

ABSTRACT
Deamidation evaluation and mitigation is an important aspect of therapeutic antibody developability assessment. We investigated the structure and function of the Asn-Gly deamidation in a human anti-CD52 IgG1 antibody light chain complementarity-determining region 1, and risk mitigation through protein engineering. Antigen binding affinity was found to decrease about 400-fold when Asn33 was replaced with an Asp residue to mimic the deamidation product, suggesting significant impacts on antibody function. Other variants made at Asn33 (N33H, N33Q, N33H, N33R) were also found to result in significant loss of antigen binding affinity. The co-crystal structure of the antigen-binding fragment bound to a CD52 peptide mimetic was solved at 2.2Å (PDB code 6OBD), which revealed that Asn33 directly interacts with the CD52 phosphate group via a hydrogen bond. Gly34, but sits away from the binding interface, rendering it more amendable to mutagenesis without affecting affinity. Saturation mutants at Gly34 were prepared and subjected to forced deamidation by incubation at elevated pH and temperature. Three mutants (G34R, G34K and G34Q) showed increased resistance to deamidation by LC-MS peptide mapping, while maintaining high binding affinity to CD52 antigen measured by Biacore. A complement -dependent cytotoxicity assay indicated that these mutants function by triggering antibody effector function. This study illustrates the importance of structure-based design and extensive mutagenesis to mitigate antibody developability issues.
KEYWORDS: Antibody engineering, deamidation, mutagenesis, developability, structure-function
Introduction
The successful development of therapeutic antibodies depends on the evaluation of many factors, including potential physical and chemical degradations, as part of a developability assessment and any subsequent mitigation.1 Identifying and engineering antibodies that are stable throughout expression, purification, formulation, storage and distribution are critical for cost reduction and quality control of therapeutic manufacturing and in vivo pharmacokinetics and potency.2–5 The concept of developability assessments for therapeutic proteins has gained attention in recent years, with substantial effort focused on the identification, understanding, and mitigation of specific liabilities. These liabilities include chemical degradations such as oxidation, deamidation, isomerization, and fragmentation.6–10
Deamidation of asparagine (Asn) residues is a major post-translational modification that can significantly impact protein structure and function.11,12 The non-enzymatic modification proceeds via formation of a five-member ring succinimide intermediate, which is subsequently hydrolyzed into a mixture of isoaspartate (isoAsp) and aspartate (Asp). The intermediate and the final products formed in the deamidation process lead to mass change, charge heterogeneity, and structural alteration of the peptide backbone in the case of isoAsp formation.13,14 Multiple factors have been identified that affect the rate of asparagine deamidation. With regard to the primary sequence, deamidation rates depend on the amino acid residues adjacent to Asn in the peptide chain, with Gly and Ser the most destabilizing C-terminal amino acids.12 The rate of deamidation also depends on external factors, increasing with elevated temperature and pH in general. More evidence suggests that deamidation can also happen at non-canonical sequences, when asparagine is followed by other amino acids.7,15 The three-dimensional structure has been found to be another major determining factor,12,14,16 but a detailed understanding of the correlation has been largely lacking. Protein interaction is also found to affect deamidation. For example, an isoaspartate product has been detected in the complementarity-determining region (CDR) region of a free antibody, but was completely missing in the antibody-antigen complex.17
Potential deamidation sites in the CDR regions in recombinant monoclonal antibodies have been the subject of many studies, due to continuous demonstration of its impact on antibody functions.17,18 The loops in antibody CDRs are typically flexible, and thus could be subject to deamidation. Multiple reports have described Asn deamidation in CDRs of monoclonal antibodies and their impacts in detail19–26 Sydow et al. examined a collection of 37 different therapeutic IgG1, IgG2 and IgG4 mAbs and found that deamidation was only detected in the CDR regions, and when Asn was followed by a Gly, Ser, Thr, or Asn.27 Recently, Lu et al. analyzed the deamidation and isomerization liability of 131 clinical-stage antibodies and further established that deamidation occurs in CDR regions at non-canonical motifs including NN, NF, NQ, NH, NW, NY and NF.7 It remains unclear as to why deamidation frequently occurs in these non-canonical motifs, but the authors suggest that protein conformation and local structural context could be major contributors. It further emphasizes the challenges of sequence-based approaches for liability prediction, and highlights a knowledge gap in the field.
Robinson et al. have developed a computational method for the quantitative estimation of the deamidation rates for any protein of which the three-dimensional structure is known.15,28 They concluded that most deamidation rates are determined by the primary sequence and modulated by three-dimensional structures. Sinha et al. demonstrated experimentally that the higher-order structure influences both the rate of Asn-deamidation and the product distribution in antibody crystallizable fragments (Fcs).29
This work addresses the structure and functional aspects of an Asn-Gly (NG) deamidation site found in the light chain CDR1 region of an anti-CD52 antibody (MAB1). We studied the effects of deamidation at this site, and determined the X-ray structure of a peptide antigen mimic complexed with the antibody antigen-binding fragment (Fab) to examine the structure-function correlations. Engineered variants were generated based on the structure at the Asn-Gly deamidation site, and characterized for their stability, antigen binding affinity, and cell-based effector function. This led to successful removal of the deamidation site while retaining its biological function, and elucidated the structure-based impact and mechanisms of deamidation.
Results
MAB1 loss of binding paralleled the level of deamidation
Peptide mapping was performed in parallel with Biacore binding for anti-CD52 antibody MAB1 to monitor the impact of deamidation. This was done after incubating the antibody at elevated pH (7.0, 7.5 and 8.0) and temperature (37°C) to monitor deamidation and its impact on function. Site-specific deamidation in the peptide containing the light chain CDR1 Asn33-Gly34 site was found to increase with incubation time and elevated pH (Figure 1A). Size-exclusion chromatography- high performance liquid chromatography (SEC-HPLC) analysis suggests MAB1 remained monomeric, and there was no detectable aggregation during the incubation. Deamidation was also observed at the Asn 381 position on the PENNY peptide in the Fc domain (data not shown). Biacore analysis was performed to determine the binding affinity of the incubated samples, which demonstrated that the loss of MAB1 binding paralleled the level of deamidation at this Asn-Gly site (Figure 1B), suggesting a possible connection of CDR deamidation with affinity loss.
Figure 1.
