

Abstract
Little is known on maize germplasm adapted to the African highland agro-ecologies. In this study, we analyzed high-density genotyping by sequencing (GBS) data of 298 African highland adapted maize inbred lines to (i) assess the extent of genetic purity, genetic relatedness, and population structure, and (ii) identify genomic regions that have undergone selection (selective sweeps) in response to adaptation to highland environments. Nearly 91% of the pairs of inbred lines differed by 30–36% of the scored alleles, but only 32% of the pairs of the inbred lines had relative kinship coefficient <0.050, which suggests the presence of substantial redundancy in allelic composition that may be due to repeated use of fewer genetic backgrounds (source germplasm) during line development. Results from different genetic relatedness and population structure analyses revealed three different groups, which generally agrees with pedigree information and breeding history, but less so by heterotic groups and endosperm modification. We identified 944 single nucleotide polymorphic (SNP) markers that fell within 22 selective sweeps that harbored 265 protein-coding candidate genes of which some of the candidate genes had known functions. Details of the candidate genes with known functions and differences in nucleotide diversity among groups predicted based on multivariate methods have been discussed.
Subject terms: Genetic variation, Plant breeding, Plant stress responses
Introduction
Maize (Zea mays ssp. mays L.) is one of the top three crops globally in total production and is cultivated as a multi-purpose crop for food, feed, biofuel, and raw material for synthesis of various industrial products1. In Africa, maize is produced on a total area of nearly 37 million hectares, which is about 20% of the total maize area of the world. However, the total maize production for the continent is 70.6 million metric tons, which accounts only for 7% of the global production (http://www.fao.org). Lack of congruence between the proportion of production and the cultivated area is due to the low productivity of maize in Africa (<2.0 t ha−1) as compared to a global average of 5.6 t ha−1. In Sub-Sahara Africa (SSA), maize is the primary source of calories (466.5 kcal/capita/day) and is the second most important source of protein (12 g/capita/day) only after wheat. In Ethiopia, maize is the second most popular staple crop after tef (Eragrostis tef (Zucc.) Trotter)2 with huge potential to feed over 100 million people in the country. Between 2008 and 2017, the total maize production and average grain yield in Ethiopia have increased from 3.8 to 8.1 million tons and from 2.1 to 3.7 t ha−1, respectively (http://www.fao.org).
Maize is broadly divided into temperate, subtropical and tropical germplasm depending on latitudinal variations and environmental characteristics3. Tropical maize is further classified into lowland, midaltitude and highland. Highland maize germplasm encompasses a wide range of cold tolerant genotypes evolved in Mexico, Guatemala, the Andean highlands and other small patches of cold valleys and mountains4. However, they tend to be susceptible to lodging, have taller plants as well as ear heights (the height of a plant from the ground level to the upper most ear-bearing node), sensitive to deep planting, susceptible to inbreeding depression, slow grain drying after harvest for storage and have low harvest index. The International Maize and Wheat Improvement Center (CIMMYT) started highland maize breeding program in Mexico in the 1970s with the intention of developing high yielding and cold tolerant improved germplasm from pools and populations carrying tropical and subtropical genetic backgrounds5.
Although maize breeding for the east African highlands started in the 1950s by assembling locally available germplasm and making synthetic populations, the introduction of Ecuador 573 in 1959 to the region significantly impacted highland maize improvement4. Ecuador 573 together with Kitale Synthetic II (Kitale-SYN) were used in the reciprocal recurrent selection for genetic improvement and variety development in the African highlands4,6. In the 1980s, CIMMYT introduced a tropical highland transition zone adapted pool (Pool 9A) to Africa. The pool was not only made available to farmers as an open-pollinated variety but also was used in various breeding programs4. The original Pool 9A was improved for maize streak virus (MSV) resistance at CIMMYT breeding hub in Harare (Zimbabwe), which was then extensively used in highland maize breeding in Africa. The initiation of Highland Maize Genepool Project in 1997 by CIMMYT, in collaboration with the National Agricultural Research Systems (NARS) in eastern Africa, further strengthened the highland maize breeding efforts in the region through introduction and improvement of highland adapted maize germplasm6.
Various studies were conducted to determine the genetic diversity, relationship, population structure and heterotic grouping of maize inbred lines developed by CIMMYT7–13 and International Institute of Tropical Agriculture (IITA)9,14–16 using different genotyping platforms and marker density. Recently, Ertiro et al. (2017) genotyped 265 inbred lines developed by EIAR, CIMMYT, and IITA that are widely used in the mid-altitude sub-humid maize agro-ecology of Ethiopia with 220,878 SNPs. The authors reported that only 22% of the inbred lines were considered genetically pure with >95% homogeneity (genetic purity), which requires purification or further inbreeding except those lines deliberately maintained at early inbreeding level to avoid inbreeding depression. Pairwise genetic distances among the 265 inbred lines varied from 0.011 to 0.345, with only <1% of the pairs of lines differing by less than 20% of the total number of scored alleles. Finally, the different multivariate methods consistently suggested the presence of three groups, which generally agreed with pedigree information (genetic background). However, little is known about the genetic purity, variation and population structure of the maize germplasm adapted to the African highlands, which is widely used in eastern Africa. Previous genetic diversity studies conducted on highland maize inbred lines adapted to the African ecology were based on a small number of samples and low marker density17–21. For example, Beyene et al. (2006 a, b) studied genetic diversity and relationships among 62 Ethiopian highland maize collections using only 20 simple sequence repeat (SSR) markers and eight amplified fragment length polymorphism (AFLP) primers. Legesse et al. (2007) assessed the genetic diversity of 35 highland inbred lines from CIMMYT-Ethiopia and 21 mid-altitude inbred lines from CIMMYT-Zimbabwe using 27 SSR markers and nine AFLP primers. Abakemal et al. (2015) studied genetic purity and patterns of relationships among 36 maize inbred lines adapted to African highland agro-ecology using 25 SSR markers.
Selective sweeps leave distinct signatures in genomes, which are indicative of loci that have undergone selection22–24. Selection increases the frequency of a beneficial allele within a group or population and may even lead to fixation, which then increases the fitness of the individuals carrying it but reduces overall genetic diversity in specific regions that undergone selection25–27. Although all the highland-adapted inbred lines have undergone selection for better adaptation to the highland agro-ecology, we expect differential selection in response to target traits, including germplasm type (normal vs. quality protein maize, QPM), heterotic grouping, and abiotic and biotic stresses. Different statistical methods are available to identify genomic regions that have undergone selective sweep25,28,29. Therefore, the present study was carried out to (i) assess the genetic purity, genetic relationship and population structure of African highland adapted maize inbred lines using high-density genotyping by sequencing (GBS); (ii) identify genomic regions that have undergone selective sweeps, and examine if those selective sweeps showed greater reduction of nucleotide diversity in specific categorical variables (groups or populations) than others; and (iii) compare the extent of molecular diversity indices and genetic differentiation among different groups of highland maize germplasm.
Methods
Plant materials and genotyping
A total of 298 white-grained inbred lines from CIMMYT and Ethiopian Institute of Agricultural Research (EIAR) collaborative highland maize breeding program were used in the study (Supplementary Table S1). These inbred lines are currently widely used in maize breeding programs in the high-altitude sub-humid maize growing areas of eastern and southern Africa (ESA). Early generation lines were originally introduced from CIMMYT-Mexico highland breeding program and CIMMYT-Zimbabwe mid-altitude breeding program, screened under the local environments, and advanced through generations at the EIAR experimental station in Ambo, Ethiopia. Extensive field evaluations were then conducted on advanced generation lines in collaboration with NARS in Kenya, Tanzania, Uganda, Rwanda and Burundi6. The lines were selected for desirable agronomic performances, resistance to common leaf rust, Turcicum leaf blight, gray leaf spot, and germplasm type (normal or QPM). The inbred lines used in our study comprised of 111 normal endosperm (non-QPM) lines derived primarily from Kitale Synthetic II (Kitale-SYN), Ecuador 573, and Pool 9A. The remaining 187 samples were QPM lines that were either developed through backcross breeding30 or extracted from adapted QPM populations. CML144, CML159, and CML176 were the QPM donor parents. Heterotically, 123, 95 and 11 inbred lines belong to groups A, B and AB, respectively, while the remaining 69 inbred lines are not yet assigned.
For each inbred line, seed samples were obtained from Ambo Research Center, Ethiopia. The detailed procedures on genomic DNA extraction, SNP genotyping using GBS31 and data filtering were described in a previous study32. The 298 inbred lines were genotyped with 955,690 SNPs by the Institute of Biotechnology, Cornell University, the USA, of which 237,018 SNPs (hereafter referred as Dataset-1) with a minor allele frequency (MAF) of ≥0.05 and a maximum missing data of 20% (Table 1) were selected. Dataset-1 was imputed using Beagle V4.233 with the default parameters (i.e., window = 50,000, overlap = 3,000; niterations = 15, and cluster = 0.0) and then filtered if there were SNPs with a MAF less than 0.05, which resulted in 235,019 SNPs (Dataset-2) for further statistical analyses.
Table 1.
