 11), the syn-triazole derivative, exhibited decent binding affinity to AchE with a Kd value of 99 nM, while compound 96 (Chart 11) of the S-isomers of syn-triazole demonstrated much lower affinity to AChE, having a Kd value of 33 nM. Fokin et al. (2012) described the combination of CuAAC and in situ click chemistry for the synthesis of a highly potent and selective ligand (97, Chart 11) for Lymnaea stagnalis acetylcholine binding protein (AChBPs).188 This syn-regioisomer of the triazole displayed an exceptional affinity to AChBPs due to a planned modification in conformation that boosted the aromatic π–π connotation at the outlying site.
Chart 11. In situ enzyme inhibitors based on 1,4-disubstituted-1,2,3-triazole.

4.2. In situ inhibitors of carbonic anhydrase (CA)
Carbonic anhydrases (CA) are a family of metalloenzymes which catalyze the interconversion of bicarbonate ion and CO2, thereby playing vital roles in respiration, pH balancing of blood and other tissues, transport of CO2 and H+ and tumorigenicity.189 Mocharla et al. (2005)190 used in situ click chemistry with acetylenic benzenesulfonamide (98, Chart 11, Scheme 25) as a reactive scaffold and several different azides to screen the lead inhibitors of CA (99, Chart 11, Scheme 25). Examination of the unfinished reaction mixture using LC/MS-SIM discovered that all of the observed triazoles were of an anti-regiochemistry, which relates to the inhibitor selection by the enzyme active site with a Ki value of 0.2 ± 0.3 nM.190
4.3. In situ inhibitors of cyclooxygenase-2 (COX-2)
Cyclooxygenase-2 isozyme is an induced isoform of the cyclooxygenase family of enzymes. Its overexpression is associated with several cancers and neurodegenerative diseases.191 The rationally designed cyclooxygenase-2 inhibitors face significant challenges due to the amino-acid sequence and structural analogy between inducible cyclooxygenase-2 and housekeeping cyclooxygenase-1 isoforms. Bhardwaj et al. (2017)192 described the utility of cyclooxygenase-2 active site as a reaction vessel for the in situ generation of its own inhibitors. Compounds 101a–b (Chart 11, Scheme 26) obtained from rationally designed pts-substituted alkynyls (100) displayed substantial in vivo anti-inflammatory activity compared to commercially used selective cyclooxygenase-2 inhibitors. Additionally, SAR investigations and computational studies recommend that size, the presence of a SO2Me-based COX-2 pharmacophore and proper orientation of the azide nucleus inside the COX-2-binding site collectively contribute to the in situ click chemistry synthesis of highly potent and selective COX-2 isozyme inhibitors.192
4.4. In situ probing of Abl tyrosine kinase binding site
Peruzzotti et al. (2013) investigated the potency of Abl in the selected target-catalyzed formation of FA030 from a library of building blocks simultaneously present in a single reaction mixture.193 A solution of azides (102, Chart 11, Scheme 27) and alkynes was incubated for 4 days at 37 °C in the presence of Abl (0.37 μM) in a suitable buffer solution to achieve the compounds 103a–c (Chart 11, Scheme 27). The control solution was prepared without tyrosine kinase. The investigation by HPLC and ESI-HRMS displayed only the peak generated by FA030. The peaks were absent in the control solution, confirming that Abl selectively favors the formation of the best inhibitor. The studies also validated the relevance of in situ click reaction for the discovery of new scaffolds convenient for inhibition of the large family of tyrosine kinases.
4.5. In situ inhibitors of HIV-1 protease
Aspartic protease of HIV-1 is the foremost therapeutic target against AIDS. A high rate of mutational adaptation by the HIV virus poses challenges for the discovery of new protease inhibitors. Whiting et al. (2006)109 developed hydroxyethylamine peptide isostere 106 (Chart 11, Scheme 28) by uniting the azide-bearing scaffold 105 (Chart 11, Scheme 28) with acetylenes 104 (Chart 11, Scheme 28) via the copper-catalyzed process. On screening against wild type HIV-1 protease and three mutants (G48V, V82F, V82A), compound 106 demonstrated a strong inhibition profile against all four proteases194 with activities in the low nanomolar range with Ki = 1.7 nM.
4.6. In situ inhibitors of chitinase
Chitinase inhibitors present commendable chemotherapeutic potential in the form of pesticides, fungicides, and anti-asthmatics. Hirose et al. (2016) developed a chitinase inhibitor based on an argin fragment appended with an azide group (107, Chart 12, Scheme 29) and subjected it to in situ click chemistry with a library of alkynes (108, Chart 11, Scheme 29) in the presence of chitinase template to procure 109 (Chart 12, Scheme 29), which inhibits Serratia marcescens chitinase (SmChi) B.195 The inhibitor presented around a 300-fold escalation of inhibition profile against SmChiB compared to argin, a naturally produced cyclopentapeptide based inhibitor. The crystal structures of the inhibitor-chitinase complex and an O-allyl oxime fragment, as a mimic of a click partner, revealed a sophisticated mechanism for hastening in situ syn-triazole formation by click chemistry, thus generating its own distinct inhibitor.
Chart 12. In situ enzyme inhibitors based on 1,4-disubstituted-1,2,3-triazole.

4.7. In situ inhibitors of biotin protein ligase
Tieu et al. (2013)196 utilized SaBPL-R122G, termed a ‘leaky mutant’ of Staphylococcus aureus biotin protein ligase (SaBPL) to augment the turnover rate for the reaction of biotin alkyne 110 (Chart 12, Scheme 30) with azides 111 and 112 (Chart 12, Scheme 30) to give triazoles 113 and 114 (Chart 12, Scheme 30). The strategy allows the enzyme to select the finest triazole-based inhibitors using a collection of azides in a single experiment with significantly upgraded effectiveness and sensitivity of detection.196 The leaky mutant improved the rate of product dissociation relative to the target enzyme (SaBPL) by approximately 40-fold. The investigations revealed the dormancy of ribose sugar present in inhibitor 113 (Chart 12, Scheme 30) for optimum activity.
4.8. In situ inhibitors of d-amino acid oxidase
The hDAO inhibitors display admirable chemotherapeutic potential as antipsychotic agents. Toguchi et al. (2016)197 reported in situ click chemistry between alkyne 115 (Chart 12) and azide 116 (Chart 12, Scheme 31) in the presence of human d-amino acid oxidase (hDAO) as the reaction vessel to procure potent hDAO inhibitor 117 (Chart 12, Scheme 31) with a Ki value of 1.0 mM. The novel inhibitor presented competitive inhibition of hDAO and substantial augmentation in inhibitory potency against hDAO compared to an anchor molecule of in situ click chemistry.