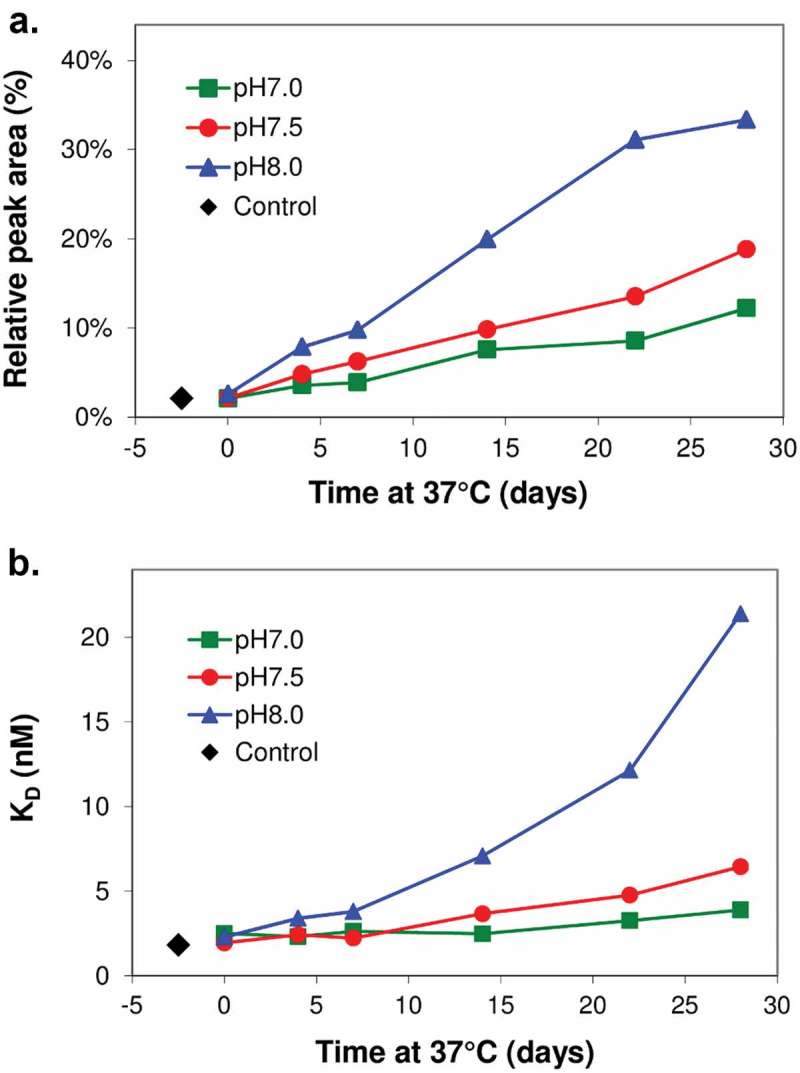
Loss of binding paralleled the level of deamidation. MAB1 was dialyzed into buffers at pH 7.0, 7.5 and 8.0 (3 component buffer containing 10 mM succinate, 10 mM histidine, and 10 mM sodium phosphate), diluted to 0.5 mg/mL and incubated at 37°C for 4 weeks. Aliquots were taken at various time points as for deamidation analysis by peptide mapping with mass spectrometry analysis (A) and Biacore binding affinity (B).
N33D mutant to mimic the deamidation product
The Asn33 to Asp33 mutation (N33D) in the CDR1 region light chain was created as a mimic of the deamidation product to assess the effect of deamidation at this site. The purified N33D mutant eluted as a single peak by SEC-HPLC, and had the correct size by SDS-PAGE, which was confirmed by mass spectrometry (MS). Biacore binding to the CD52 peptide was then performed to assess the function of the mutant. No detectable binding signal was observed for the N33D mutant at 7.5 nM antibody concentration, where wild-type antibody displayed high binding affinity (Figure 2A). Higher concentrations of N33D mutant (up to 1920 nM) were used in an effort to determine quantitatively the loss of binding affinity of the mutant. Kinetic and equilibrium fit of the binding data resulted in a KD of 1.25 μM, which represents more than a 400-fold drop in affinity compared to wild-type (Figure 2B and C). The decrease in affinity is reflected in both a reduced on-rate and an increased off-rate in the binding kinetics. This result indicates that deamidation at this light chain CDR1 site leads to significant loss of its biological function as measured by its antigen binding affinity.
Figure 2.
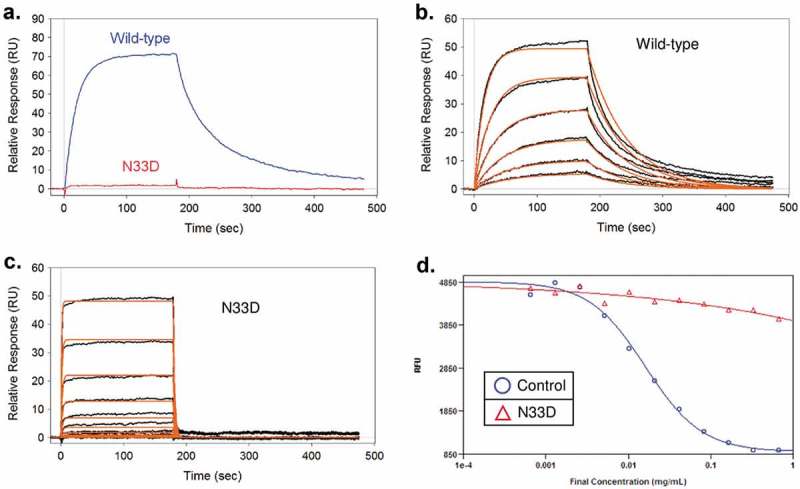
N33D mutation has significantly reduced antigen binding affinity and CDC effector function. (A) Biacore sensorgrams of 7.5 nM wild-type and N33D mutant binding to immobilized CD52 peptide mimic. (B and C) Binding sensorgrams and global fit using a 1:1 binding model to obtain the binding constants. For wild-type (panel B), 0.2–7.5 nM antibody were used; for N33D (panel C), 0.2–1920 nM antibody were used. (D) CDC assay to test the effector function of the N33D mutant.
The complement-dependent cytotoxicity (CDC) assay was performed to establish the link between the affinity loss of the N33D mutant to its peptide antigen and its biologic function. This assay measures the ability of an antibody to induce lysis of the receptor-positive lymphocytes. Complement-initiated cell lysis of CD52-expressing Pfeiffer B-lymphocytes was monitored with increasing concentration of anti-CD52 wild-type and mutant antibodies. The result (Figure 2D) indicated that the N33D mutant is deficient in the CDC assay. While the wild-type antibody control effectively induces cell lysis with an EC50 of 10–20 µg/ml concentration, no significant cell lysis was observed at the highest concentration of N33D mutant tested (1 mg/mL).