Summary of the different datasets, chromosomal distribution and physical map length of SNP markers used in the present study.
| Chromosome | Dataset-1 (unimputed) | Dataset-2 (Imputed) | Dataset-3 | Dataset-4 | |||||
|---|---|---|---|---|---|---|---|---|---|
| No. of SNPs | Proportion of SNPs (%) | Map length (Mb) | Proportion of missing data | No. of SNPs | Proportion of SNPs (%) | Map length (Mb) | No. of SNPs | No. of SNPs | |
| Chr 1 | 36,988 | 15.6% | 301.3 | 6.8% | 36,694 | 15.6% | 301.3 | 3,516 | 133 |
| Chr 2 | 28,793 | 12.1% | 237.0 | 7.0% | 28,567 | 12.2% | 237.0 | 2,604 | 0 |
| Chr 3 | 27,156 | 11.5% | 232.1 | 7.2% | 26,917 | 11.5% | 232.1 | 2,463 | 73 |
| Chr 4 | 22,037 | 9.3% | 241.4 | 7.3% | 21,847 | 9.3% | 241.4 | 2,458 | 75 |
| Chr 5 | 27,767 | 11.7% | 217.7 | 7.3% | 27,483 | 11.7% | 217.7 | 2,413 | 46 |
| Chr 6 | 19,305 | 8.1% | 169.1 | 7.5% | 19,137 | 8.1% | 169.1 | 1,813 | 55 |
| Chr 7 | 20,339 | 8.6% | 176.8 | 7.4% | 20,170 | 8.6% | 176.8 | 1,892 | 318 |
| Chr 8 | 20,730 | 8.7% | 175.7 | 7.3% | 20,566 | 8.8% | 175.7 | 2,006 | 151 |
| Chr 9 | 17,783 | 7.5% | 156.5 | 7.2% | 17,641 | 7.5% | 156.5 | 1,734 | 59 |
| Chr 10 | 16,120 | 6.8% | 150.1 | 7.4% | 15,997 | 6.8% | 150.1 | 1,601 | 34 |
| Total | 237,018 | 100.0% | 2057.6 | — | 235,019 | 100.0% | 2,057.6 | 22,500 | 944 |
| Mean | 23,702 | 10.0% | 205.8 | 7.2% | 23,502 | 10.0% | 205.8 | 2,250 | 94 |
Statistical analyses
We first computed identity-by-state (IBS)-based genetic distance matrices from both Dataset-1 (unimputed) and Dataset-2 (imputed) and compared the two distance matrices using Mantel test34, which showed perfect positive correlation (r = 0.999). All statistical analyses except the model-based population structure were, therefore, computed on Dataset-2. The proportion of heterogeneity, relative kinship coefficients, IBS-based genetic distance matrices, and principal component analysis (PCA) were computed (from Dataset-2) using TASSEL v.5.2.51. Cluster analysis was performed on the genetic distance matrix using the neighbor-joining algorithm implemented in molecular evolutionary genetics analysis (MEGA) v.7.035. The first two principal components (PCs) from the PCA were plotted for visual examination in XLSTAT 2012 (Addinsof, New York, USA; www.xlstat.com) using categorical variables, which include heterotic groups, germplasm type (QPM vs. non-QPM), genetic background and group membership predicted both from population STRUCTURE and cluster analyses.
HapMap format of Dataset-2 was exported to PHYLIP interleaved format using TASSEL v.5.2.51, which was then converted to both MEGA36 and ARLEQUIN v.3.5.2.237 formats using PGDSpider v.2.1.1.338. We used MEGA X36 to estimate the number of segregating sites (S), the proportion of polymorphic sites (Ps), Theta (θS), nucleotide diversity (θπ) and Tajima’s D test statistic39. Analysis of molecular variance (AMOVA)40 and FST-based pairwise genetic distance matrices41 were computed among categorical variables using ARLEQUIN v.3.5.2.237. FST values between pairs of populations or groups are indicative of the evolutionary processes that influence the structure of genetic variation with <0.05, 0.05–0.15, 0.15–0.25 and >0.25 indicating little, moderate, great and very great genetic differentiation, respectively42. To minimize the computational requirement in population structure analyses, the 235,019 SNPs in Dataset-2 were further filtered using a MAF of 0.10 and a minimum physical distance of 10-kb between adjacent markers, which resulted in 22,500 SNPs (hereafter referred as Dataset-3). Population structure was analyzed using Dataset-3 and the model-based method implemented in the software package STRUCTURE v.2.3.443 as described in our previous studies32,44. Inbred lines with membership probabilities >60% were assigned to the same clusters, while those with probabilities <60% in any group were assigned to a “mixed” group.
SweeD v.4.0.045 was used to detect selective sweeps that may have undergone selection during breeding process. For this purpose, Dataset-2 was converted into reference and alternative alleles using the variant call format (VCF) option in TASSEL v.5.2.51, which corresponds to the major and minor alleles, respectively. SweeD v.4.0.0 was run on the VCF input file as described in a previous study45 to evaluate a grid of 10,000 equidistant physical locations. The threshold score for declaring selective sweeps was set as the 99.9%, so the 0.1% with likelihood scores >3.1 were retained to represent a candidate selective sweep. The start and end of physical positions of each selective sweep region were used to search for candidate genes and their predicted functions46 at the Gramene Genome Brower (http://ensembl.gramene.org/Zea_mays/Info/Index).
Results
Marker summary and genetic purity
Among the 955,690 SNPs initially generated through GBS, about 25% of the SNPs were used for statistical analyses. The 235,019 SNPs in Dataset-2 were distributed across all 10 chromosomes, which varied from 15,997 on chromosome 10 to 36,694 SNPs on chromosome 1 (Table 1). Minor allele frequency per SNP ranged from 0.05 to 0.50, with an overall average of 0.233 (Supplementary Table S2a). Genetic purity estimated per inbred line ranged from 67.9% to 99.8% (Supplementary Fig. S1, Supplementary Table S1), with a mean of 88.9%. Because of the low genetic purity previously reported in most inbred lines adapted to the Ethiopian mid-altitude sub-humid maize agro-ecology of Ethiopia32, we increased the threshold value from 5.0% to 6.25%, which is the expected average residual heterozygosity (heterogeneity) for lines developed after four generation of inbreeding. Using this threshold criterion, only 34 of the 298 inbred lines (11.4%) were considered fixed with a heterogeneity of ≤6.25, while 57.7% and 30.8% of the inbred lines had heterogeneity varying from 6.26 to 12.50 and from 12.51 to 32.10%, respectively (Supplementary Table S1).
Genetic relatedness and distance
Kinship coefficients between pairs of the 298 inbred lines ranged between 0.00 and 1.98 (on a scale of 0 to 2). Nearly 32% of the pairwise relative kinship values were close to zero (<0.05), 66% were between 0.051 and 0.500 and the remaining 3% between 0.501 to 1.98 (Supplementary Table S3a). When kinship values were compared among groups predicted based on cluster analysis and the model-based STRUCTURE (see below), only 20.3% of the pairs of inbred lines in Group-2 had values close to zero as compared to 34.6% in Group-3 and 55.7% in Group-1 (Fig. 1, Table S3). Genetic distance between pairs of inbred lines ranged from 0.010 to 0.360 (Supplementary Table S4), and the overall mean was 0.323. Nearly 91% of the pairs of 298 lines had genetic distance values between 0.301 and 0.360 as compared to just 0.3% of the pairs that differed by <10% of the scored alleles. About 58.4% and 65.4% of the pairs of inbred lines belonging to Group-2 and Group-3, respectively, differed by >30% of the scored alleles (0.301–0.400) as compared to the 94.6–97.0% pairs in Group-1 predicted based on cluster analysis and model-based population structure (Fig. 1, Supplementary Table S4).
Figure 1.

Frequency distribution of (a) relative kinship and (b) pairwise genetic distance matrices computed using SNPs that were polymorphic within a given number of inbred lines, each with a minor allele frequency >0.05: (i) all the 298 inbred lines using 235,019 SNPs; (ii) 88 inbred lines that belong to Group-1A (G1-A) using 218,208 of 235,019 SNPs; (iii) 69 inbred lines in G1-B using 214,566 of 235,019 SNPs; (iv) 32 inbred lines in G1-C using 200,864 of 235,019 SNPs; (v) 71 inbred lines in G2 using 129,031 of 235,019 SNPs; (vi) 36 inbred lines in G3 using 171,163 of 235,019 SNPs.
Population structure and genetic relationship
The log probability of the data (LnP(D)) and ad hoc statistics ∆K obtained from the model-based population structure analysis suggested that the 298 lines can be divided into two or three possible groups or sub-populations (Fig. 2). However, when the results at various K values were compared with their pedigree information and breeding history, the groups obtained at K = 3 were considered as the best possible number of groups. The proportion of inbred lines assigned to Group-1, Group-2, and Group-3 was 64%, 23%, and 12%, respectively, with only two lines belonging to a mixed group (Table 2, Supplementary Table S1). The first group consisted of 192 inbred lines with mixed heterotic groups, genetic background, and endosperm modification. The second group consisted of 69 QPM inbred lines from heterotic group A (68 lines) and B (1 line) that were developed using CML144 as donor parent and Ecuador-573 (55 lines), Pool 9A-SR (13 lines) and Kitale-SYN (1 line) as recurrent parents. The third group consisted of 35 non-QPM inbred lines extracted from Pool 9 A.
Figure 2.
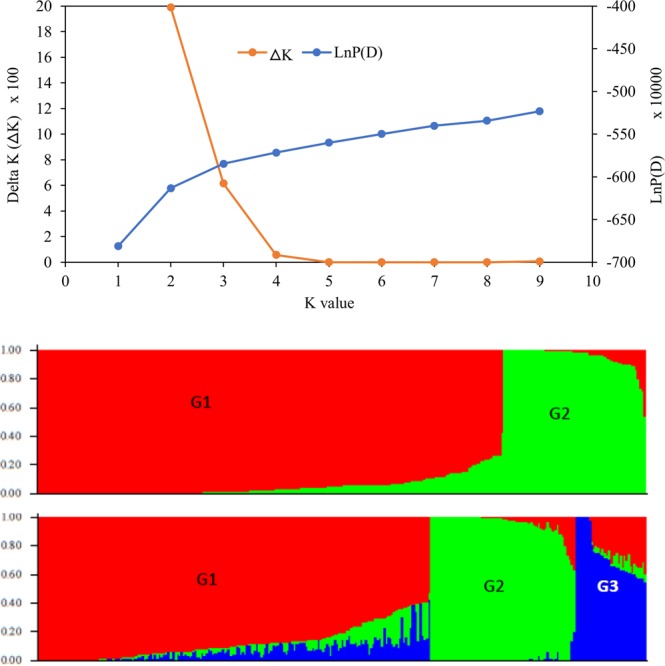
Population structure of 298 maize inbred lines based on 22,500 SNPs in Dataset-3: (a) plot of LnP(D) and a ΔK calculated for K ranging from 1 to 10, with each K repeated thrice; (b) population structure of the 298 inbred lines at K = 2 and K = 3. Every line is represented by a single vertical line that is partitioned into K colored segments on the x-axis, with lengths proportional to the estimated probability membership (y-axis) to each of the K inferred clusters. For membership of each line, see Supplementary Table S1.