5. Bioorthogonal profiling of proteins with 1,2,3-1H-triazoles
Bioorthogonal reactions present a robust approach for profiling and labeling vital proteins by integrating the bioorthogonal groups into the target protein, followed by desired chemoselective modifications.198 Since proteins carry multiple prints of diverse functional groups, their labelling at a distinct site becomes necessary. Optimum functioning of cellular proteins is obligatory to sustain tissue homeostasis.199 The methodologies facilitating in vivo visualization of proteins in their cellular environment rationalize the cellular functioning and dynamics regulated by these proteins.200 However, the labeling conditions should be mild and reactions should preferably happen in aqueous medium. Importantly, the labeling reagents should also be orthogonal and physiologically labile.201 These limitations pose a significant challenge for labelling proteins in a chemoselective and site-specific manner.202 Prevalent approaches for visualizing proteins in their native environments depend solely on genetically encoded fluorescent proteins (GFP). GFPs enable the visualization of a protein of interest in live cellular environment or organisms by fusing a target protein to GFP. However, the large size of GFP may interfere with the structure and function of the fused protein. In this case, a fitting peptide sequence harboring an explicit site for bioorthogonal conjugation is employed to tailor a target protein for in vivo imaging.
5.1. Bioconjugated triazole-based probes for profiling cell surface proteins
Treatment of the tagged protein present on the cell surface with a suitable enzyme (biotin ligase, transglutaminase, formylglycine-generating enzyme)203–205 to add a synthetic prosthetic group analog carrying the desired orthogonal functionality enables labeling of a desired protein with an imaging probe through bioorthogonal click reactions. Lin et al. (2006) exploited microbial lipoic acid ligase (LplA) enzyme to specifically attach an alkyl azide with a tailored LplA acceptor peptide (LAP) tethered to cell-surface proteins in live mammalian cells.206 Selective labelling of this alkyl azide with fluorescently conjugated cyclooctyne probes (Fig. 3) assisted in imaging the dynamics of the labeled proteins.207 This protocol optimized the studies of the dynamics and distribution of the desirable preselected proteins. However, this method loses validity when monitoring the newly synthesized proteins because, to tag a fluorescent protein or a peptide to the protein of interest, genetic manipulations are required. Dieterich et al. (2010) developed the novel method ‘bioorthogonal noncanonical amino acid tagging’ for labelling proteins on the cellular surface.208 The method included the metabolic incorporation of methionine analogue azidohomoalanine (AHA) or homopropargylglycine (HPG) into the newly synthesized proteins, followed by a TBTA-mediated CuAAC. The fluorophore tagged to the alkyne or azide eventually detects these proteins in vivo in fixed cells. The method has been quite successful for visualizing newly synthesized proteins in rat hippocampal neurons and in spine-like protrusions from dendrites.209
Fig. 3. Probing of cellular protein function by bioorthogonal 1,2,3-triazoles.

5.2. Bioconjugated triazole-based probes for profiling PTM in proteins
The identification of post translational modifications (PTM) in proteins advocates that the structure and function of proteins are not merely dictated by progenitor nucleic acids and amino acid building blocks, but are also governed by several other factors.210,211 The quantization of these associated parameters provides useful tools for revealing the expressional behavior and functions of target genes; however, these tools fail to rationalize some crucial cellular processes that modulate protein folding, functional states, and their subcellular localizations.212–215 A typical bioconjugation method for profiling protein PTMs is comprised of a preliminary appending of an alkyne or azide functionality onto a natural substrate of PTM enzyme which then acts upon this engineered substrate, thereby modifying the substrate proteins in their native environment. Afterward, the introduction of an imaging probe assists in visualizing the labeled biomolecules in the living systems. Jiang et al. (2010)216 designed novel substrates of poly(ADP-ribose) polymerases (PARPs) from clickable nicotinamide adenine dinucleotide (NAD) analogues. PARPs reportedly add ADP-ribose units to the proteins formed after translation and thereby regulate their poly-ADP-ribosylation. Additionally, PARPs play important roles in DNA repair, transcription, and mitosis. Therefore, identifying the substrate proteins on which a PARP acts validates the regulatory functions of individual PARPs found in humans. Jiang and coworkers utilized PARP-1 specific alkyne-tagged NAD analogue by facilitating the conjugation of affinity tags to the substrates of PARP-1 and performed affinity purification under denaturing conditions (Fig. 4).216 The affinity purification enabled identification of seventy candidate substrates for PARP-1 by tandem mass spectrometry technique. In a similar approach, Wilson et al. (2011) successfully recognized sixty known fatty-aceylated proteins and around a hundred new candidate proteins, including histone H3 variants from Jurkat cells.217 This methodology also assisted in profiling palmitoylated proteins in dendritic cells.218S-Palmitoylated proteins profiled in dendritic cell line DC2.4 validated the palmitoylation of interferon-induced transmembrane protein 3 (IFITM3), which is also associated with the host immune response to microbial infection. Additionally, the S-palmitoylation of IFITM3 led to the clustering of protein in the membrane of the endoplasmic reticulum in the host cell.219
Fig. 4. Probing of protein PTMs by bioorthogonal 1,2,3-triazoles.

5.3. Bioconjugated triazole-based probes for activity based protein profiling
Click chemistry is quickly emerging as an admirable route for ABPP (activity based protein profiling) by employing active site-directing probes to study the dynamic expressions and functions of mechanistically similar enzymes.220,221 A conventional activity-based probe carries three moieties: an electrophilic reactive group to selectively bind and covalently modify the catalytically relevant and reactive nucleophiles, a high-affinity binding group to direct the activity based protein to structurally conserved features in the active sites of the target enzymes, and, essentially, an analytical switch to visualize and segregate the probe-labeled targets.222,223 Identification of the target protein of FR182877 presents an early success story of ABPP coupled with click chemistry as it reportedly prompts in vitro microtubule assembly and in vivo antitumor activity.224 FR182877 contains two electrophilic sites and in vivo covalent modifications of specific proteins by affinity tag conjugation validate its biological activity. Structural complexity coupled with the nucleophilic sensitivity of FR182877 facilitate several side reactions, thereby discouraging conventional methods for its functionalization. However, a rhodamine- and biotin-tagged short peptide appended to FR182877 by Cu(i)-catalyzed triazole formation generated a trifunctional probe.225 This probe, on in vitro incubation with mouse heart proteome followed by avidin chromatography, directed isolation of the biotin-labeled protein identified carboxylesterase-1 (CE-1) as a specific target of this natural product. Additionally, the competition experiments with the probe fluorophosphonate rhodamine directed on serine hydrolase validated (–) FR182877 (118) as a highly potent and selective inhibitor of CE-1 with IC50 = 34 nM. Subsequent developments led to the design of second-generation probes with small, bioorthogonal chemical handles (i.e., an alkyne or an azide) replacing large reporter tags.226,227 In designing an azide-bearing active-site probe, azidohexyl benzenesulfonate and a rhodaminealkyne detection probe led to the identification of a number of serine hydrolases, including ECH-1, in the human breast cancer cell line MDA-MB-435.