Structural analysis of antibody antigen interactions by x-ray crystallography
An X-ray crystallographic study was performed to understand the structural basis for the antibody–antigen interaction. Wild-type Fab was prepared by overnight papain digestion of the MAB1 IgG1 followed by Mono S cation exchange chromatography. The concentrated protein was mixed with a 10-fold molar excess of phosphoethanolamine (ETP) CD52 peptide mimic (GQNDTSQTSSPS-Eta-PO4). The CD52 peptide mimetic contains the entire human CD52 peptide sequence, as well as the ethanolamine phosphate moiety to mimic the beginning part of the glycosylphosphatidylinositol (GPI) anchor present in the native glycopeptide on immune cells. The crystal structure of MAB1 Fab complexed to CD52-ETP peptide was solved at 2.2Å by molecular replacement using PDB code 1FL5 as a search model (Figure 3 and Supplementary Table 1). The CD52 peptide was manually built into the density and the complete model was refined. The structure identified the residues on the antibody CDRs that are in direct contact with the CD52 peptide. CDR1 and 3 of the heavy chain and CDR1 and 2 of the light chain are involved in CD52 binding. On the antigen side, the CD52-ETP peptide was bent 180° at the “SSP” region and formed an extensive hydrogen bonding network with MAB1 at the “QTS” sequence and the phosphate group of the GPI anchor. At the CDR1 deamidation site, the structure reveals two direct interactions between the light chain CDR1 of the antibody and the phosphate group of CD52. The Asn33 of light chain CDR1 forms hydrogen bonds with the phosphate oxygen and Lys35 forms ionic interaction with another phosphate oxygen. This newly-identified interaction between Asn33 and CD52 provides an explanation for the observation that when Asn33 is substituted with an Asp (N33D) to mimic the deamidation product, the affinity is almost completely lost due to charge repulsion (Figure 2). Another important observation from the structure is that, while Asn33 is making critical interactions with CD52 peptide, Gly34 sits away from the interaction interface and could be much more amendable to mutagenesis without affecting the interaction.
Figure 3.
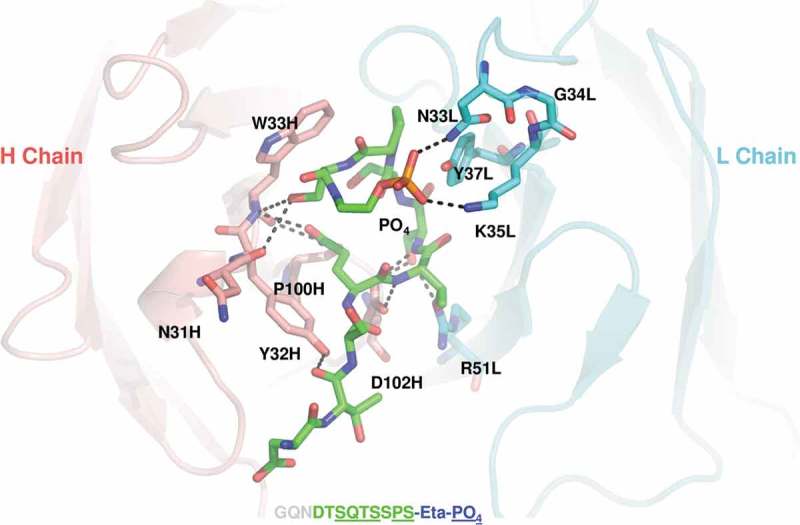
Crystal structure of the antigen-binding fragment of wild-type antibody complex to a CD52 peptide mimic. Detailed view shows interface of peptide (green carbon atoms, orange phosphorus) and CDRs in heavy chain (Salmon) and light chain (Cyan), with all visible peptide residues and paratope residues in sticks. Two polar interactions between phosphate on peptide and N33 and K35 in CDRL1 are labeled. Paratope residues are annotated as residue name, residue number, followed by chain name.
Asn33 mutants to remove deamidation sequence
In parallel with structural determination, multiple mutants were designed and prepared to replace Asn33 to remove the deamidation site and to assess the impact on antigen binding. These mutants include N33Q, N33H, N33R, N33T, N33R, N33K, and N33Y. The mutants are either consevative (N33Q), or have side chains that could potentially replace the hydrogen bonding between Asn33 and the peptide antigen observed in the crystal structure. One mutant substituting the wild-type Asn33-Gly34-Lys35(NGK) with a reversed Lys33-Gly34-Asn35 (designated as “KGN” mutant) was also prepared and evaluated to see whether changing the amino acid sequence order can retain the binding in the tightly-knitted binding interface. Conditioned HEK293 media from transient transfection were checked for expression with protein A biosensors using Octet, and their CD52 binding was measured by Biacore (Supplementary Figure 1). The result suggests that all substitutions at Asn33, including the conservative N33Q substitution and the sequence reversed KGN mutant, lead to significantly reduced CD52 binding affinity. This is consistent with the observation in the crystal structure that Asn30 makes critical interactions with CD52 peptide.
Mutation design and preparation at the light chain Gly34
Light chain Gly34 was subjected to mutagenesis based on the structural studies. In order to obtain light chain mutants with maximum amino acid representation at this position, a randomized NNK oligonucleotide mixture was used for saturation mutagenesis to generate all possible variants at the Gly34 position. The conditioned media of transiently expressed Gly34 mutants was screened for expression level and CD52 binding affinity. All of these mutants were found to express at similar levels. The antigen binding affinity was semi-quantitatively ranked from the media using the rapid transfection and screening procedure. The affinities of the G34 mutants were ranked in the following order: G34K ≈ G34R > Wild-type > (G34S, G34Q, G34H, G34D, G34Y, G34A) > other mutants (Figure 4). Mutants with the most-preserved affinity (solid bars in Figure 4A) were scaled up for transfection and purified to homogeneity for further characterization. The CD52 antigen-binding affinity of the purified Gly34 mutants was determined by Biacore and are summarized in Table 1. The rank order of the affinity agrees with the result from the media screening: G34K and G34R mutants have higher affinity than the wild-type; and the rest of the selected mutants (G34S, G34Q, G34H, G34K, G34Y, and G34A) have comparable or slightly lower affinity.
Figure 4.
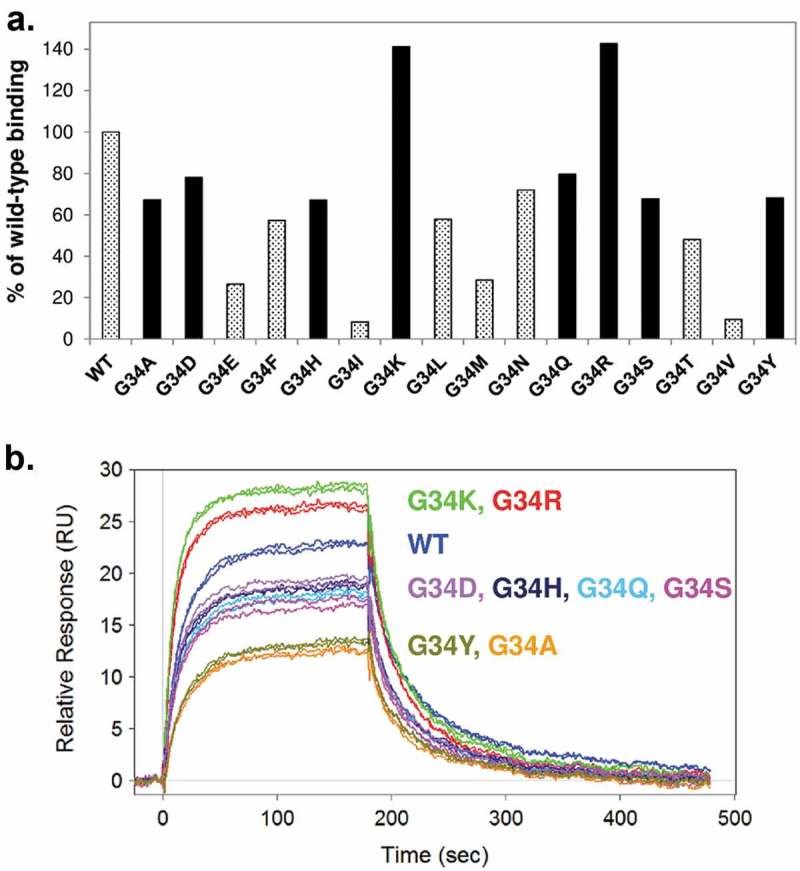
Affinity characterization of Gly34 mutants. (A) HEK293 conditioned media expressing the Gly34 mutants were first screened by Octet and Biacore. Binding response of 0.5 nM mutants were compared to wild-type and presented as % of wild-type binding at the end of the 3-minute injection. Mutants with highest affinity (solid bars) were selected for further analysis. (B) Biacore binding sensorgrams of selected purified mutants. Duplicate injections of 7.5 nM antibody are presented for comparisons.