Table 2.
Summary of the 298 inbred lines assigned to the three groups predicted based on the model-based population structure analysis by heterotic grouping, endosperm modification (kernel type) and genetic backgrounds.
| Category | Group | Group-1 | Group-2 | Grop-3 | Mixed | Sub-total |
|---|---|---|---|---|---|---|
| Heterotic groups | A | 53 | 68 | 2 | 123 | |
| AB | 11 | 11 | ||||
| B | 92 | 1 | 2 | 95 | ||
| Unknown | 36 | 33 | 69 | |||
| Total | 192 | 69 | 35 | 2 | 298 | |
| Neighbor-joining cluster analysis | G1-A | 88 | 88 | |||
| G1-B | 69 | 69 | ||||
| G1-C | 32 | 32 | ||||
| G2 | 69 | 2 | 71 | |||
| G3 | 1 | 35 | 36 | |||
| Ungrouped | 2 | 2 | ||||
| Total | 192 | 69 | 35 | 2 | 298 | |
| Germplasm type | Non-QPM | 75 | 35 | 1 | 111 | |
| QPM | 117 | 69 | 1 | 187 | ||
| Total | 192 | 69 | 35 | 2 | 298 | |
| Genetic background of recurrent parents | Ecuador-573 | 14 | 55 | 69 | ||
| Kitale-SYN | 29 | 1 | 30 | |||
| Others | 11 | 11 | ||||
| Pool-9A | 9 | 35 | 44 | |||
| Pool-9A-SR | 65 | 13 | 2 | 80 | ||
| Pop-502-SR | 17 | 17 | ||||
| SADVLA | 10 | 10 | ||||
| SUSUMA | 24 | 24 | ||||
| Tuxpeno | 13 | 13 | ||||
| Total | 192 | 69 | 35 | 2 | 298 | |
| Genetic background by donor parents | CML144 | 29 | 69 | 1 | 99 | |
| CML159 | 15 | 15 | ||||
| CML176 | 46 | 46 | ||||
| Non-CML | 102 | 35 | 1 | 138 | ||
| Total | 192 | 69 | 35 | 2 | 298 |
The neighbor-joining (NJ) tree constructed from the genetic distance matrix grouped 296 of the 298 inbred lines into three major groups as the model-based STRUCTURE and five sub-groups; two inbred lines were not assigned into any of the sub-groups (Fig. 3, Supplementary Table S1). Nearly all inbred lines that belong to Group-2 and Group-3 remained the same as the group membership predicted based on the model-based population structure analysis. On the other hand, lines belonging to the first group in the model-based population structure were further divided into three subgroups (Group-1A, Group-1B, and Group-1C) in the cluster analysis (Supplementary Table S1). Group-1A consisted of a total of 88 inbred lines of mixed heterotic groups, germplasm type (both QPM and non-QPM), and genetic backgrounds. Group-1B had 69 inbred lines, which are primarily QPM (65 lines) with mixed heterotic groups and diverse genetic backgrounds, while Group-1C consisted of 32 inbred lines that were primarily non-QPM with both Ecuador-573 and Kitale-SYN genetic background, but mixed heterotic groups (Supplementary Table S1). As shown in Fig. 3, however, the three sub-groups clustering pattern based on the model-based population structure analysis does not fully match the pattern obtained in NJ analysis.
Figure 3.

Neighbor-joining tree of 298 inbred lines based on identity-by-state genetic distance matrix computed from 235,019 SNPs, each with minor allele frequency >0.05. Line colors are as follows: Group-1A (black); Group-1B (red), Group-1C (blue), Group-2 (green), Group-3 (pink) and ungrouped (orange). Group-1, Group-2, and Group-3 were obtained based on the model-based STRUCTURE. See Supplementary Table S1 for details of each group membership.
To get insight on the patterns of relationship among the 298 inbred lines, we constructed various phylogenetic trees (Fig. 3, Supplementary Fig. S2) and also plotted PC1 (16.9%) against PC2 (8.3%) from PCA using diverse categorical variables (Fig. 4, Supplementary Fig. S3), including heterotic grouping, germplasm type (QPM vs. non-QPM), genetic backgrounds and predicted group memberships based on both cluster and model-based STRUCTURE. The different plots clearly demonstrated three distinct groups, which was consistent with the group membership of the model-based STRUCTURE at K = 3 than any of the other categorical variables. Nearly 77% of the 298 inbred lines have already been assigned to heterotic groups A (123 lines), B (95 lines) and AB (11 lines) by breeders based on combining ability tests, mainly using diallel and line-by-tester analyses. As shown in Supplementary Figs S2 and S3, lines belonging to the same heterotic group did not necessarily clustered together. Nearly 95% of the inbred lines belonging to heterotic group B showed clear population structure as compared to those in heterotic group A that were divided into two subgroups.
Figure 4.
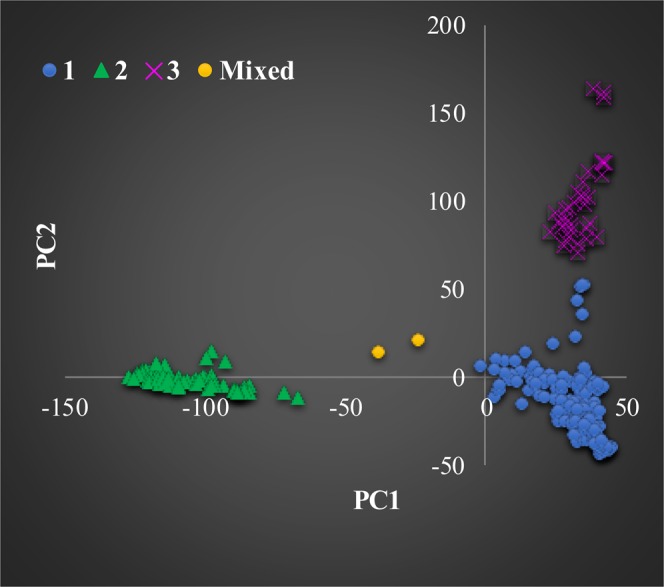
Plot of PC1 (11.3%) and PC2 (5.4%) from a principal component analysis of 298 inbred lines using 235,019 SNPs, each with minor allele frequency >0.05. Group-1 (blue), Group-2 (green), Group-3 (pink) and mixed (orange) were obtained from the model-based STRUCTURE at K = 3. See Supplementary Table S1 for details of each group membership.
Genetic differentiation
Results from the partitioning of the molecular variance by different categorical variables revealed that differences in heterotic groups (A vs. B) and germplasm type (QPM vs. non-QPM) accounted for 12.0% and 8.1% of the genetic variation, respectively, which both fell under moderate genetic differentiation. On the other hand, the differentiation among groups based on genetic background (pedigree information), groups predicted based on cluster analysis and the model-based population structure accounted for 18.8–21.6% and 25.3–29.6% of the total molecular variation, respectively (Table 3), which suggest great and very great genetic differentiation. When pairwise FST values between groups were compared (Supplementary Table S5), the values among groups predicted from the model-based STRUCTURE was the highest between Group-2 and Group-3 (0.498) and the lowest between the Group-1 and Group-3 (0.221), which is also evident in the PCA plot (Fig. 4). FST values of the 21 possible pairwise comparisons based on the genetic backgrounds of the recurrent parents varied from 0.086 between Kitale-SYN and Pool-9A to 0.368 between Ecuador-573 and Pop-502-SR with most pairs showing either moderate (0.05–0.15) or great (0.15–0.25) genetic differentiation.
Table 3.