6. Conclusion
High selectivities, optimum reaction conditions and no side products, in addition to excellent yields, make azide–alkyne click-cycloaddition the most exploitable reaction in contemporary drug development. The triazole nucleus being an amide bond bioisostere plays a crucial role in the development of peptide-based therapeutics. Their planar nature makes them a desirable scaffold for developing DNA-mimics to effect gene expression and peptide transcription. The ability of the clickable synthons to identify each other's proximity led to the development of the magnificent ‘kinetic target guided synthesis’ for in situ inhibitor synthesis in the active site vessel of the target enzyme. The novel bioorthogonal developments in proteomics have contributed to successful protein profiling and identifying the PTMs revolutionized chemical biology by assisting in the development of new drug targets. Click-chemistry-synthesized 1,2,3-1H-triazoles, hence, serve as an indispensable tool in the current drug development paradigm.
Conflicts of interest
The authors declare that they do not have any conflict of interest.
Acknowledgments
PP and MS duly acknowledge Department of Chemistry, University of Petroleum & Energy Studies and Guru Nanak Dev University.
Biographies

Parteek Prasher
Dr. Parteek Prasher is currently an Assistant Professor at the Department of Chemistry, University of Petroleum & Energy Studies, Dehradun. He obtained his MSc. Hons. and PhD. in medicinal chemistry from Guru Nanak Dev University. His research interests include bioorthogonal chemistry and therapeutic applications of tailored bioconjugated metal nanoparticles.

Mousmee Sharma
Dr. Mousmee Sharma did her doctorate from Guru Nanak Dev University, India in the year 2019. She obtained her PhD. from Guru Nanak Dev University in Physical Chemistry. A Gold Medalist in MSc. Chemistry, her research interests include the investigations of physicochemical interactions at the bio-membranes interface. Her hobbies include reading books and playing badminton.
References
- Hughes J. P., Rees S., Kalindjian S. B., Philpott K. L. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krska S. W., DiRocco D. A., Dreher S. D., Shevlin M. Acc. Chem. Res. 2017;50:2976–2985. doi: 10.1021/acs.accounts.7b00428. [DOI] [PubMed] [Google Scholar]
- Broach J. R., Thorner J. Nature. 1996;384:14–16. [PubMed] [Google Scholar]
- Li F., Mahato R. I. Mol. Pharmaceutics. 2017;14:1321–1324. doi: 10.1021/acs.molpharmaceut.7b00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. W., Rees D. C. Nat. Chem. 2009;1:187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- Hu X., Manetsch R. Chem. Soc. Rev. 2010;39:1316–1324. doi: 10.1039/b904092g. [DOI] [PubMed] [Google Scholar]
- Nicalaou K. C. Angew. Chem., Int. Ed. 2014;53:9128–9140. doi: 10.1002/anie.201404761. [DOI] [PubMed] [Google Scholar]
- Bylinsky G. Fortune. 1981;104:106–114. [Google Scholar]
- Gund P., Grabowski E. J. J., Hoff D. R., Smith G. M., Andose J. D., Rhodes J. B., Wipke W. T. J. Chem. Inf. Comput. Sci. 1980;20:88–93. [Google Scholar]
- Gund P., Andose J. D., Rhodes J. B., Smith G. M. Science. 1980;208:1425–1431. doi: 10.1126/science.6104357. [DOI] [PubMed] [Google Scholar]
- Brown F. K., Sherer E. C., Johnson S. A., Holloway M. K., Sherborne B. S. J. Comput.-Aided Mol. Des. 2017;31:255–266. doi: 10.1007/s10822-016-9993-1. [DOI] [PubMed] [Google Scholar]
- Macarron R., Banks M. N., Bojanic D., Burns D. J., Cirovic D. A., Garyantes T., Green D. V. S., Hertzberg R. P., Janzen W. P., Paslay J. W., Schopfer U., Sittampalam G. S. Nat. Rev. Drug Discovery. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Rademacher C. and Seeberger P. H., Handbook Exp. Pharmacol., ed. U. Nielsch, U. Fuhrmann and S. Jaroch, Springer, Cham, 2016, vol. 232, pp. 73–89. [Google Scholar]
- Kerns E. H. J. Pharm. Sci. 2001;90:1838–1858. doi: 10.1002/jps.1134. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E. Future Med. Chem. 2009;1:11–15. doi: 10.4155/fmc.09.2. [DOI] [PubMed] [Google Scholar]
- Jhoti H., Rees S., Solari R. Expert Opin. Drug Discovery. 2013;8:1449–1453. doi: 10.1517/17460441.2013.857654. [DOI] [PubMed] [Google Scholar]
- Bibette J. Proc. Natl. Acad. Sci. U. S. A. 2012;109:649–650. doi: 10.1073/pnas.1119350109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittker J. A. and Ross N. T., High Throughput Screening Methods: Evolution and Refinement, Royal Society of Chemistry, United Kingdom, 2016. [Google Scholar]
- Janzen W. P. and Bernasconi P., High Throughput Screening, Humana Press, Totowa, New Jersey, 2009. [Google Scholar]
- Malo N., Hanley J. A., Cerquozzi S., Pelletier J., Nadon R. Nat. Biotechnol. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- Sink R., Gobec S., Pecar S., Zega A. Curr. Med. Chem. 2010;17:4231–4255. doi: 10.2174/092986710793348545. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Drug Discovery Today. 1997;2:382–384. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- Zhang J. H., Chung T. D. Y., Oldenburg K. R. J. Biomol. Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang J. H., Chung T. D. Y., Oldenburg K. R. J. Comb. Chem. 2000;2:258–265. doi: 10.1021/cc9900706. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. J. Med. Chem. 2006;49:7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- Walters W., Namchuk M. Nat. Rev. Drug Discovery. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- Parham F., Austin C., Southall N., Huang R., Tice R., Portier C. J. Biomol. Screening. 2009;14:1216–1227. doi: 10.1177/1087057109349355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishton G. M. Drug Discovery Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- Hann M. M., Oprea T. I. Curr. Opin. Chem. Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Thorne N., Auld D. S., Inglese J. Curr. Opin. Chem. Biol. 2010;14:315–324. doi: 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftsson T., Brewster M. E. J. Pharm. Pharmacol. 2010;62:1607–1621. doi: 10.1111/j.2042-7158.2010.01030.x. [DOI] [PubMed] [Google Scholar]