Table 1.
Biacore CD52 binding affinity (KD, nM) of wild-type and mutant antibodies before and after forced deamidation incubation.
| WEEK 0 | WEEK 2 | WEEK 4 | |
|---|---|---|---|
| 37°C | |||
| Wild-type | 4.20 | 8.9 | 18.3 |
| G34H | 6.20 | 11.3 | 15.6 |
| G34R | 3.30 | 4.6 | 5.1 |
| G34Q | 6.10 | 8.8 | 10.2 |
| G34K | 3.30 | 4.5 | 4.8 |
| G34A | 7.40 | 17.7 | 25.6 |
| G34Y | 7.00 | 10.5 | 11.3 |
| G34S | 7.30 | 20.5 | 42.5 |
| 45°C | |||
| Wild-type | 4.30 | 28 | 1230 |
| G34K | 3.30 | 5.3 | 8.9 |
| G34R | 3.30 | 5.8 | 9.3 |
| G34Q | 6.10 | 11.3 | 16.1 |
Stability characterization of G34 mutants after accelerated deamidation conditions
Selected Gly34 mutants were purified and further studied with elevated temperature (37°C or 45°C) and pH (pH 7.5) to test their deamidation susceptibility. Aliquots of the mutant antibodies were taken before the incubation (T0) and at week 2 and week 4 (T2 and T4) to assess their affinities for CD52 peptide by Biacore and its correlation with deamidation level. An ~ 3–4-fold decrease in affinity at 37°C was observed for the wild-type antibody (Figure 5), presumably due to deamidation at the light chain CDR deamidation site. The G34A and G34S mutants also demonstrated a large decrease in affinity after the 4-week incubation at 37°C (3.5-fold and 5.8-fold, respectively, see Table I). G34R, G34K, and G34Q, however, largely retained their antigen binding affinity after the 4-week incubation (less than 1.5-fold change in KD). These three stable mutants were further tested for accelerated deamidation at 45°C in the same buffer (Figure 5). This result confirms the stability of these three mutants, which all retained their binding affinity with KD ˂10 nM after the incubation. The wild-type control, on the other hand, lost most of its binding affinity for CD52 peptide by Biacore under the forced deamidation condition, with a KD change from 4.3 nM to 1230 nM (Table 1). The degree of stabilization seems to be comparable among the three mutants, although the G34Q mutant has a lower baseline affinity. SEC-HPLC analysis of the post-incubation samples suggests no sign of aggregation or degradation of either the wild-type or mutant antibodies (data not shown).
Figure 5.
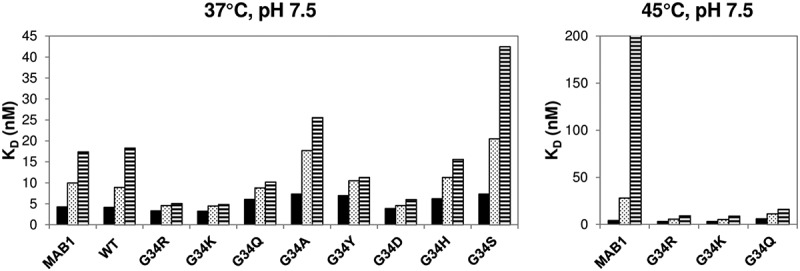
Prolonged incubation at elevated pH and temperature was performed to test deamidation stability by selected mutants. The result of incubation at 37°C is presented in the left panel with the three most stable mutants highlighted with arrows. The result at 45°C is presented in the right panel.
Selected incubation samples at time 0 and 4 weeks at 45°C were analyzed by LC-MS peptide mapping to monitor the deamidation level on the peptide corresponding to the light chain CDR1 Asn33Gly34 deamidation site. The three mutants that retained affinity by Biacore analysis (G34R, G34K, and G34Q) were tested and compared with the wild-type control. The samples were digested using trypsin and Endo Lys C, and the digested samples were subjected to LC-MS peptide mapping. The peptide containing the light chain deamidation site, SSQSLLYSNGK (designated as peptide LT2), was identified for detailed analysis. The isoDG LT2 peptide, one of the deamidation products, was found to co-elute with the wild-type NG LT2 peptide. Only the DG LT2 peptide, which is the other deamidation product, elutes at a different retention time. Since deamidation of NG to both isoDG and DG leads to a one Dalton increase in mass, isotopic mass distribution was used to estimate the amount of deamidated LT2 peptides (DG LT2 + isoDG LT2) in the samples by comparing the shift from the first isotope N to the second N + 1 peak (Figure 6). In the case of wild-type, this corresponds to the shift of the 592 to the 593 m/z. We estimated that greater than 90% deamidation occurred on wild-type LT2 peptide. The G34R, G34Q and G34K mutants, on the other hand, showed a significantly reduced amount of deamidation at Asn33 under this accelerated deamidation condition, as indicated by the reduced shift to N + 1 peak. There is an additional Lys C cleavage site introduced by the G34K mutation in the middle of the original LT2 peptide to generate LT2′ and the incompletely digested LT2′-LT3′, which were both monitored (Figure 6B). The level of observed deamidation in these force-deamidated samples can be ranked as follows: wild-type ⋙ G34K > G34Q > G34R. We conclude from these sets of experiments that these three engineered variants are much more stable compared to wild-type MAB1, due to increased resistance to deamidation.
Figure 6.