Analysis of molecular variance (AMOVA) of 298 inbred lines grouped on different categorical variables for the extraction of SNP variation among and within groups (populations) based on 235,019 SNPs.
| Category | Source of variation | Degree of freedom | Sum of squares | Variance components | Percentage of variation |
|---|---|---|---|---|---|
| Groups based on STRUCTURE at K = 3 | Among groups | 2 | 1,396,725.3 | 8,954.0 | 29.6 |
| Within groups | 293 | 6,233,282.8 | 21,274.0 | 70.4 | |
| Total | 295 | 7,630,008.0 | 30,228.0 | 100.0 | |
| Groups based on neighbor-joining method | Among groups | 4 | 1,676,030.9 | 6,964.1 | 25.3 |
| Within groups | 291 | 5,969,328.7 | 20,513.2 | 74.7 | |
| Total | 295 | 7,645,359.6 | 27,477.3 | 100.0 | |
| Groups based on genetic backgrounds of recurrent parents | Among groups | 6 | 1,194,314.2 | 5,066.3 | 18.8 |
| Within groups | 280 | 6,144,305.6 | 21,943.9 | 81.2 | |
| Total | 286 | 7,338,619.8 | 27,010.2 | 100.0 | |
| Groups based on genetic backgrounds of recurrent parents, excluding “Others” in Table 2 | Among groups | 7 | 1,495,876.8 | 5,759.3 | 21.6 |
| Within groups | 279 | 5,842,743.0 | 20,941.7 | 78.4 | |
| Total | 286 | 7,338,619.8 | 26,701.0 | 100.0 | |
| Groups based on genetic backgrounds of CML donor parents | Among groups | 2 | 509,151.8 | 5,532.8 | 20.2 |
| Within groups | 157 | 3,436,444.4 | 21,888.2 | 79.8 | |
| Total | 159 | 3,945,596.1 | 27,421.0 | 100.0 | |
| Heterotic groups | Among groups | 1.0 | 371,556.3 | 3,244.0 | 12.0 |
| Within groups | 216.0 | 5,139,101.0 | 23,792.1 | 88.0 | |
| Total | 217.0 | 5,510,657.3 | 27,036.1 | 100.0 | |
| Groups based on germplasm type | Among groups | 1 | 330,070.6 | 2,191.0 | 8.1 |
| Within groups | 296 | 7,353,918.8 | 24,844.3 | 91.9 | |
| Total | 297 | 7,683,989.5 | 27,035.3 | 100.0 |
Diversity indices and selective sweeps
Table 4 summaries the marker polymorphism, diversity indices, and Tajima’s D computed for inbred lines belonging to the same categorical variables (heterotic groups, germplasm type, genetic backgrounds and predicted group membership based on NJ cluster analysis and the model-based STRUCTURE). Of the 235,019 segregating sites across the 298 inbred lines, the number of segregating sites, proportion of polymorphic sites and nucleotide diversity (π) observed within Group-2 and Group-3 predicted based on the model-based STRUCTURE and NJ cluster analyses were much smaller than Group-1, which all indicate reduction in diversity in the former two groups. Inbred lines with Pop-502-SR, SADVLA, SUSUMA, and Tuxpeno genetic backgrounds showed smaller diversity indices than those lines derived from Ecuador-573, Kitale-SYN, Pool-9A and Pool-9A-SR. However, we did not observe obvious differences when the analyses were conducted using the two heterotic groups (A vs. B) and germplasm type (QPM vs. non-QPM) as categorical variables (Table 4). Tajima’s D values computed from Dataset-2 were negative in both Group-2 and Group-3, which is an indication for stronger positive selection in these two groups than Group-1. SweeD45 identified 22 candidate selective sweep regions distributed across all chromosomes except chromosome 2 (Table 5, Fig. 5). The selective sweep regions spanned from 6-kb to 4,229-kb and consisted of clusters of markers that varied from 8 to 125 SNPs, except one region on chromosome 8 (Chr8-Reg-02) that had just one SNP (Table 5). Overall, a total of 944 SNPs were mapped within the 22 selective sweep regions (Dataset-4). Selective sweeps increase the frequency of beneficial alleles and surrounding variants and may eventually lead to fixation, while recombination and mutation introduce new alleles that are rare (causing alleles of very low frequency), which are evident in Supplementary Table S2.
Table 4.
Summary of the molecular diversity indices for different categorical variables based on Dataset-2 (235,019 SNPs) and Dataset-4 (944 SNPs that fell within 22 selective sweeps identified using SweeD). Dataset-4 was used to assess reduction in diversity indices within each group (but not among groups) as compared to the genome-wide SNPs in Dataset-2.
| Groups | N* | Dataset-2 (235,019 genome-wide SNPs)** | Dataset-4 (944 SNPs with 22 selective sweeps)** | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S | Ps | Θ S | Θ π | D | S | Ps | Θ S | Θ π | D | ||
| Groups based on TRUCTURE at K = 3 | |||||||||||
| Group-1 | 192 | 234,655 | 0.998 | 0.171 | 0.212 | 0.765 | 942 | 0.998 | 0.171 | 0.140 | −0.595 |
| Group-2 | 69 | 133,423 | 0.568 | 0.118 | 0.114 | −0.119 | 283 | 0.300 | 0.062 | 0.052 | −0.570 |
| Group-3 | 35 | 172,317 | 0.733 | 0.178 | 0.143 | −0.747 | 442 | 0.468 | 0.114 | 0.069 | −1.492 |
| Groups based on NJ cluster analysis | |||||||||||
| Group-1A | 88 | 226,991 | 0.966 | 0.191 | 0.210 | 0.340 | 865 | 0.916 | 0.181 | 0.131 | −0.965 |
| Group-1B | 69 | 221,508 | 0.943 | 0.196 | 0.185 | −0.202 | 860 | 0.911 | 0.190 | 0.121 | −1.269 |
| Group-1C | 32 | 204,603 | 0.871 | 0.216 | 0.215 | −0.020 | 761 | 0.806 | 0.200 | 0.170 | −0.590 |
| Group-2 | 71 | 145,321 | 0.618 | 0.128 | 0.119 | −0.258 | 305 | 0.323 | 0.067 | 0.054 | −0.661 |
| Group-3 | 36 | 173,390 | 0.738 | 0.178 | 0.142 | −0.770 | 462 | 0.489 | 0.118 | 0.069 | −1.573 |
| Groups based on endosperm modification | |||||||||||
| QPM | 187 | 233,320 | 0.993 | 0.171 | 0.211 | 0.754 | 937 | 0.993 | 0.171 | 0.113 | −1.097 |
| Non-QPM | 111 | 229,277 | 0.976 | 0.185 | 0.213 | 0.514 | 878 | 0.930 | 0.176 | 0.131 | −0.854 |
| Groups based on genetic background of QPM donor parents | |||||||||||
| CML144 | 99 | 205,635 | 0.875 | 0.169 | 0.179 | 0.201 | 638 | 0.676 | 0.131 | 0.089 | −1.097 |
| CML159 | 15 | 170,423 | 0.725 | 0.223 | 0.170 | −1.067 | 487 | 0.516 | 0.159 | 0.108 | −1.425 |
| CML176 | 46 | 218,615 | 0.930 | 0.212 | 0.207 | −0.088 | 825 | 0.874 | 0.199 | 0.133 | −1.217 |
| Non-CML | 27 | 184,241 | 0.784 | 0.203 | 0.156 | −0.927 | 628 | 0.66525 | 0.1726 | 0.10821 | −1.4805 |
| Groups based on genetic background of recurrent parents | |||||||||||
| Ecuador-573 | 69 | 197,259 | 0.839 | 0.175 | 0.148 | −0.532 | 653 | 0.692 | 0.144 | 0.074 | −1.714 |
| Kitale-SYN | 30 | 206,194 | 0.877 | 0.221 | 0.226 | 0.084 | 739 | 0.783 | 0.198 | 0.158 | −0.775 |
| Pool-9A | 44 | 191,329 | 0.814 | 0.187 | 0.167 | −0.407 | 596 | 0.631 | 0.145 | 0.092 | −1.365 |
| Pool-9A-SR | 80 | 223,315 | 0.950 | 0.192 | 0.209 | 0.309 | 850 | 0.900 | 0.182 | 0.122 | −1.135 |
| Pop-502-SR | 17 | 153,519 | 0.653 | 0.193 | 0.162 | −0.711 | 470 | 0.498 | 0.147 | 0.107 | −1.172 |
| SADVLA | 10 | 140,062 | 0.596 | 0.211 | 0.160 | −1.217 | 398 | 0.422 | 0.149 | 0.102 | −1.575 |
| SUSUMA | 24 | 175,493 | 0.747 | 0.200 | 0.151 | −0.998 | 594 | 0.629 | 0.169 | 0.106 | −1.499 |
| Tuxpeno | 13 | 156,379 | 0.665 | 0.214 | 0.159 | −1.201 | 426 | 0.451 | 0.145 | 0.096 | −1.565 |
| Groups based on heterotic grouping | |||||||||||
| Heterotic group A | 123 | 226,605 | 0.964 | 0.179 | 0.199 | 0.369 | 848 | 0.898 | 0.167 | 0.103 | −1.273 |
| Heterotic group B | 95 | 232,840 | 0.991 | 0.193 | 0.207 | 0.250 | 921 | 0.976 | 0.190 | 0.132 | −1.045 |
| Heterotic group AB | 11 | 171,143 | 0.728 | 0.249 | 0.231 | −0.339 | 489 | 0.518 | 0.177 | 0.158 | −0.509 |
| All inbred lines without groups | |||||||||||
| All samples | 298 | 234,956 | 1.000 | 0.159 | 0.220 | 1.202 | 943 | 0.999 | 0.159 | 0.125 | −0.682 |
*In cases where the sample size (N) do not add up to 298, some lines were excluded from the selective sweep analyses, which included the following: (i) two lines assigned to a “mixed” group at K = 3 and those remained unassigned to any of the sub-groups in the NJ cluster analysis; (ii) 69 lines with yet unknown heterotic groups; (iii) 11 lines with uncertain recurrent parent genome and (iv) 26 lines with uncertain genetic background of QPM donor parents. See Supplementary Table S1 for details.
**Number of segregating sites (S); Proportion of polymorphic sites (Ps); Theta (θS); Nucleotide diversity (Θπ); Tajima’s D test statistic (D).
Table 5.