- Hodgson J. Nat. Biotechnol. 2001;19:722–726. doi: 10.1038/90761. [DOI] [PubMed] [Google Scholar]
- Kalia J., Raines R. T. Curr. Org. Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Mol. Pharmaceutics. 2017;14:1321–1324. doi: 10.1021/acs.molpharmaceut.7b00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam M. and Dent A., Bioconjugation: Protein Coupling Techniques for the Biomedical Sciences, Macmillan Reference Ltd, London, 1998. [Google Scholar]
- Lundblad R. L., Chemical Reagents for Protein Modification, 3rd edn., CRC Press, Boca Raton, FL, 2005. [Google Scholar]
- Hermanson G. T., Bioconjugate Techniques, 2nd edn, Academic Press, San Diego, CA, 2008. [Google Scholar]
- Kopecek J., Kopeckova P., Minko T., Lu Z. R. Eur. J. Pharm. Biopharm. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- Shiah J. G., Dvorak M., Kopeckova P., Sun Y., Peterson C. M., Kopecek J. Eur. J. Cancer. 2001;37:131–139. doi: 10.1016/s0959-8049(00)00374-9. [DOI] [PubMed] [Google Scholar]
- Maeda H., Seymour L. M., Miyamoto Y. Bioconjugate Chem. 1992;3:351–362. doi: 10.1021/bc00017a001. [DOI] [PubMed] [Google Scholar]
- Lu Z.-R., Shiah J.-G., Sakuma S., Kopeckova P., Kopecek J. J. Controlled Release. 2002;78:165–173. doi: 10.1016/s0168-3659(01)00495-3. [DOI] [PubMed] [Google Scholar]
- Stephanopoulos N., Francis M. B. Nat. Chem. Biol. 2011;7:876–884. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- He X.-P., Hu X.-L., James T. D., Yoon J., Tian H. Chem. Soc. Rev. 2017;46:6687–6696. doi: 10.1039/c6cs00778c. [DOI] [PubMed] [Google Scholar]
- Yu C., Xi J., Li M., An M., Liu H. Bioconjugate Chem. 2018;29:719–732. doi: 10.1021/acs.bioconjchem.7b00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kairdolf B. A., Qian X., Nie S. Anal. Chem. 2017;89:1015–1031. doi: 10.1021/acs.analchem.6b04873. [DOI] [PubMed] [Google Scholar]
- Liu Z., Chen X. Chem. Soc. Rev. 2016;45:1432–1456. doi: 10.1039/c5cs00158g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. M., Nazarova L. A., Prescher J. A. ACS Chem. Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Jiang H., Zheng T., Lopez-Aguilar A., Feng L., Kopp F., Marlow F. L., Wu P. Bioconjugate Chem. 2014;25:698–706. doi: 10.1021/bc400502d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister D., Ulmer N., Klaue A., Ingold O., Morbidelli M. Chem. Ing. Tech. 2016;88:1598–1608. [Google Scholar]
- Patterson D. M., Nazarova L. A., Prescher J. A. ACS Chem. Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Ramil C. P., Lin Q. Chem. Commun. 2013;49:11007–11022. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer A. N. Annu. Rev. Biochem. 1970;39:101–130. doi: 10.1146/annurev.bi.39.070170.000533. [DOI] [PubMed] [Google Scholar]
- Kaiser E. T., Lawrence D. S., Rokita S. E. Annu. Rev. Biochem. 1985;54:565–595. doi: 10.1146/annurev.bi.54.070185.003025. [DOI] [PubMed] [Google Scholar]
- Tsuchikama K., An Z. Protein Cell. 2018;9:33–46. doi: 10.1007/s13238-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A. M., Senter P. D. Nat. Biotechnol. 2005;23:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Unver M. Y., Gierse R. M., Ritchie H., Hirsch A. K. H. J. Med. Chem. 2018;61:9395–9409. doi: 10.1021/acs.jmedchem.8b00266. [DOI] [PubMed] [Google Scholar]
- Huang R., Leung I. K. H. Molecules. 2016;21:910. doi: 10.3390/molecules21070910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosc D., Jakhlal J., Deprez B., Deprez-Poulain R. Future Med. Chem. 2016;8:381–404. doi: 10.4155/fmc-2015-0007. [DOI] [PubMed] [Google Scholar]
- Oueis E., Sabot C., Renard P.-Y. Chem. Commun. 2015;51:12158–12169. doi: 10.1039/c5cc04183j. [DOI] [PubMed] [Google Scholar]
- Cheezeman J. D., Corbett A. D., Gleason J. L., Kazlauskas R. J. Chem. – Eur. J. 2005;11:1708–1716. doi: 10.1002/chem.200400371. [DOI] [PubMed] [Google Scholar]
- Miller B. L., Dynamic Combinatorial Chemistry: In Drug Discovery, Bioorganic Chemistry and Materials Science, Wiley, 2009. [Google Scholar]
- Cuccia L. A. J. Am. Chem. Soc. 2011;133:7236–7236. [Google Scholar]
- Otto S., Furlan R. L. E., Sanders J. K. M. Drug Discovery Today. 2002;7:117–125. doi: 10.1016/s1359-6446(01)02086-4. [DOI] [PubMed] [Google Scholar]