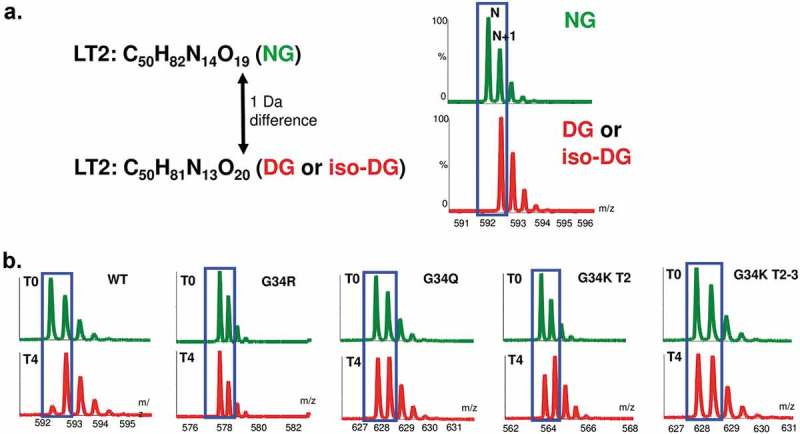
LC-MS peptide mapping experiments to monitor specifically the peptide corresponding to the light chain CDR1 Asn33Gly34 deamidation site. The peptide containing the light chain deamidation site -SSQSLLYSNGK-(designated as LT2 peptide) was identified for detailed analysis. (A) Deamidation of LT2 NG to both isoDG and DG leads to one Dalton increase in mass. (B) Isotopic mass distribution was used to estimate the amount of deamidated LT2 peptides (DG LT2 + isoDG LT2) in the samples by comparing the shift from the first isotope N to the second N + 1 peak. Mutants were found to be much more resistant to deamidation than the control.
Stability mutants are biologically functional in a complement-dependent cytotoxicity assay
The three mutants (G34 R, G34Q and G34K) with increased stability were further evaluated in the CDC assay to study whether the amino acid changes in the CDR region could affect the binding to CD52 on the cell surface, and thus impact their effector function. The results show concentration-dependent cell lysis for wild-type and the three selected mutants (Supplementary Figure 2). G34Q had slightly reduced potency in this assay, which is in agreement with the lower antigen binding affinity observed in the Biacore binding assay. The potency of G34R and G34K were comparable to the wild-type control.
DISCUSSION
Deamidation in the CDR regions of an antibody could lead to loss of function, and thus has been a subject of many studies in the literature.7,31,32 These reports have begun to elucidate the mechanisms underlying deamidations and chemical modifications, as well as their effects. However, substantial gaps still exist in the understanding of the determinants of deamidation, and how the change will affect antibody structure and function. The mutagenesis, structure and function studies described here represent how we elucidated the critical functional residues in our mAb, and used this information to engineer out the liability in the CDR. This approach may have application for other antibodies where the crystal structure can be determined and similar mutagenesis studies carried out.
The asparagine deamidation in the light chain CDR1 (L1) of MAB1 reported here is consistent with the recent report by Xu et al. that examined the deamidation liability of 131 clinical-stage mAbs.7 They found that, among this large set of mAbs, relatively high frequencies of deamidation events were observed in CDRs H2 and L1, with CDR L1 accounting for 81.8% (18/22) of all observed light chain deamidation events. Our work, together with other reports,7,27,33 indicates that asparagines in the light chain CDR1 region have a higher risk of deamidation, likely due to local structure and conformation.
To better understand this observation, we have reviewed both the sequences of human Vkappa and Vlambda germlines and CDRL1 conformations in published antibody structures (MOE, version 2018.0101). The length of CDRL1 from 1920 non-redundant structurally known antibodies appears bimodal, featuring 11 amino acids (44.7%) and 16 amino acids (13.9%) as the two most popular CDRL1 lengths (Figure 7A). In addition, CDRL1 is commonly stabilized by a hydrophobic residue (I, V, L) that inserts into the hydrophobic core between the two beta sheets in VL domain, and its conformation can be grouped to two major populations, with shorter loops simply spanning the Ig domain and longer loops forming an additional hairpin structure after the stabilizing hydrophobic residues (Figure 7B). CDRL1 in the MAB1 structure has 16 amino acids, and the NG motif sits at the tip of the hairpin. A 50 ns molecular dynamics of MAB1 Fab in the absence of peptide shows NG motif has highest flexibility in the VL domain. As a result, we suspect the structural flexibility of the NG motif in the long CDRL1 loop in MAB1 is the root cause of deamidation observed in stress studies. Furthermore, it is not rare to find long CDRL1 in many kappa and lambda germlines with a NG, NA, or NT motif in the hairpin structured region, which poses a risk of deamidation-based liability. This provides the structural explanation of the deamidation liability report of 131 mAbs where CDR L1 accounted for the majority of their observed light chain deamidation events and 6 of 8 NG motifs were deamidated.7 It is worth noting that all these 8 NG motifs are in a 16-amino acid CDRL1 and position equivalent to Asn33Gly34 in MAB1. It would be interesting to map each count of deamidation onto corresponding CDRL1 sequences and explore descriptors for any potential correlation.
Figure 7.
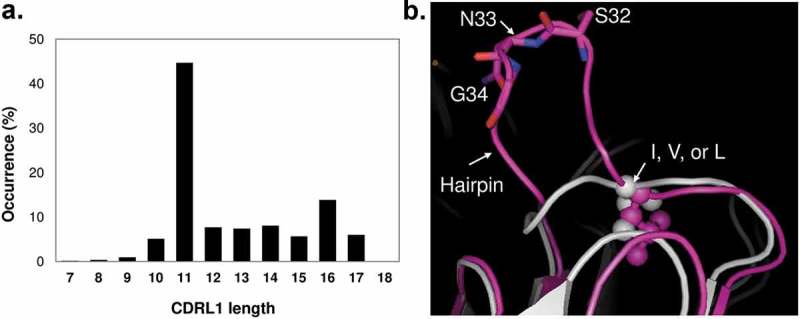
Length distribution of CDRL1 in 1920 non-redundant published antibody structures and representative conformations of CDRL1 in two most popular lengths. CDRL1 in MAB1 in pink represents the 16 amino acid population, and CDRL1 from PDB 3SOB in light grey for 11 amino acid. The position conserved hydrophobic residues, L in MAB1 and V in 3SOB, are shown in stick and sphere.