Summary of the 22 selective sweeps identified using SweeD, including chromosomal position, number of SNPs that fell within each region and number of candidate genes. See Supplementary Table S6 for details on candidate genes identified in each region.
| Selective sweep name | Selective sweep interval | Chrom. | Minimum likelihood | Maximum likelihood | Minimum Alpha* | Maximum Alpha* | Start position (bp) | End position (bp) | Interval (kb) | No. of SNPs within the selective sweep interval (Dataset-4) | No. of candidate genes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr1-Reg-01 | 1:106060208-110288743 | 1 | 3.2 | 5.9 | 3.9E-06 | 4.5E-05 | 106,060,208 | 110,288,743 | 4229 | 125 | 45 |
| Chr1-Reg-02 | 1:218716421-219612163 | 1 | 3.3 | 4.1 | 1.6E-05 | 3.9E-04 | 218,716,421 | 219,612,163 | 896 | 8 | 9 |
| Chr3-Reg-01 | 3:25123538-25922284 | 3 | 3.6 | 8.5 | 2.5E-05 | 1.1E-04 | 25,123,538 | 25,922,284 | 799 | 22 | 14 |
| Chr3-Reg-02 | 3:127512961-127518662 | 3 | 3.1 | 3.1 | 1.7E-04 | 1.7E-04 | 127,512,961 | 127,518,662 | 6 | 14 | 2 |
| Chr3-Reg-03 | 3:215786813-216005903 | 3 | 4.8 | 4.8 | 9.9E-05 | 9.9E-05 | 215,786,813 | 216,005,903 | 219 | 37 | 4 |
| Chr4-Reg-01 | 4:149724977-149899895 | 4 | 4.2 | 4.2 | 6.4E-05 | 6.4E-05 | 149,724,977 | 149,899,895 | 175 | 34 | 1 |
| Chr4-Reg-02 | 4:186912590-187083281 | 4 | 4.3 | 4.8 | 1.1E-04 | 1.4E-04 | 186,912,590 | 187,083,281 | 171 | 41 | 6 |
| Chr5-Reg-01 | 5:49802255-49836436 | 5 | 3.2 | 3.2 | 1.3E-04 | 1.3E-04 | 49,802,255 | 49,836,436 | 34 | 11 | 0 |
| Chr5-Reg-02 | 5:179785894-179940120 | 5 | 3.3 | 5.4 | 9.5E-05 | 5.5E-04 | 179,785,894 | 179,940,120 | 154 | 9 | 1 |
| Chr5-Reg-03 | 5:184394549-184699627 | 5 | 3.2 | 3.2 | 9.8E-05 | 9.8E-05 | 184,394,549 | 184,699,627 | 305 | 26 | 6 |
| Chr6-Reg-01 | 6:140675365-140726905 | 6 | 3.2 | 3.3 | 8.4E-05 | 2.6E-04 | 140,675,365 | 140,726,905 | 52 | 17 | 1 |
| Chr6-Reg-02 | 6:150939481-151135404 | 6 | 3.4 | 4.3 | 4.7E-05 | 6.8E-04 | 150,939,481 | 151,135,404 | 196 | 38 | 4 |
| Chr7-Reg-01 | 7:32523-1269208 | 7 | 3.2 | 3.4 | 1.8E-05 | 2.2E-05 | 32,523 | 1,269,208 | 1237 | 119 | 32 |
| Chr7-Reg-02 | 7:11953168-13065716 | 7 | 3.3 | 3.6 | 2E-05 | 2.4E-05 | 11,953,168 | 13,065,716 | 1113 | 28 | 23 |
| Chr7-Reg-03 | 7:120966174-121400969 | 7 | 3.3 | 3.6 | 5.8E-05 | 1.2E-04 | 120,966,174 | 121,400,969 | 435 | 71 | 9 |
| Chr7-Reg-04 | 7:131185931-132315208 | 7 | 3.2 | 3.3 | 2E-05 | 8.2E-05 | 131,185,931 | 132,315,208 | 1129 | 100 | 20 |
| Chr8-Reg-01 | 8:27653451-28591523 | 8 | 3.4 | 3.9 | 2.5E-05 | 1.0E-04 | 27,653,451 | 28,591,523 | 938 | 35 | 15 |
| Chr8-Reg-02 | 8:65750670-67244274 | 8 | 3.2 | 5.0 | 1E-05 | 3.0E-04 | 65,750,670 | 67,244,274 | 1494 | 1 | 25 |
| Chr8-Reg-03 | 8:111704655-112150258 | 8 | 3.5 | 3.9 | 2.9E-05 | 9.8E-05 | 111,704,655 | 112,150,258 | 446 | 28 | 3 |
| Chr8-Reg-04 | 8:157235205-157836943 | 8 | 3.3 | 3.7 | 3.2E-05 | 3.8E-05 | 157,235,205 | 157,836,943 | 602 | 87 | 21 |
| Chr9-Reg-01 | 9:7105365-7663379 | 9 | 3.2 | 3.9 | 2E-05 | 6.3E-05 | 7,105,365 | 7,663,379 | 558 | 59 | 15 |
| Chr10-Reg-01 | 10:29222096-30868221 | 10 | 3.2 | 4.1 | 1E-05 | 7.9E-05 | 29,222,096 | 30,868,221 | 1646 | 34 | 9 |
*The minimum and maximum alpha values are given in exponentials (e.g., 3.9E-06 = 3.9 × 10−6).
Figure 5.

Manhattan plot showing the 22 selective sweep regions detected using SweeD v.4.0.0. The horizontal solid line indicates the threshold value of 3.1 for declaring candidate selective sweeps.
In dataset-4, the major and minor allele frequency of the 944 SNPs that fell within the selective sweeps were 0.800-0.950 and 0.05-0.200, respectively. Nearly 3%, 72%, 53% and 37% of the 944 SNPs had major allele frequency greater than 0.950 in Group-1, Group-2, Group-3 and Group-2 and Group-3 combined, respectively. In fact, 32% and 17% of the 944 SNPs were fixed in Group-2 and Group-3, respectively, as compared to none in Group-1. Such results suggest that most SNPs that fell within the 22 selective sweep regions showed a reduction in diversity in Group-2 and Group-3, which is likely due to selection for better adaptation to specific traits that may not be the case in Group-1. Comparisons of Ps and θS computed from the 944 SNPs (Dataset-4) that fell within the 22 selective sweeps with the genome-wide SNPs (Dataset-2) showed reduction in both Group-2 and Group-3 than Group-1. A less obvious reduction were noted among inbred lines belonging to the different heterotic groups, germplasm type and genetic backgrounds (Table 4). To gain insight into possible roles of each of the selective sweep region, we compiled a list of 265 protein-coding candidate genes that fell within the 21 of 22 selective sweep regions (Supplementary Table S6). Each of the 21 selective sweeps consisted of one to forty-five protein-coding genes. Some of the candidate genes had known functions, which are summarized in Supplementary Table S6 and partly discussed in the next section.
Discussion
Genetic purity in African highland maize inbred lines
Maintenance of genetic purity in inbred lines by minimizing residual heterozygosity (heterogeneity) is important for quality seed production32,47,48. The threshold value may vary depending on the purpose of the line development program and level of inbreeding. In the current study, only 34 of the 298 inbred lines (~11%) were found to be genetically homogeneous (Supplementary Table S1), which agrees with another recent study on maize inbred lines adapted to the mid-altitude sub-humid maize ecology in Ethiopia32. In that previous study, about 53% of the maize inbred lines developed by EIAR showed higher than expected level of genetic heterogeneity as compared to 13% and 8% of the inbred lines developed by CIMMYT and IITA breeding programs, respectively, which may be due to one or more of the following reasons. First, the three institutions use different methods for line maintenance, besides the source germplasm. EIAR breeders often use sib-mating (by bulking pollens of multiple plants from the same entry) during line development, which is less common both at CIMMYT and IITA. In addition, the high level of genetic heterogeneity within EIAR maize inbred lines could also be due to human errors (e.g., contamination by off types, stray pollens and/or seed admixture) during line development and/or line maintenance. If such types of errors occur, the sib-mating method is more prone to introducing new sources of genetic variability that in turn reduces genetic purity than selfing of individual plants. Because of the extensive collaboration between CIMMYT-Ethiopia and EIAR, including sharing nurseries, most of the inbred lines analyzed in the present study could have resulted from a combination of sib-mating and selfing.
Second, most of the source germplasm used for developing the inbred lines in the current study were composites, pools, and synthetics49, which are suitable for developing open-pollinated varieties (OPVs) but may not be suitable for extracting genetically pure inbred lines. Third, some of the inbred lines were deliberately extracted from early generation (such as S3) lines and maintained by sib-mating to avoid severe inbreeding depression upon continuous self-pollination4. Although such approach is useful to attain higher seed yield per unit area, which in turn decreases the cost of seed production and increases access to seed by small-scale farmers50, it would be very challenging in terms of line maintenance. However, the third case is a less likely scenario as there are multiple lines with heterogeneity greater than 12.5%, which is the expected average heterogeneity among lines extracted at S3 generation.
Currently, there is more demand in developing uniform hybrids using genetically pure parental lines, especially doubled haploid lines, as this has several advantages, including better heterosis, simplicity in parental line maintenance and implementing quality control during hybrid seed production32,47,48,51. As a result, maize breeders are using fixed lines in their new pedigree starts up and advance each generation through selfing than sib-mating. One of the immediate solutions for improving genetic purity of the inbred lines used in the present study may be to purify seed stocks of those lines with higher than expected heterogeneity by rouging off-types in seed maintenance and production plots, but such method requires enormous efforts and incurs additional costs. The long-term solution is to use doubled haploid (DH) technology in developing genetically pure doubled haploid (DH) lines that can be derived in a short period of time52–54. In partnership with the Kenya Agriculture and Livestock Research Organization (KALRO), a state-of-the-art maize DH facility for Africa has been established in 2013 by CIMMYT at Kiboko station, Kenya, which is annually producing nearly 70,000 DH lines from African-adapted maize source germplasm.