- Schmidt M. F., Rademann J. R. Trends Biotechnol. 2009;27:512–521. doi: 10.1016/j.tibtech.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgürtel M., Kroth H., Piecha D., Hofman M. W., Nicolau C., Krause S., Schaaf O., Sonnenmoser G., Aliseev A. V. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3382–3387. doi: 10.1073/pnas.052703799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidyala S. K., Finn M. G. Chem. Soc. Rev. 2010;39:1252–1261. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- Antti H., Sellstedt M. ACS Med. Chem. Lett. 2018;9:351–353. doi: 10.1021/acsmedchemlett.7b00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W. G., Green L. G., Grynszpan F., Radic Z., Carlier P. R., Taylor P., Finn M. G., Sharpless K. B. Angew. Chem., Int. Ed. 2002;41:1053–1056. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Sharpless K. B., Manetsch R. Expert Opin. Drug Discovery. 2006;1:525–538. doi: 10.1517/17460441.1.6.525. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A., Wells J. A., Braisted A. C. Annu. Rev. Biophys. Biomol. Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A., McDowell R. S., He M. M., Rendal M., Simmons R. L., Kung J., Waight A., Hansen S. K. J. Am. Chem. Soc. 2003;125:5602–5603. doi: 10.1021/ja034440c. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A., Lam J. W., Wiesmann C., Luong T. N., Simmons R. L., DeLano W. L., Choong I. C., Burdett M. T., Flanagan W. M., Lee D., Gordon E. M., O'Brien T. Nat. Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A., Braisted A. C., Raphael D. R., Randal M., Stroud R. M., Gordon E. M., Wells J. A. Proc. Natl. Acad. Sci. U. S. A. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslob J. D., Erlanson D. A. Drug Discovery Today: Targets. 2004;3:143–150. [Google Scholar]
- Dettmer K., Aronov P. A., Hammock B. D. Mass Spectrom. Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O., Kopka J., Dormann P., Altmann T., Trethewey R. N., Willmitzer L. Nat. Biotechnol. 2000;18:1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- Lu W., Bennett B. D., Rabinowitz J. D. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2008;871:236–242. doi: 10.1016/j.jchromb.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. J., Lin W. Y., Liu K., Lin R. J., Selke M., Kolb H. C., Zhang N. G., Zhao X. Z., Phelps M. E., Shen C. K. F., Faull K. F., Tseng H. R. Lab Chip. 2009;9:2281–2285. doi: 10.1039/b907430a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelke S. V., Cutting B., Jiang X., Koliwer-Brandl H., Strasser D. S., Schwardt O., Kelm S., Ernst B. A. Angew. Chem., Int. Ed. 2010;49:5721–5725. doi: 10.1002/anie.200907254. [DOI] [PubMed] [Google Scholar]
- Kolb H. C., Finn M. G., Sharpless K. B. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kappe C. O., Van der Eycken E. Chem. Soc. Rev. 2010;39:1280–1290. doi: 10.1039/b901973c. [DOI] [PubMed] [Google Scholar]
- Wu P., Feldman A. K., Nugent A. K., Hawker C. J., Scheel A., Voit B., Pyun J., Fréchet J. M. J., Sharpless K. B., Fokin V. V. Angew. Chem., Int. Ed. 2004;43:3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B. Angew. Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bouillon C., Meyer A., Vidal S., Jochum A., Chevolot Y., Cloarec J.-P., Praly J.-P., Vasseur J.-J., Morvan F. J. Org. Chem. 2006;71:4700–4702. doi: 10.1021/jo060572n. [DOI] [PubMed] [Google Scholar]
- Thirumurugan P., Matosiuk D., Jozwiak K. Chem. Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- Heravi M. M., Tamimi M., Yahyavi H., Hosseinnejad T. Curr. Org. Chem. 2016;20:1591–1647. [Google Scholar]
- (a) Emamian S. R., Ali-Asgari S., Zahedi E. J. Chem. Sci. 2014;126:293–302. [Google Scholar]; (b) Singh M. S., Chowdhury S., Koley S. Tetrahedron. 2016;72:5257–5283. [Google Scholar]
- Huisgen R. J. Org. Chem. 1968;33:2291–2297. [Google Scholar]
- Firestone R. J. Org. Chem. 1968;33:2285–2290. doi: 10.1021/jo01267a059. [DOI] [PubMed] [Google Scholar]
- Freindorf M., Sexton T., Kraba E., Cremer D. Theor. Chem. Acc. 2014;133:1423. [Google Scholar]
- Kolb H. C., Sharpless K. B. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- Meldar M., Tornoe C. W. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Presolski S. I., Hong V. P., Finn M. G. Curr. Protoc. Chem. Biol. 2011;3:153–162. doi: 10.1002/9780470559277.ch110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V., Presolski S. I., Ma C., Finn M. G. Angew. Chem., Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V., Steinmetz N. F., Manchester M., Finn M. G. Bioconjugate Chem. 2009;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel Jr G. R., Calabrese Z. A., Ayco J., Hein J. E., Ye T. Bioconjugate Chem. 2016;27:698–704. doi: 10.1021/acs.bioconjchem.5b00665. [DOI] [PubMed] [Google Scholar]
- (a) Li L., Zhang Z. Molecules. 2016;21:1393. [Google Scholar]; (b) Guezguez R., Bougrin K., El Akri K., Benhida R. Tetrahedron Lett. 2006;47:4807–4811. [Google Scholar]; (c) Thanh N. D., Hai D. S., Ha N. T. T., Tung D. T., Le C. T., Van H. T. K., Toan V. N., Toan D. N., Dang L. H. Bioorg. Med. Chem. Lett. 2019;29:164–171. doi: 10.1016/j.bmcl.2018.12.009. [DOI] [PubMed] [Google Scholar]; (d) Alonso F., Moglie Y., Radivoy G., Yus M. J. Org. Chem. 2011;76:8394–8405. doi: 10.1021/jo2016339. [DOI] [PubMed] [Google Scholar]
- Jewett J. C., Bertozzi C. R. Chem. Soc. Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ess D. H., Jones G. O., Houk K. N. Org. Lett. 2008;10:1633–1636. doi: 10.1021/ol8003657. [DOI] [PubMed] [Google Scholar]
- Debets M. F., van Berkel S. S., Schoffelen S., Rutjes F. P. J. T., van Hest J. C. M., van Delft F. L. Chem. Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- Kuzmin A., Poloukhtine A., Wolfert M. A., Popik V. V. Bioconjugate Chem. 2010;21:2076–2085. doi: 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- Yao J. Z., Uttamapinant C., Poloukhtine A., Baskin J. M. J. Am. Chem. Soc. 2012;134:3720–3728. doi: 10.1021/ja208090p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Mackey J. L., Lopez S. A., Liu F., Houk K. N. J. Am. Chem. Soc. 2012;134:17904–17907. doi: 10.1021/ja309241e. [DOI] [PubMed] [Google Scholar]