Although often considered conservative, the deamidation rate of Gln is much slower than that of Asn.33 It was therefore a common practice to remove a deamidation site by mutating the Asn to an Gln, which is otherwise a conservative mutation.34 However, in our case, the mutagenesis and binding results indicate that Gln or Asp substitution of Asn33 abolishes its antigen binding affinity and CDC effector function (Figure 2, Supplementary Figure 1). The structure of the antibody-antigen complex (Figure 3) provided a clear explanation as to why it is an exception (due to steric clash with one carbon longer on the side chain). This also explains why deamidation products at Asn33 positions (isoAsp and Asp) have significantly reduced affinity. A change of Asn to Asp at this position would not only disrupt the hydrogen bonding between Asn33 and the phosphate on the peptide, but also create charge repulsion between Asp33 in the mutant and the phosphate group on CD52 at the binding site. In addition, the side chain of Lys35, two amino acids after which forms a salt bridge with the phosphate group in the co-crystal structure, would potentially form a salt bridge with the engineered Asp33 in the mutant, and thus lose the interaction with CD52. Taken together, these data may explain the more than 400-fold decrease in affinity by N33D compared to wild-type (Figure 2). The structure would predict that an isoAsp substitution at this position, which is the other potential deamidation product, would also destroy this antigen-binding interface due to this charge repulsion. In addition, a kink would be created in the peptide backbone, which would disrupt the conformational integrity of this critical binding region. N33R and N33K mutants appear to retain some residual binding affinity. One possible explanation for this is that the positively charged and flexible side chains in these two mutants help to preserve some binding by ionic interactions with the phosphate groups in the peptide mimic. However, the Lys33Gly34Asn35 (KGN) mutant, which has the same amino acids in the binding site but with reversed order, has little to no binding by Biacore, again indicating this binding interface is a very tight fit.
The amino acids that immediately follow an Asn, within the primary sequence, are considered an important determining factor for the propensity to deamidate and, accordingly, NG, NS, and NT are commonly recognized as consensus deamidation sites that enable the flexibility needed for the formation of the deamidation intermediate. Replacing the Gly with amino acids with larger side chains would result in a less flexible peptide bond between amino acid 33 and 34, and thus would be more resistant to deamidation. All Gly34 mutants retain some degree of CD52 antigen binding affinity (Figure 4), which is consistent with the observation that the Cα of Gly34 faces away from the CD52 peptide in the crystal structure. It is not clear, however, how the nature of amino acid substitution at this position would affect the local structure at the binding site and the binding affinity. Two Gly34 substitutions with bulky and hydrophobic side chains, valine and isoleucine (G34V and G34I), which are favored substitutions for removing a deamidation site based on peptide-based deamidation studies, lead to significantly reduced binding affinity in our case (Figure 4).35 The G34K and G34R mutants, however, were found to have moderately increased the binding affinity as measured by binding to immobilized CD52 peptide on Biacore. We suspected that the negatively charged carboxydextran (CM) Biacore chip surface may contribute partially to the high affinities of these two mutants with artificial charge-charge attractions. However, the CDC effector function assay result (Supplementary Figure 2) indicated that these two mutants have potency comparable to or slightly higher than the wild-type antibody, which agrees with the Biacore measurement. This confirms that the increased affinity and potency is caused by the Gly34→Arg34 and Gly34→Lys34 substitutions, respectively.
Nakana et al. reported a similar deamidation site at an Asn-Gly sequence in light chain CDR1 in their humanized anti-glypican 3 antibody.22 The authors found that substitution of Asn diminished the antigen binding, and they used a Gly to Arg mutation to address the deamidation issue. Since glypican 3 and CD52 both have a GPI group, it is possible that the Asn residue in the light chain CDR1 of the reported anti-glypican 3 antibody is involved in binding to the phosphate group of glypican 3, with a similar hydrogen binding and hydrophobic interaction network observed in our structural studies (Figure 3). It is interesting that Nakano et al. saw no reduction in the binding activity of the wild-type glypican 3 antibody, even with incubation at 60°C for 2 weeks.22 This may suggest that the stability of the two CDR1 loops, and the Asn-Gly peptide bonds in particular, could be different between the two antibodies. Moreover, the interaction interface reported in this work contrasts sharply with the charge-based interaction network commonly reported for phosphate binding in antibodies as shown previously with anti-CD52 Campath and anti-phosphocholine McPC603 antibodies.36,37
Deamidation events are increasingly observed in proteins and antibodies containing an Asn followed by other amino acids, the so called “non-canonical deamidation sites”.7,12 In our study, we found that G34R, G34K, and G34Q variants showed significantly increased resistance to deamidation, and performed better in our assays than substitutions with bulky and hydrophobic side chains valine and isoleucine (G34V and G34I). This contradicts what has been observed in previous peptide-based deamidation studies, but there may be additional factors at play, as the structure may be otherwise altered by the substitutions, and the bulkiness of the side chain may only be one influencing factor.35
Sequence-based developability prediction and mitigation is becoming a rather challenging practice, as consensus sequences are not always labile, while some labile sites exist that are not predicted based solely on primary sequence. Considerations for contributions from structure and local conformation thus become increasingly important. Our work provides a structural demonstration of how deamidation could affect antibody function. Furthermore, the saturation mutagenesis study results, together with other reports on surprising findings with mutants for deamidation stabilization,8 indicates that extensive screening of stabilization mutants may be necessary to understand the structure-function relationship of the CDR deamidation sites, and ultimately obtain variants with increased resistance to deamidation for improved developability.
Materials and methods
Mutant design and preparation
Mutants were designed at the antibody’s light chain CDR1 Asn33-Gly34 site. The mutant DNAs were synthesized in pDONR221entry vector by DNA2.0 in the light chain backbone. A DNA library was also created where the DNA sequence encoding the Gly34 position was randomized to create a small NNK library, where N is either G, A, T or C; and K is either T or G. They were subsequently subcloned into the HEK293 expression vector pCEP4(-E + I)Dest by Gateway cloning. The conditioned media were subjected to screening of CD52 binding affinity, or loaded to a HiTrap Protein A column (GE) column for further purification and characterization for large-scale preparation. Selected mutants were also prepared in Chinese hamster ovary cells for detailed characterization.
CD52 binding by biacore
CD52 binding affinity was determined by Biacore. Low level of a CD52 peptide mimotope (CGQNDTSQTSSPSAD) was immobilized on a CM5 chip via thiol chemistry using an N-terminal Cys. A new surface was immobilized on each assay occasion and the level of binding was checked with an internal reference standard (wild-type). Antibodies were diluted in HBS-EP running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% Tween-20 pH 7.4) to either a single concentration for relative binding comparison or several concentrations for kinetic analysis. The diluted antibodies were injected for 3 min over the surface to monitor the binding at 30 μL/min flow-rate. Either a report point was taken at the end of injection to calculate % of wild-type binding or kinetic analysis was performed with a 1:1 binding model using the Scrubber2 software. In early studies, binding measurements were performed a single time in “screening mode,” but for the final leads, KD was carefully determined using kinetic mode.