Genetic relationship and population structure
Relative kinship coefficients are widely used as an indicator of the genetic relationship between pairs of genotypes, where values close to zero indicate a lack of relationship, while higher values indicating stronger relationship. About sixty-nine percent of the pairwise comparisons of the 298 inbred lines had kinship values ranging from 0.05 to 1.98 as compared to just 32% that had kinship values close to zero, suggesting presence of high level of genetic similarity that may be due to the use of closely related parents that tend to introduce redundant alleles in a breeding program. Similar results were reported in previous studies in maize inbred lines from different breeding programs7–10,32. The 32% pairs of highland maize inbred lines with kinship coefficients close to zero was six-fold greater than the 5% reported in maize inbred lines originated from CIMMYT ESA breeding programs7, but nearly half of the values reported for maize inbred lines adapted to the mid-altitude ecologies of Ethiopia32, the global maize collection10, inbred lines from INERA and IITA14, inbred lines from CIMMYT and IITA9 and CIMMYT maize inbred lines8. On the other hand, nearly 91% of the pairs of 298 inbred lines differed by 30–36% of the scored alleles (of 235,019 SNPs in Dataset-2) as compared with just 10% of the pairs that differed by ≤30% of the scored alleles (Supplementary Table S4 and Fig. 1).
The high genetic differences among most pairs of inbred lines agrees with pedigree information and breeding history, as have been reported in other studies11,14,17,21,32. Of the inbred lines assigned into heterotic groups based on combining ability tests through diallel and line-by-tester analyses6, only part of them showed clear population structure which is expected due to their genetic backgrounds (composites, pools, synthetics). Several previous studies reported the lack of consistencies between heterotic classification based on genotype data and combining ability or pedigree information in tropical maize germplasm7,8,14,32. The broad genetic base of the germplasm, lack of clear information on origin and heterotic background, inadequate pedigree information, short breeding history, and use of variable testers and testcross evaluation for assigning lines to heterotic groups have been frequently cited as possible reason for disagreement between markers-based heterotic grouping and combining ability and pedigree-based heterotic assignment.
Role of candidate genes in selective sweeps
As shown in the Manhattan plot in Fig. 5, the highest SweeD score was 8.5, which was observed within 799-kb interval on chromosome 3 (3:25123538-25922284). This region harbored 14 protein-coding genes, including glycosyl transferases in family 61 protein that mediate arabinofuranosyl transfer onto xylan in grasses55,56, which plays an essential structural role in cell walls of all plants and valuable components of human and animal nutrition due to its major dietary fiber composition in cereals57,58. One of the selective sweeps on chromosome 1 (1:106060208-110288743) consisted of 45 candidate genes, including (i) the WRKY-transcription factors that play crucial roles in plant growth and development, defense regulation and response to different biotic and abiotic stresses59,60; (ii) roothairless6 (rth6), which is one of the genes that control root hairs formation and facilitates nutrient uptake and optimal development61,62; (iii) JUMONJI-transcription factor 14 (JMJ14) and CCAAT-binding transcription factor that control flowering time63–65. The selective sweep on chromosome 5 (5:184394549-184699627) harbors the maize red aleurone1 (pr1) that encodes a CYP450-dependent flavonoid 3′-hydroxylase, which is required for the biosynthesis of purple and red anthocyanin pigments. Anthocyanins accumulate in maize pericarps, cob glumes, and silks66 and believed to have a protective role in plants against extreme temperatures. The pr1 locus has also been extensively used as a phenotypic marker in determining kernel aleurone color by hydroxylation of anthocyanin compounds67. Different studies have implicated members of the bZIP family of transcription factors (proteins that bind to the G-box) as mediators of abscisic acid dependent gene expression68,69 of which bZIP transcription factor 1 (bzip1) is located in one of the selective sweep regions (3:215786813-216005903) identified in the present study. Abscisic acid plays a central role in plants abiotic stress resistance by regulating a large number of stress-responsive genes to confer abiotic stress tolerance in plants70. The candidate genes mentioned above could be potentially influencing traits of adaptation to highland agro-ecologies in Africa, and the observed selective sweeps might be due to positive natural selection or deliberate selection during the development of inbred lines or source populations.
Multiple candidate genes of known function have been identified on chromosome 6, which include (a) elongation factors that are highly correlated with total lysine content of the endosperm71,72; (b) glutathione transferases that catalyze the conjugation of glutathione to xenobiotic compounds in the detoxification process73; (c) G2-like transcription factors that play a central role in regulating chloroplast development, which contain the green pigment chlorophyll and are responsible for the light-powered reactions of photosynthesis (Liu et al. 2016); and (d) basic leucine zipper (bZIP) gene family that play important roles in multiple biological processes, such as light signaling, seed maturation, flower development as well as abiotic and biotic stress responses74. The four selective sweep regions identified on chromosome 7 consisted of multiple candidate genes, including Kinesin-related proteins (KRPs) that play central roles in the transport of various vesicles and organelles in eukaryotic cells75; gibberellin 2-oxidases (GA2oxs) that regulates plant growth by inactivating endogenous bioactive Gibberellins76; the maternally expressed gene (Meg) family, which encodes cysteine-rich proteins (CRPs)77 that are involved in both cell-signaling and antimicrobial processes78,79; the Endosperm5 (o5) showed moderate correlation (R2 = 0.66) with Opaque 2 (o2) and affect different aspects of storage protein synthesis in maize80; the cellulose synthase (CesA) gene family that are primary determinant of wall formation, stalk strength and improve harvest index81; carbonic anhydrase (CA) that catalyzes the reversible hydration of CO2 into bicarbonate82, and implicated in photosynthesis83, stomatal conductance and guard cell movement in C3 plants84, and providing bicarbonate to the initial carboxylating enzyme phosphoenolpyruvate carboxylase in C4 plants85; the basic helix-loop-helix (bHLH) transcription factors that play key roles in diverse biological processes, including seed germination, shade avoidance response, flowering time regulation, stress responses and anthocyanins synthesis86–88; pentatricopeptide repeat (PPR) proteins that have been implicated in RNA editing, RNA processing, translation, photosynthesis, respiration and kernel development89,90; the maize D-cyclin gene asceapen1 (asc1) that plays a role in leaf and shoot development91 and regulates progression through the G1 phase of the cell cycle92.
On chromosome 8, the selective sweeps consisted of several AP2/EREBP (APETALA2/ethylene responsive element-binding protein) transcription factors that are involved in many different pathways, including drought and high salt concentration93, low temperature94, diseases95,96 and the control of flowering97; receptor for activated C kinase that plays a role in plant response to fungal phytopathogens98, affect different signal transduction pathways and multiple developmental processes in plants99; MYB transcription factors that are involved in controlling responses to biotic and abiotic stresses, development, differentiation, metabolism, hormone signal transduction100,101; and AP kinase kinase kinase 18 (MAPKKK18) that controls plant growth by adjusting the timing of senescence via its protein kinase activity102.
Conclusions
Most of the 298 inbred lines adapted to the African highland ecology showed high level of genetic heterogeneity than expected for lines extracted from S4 or later generations, which suggests the need for revising the line development strategy, including line finishing and use of genetically pure parental lines for line development (as compared to landraces, composites and pools that were used in the past); generating reference genotype data as one of the requirements for releasing lines; implementing quality assurance (QA) and quality control (QC) genotyping methods to regularly check genetic purity of key inbred lines during line maintenance; and more frequent use of DH technology in developing breeding lines. The germplasm used in the current study showed clear population structure, primarily by pedigree information and breeding history, and less so by heterotic groups and germplasm type. There was a high level of genetic difference among most pairs of inbred lines although they have a large proportion of alleles in common, which is expected when a limited number of parental lines are used for line development. We identified 944 SNPs that fell within 22 selective sweep regions, which harbored 265 annotated genes whose functions provide clues on the adaptation of the tropical maize to the African highlands. Molecular diversity indices computed across multiple categorical variables using SNPs that fell within the selective sweeps showed a two-fold reduction on polymorphic sites and nucleotide diversity in two of the three groups predicted based on the model-based STRUCTURE as compared to the genome-wide SNPs. Such thorough analyses on the genotypic data depict a significant contribution of this study to the available knowledge on selective sweeps in maize. Results from this study provide valuable information for further improvement of highland maize breeding programs in Africa, including the need for revising the line development strategy, diversifying parental lines for developing new inbred lines, and verifying genetic purity of newly fixed inbred lines using QC genotyping.
Supplementary information
Acknowledgements
The authors wish to thank CIMMYT and NARS field technicians at various research stations who were involved in the development of inbred lines used in this study. The management of Ethiopian Institute of Agricultural Research Institute (EIAR) is acknowledged for giving us access to the research centers in the country. The kind support of Dr Huihui Li, CIMMYT-China office, in imputing the GBS data is dully acknowledged. This study was supported by the CGIAR Research Program on Maize (MAIZE); Nutritious Maize for Ethiopia (NuME) project funded by the Global Affairs Canada (GAC); Stress Tolerant Maize for Africa (STMA) project funded by the Bill & Melinda Gates Foundation (BMGF), and the U.S. Agency for International Development (USAID).
Author Contributions
D.W. and A.T. conceived the experiment and drafted the paper; K.S. and B.T.E. conducted data analyses and edited the paper; D.N. and G.A. were responsible for seed sample preparation and compilation of passport information; V.O. was responsible for sample preparation, raising seedlings and DNA extraction; D.A., M.G. and B.M.P. contributed to and edited the paper; and N.A. conducted the selective sweep analyses.
Data Availability
All relevant data are within the paper and its Supporting Information Files.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adefris Teklewold, Email: a.teklewold@cgiar.org.
Kassa Semagn, Email: k.semagn@gmail.com.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-49861-z.
References
- 1.Zhou S, et al. A single molecule scaffold for the maize genome. PLoS Genet. 2009;5:e1000711. doi: 10.1371/journal.pgen.1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abate T, et al. Factors that transformed maize productivity in Ethiopia. Food Security. 2015;7:965–981. doi: 10.1007/s12571-015-0488-z. [DOI] [Google Scholar]
- 3.Paliwal, R. L., Grandos, G., Lafitte, H. R. & Marathee, J. P. Tropical Maize: Improvement and Production. (Food and Agricultural Organization of the United Nations, 2000).