- Nicholas J. A., Prescher J. A., Bertozzi C. R. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Bonandi E., Christodoulou M. S., Fumagalli G., Perdicchia D., Rasteli G., Passarella D. Drug Discovery Today. 2017;22:1572–1581. doi: 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Tron G. C., Pirali T., Billington R. A., Conanico P. L., Sorba G., Genazzani A. A. Med. Res. Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- Ferreira S. B., Sodero A. C., Cardoso M. F., Lima E. S., Kaiser C. R., Silva F. P., Ferreira V. F. J. Med. Chem. 2010;53:2364–2375. doi: 10.1021/jm901265h. [DOI] [PubMed] [Google Scholar]
- Bourne Y., Kolb H. C., Radic Z., Sharpless K. B., Taylor P., Marchot P. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1449–1454. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting M., Muldoon J., Lin Y. C., Silverman S. M., Lindstron W., Olson A. J., Kolb H. C., Finn M. G., Sharpless K. B., Elder J. H., Fokin V. Angew. Chem., Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- Tron G. C., Pirali T., Billington R. A. Med. Res. Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- Mohammed I., Parai M. K., Jiang X., Sharova N., Singh G. K., Stevenson M., Rana T. M. ACS Med. Chem. Lett. 2012;3:465–469. doi: 10.1021/ml300037k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed I., Kummetha I. R., Singh G. K., Sharova N., Lichinchi G., Dang J., Stevenson M., Rana T. M. J. Med. Chem. 2016;59:7677–7682. doi: 10.1021/acs.jmedchem.6b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell S. M., Lee Y. S., Cain J. P., Hruby V. J. Curr. Med. Chem. 2004;11:2785–2798. doi: 10.2174/0929867043364270. [DOI] [PubMed] [Google Scholar]
- Yan H., Zhou M., Bhattarai U. Mol. Pharmaceutics. 2019;16:914–920. doi: 10.1021/acs.molpharmaceut.8b01247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagner J., Qu H., Hruby V. J. Curr. Opin. Chem. Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch B. D., Van Demark A. P., Heroux A., Hill C. P., Kay M. S. Proc. Natl. Acad. Sci. U. S. A. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Thomas R. M., Suzuki H., De Brabander J. K., Wang X., Harran P. G. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- Bock V. D., Speijer D., Hiemstra H., van Maarseveen J. H. Org. Biomol. Chem. 2007;5:971–975. doi: 10.1039/b616751a. [DOI] [PubMed] [Google Scholar]
- Horne W. S., Olsen C. A., Beierle J. M., Montero A., Ghadiri M. R. Angew. Chem., Int. Ed. 2009;48:4718–4724. doi: 10.1002/anie.200805900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz M., Murray P. J., Taylor S., Upton R. J., Clegg W., Elsegood M. R. J. J. Pept. Sci. 2006;12:383–388. doi: 10.1002/psc.738. [DOI] [PubMed] [Google Scholar]
- Singh S. B., Zink D. L., Polishook J. D., Dombrowski A. W., Darkin–Rattray S. J., Schmatz D. M., Goetz M. A. Tetrahedron Lett. 1996;37:8077–8080. [Google Scholar]
- (a) Oh K., Guan Z. Chem. Commun. 2006:3069–3071. doi: 10.1039/b606185k. [DOI] [PubMed] [Google Scholar]; (b) Beierle J. M., Horne W. S., van Marseveen J. H., Waser B., Reubi J. C., Ghadiri M. R. Angew. Chem., Int. Ed. 2009;48:4725–4729. doi: 10.1002/anie.200805901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z., Cabell L. A., Schaefer T. S., McMurray J. S. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- Chen J. Y., Nikolovska-Coleska Z., Yang C. Y., Gomez C., Gao W., Krajewski K., Jiang S., Roller P., Wang S. M. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- Wang Z. A., Ding X. Z., Tian C.-L., Zheng J.-S. RSC Adv. 2016;6:61599–61609. [Google Scholar]
- Sawyer N., Watkins A. M., Arora P. S. Acc. Chem. Res. 2017;50:1313–1322. doi: 10.1021/acs.accounts.7b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin W. A., Tyndall J. D. A., Glenn M. P., Fairlie D. P. Chem. Rev. 2004;104:6085–6118. doi: 10.1021/cr040648k. [DOI] [PubMed] [Google Scholar]
- Venkatraman J., Shankaramma S. C., Balaram P. Chem. Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- Cheng R. P., Gellman S. H., DeGrado W. F. Chem. Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- Angelo N. G., Arora P. S. J. Am. Chem. Soc. 2005;127:17134–17135. doi: 10.1021/ja056406z. [DOI] [PubMed] [Google Scholar]
- Angelo N. G., Arora P. S. J. Org. Chem. 2007;72:7963–7967. doi: 10.1021/jo701292h. [DOI] [PubMed] [Google Scholar]
- Jochim A. L., Miller S. E., Angelo N. G., Arora P. S. Bioorg. Med. Chem. Lett. 2009;19:6023–6026. doi: 10.1016/j.bmcl.2009.09.049. [DOI] [PubMed] [Google Scholar]
- Bone R., Vacca J. P., Anderson P. S., Holloway M. K. J. Am. Chem. Soc. 1991;113:9382–9384. [Google Scholar]
- Montagnat O. D., Lessene G., Hughes A. B. Tetrahedron Lett. 2006;47:6971–6974. [Google Scholar]
- Montagnat O. D., Lessene G., Hughes A. B. J. Org. Chem. 2010;75:390–398. doi: 10.1021/jo9021887. [DOI] [PubMed] [Google Scholar]
- Sola J., Bolte M., Alfonso I. Org. Biomol. Chem. 2015;13:10797–10801. doi: 10.1039/c5ob01461a. [DOI] [PubMed] [Google Scholar]
- Kee J. M., Villani B., Carpenter L. R., Muir T. W. J. Am. Chem. Soc. 2010;132:14327–14329. doi: 10.1021/ja104393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski M., Seaber B., Esslinger C. S. Bioorg. Med. Chem. Lett. 2007;17:4163–4166. doi: 10.1016/j.bmcl.2007.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley N. J., Pedersen D. S., Nielsen B., Kvist T., BräunerOsbrone H., Taylor D. K., Abell A. D. Bioorg. Med. Chem. Lett. 2010;20:7512–7515. doi: 10.1016/j.bmcl.2010.09.139. [DOI] [PubMed] [Google Scholar]
- Lipps H. J., Gruissem W., Prescott D. M. Proc. Natl. Acad. Sci. U. S. A. 1982;79:2495–2499. doi: 10.1073/pnas.79.8.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M., Lipsett M. N., Davies D. R. Proc. Natl. Acad. Sci. U. S. A. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. T. Angew. Chem., Int. Ed. 2004;43:668–698. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- Sundquist W. I., Klug A. Nature. 1989;342:825–829. doi: 10.1038/342825a0. [DOI] [PubMed] [Google Scholar]