Peptide map/LC-MS
Wild-type and mutant antibodies were reduced and alkylated prior to a double enzyme digestion with trypsin and Lys C in a low pH (7.1) buffer to minimize method-induced deamidation. The peptide map was separated on an Acquity UPLC with ESI-TOF MS (LCT Premier) detection online. Extracted ion chromatograms of the isotope cluster were used to generate the peak area. In the wild type analysis, where baseline separation was optimized for the Asp, Iso-Asp, and Asn separation, the peak areas can be used for quantitation of deamidation. As the primary sequence was different for each variant, the retention time of the variant and degree of chromatographic separation for the Asp-, IsoAsp-, and Asn-containing peptides varied. As chromatographic baseline separation of the peaks was not achieved in all cases, MS peak area ratios were used to calculate the relative amounts of the various peptides for all sequences. The amount of deamidation is approximated from the shift of first isotope to the second, caused by 1 Dalton increase after deamidation. In the case of the wild-type antibody, this corresponds to the ratio of the 592.3 versus the 592.8 m/z. This methodology cannot differentiate between Asp and Iso- asp, but will approximate the amount of Asn conversion to Asp/IsoAsp. Due to the fact that deamidation may be induced by analytical method conditions,38–40 we specifically monitored the amount of method-induced deamidation in our analyses. A time zero sample for each variant was used as a control to monitor deamidation resulting from the analysis. The conditions used (pH 7.1, short, optimized digestion time) minimized the amount of method-induced deamidation to a level significantly less than the deamidation caused by the forced deamidation conditions of the study.
Forced deamidation
Antibodies were dialyzed into buffers at pH 7.0, 7.5 and 8.0, diluted to 0.5 mg/mL, and incubated at 37°C and 45°C. The three-component buffer contained 10 mM succinate, 10 mM histidine, and 10 mM sodium phosphate. It should be noted that we showed independently through crystallography studies that Asn33 interacts with the phosphate moiety on the GPI anchor. Therefore, there is a potential that the presence of phosphate in the buffer used for the forced deamidation studies may have influenced deamidation by possibly exerting a protective effect. However, we have shown that we were able to achieve significant deamidation under our study conditions, despite potential influences of the buffer system, suggesting the influence was not impactful to the studies described. Aliquots were taken at various time points and frozen at −80°C for later analysis.
Complement-dependent cytotoxicity assay
All mutant, control, and wild-type materials were serially diluted 1:2 across one solid black 96-well plate from 2 mg/mL to 0.002 mg/mL in assay medium (phenol-red free IMDM medium + 0.1% bovine serum albumin). Normal human serum complement (Quidel Corporation) was added to all wells at a final concentration of 5% (v/v). Pfeiffer B-lymphocytes (ATCC CRL-2632) were then added at a final concentration of 0.6 × 106 cells/mL. A negative cell lysis control (assay medium + cells), a positive cell lysis control (assay medium + cells + 2% (w/v) Triton X-100), and a positive dose-response control (4 mg/mL control material) were included on the same plate. The reactions were incubated for one hour in a humidified, 37°C, 5% CO2 incubator. Fifty μL of pre-warmed alamarBlue® detection reagent (Life Technologies) were then added to all wells, followed by incubation in reduced light for four hours. The relative reduction of alamarBlue® was measured using a fluorescent plate reader (ex: 530 nm, em: 590, cutoff: 570 nm). Softmax Pro, v. 5.3 (Molecular Devices) was used to generate dose-response curves fit to a four-parameter model.
MAB1 fab preparation and crystallization
MAB1 Fabs were liberated from the full-length IgG by immobilized papain digestion (Thermo-Fisher 20341) following the manufacture’s recommendations. Fabs and Fcs were separated by cation exchange on a mono S 5/50 column (GE Lifesciences 17-5166-01) with a 20 mM MES, pH 6.5, 20 mM to 500 mM NaCl gradient over 30 column volumes.
Fractions containing the MAB1 Fab were pooled and concentrated to a final concentration of 10 mg/mL. The concentrated protein was mixed with a ten-fold molar excess of phosphoethanolamine (ETP) CD52 peptide. The CD52 peptide mimetic contains the entire human CD52 sequence, as well as the ethanolamine phosphate moiety to mimic the GPI anchor present in the native glycopeptide on immune cells. The best diffracting crystal, which was used to solve the structure was obtained from 0.05M Zinc acetate, 17% PEG 3,350 and 0.1 M MES, pH 5.8.
Data collection, structure determination, and refinement
Crystals were cryo-protected with the well solution supplemented with 30% PEG400 and flash frozen in liquid nitrogen. A full data set was collected at 1.5419Å wavelength on the FRE+ superbright with Saturn 944+ CCD detector using CrystalClear2 (Rigaku Americas) with 0.5-degree oscillation. The crystal belongs to space group P212121, a = 54.4 Å, b = 131.2 Å, c = 133.8 Å, α = β = γ = 90° with two Fab molecules in each asymmetric unit.
The scaled reflection data were converted to the mtz format and a molecular replacement search model was sought out using the program BALBES in CCP4, which identified PDB code 1FL5 as a suitable search model. A molecular replacement solution was found using program Molrep with 1FL5 as the model and two copies of Fabs were produced. An electron density map was generated by first rigid body refinement using Refmac5 with a resolution cutoff of 3 Å, followed by a full resolution restrained refinement in Refmac5. The data collection and final refinement statistics were shown in Supplementary Table 1. The atomic coordinates and structural factors have been deposited to Protein Data Bank with code 6OBD.
Funding Statement
This work was supported by Sanofi.
Acknowledgments
We thank Drs. Nicholas Guziewicz, Marcella Yu, Bill Brondyk, Christine DeMaria, Francis Poulin, Anna Park, Tim Edmunds, and Robert Mattaliano for discussions and support; Joanne Cotton and Xiaoying Jin for LC-MS intact mass analysis; Laura Geagan for helps with a figure preparation; Maria Wendt and Justin Slawson for critical review of the manuscript.
Disclosure of Potential Conflicts of Interest
All work is done at Sanofi.