- 4.Lothrop, J. E. In Maize Improvement, production and protection in Eastern and Southern Africa: Proceedings of the third Eastern and Southern Africa Regional Maize Workshop. (ed. Gebrekidan, B.) 75–92 (AMREF).
- 5.Beck, D. Research on tropical highland maize. 9–17 (The Maize Program, CIMMYT, Mexico D. F., 2001).
- 6.Twumasi-Afriyie, S. et al. In Integrated Approaches to Higher Maize Productivity in the New Millennium. (eds Friesen, D. K. Palmer, A. F. E.) 80–84 (CIMMYT and KARI).
- 7.Semagn K, et al. Molecular characterization of diverse CIMMYT maize inbred lines from eastern and southern Africa using single nucleotide polymorphic markers. BMC Genomics. 2012;13:113. doi: 10.1186/1471-2164-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, et al. Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor. Appl. Genet. 2016;129:753–765. doi: 10.1007/s00122-016-2664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen W, et al. Molecular characterization of a diverse maize inbred line collection and its potential utilization for stress tolerance improvement. Crop Sci. 2011;51:2569–2581. doi: 10.2135/cropsci2010.08.0465. [DOI] [Google Scholar]
- 10.Yan J, et al. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE. 2009;4:e8451. doi: 10.1371/journal.pone.0008451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warburton ML, et al. Genetic characterization of 218 elite CIMMYT maize inbred lines using RFLP markers. Euphytica. 2005;142:97–106. doi: 10.1007/s10681-005-0817-y. [DOI] [Google Scholar]
- 12.Xia XC, et al. Genetic diversity among CIMMYT maize inbread lines investigated with SSR markers: I. Lowland tropica maize. Crop Sci. 2004;44:2230–2237. doi: 10.2135/cropsci2004.2230. [DOI] [Google Scholar]
- 13.Xia XC, et al. Genetic diversity among CIMMYT maize inbred lines investigated with SSR markers: II. Subtropical, tropical midaltitude, and highland maize inbred lines and their relationships with elite U.S. and European maize. Crop Sci. 2005;45:2573–2582. doi: 10.2135/cropsci2005.0246. [DOI] [Google Scholar]
- 14.Dao A, Sanou J, Mitchell SE, Gracen V, Danquah EY. Genetic diversity among INERA maize inbred lines with single nucleotide polymorphism (SNP) markers and their relationship with CIMMYT, IITA, and temperate lines. BMC Genet. 2014;15:127. doi: 10.1186/s12863-014-0127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adebayo MA, et al. Diversity assessment of drought tolerant exotic and adapted maize (Zea mays L.) inbred lines with microsatellite markers. J Crop Sci. Biotechnol. 2015;18:147–154. doi: 10.1007/s12892-014-0076-3. [DOI] [Google Scholar]
- 16.Lu Y, et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor. Appl. Genet. 2009;120:93–115. doi: 10.1007/s00122-009-1162-7. [DOI] [PubMed] [Google Scholar]
- 17.Legesse, B. W., Myburg, A. A., Pixley, K. V. & Botha, A. M. Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas144, 10–17, doi:HRD1921 [pii];10.1111/j.2006.0018-0661.01921.x [doi] (2007). [DOI] [PubMed]
- 18.Beyene Y, Botha AM, Myburg AA. Genetic diversity among traditional Ethiopian highland maize accessions assessed by simple sequence repeat (SSR) markers. Genet. Resour. Crop Evol. 2006;53:1579–1588. doi: 10.1007/s10722-005-8509-y. [DOI] [Google Scholar]
- 19.Beyene Y, Botha AM, Myburg AA. Genetic diversity in traditional Ethiopian highland maize accessions assessed by AFLP markers and morphological traits. Biodivers. Conserv. 2006;15:2655–2671. doi: 10.1007/s10531-005-5407-8. [DOI] [Google Scholar]
- 20.Wende A, et al. Genetic interrelationships among medium to late maturing tropical maize inbred lines using selected SSR markers. Euphytica. 2013;191:269–277. doi: 10.1007/s10681-012-0826-6. [DOI] [Google Scholar]
- 21.Abakemal D, Hussein S, Derera J, Semagn K. Genetic purity and patterns of relationships among tropical highland adapted quality protein and normal maize inbred lines using microsatellite markers. Euphytica. 2015;204:49–61. doi: 10.1007/s10681-014-1332-9. [DOI] [Google Scholar]
- 22.Alachiotis N, Pavlidis P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Communications Biology. 2018;1:79. doi: 10.1038/s42003-018-0085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen M, et al. Identification of selective sweeps reveals divergent selection between Chinese Holstein and Simmental cattle populations. Genet. Sel. Evol. 2016;48:76. doi: 10.1186/s12711-016-0254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peter BM, Huerta-Sanchez E, Nielsen R. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genet. 2012;8:e1003011. doi: 10.1371/journal.pgen.1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alachiotis, N. & Pavlidis, P. Scalable linkage-disequilibrium-based selective sweep detection: A performance guide. GigaScience5, 10.1186/s13742-016-0114-9 (2016). [DOI] [PMC free article] [PubMed]
- 26.Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annu. Rev. Genet. 2013;47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- 27.Olsen KM, et al. Selection under domestication: evidence for a sweep in the rice waxy genomic region. Genetics. 2006;173:975–983. doi: 10.1534/genetics.106.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavlidis P, Alachiotis N. A survey of methods and tools to detect recent and strong positive selection. Journal of Biological Research-Thessaloniki. 2017;24:7. doi: 10.1186/s40709-017-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crisci JL, Poh Y-P, Mahajan S, Jensen JD. The impact of equilibrium assumptions on tests of selection. Frontiers in Genetics. 2013;4:235. doi: 10.3389/fgene.2013.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivek, B. S., Krivanek, A. F., Palacios-Rojas, N., Twumasi-Afriyie, S. & Diallo, A. O. Breeding Quality Protein Maize (QPM): Protocols for Developing QPM Cultivars. 50 (International Maize and Wheat Improvemen (CIMMYT), 2008).
- 31.Elshire RJ, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ertiro BT, et al. Genetic variation and population structure of maize inbred lines adapted to the mid-altitude sub-humid maize agro-ecology of Ethiopia using single nucleotide polymorphic (SNP) markers. BMC Genomics. 2017;18:777. doi: 10.1186/s12864-017-4173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- 35.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows [electronic resource] Mol. Ecol. Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 38.Lischer HEL, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28:298–299. doi: 10.1093/bioinformatics/btr642. [DOI] [PubMed] [Google Scholar]
- 39.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nature Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright, S. Evolution and the Genetics of Populations: Variability within and among Natural Populations . Vol. Volume 4 (University of Chicago Press, 1978).
- 43.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semagn K, et al. Molecular characterization of diverse CIMMYT maize inbred lines from eastern and southern Africa using single nucleotide polymorphic markers. BMC Genomics. 2012;13:113. doi: 10.1186/1471-2164-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pavlidis P, Živković D, Stamatakis A, Alachiotis N. SweeD: Likelihood-based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 2013;30:2224–2234. doi: 10.1093/molbev/mst112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ndjiondjop Marie Noelle, Alachiotis Nikolaos, Pavlidis Pavlos, Goungoulou Alphonse, Kpeki Sèdjro Bienvenu, Zhao Dule, Semagn Kassa. Comparisons of molecular diversity indices, selective sweeps and population structure of African rice with its wild progenitor and Asian rice. Theoretical and Applied Genetics. 2018;132(4):1145–1158. doi: 10.1007/s00122-018-3268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ertiro BT, et al. Comparison of kompetitive allele specific PCR (KASP) and genotyping by sequencing (GBS) for quality control analysis in maize. BMC Genomics. 2015;16:908. doi: 10.1186/s12864-015-2180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semagn K, et al. Quality control genotyping for assessment of genetic identity and purity in diverse tropical maize inbred lines. Theor. Appl. Genet. 2012;125:1487–1501. doi: 10.1007/s00122-012-1928-1. [DOI] [PubMed] [Google Scholar]
- 49.Tolessa, B. et al. In Proceedings of the First National Maize Workshop of Ethiopia . (eds Tolesa, B. & Ranson, J. K.) 13–22 (IAR/CIMMYT).
- 50.Gebre, T. In Enhancing the Contribution of Maize to Food Security in Ethiopia. (eds Nigussie, M., Tanner, D. & Twumasi-Afriyie, S.) 232 (EIAR/CIMMYT).
- 51.Prasanna, B. M., Chaikam, V. & Mahuku, G. Doubled Haploid Technology in Maize Breeding: Theory and Practice., (International Maize and Wheat Improvement Center (CIMMYT), 2012).
- 52.Prigge V, et al. Doubled haploids in tropical maize: II. Quantitative genetic parameters for testcross performance. Euphytica. 2012;185:453–463. doi: 10.1007/s10681-012-0626-z. [DOI] [Google Scholar]
- 53.Prigge V, et al. Doubled haploids in tropical maize: I. Effects of inducers and source germplasm on in vivo haploid induction rates. Crop Sci. 2011;51:1498–1506. doi: 10.2135/cropsci2010.10.0568. [DOI] [Google Scholar]
- 54.Prigge V, et al. New insights into the genetics of in vivo induction of maternal haploids, the backbone of doubled haploid technology in maize. Genetics. 2012;190:781–793. doi: 10.1534/genetics.111.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders N, et al. Glycosyl transferases in family 61 mediate arabinofuranosyl transfer onto xylan in grasses. Proceedings of the National Academy of Sciences. 2012;109:989. doi: 10.1073/pnas.1115858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiniquy D, et al. XAX1 from glycosyltransferase family 61 mediates xylosyltransfer to rice xylan. Proceedings of the National Academy of Sciences. 2012;109:17117. doi: 10.1073/pnas.1202079109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ebringerová, A. & Heinze, T. Xylan and xylan derivatives – biopolymers with valuable properties, 1. naturally occurring xylans structures, isolation procedures and properties. Macromolecular Rapid Communications21, 542–556, doi:10.1002/1521-3927(20000601)21:9<542::AID-MARC542>3.0.CO;2-7 (2000).