- Burge S., Parkinson G. N., Hazel P., Todd A. K., Neidle S. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D. J., Phan A. T., Kuryavyi V. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui-Jain A., Grand C. L., Bearss D. J., Hurley L. H. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J. X., Dexheimer T. S., Chen D., Carver M., Ambrus A., Jones R. A., Yang D. Z. J. Am. Chem. Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J. X., Chen D., Jones R. A., Hurley L. H., Yang D. Z. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoi S., Xodo L. E. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin S., Reszka A. P., Huppert J., Zloh M., Parkinson G. N., Todd A. K., Ladame S., Balasubramanian S., Neidle S. J. Am. Chem. Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando H., Reszka A. P., Huppert J., Ladame S., Rankin S., Venkitaraman A. R., Neidle S., Balasubramanian S. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos K., Mita A., Ricart A., Hufnagel D., Northfelt D., Von Hoff D., Darjania L., Lim J., Padgett C., Marschke R. Mol. Cancer Ther. 2007;6:B93. [Google Scholar]
- Drygin D., Whitten J., Rice W. Cancer Res. 2008;68:3301. [Google Scholar]
- Reyes-Reyes E. M., Salipur F. R., Shams M., Forsthoefel M. K. Mol. Oncol. 2015;9:1392–1405. doi: 10.1016/j.molonc.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Brosh Jr R. M. Rev. Geophys. 2010;277:3470–3488. [Google Scholar]
- Saha P., Panda D., Dash J. Chem. Commun. 2019;55:731–750. doi: 10.1039/c8cc07107a. [DOI] [PubMed] [Google Scholar]
- Moorhouse A. D., Santos A. M., Gunaratnam M., Moore M., Neidle S., Moses J. E. J. Am. Chem. Soc. 2006;128:15972–15973. doi: 10.1021/ja0661919. [DOI] [PubMed] [Google Scholar]
- Moses J. E., Ritson D. J., Zhang F., Lombardo C. M., Haider S., Oldham N., Neidle S. Org. Biomol. Chem. 2010;8:2926–2930. doi: 10.1039/c005055e. [DOI] [PubMed] [Google Scholar]
- Lombardo C. M., Martinez I. S., Haider S., Gabelica V., Pauw E. D., Moses J. E., Neidle S. Chem. Commun. 2010;46:9116–9118. doi: 10.1039/c0cc02917c. [DOI] [PubMed] [Google Scholar]
- Ritson D. J., Moses J. E. Tetrahedron. 2012;68:197–203. [Google Scholar]
- Drewe W. C., Neidle S. Chem. Commun. 2008:5295–5297. doi: 10.1039/b814576h. [DOI] [PubMed] [Google Scholar]
- Methods in Molecular Biology, ed. P. Herdewijn, Oligonucleotide Synthesis: Methods and Applications, Human Press Inc, Totowa, NJ, 2005, vol. 288. [Google Scholar]
- Buchini S., Leumann C. J. Curr. Opin. Chem. Biol. 2003;7:717–726. doi: 10.1016/j.cbpa.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Luyten I., Herdewijn P. Eur. J. Med. Chem. 1998;33:515–576. [Google Scholar]
- Guianvarc'h D., Fourrey J. L., Maurisse R., Sun J. S., Benhida R. Bioorg. Med. Chem. 2003;11:2751–2759. doi: 10.1016/s0968-0896(03)00229-3. [DOI] [PubMed] [Google Scholar]
- Obika S., Hari Y., Morio K., Imanishi T. Tetrahedron Lett. 2000;41:221–224. [Google Scholar]
- Prasher P., Sharma M. Med. Chem. Commun. 2018;9:1589–1618. doi: 10.1039/c8md00384j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara M., Kuboyama T., Izawa A., Hari Y., Imanishi T., Obika S. Bioorg. Med. Chem. Lett. 2009;19:3316–3319. doi: 10.1016/j.bmcl.2009.04.063. [DOI] [PubMed] [Google Scholar]
- Chittepu P., Sirivolu V. R., Seelu F. Bioorg. Med. Chem. 2008;16:8427–8439. doi: 10.1016/j.bmc.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Nuzzi A., Massi A., Dondoni A. QSAR Comb. Sci. 2007;26:1191–1199. [Google Scholar]
- Shivalingam A., Tyburn A. E. S., El-Sagheer A. H., Brown T. J. Am. Chem. Soc. 2017;139:1575–1583. doi: 10.1021/jacs.6b11530. [DOI] [PubMed] [Google Scholar]
- El-Sagheer A. H., Brown T. Chem. Commun. 2011;47:12057–12058. doi: 10.1039/c1cc14316f. [DOI] [PubMed] [Google Scholar]
- Walczak S., Nowicka A., Kubacha D., Fac K. Chem. Sci. 2017;8:260–267. doi: 10.1039/c6sc02437h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Leonard P., Muller S. L., Daniliuc C., Seela F. Beilstein J. Org. Chem. 2018;14:2404–2410. doi: 10.3762/bjoc.14.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemamalini A., Das T. M. RSC Adv. 2014;4:34189–34198. [Google Scholar]
- Weller R. L., Rajski S. R. Org. Lett. 2005;7:2141–2144. doi: 10.1021/ol0504749. [DOI] [PubMed] [Google Scholar]
- Huggins D. J., Sherman W., Tidor B. J. Med. Chem. 2012;55:1424–1444. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Voet A., Zhang K. Y. J. Curr. Med. Chem. 2012;19:5128–5147. doi: 10.2174/092986712803530467. [DOI] [PubMed] [Google Scholar]
- Poulin-Kerstien A. T., Dervan P. B. J. Am. Chem. Soc. 2003;125:15811–15821. doi: 10.1021/ja030494a. [DOI] [PubMed] [Google Scholar]
- Shen Q., Tang S., Li W., Nie Z., Liu Z., Huang Y., Yao S. Chem. Commun. 2012;48:281–283. doi: 10.1039/c1cc16049d. [DOI] [PubMed] [Google Scholar]
- Ohkanda J. Chem. Rec. 2013;13:561–575. doi: 10.1002/tcr.201300026. [DOI] [PubMed] [Google Scholar]
- Mock W. L. J. Org. Chem. 1983;48:3619–3620. [Google Scholar]
- Mock W. L. J. Org. Chem. 1989;54:5302–5308. [Google Scholar]
- Mock W. L. Top. Curr. Chem. 1995;175:1–24. [Google Scholar]