Supplemental materials
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D.. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11:239–64. doi: 10.1080/19420862.2018.1553476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavoisier A, Schlaeppi JM.. Early developability screen of therapeutic antibody candidates using Taylor dispersion analysis and UV area imaging detection. MAbs. 2015;7:77–83. doi: 10.4161/19420862.2014.985544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobson CL, Devine PW, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6:38644. doi: 10.1038/srep38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, et al. Developability studies before initiation of process development: improving manufacturability of monoclonal antibodies. MAbs. 2013;5:787–94. doi: 10.4161/mabs.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley RF, Mehl JT, Sleczka BG, Ciccimaro EF, Kozhich AT, Gilbertson DG, Sweeney TD, Vanbever R, Diot P, Owen CA, et al. Quantification of in vivo site-specific Asp isomerization and Asn deamidation of mAbs in animal serum using IP-LC-MS. MAbs. 2016;8:1611–22. doi: 10.1080/19420862.2016.1196521. [DOI] [PubMed] [Google Scholar]
- 7.Lu X, Nobrega RP, Lynaugh H, Jain T, Barlow K. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. mAbs. 2019;11:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiCara DM, Andersen N, Chan R, Ernst JA, Ayalon G, Lazar GA, Agard NJ, Hilderbrand A, Hötzel I.. High-throughput screening of antibody variants for chemical stability: identification of deamidation-resistant mutants. MAbs. 2018;10:1073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I, Sun T, Caffry I, Brown M, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 2017;9:646–53. doi: 10.1080/19420862.2017.1290753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohli N, Geddie ML. Novel HPLC-based screening method to assess developability of antibody-like molecules. Methods Mol Biol. 2017;1575:189–96. [DOI] [PubMed] [Google Scholar]
- 11.Reissner KJ, Aswad DW. Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cell Mol Life Sci. 2003;60:1281–95. doi: 10.1007/s00018-003-2287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson NE, Robinson AB. Deamidation of human proteins. Proc Natl Acad Sci U S A. 2001;98:12409–13. doi: 10.1073/pnas.221463198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riggs DL, Gomez SV, Julian RR. Sequence and solution effects on the prevalence of d-isomers produced by deamidation. ACS Chem Biol. 2017;12:2875–82. doi: 10.1021/acschembio.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aswad DW, Paranandi MV, Schurter BT. Isoaspartate in peptides and proteins: formation, significance, and analysis. J Pharm Biomed Anal. 2000;21:1129–36. doi: 10.1016/S0731-7085(99)00230-7. [DOI] [PubMed] [Google Scholar]
- 15.Robinson NE, Robinson AB. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc Natl Acad Sci U S A. 2001;98:4367–72. doi: 10.1073/pnas.071066498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kossiakoff AA. Tertiary structure is a principal determinant to protein deamidation. Science. 1988;240:191–94. [DOI] [PubMed] [Google Scholar]
- 17.Nowak C, Tiwari A, Liu H. Asparagine deamidation in a complementarity determining region of a recombinant monoclonal antibody in complex with antigen. Anal Chem. 2018;90:6998–7003. doi: 10.1021/acs.analchem.8b03233. [DOI] [PubMed] [Google Scholar]
- 18.Corn JE, Maass KF, Wang K, Dion MZ, Andersen N, Loyet KM, Mathieu M, Yi L, Corn JE, Maass KF, et al. Protein engineering to increase the potential of a therapeutic antibody Fab for long-acting delivery to the eye. mAbs. 2017;9:1297–305. doi: 10.1080/19420862.2017.1372078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752:233–45. doi: 10.1016/S0378-4347(00)00548-X. [DOI] [PubMed] [Google Scholar]
- 20.Huang L, Lu J, Wroblewski VJ, Beals JM, Riggin RM. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem. 2005;77:1432–39. doi: 10.1021/ac0494174. [DOI] [PubMed] [Google Scholar]
- 21.Lyubarskaya Y, Houde D, Woodard J, Murphy D, Mhatre R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Anal Biochem. 2006;348:24–39. doi: 10.1016/j.ab.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Nakano K, Ishiguro T, Konishi H, Tanaka M, Sugimoto M, Sugo I, Igawa T, Tsunoda H, Kinoshita Y, Habu K, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization. Anticancer Drugs. 2010;21:907–16. [DOI] [PubMed] [Google Scholar]
- 23.Qi P, Volkin DB, Zhao H, Nedved ML, Hughes R, Bass R, Yi SC, Panek ME, Wang D, DalMonte P, et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J Pharm Sci. 2009;98:3117–30. doi: 10.1002/jps.21617. [DOI] [PubMed] [Google Scholar]
- 24.Zheng JY, Janis LJ. Influence of pH, buffer species, and storage temperature on physicochemical stability of a humanized monoclonal antibody LA298. Int J Pharm. 2006;308:46–51. doi: 10.1016/j.ijpharm.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 25.Yan B, Steen S, Hambly D, Valliere-Douglass J, Vanden Bos T, Smallwood S, Yates Z, Arroll T, Han Y, Gadgil H, et al. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J Pharm Sci. 2009;98:3509–21. doi: 10.1002/jps.21655. [DOI] [PubMed] [Google Scholar]
- 26.Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, Kirchmeier M, Corvaïa N, Ionescu R, Beck A.. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009;392:145–54. doi: 10.1016/j.ab.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 27.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Molhoj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9:e100736. doi: 10.1371/journal.pone.0100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson NE. Protein deamidation. Proc Natl Acad Sci U S A. 2002;99:5283–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha S, Zhang L, Duan S, Williams TD, Vlasak J, Ionescu R, Topp E M.. Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Protein Sci. 2009;18:1573–84. doi: 10.1002/pro.v18:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Lin C, O’Connor PB. Glutamine deamidation: differentiation of glutamic acid and gamma-glutamic acid in peptides by electron capture dissociation. Anal Chem. 2010;82:3606–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gamage CLD, Hageman TS, Weis DD. Rapid prediction of deamidation rates of proteins to assess their long-term stability using hydrogen exchange-mass spectrometry (HX-MS). J Pharm Sci. 2019;108(6):1964-1972. [DOI] [PubMed] [Google Scholar]
- 32.Xu A, Kim HS, Estee S, ViaJar S, Galush WJ, Gill A, Hötzel I, Lazar GA, McDonald P, Andersen N, et al. Susceptibility of antibody CDR residues to chemical modifications can be revealed prior to antibody humanization and aid in the lead selection process. Mol Pharm. 2018;15:4529–37. [DOI] [PubMed] [Google Scholar]
- 33.Capasso S, Salvadori S. Effect of the three-dimensional structure on the deamidation reaction of ribonuclease A. J Pept Res. 1999;54:377–82. doi: 10.1111/jpp.1999.54.issue-5. [DOI] [PubMed] [Google Scholar]
- 34.Verma A, Burns DL. Improving the stability of recombinant anthrax protective antigen vaccine. Vaccine. 2018;36:6379–82. doi: 10.1016/j.vaccine.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 35.Aswad DW, editor. Deamidation and isoaspartate formation in peptides and proteins. Boca Raton, FL: CRC press; 1994. [Google Scholar]
- 36.James LC, Hale G, Waldmann H, Bloomer AC. 1.9 A structure of the therapeutic antibody CAMPATH-1H fab in complex with a synthetic peptide antigen. J Mol Biol. 1999;289:293–301. [DOI] [PubMed] [Google Scholar]
- 37.Satow Y, Cohen GH, Padlan EA, Davies DR. Phosphocholine binding immunoglobulin Fab McPC603. An X-ray diffraction study at 2.7 A. J Mol Biol. 1986;190:593–604. [DOI] [PubMed] [Google Scholar]
- 38.Cao M, Mulagapati SHR, Vemulapalli B, Wang J, Saveliev SV, Urh M, Hunter A, Liu D.. Characterization and quantification of succinimide using peptide mapping under low-pH conditions and hydrophobic interaction chromatography. Anal Biochem. 2019;566:151–59. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Moulton KR, Auclair JR, Zhou ZS. Mildly acidic conditions eliminate deamidation artifact during proteolysis: digestion with endoprotease Glu-C at pH 4.5. Amino Acids. 2016;48:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klaene JJ, Ni W, Alfaro JF, Zhou ZS. Detection and quantitation of succinimide in intact protein via hydrazine trapping and chemical derivatization. J Pharm Sci. 2014;103:3033–42. doi: 10.1002/jps.24074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.