- 58.Grabber JH, Ralph J, Hatfield RD. Cross-linking of maize walls by ferulate dimerization and incorporation into lignin. J. Agric. Food Chem. 2000;48:6106–6113. doi: 10.1021/jf0006978. [DOI] [PubMed] [Google Scholar]
- 59.Wang C-T, et al. Maize WRKY transcription factor ZmWRKY106 confers drought and heat tolerance in transgenic plants. International Journal of Molecular Sciences. 2018;19:3046. doi: 10.3390/ijms19103046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei K-F, Chen J, Chen Y-F, Wu L-J, Xie D-X. Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes. 2012;19:153–164. doi: 10.1093/dnares/dsr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilroy SS, Jones DLDL. Through form to function: root hair development and nutrient uptake. Trends Plant Sci. 2000;5:56–60. doi: 10.1016/S1360-1385(99)01551-4. [DOI] [PubMed] [Google Scholar]
- 62.Li L, et al. Characterization of maize roothairless6 which encodes a D-type cellulose synthase and controls the switch from bulge formation to tip growth. Scientific Reports. 2016;6:34395. doi: 10.1038/srep34395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu F, Cui X, Zhang S, Liu C, Cao X. JMJ14 is an H3K4 demethylase regulating flowering time in arabidopsis. Cell Res. 2010;20:387. doi: 10.1038/cr.2010.27. [DOI] [PubMed] [Google Scholar]
- 64.Ning Y-Q, et al. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res. 2015;43:1469–1484. doi: 10.1093/nar/gku1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai X, et al. A putative CCAAT-binding transcription factor is a regulator of flowering timing in arabidopsis. Plant Physiol. 2007;145:98–105. doi: 10.1104/pp.107.102079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma M, et al. Expression of flavonoid 3′-hydroxylase is controlled by P1, the regulator of 3-deoxyflavonoid biosynthesis in maize. BMC Plant Biol. 2012;12:196. doi: 10.1186/1471-2229-12-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma M, et al. Identification of the <em>Pr1</em> gene product completes the anthocyanin biosynthesis pathway of maize. Genetics. 2011;188:69. doi: 10.1534/genetics.110.126136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nieva C, et al. Isolation and functional characterisation of two new bZIP maize regulators of the ABA responsive gene rab28. Plant Mol. Biol. 2005;58:899–914. doi: 10.1007/s11103-005-8407-x. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Misztal I, Aguilar I, Legarra A, Muir WM. Genome-wide association mapping including phenotypes from relatives without genotypes. Genetics Research. 2012;94:73–83. doi: 10.1017/S0016672312000274. [DOI] [PubMed] [Google Scholar]
- 70.Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annu. Rev. Plant Biol. 2010;61:651–679. doi: 10.1146/annurev-arplant-042809-112122. [DOI] [PubMed] [Google Scholar]
- 71.Habben JE, Moro GL, Hunter BG, Hamaker BR, Larkins BA. Elongation factor 1 alpha concentration is highly correlated with the lysine content of maize endosperm. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8640–8644. doi: 10.1073/pnas.92.19.8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun Y, et al. Characterization of maize elongation factor 1A and its relationship to protein quality in the endosperm. Plant Physiol. 1997;115:1101. doi: 10.1104/pp.115.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gonzalez D, et al. Characterization of a drosophila glutathione transferase involved in isothiocyanate detoxification. Insect Biochem. Mol. Biol. 2018;95:33–43. doi: 10.1016/j.ibmb.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y, et al. Identification and characterization of the bZIP transcription factor family and its expression in response to abiotic stresses in sesame. PLOS ONE. 2018;13:e0200850. doi: 10.1371/journal.pone.0200850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 76.Lo S-F, et al. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. The Plant Cell. 2008;20:2603–2618. doi: 10.1105/tpc.108.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiong Y, et al. Adaptive expansion of the maize maternally expressed gene (Meg) family involves changes in expression patterns and protein secondary structures of its members. BMC Plant Biol. 2014;14:204–204. doi: 10.1186/s12870-014-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tailor RH, et al. A novel family of small cysteine-rich antimicrobial peptides from seed of impatiens balsamina is derived from a single precursor protein. J. Biol. Chem. 1997;272:24480–24487. doi: 10.1074/jbc.272.39.24480. [DOI] [PubMed] [Google Scholar]
- 79.Wheeler MJ, Vatovec S, Franklin-Tong VE. The pollen S-determinant in papaver: comparisons with known plant receptors and protein ligand partners. J. Exp. Bot. 2010;61:2015–2025. doi: 10.1093/jxb/erp383. [DOI] [PubMed] [Google Scholar]
- 80.Hunter BG, et al. Maize opaque endosperm mutations create extensive changes in patterns of gene expression. The Plant Cell. 2002;14:2591. doi: 10.1105/tpc.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Appenzeller L, et al. Cellulose synthesis in maize: isolation and expression analysis of the cellulose synthase (CesA) gene family. Cellulose. 2004;11:287–299. doi: 10.1023/B:CELL.0000046417.84715.27. [DOI] [Google Scholar]
- 82.Moroney JV, Bartlett SG, Samuelsson G. Carbonic anhydrases in plants and algae. Plant, Cell Environ. 2001;24:141–153. doi: 10.1111/j.1365-3040.2001.00669.x. [DOI] [Google Scholar]
- 83.Ludwig M. Carbonic anhydrase and the molecular evolution of C4 photosynthesis. Plant, Cell Environ. 2012;35:22–37. doi: 10.1111/j.1365-3040.2011.02364.x. [DOI] [PubMed] [Google Scholar]
- 84.Hu H, et al. Carbonic anhydrases are upstream regulators of CO2-controlled stomatal movements in guard cells. Nat. Cell Biol. 2009;12:87. doi: 10.1038/ncb2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Studer AJ, et al. A limited role for carbonic anhydrase in C4 photosynthesis as revealed by a ca1ca2 double mutant in maize. Plant Physiol. 2014;165:608–617. doi: 10.1104/pp.114.237602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ludwig SR, Wessler SR. Maize R gene family: tissue-specific helix-loop-helix proteins. Cell. 1990;62:849–851. doi: 10.1016/0092-8674(90)90259-H. [DOI] [PubMed] [Google Scholar]
- 87.Heim MA, et al. The basic helix–loop–helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003;20:735–747. doi: 10.1093/molbev/msg088. [DOI] [PubMed] [Google Scholar]
- 88.Duek PD, Fankhauser C. bHLH class transcription factors take centre stage in phytochrome signalling. Trends Plant Sci. 2005;10:51–54. doi: 10.1016/j.tplants.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 89.Manavski N, Guyon V, Meurer J, Wienand U, Brettschneider R. An essential pentatricopeptide repeat protein facilitates 5′ maturation and translation initiation of rps3 mRNA in maize mitochondria. The Plant Cell. 2012;24:3087–3105. doi: 10.1105/tpc.112.099051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hammani K, Takenaka M, Miranda R, Barkan A. A PPR protein in the PLS subfamily stabilizes the 5′-end of processed rpl16 mRNAs in maize chloroplasts. Nucleic Acids Res. 2016;44:4278–4288. doi: 10.1093/nar/gkw270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brooks L, III, et al. Microdissection of shoot meristem functional domains. PLoS Genet. 2009;5:e1000476. doi: 10.1371/journal.pgen.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G<inf>1</inf>-phase progression. Genes and Development. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 93.Dubouzet JG, et al. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. The Plant Journal. 2003;33:751–763. doi: 10.1046/j.1365-313X.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 94.Qin Q-l, et al. Isolation, optimization, and functional analysis of the cDNA encoding transcription factor OsDREB1B in Oryza Sativa L. Mol. Breed. 2007;19:329–340. doi: 10.1007/s11032-006-9065-7. [DOI] [Google Scholar]
- 95.Gutterson N, Reuber TL. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 2004;7:465–471. doi: 10.1016/j.pbi.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 96.Agarwal PK, Agarwal P, Reddy MK, Sopory SK. Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep. 2006;25:1263–1274. doi: 10.1007/s00299-006-0204-8. [DOI] [PubMed] [Google Scholar]
- 97.Elliott RC, et al. AINTEGUMENTA, an APETALA2-like gene of arabidopsis with pleiotropic roles in ovule development and floral organ growth. The Plant Cell. 1996;8:155. doi: 10.1105/tpc.8.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Baosheng, Yu Jingjuan, Zhu Dengyun, Chang Yujie, Zhao Qian. Maize ZmRACK1 Is Involved in the Plant Response to Fungal Phytopathogens. International Journal of Molecular Sciences. 2014;15(6):9343–9359. doi: 10.3390/ijms15069343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Islas-Flores T, Rahman A, Ullah H, Villanueva MA. The receptor for activated C kinase in plant signaling: tale of a promiscuous little molecule. Frontiers in Plant Science. 2015;6:1090–1090. doi: 10.3389/fpls.2015.01090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: an overview. Physiology and Molecular Biology of Plants: An International Journal of Functional Plant Biology. 2013;19:307–321. doi: 10.1007/s12298-013-0179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Katiyar A, et al. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genomics. 2012;13:544–544. doi: 10.1186/1471-2164-13-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuoka D, Yasufuku T, Furuya T, Nanmori T. An abscisic acid inducible arabidopsis MAPKKK, MAPKKK18 regulates leaf senescence via its kinase activity. Plant Mol. Biol. 2015;87:565–575. doi: 10.1007/s11103-015-0295-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are within the paper and its Supporting Information Files.