- Lewis W. G., Green L. G. Angew. Chem., Int. Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Krasiński A., Radić Z., Manetsch R., Raushel J., Taylor P., Sharpless K. B., Kolb H. C. J. Am. Chem. Soc. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- Bourne Y., Radić Z., Taylor P., Marchot P. J. Am. Chem. Soc. 2010;132:18292–18300. doi: 10.1021/ja106820e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimster N. P., Stump B., Fokin V. V. J. Am. Chem. Soc. 2012;134:6732–6740. doi: 10.1021/ja3001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M., Salmon A., Supuran C., Poulsen S. A. Curr. Pharm. Des. 2010;16:3277–3287. doi: 10.2174/138161210793429869. [DOI] [PubMed] [Google Scholar]
- Mocharla V. P., Colasson B., Lee L. V., Röper S., Sharpless K. B., Wong C. H., Kolb H. C. Angew. Chem., Int. Ed. 2005;44:116–120. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- Prasher P., Mudila H., Sharma M., Khati B. Med. Chem. Res. 2019;28:417–449. [Google Scholar]
- Bhardwaj A., Kaur J., Wuest M., Wuest F. Nat. Commun. 2017;8:1. doi: 10.1038/s41467-016-0009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzotti C., Borrelli S., Ventura M. ACS Med. Chem. Lett. 2013;4:274–277. doi: 10.1021/ml300394w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting M., Muldoon J. Angew. Chem., Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- Hirose T., Sunazuka T., Omura S. Yuki Gosei Kagaku Kyokaishi. 2016;74:1090–1097. [Google Scholar]
- Tieu W., da Costa T. P. S., Yap M. Y. Chem. Sci. 2013;4:3533–3537. [Google Scholar]
- Toguchi S., Hirose T., Yorita K., Fukui K. Chem. Pharm. Bull. 2016;64:695–703. doi: 10.1248/cpb.c15-00867. [DOI] [PubMed] [Google Scholar]
- Spasser L., Brik A. Angew. Chem., Int. Ed. 2012;51:6840–6862. doi: 10.1002/anie.201200020. [DOI] [PubMed] [Google Scholar]
- Lang K., Chin J. W. Chem. Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Marks K. M., Nolan G. P. Nat. Methods. 2006;3:591–596. doi: 10.1038/nmeth906. [DOI] [PubMed] [Google Scholar]
- Vila-Perello M., Muir T. W. Cell. 2010;143:191–200. doi: 10.1016/j.cell.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing C. R., Cornish V. W. Acc. Chem. Res. 2011;44:784–792. doi: 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I., Howarth M., Lin W., Ting A. Y. Nat. Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- Carrico I. S., Carlson B. L., Bertozzi C. R. Nat. Chem. Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- Lin C. W., Ting A. Y. J. Am. Chem. Soc. 2006;128:4542–4543. doi: 10.1021/ja0604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. W., Ting A. Y. J. Am. Chem. Soc. 2006;128:4542–4543. doi: 10.1021/ja0604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Suarez M., Baruah H., Martinez-Hernandez L., Xie K. T., Baskin J. M., Bertozzi C. R., Ting A. Y. Nat. Biotechnol. 2007;25:1483–1487. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich D. C., Hodas J. J., Gouzer G., Shadrin I. Y., Ngo J. T., Triller A., Tirrell D. A., Schuman E. M. Nat. Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty K. E., Fisk J. D., Smart B. P., Lu Y. Y., Szychowski J., Hangauer M. J., Baskin J. M., Bertozzi C. R., Tirrell D. A. ChemBioChem. 2010;11:2092–2095. doi: 10.1002/cbic.201000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Dove A. Nat. Biotechnol. 1999;17:233–236. doi: 10.1038/6972. [DOI] [PubMed] [Google Scholar]
- Pandey A., Mann M. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- Jessani N., Liu Y., Humphrey M., Cravatt B. F. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10335–10340. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J., Lee K.-J. J. Biochem. Mol. Biol. 2004;37:35–44. doi: 10.5483/bmbrep.2004.37.1.035. [DOI] [PubMed] [Google Scholar]
- Kobe B., Kemp B. E. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- Jiang H., Kim J. H., Frizzell K. M., Kraus W. L., Lin H. J. Am. Chem. Soc. 2010;132:9363–9372. doi: 10.1021/ja101588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. P., Raghavan A. S., Yang Y. Y., Charron G., Hang H. C. Mol. Cell. Proteomics. 2011;10:M110.001198. doi: 10.1074/mcp.M110.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M. E., Deschenes R. J. Nat. Rev. Mol. Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Yount J. S., Moltedo B., Yang Y. Y., Charron G., Moran T. M., Lopez C. B., Hang H. C. Nat. Chem. Biol. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Patricelli M. P., Cravatt B. F. Proc. Natl. Acad. Sci. U. S. A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields D. J., Niessen S., Murphy E. A., Mielgo A., Desgrosellier J. S., Lau S. K. M., Barnes L. A., Lesperance J., Bouvet M., Tarin D., Cravatt B. F., Cheresh D. A. Proc. Natl. Acad. Sci. U. S. A. 2010;107:2189–2194. doi: 10.1073/pnas.0911646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N., Cravatt B. F. Curr. Opin. Chem. Biol. 2004;8:54–59. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Speers A. E., Cravatt B. F. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Sato B., Muramatsu H., Miyauchi M., Hori Y., Takase S., Hino M., Hashimoto S., Terano H. J. Antibiot. 2000;53:123–130. doi: 10.7164/antibiotics.53.123. [DOI] [PubMed] [Google Scholar]
- Adam G. C., Vanderwal C. D., Sorensen E. J., Cravatt B. F. Angew. Chem., Int. Ed. 2003;42:5480–5484. doi: 10.1002/anie.200352576. [DOI] [PubMed] [Google Scholar]
- Speers A. E., Adam G. C., Cravatt B. F. J. Am. Chem. Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- Ovaa H., van Swieten P. F., Kessler B. M., Leeuwenburgh M. A., Fiebiger E., van den Nieuwendijk A. M., Galardy P. J., van der Marel G. A., Ploegh H. L., Overkleeft H. S. Angew. Chem., Int. Ed. 2003;42:3626–3629. doi: 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]