

Abstract
Guanine-rich single-stranded DNAs and RNAs that fold into G-quadruplexes (GQs) are known to complex tightly with FeII-heme and FeIII-heme (hemin), ubiquitous cellular cofactors. Heme–GQ (DNA) complexes, known as heme·DNAzymes, are able to utilize hydrogen peroxide as an oxidant to vigorously catalyze a variety of one-electron (peroxidase) and two-electron (peroxygenase) oxidation reactions. Herein, we show that complexes of FeII-heme with GQs also robustly catalyze a mechanistically distinct reaction, carbene transfer to an alkene substrate. Significant enhancements were seen in both reaction kinetics and product turnover (∼180) relative to disaggregated FeII-heme in the absence of DNA or in the presence of other DNA folds, such as single-stranded or double-stranded DNA. Heme binds to GQs by end-stacking. Simple, intramolecularly folded GQs are unable to provide a complexly structured “distal side” environment to the bound heme; therefore, such DNAzymes do not display significant product stereoselectivity. However, intermolecular GQs with multiple pendant nucleotides show increasing stereoselectivity in addition to their enhanced catalytic rates. These results recapitulate the unique functional synergy and highlight the surprising catalytic versatility of complexes formed between heme and DNA/RNA GQs. Our findings suggest that heme·DNAzymes and heme·ribozymes may prove to be useful reagents for carbon–carbon bond forming “green” reactions carried out in vitro and likely within living cells.
Introduction
Heme (Fe–protoporphyrin IX), in its FeII and FeIII oxidation states, is a ubiquitous cellular cofactor, responsible for a variety of metabolic functions. These multitudinous functions of heme occur within complexes formed by it with various cellular apoproteins, to give diverse hemoproteins such as the various cytochromes, peroxidases, gas transfer proteins such as myoglobin and hemoglobin, and nitric oxide synthase. Recently, however, DNA and RNA, anionic biopolymers, have been found to have unexpected interactions with heme in vitro and possibly in vivo.1−3 Notably, single-stranded RNA and DNA molecules that fold, under physiological conditions, into guanine-quadruplexes (G-quadruplexes, or “GQ”), held together by guanine base-quartets (G-quartets) (Figure 1), have been shown to specifically and strongly bind heme in both of its iron oxidation states. GQs can be formed in a number of different ways, such as from the intramolecular folding of a single DNA/RNA strand containing four stretches of guanines; or intermolecular GQs, assembled from four separate and guanine-rich DNA/RNA single strands. GQs are specifically stabilized by the binding within the spatial cavity that exists within as well as in between successive G-quartets of Na+ or K+ cations.
Figure 1.

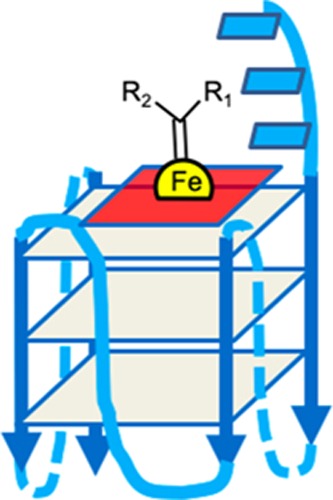
(a) Reaction of styrene with EDA to produce cyclopropane compounds (“cPr”), stereoisomers of 1-carboxyethyl-2-phenylcyclopropane. (b) A guanine base quartet. (c) Heme. (d) Schematic diagram showing a parallel-stranded intramolecular GQ DNA (GQ) complexed with heme. The blue rectangles represent guanine-quartets, and the red square a heme molecule end-stacked on a terminal G-quartet.
The tight complexes of FeIII-heme with GQs have been shown to powerfully catalyze one-electron (peroxidase) as well as two-electron (peroxygenase) oxidation reactions, this activity exceeding by orders of magnitude those of either uncomplexed heme in the presence of noninteracting DNAs or RNAs, or of heme itself, disaggregated by a variety of anionic, cationic, and charge-neutral surfactants.1,4 Heme (Figure 1c) has been shown to end-stack (Figure 1d) upon a terminal G-quartet of the GQ;5−7 FeIII-heme within such a complex is a six-coordinated complex at neutral pH,1,5−7 with both its proximal and distal axial ligands at neutral pH being water. Recent NMR studies have revealed the bound heme’s proximal axial ligand (i.e., the one pointing toward the DNA) is a water molecule positioned within the natural cavity of the most heme-proximal G-quartet.8
The UV–vis spectra of a GQ·FeIII-heme complex (Figure S1a) resembles that of hemoproteins such as metmyoglobin.1 To date, the GQ·FeIII-heme catalytic complexes (generically termed “heme·DNAzymes” and “heme·ribozymes” or “peroxidase-mimicking ribozymes/DNAzymes”) have found broad utility as reporters and reagents in both bioanalytical chemistry and bionanotechnology (reviewed in refs2,9). What is particularly interesting about this catalytic system is that it is highly specific to the GQ and to no other fold of natural RNA and DNA; and, furthermore, it is capable of efficiently replicating the catalytic activities of the peroxidase, oxygenase, and peroxygenase classes of hemoproteins. Relative to hemoproteins, heme·DNAzymes offer numerous advantages as oxidative catalysts, such as their small size, inexpensiveness, chemical robustness, ease of storage, facile synthesis and manipulation, and amenability to rational design and allosteric control. Such a combination of useful properties provides a powerful catalytic toolkit for biosensing, for the design of new biomaterials, and as reporters for a variety of analytical and other biomolecular devices.
We have been interested in exploring whether beyond the abovementioned oxidative activities, heme·DNAzymes have a larger, unexplored catalytic repertoire. Most intriguing to test for is the potential catalysis of carbene transfer reactions [such as to recipient alkenes from a diazo donor compound such as ethyl diazoacetate (EDA)—Figure 1a]. Transition-metal-mediated carbene insertion reactions have received a great deal of attention recently; it has been reported that FeII–porphyrin complexes are efficient catalysts for this reaction.10 Indeed, proteinaceous heme enzymes such as myoglobin and the cytochrome P450 P450CAM have been subjected to intensive mutagenesis and directed evolution. Mutant forms of these hemoproteins have been generated, which show superior catalysis of carbene transfer reactions compared to the wild-type proteins, with good catalytic enhancement, substrate turnover, as well as enhanced regio- and stereoselectivity.11,12
We therefore wished to test whether heme·DNAzymes and ribozymes, composed wholly of biological and bioavailable materials (heme; G-rich RNAs and DNAs), could be applied to the efficient catalysis of carbene insertion chemistries. If this were found to be so, it would not only broaden our conception of the biocatalytic repertoire available to these unique ribozymes and DNAzymes, but also, comparably to the engineered hemoproteins referred to above,10−12 these ribozymes and DNAzymes could find potential large-scale application for the “green” biosynthesis of novel organic compounds within living cells, perhaps on an industrial scale.
Roelfes and colleagues have reported the interesting utility of certain synthetic, cationic iron porphyrins, complexed to double-helical DNA, to catalyze carbene insertion reactions, often with notable stereoselectivity.13 In these systems, the second coordination sphere supplied by the DNA to the iron of the metalloporphyrin presents a complex and likely chiral environment toward this catalysis.14
However, given that the bioavailable cofactor heme is our chosen metalloporphyrin, we wished to test, first, whether different DNA folds (such as single strands, double helices and GQs) were equally or differentially capable of activating heme toward enhanced catalysis of carbene transfer in vitro, paying particular attention toward whether some of the attractive catalytic features [enhancements in product yield, turnover number (TON), and stereoselectivity] achieved by the engineered and evolved hemoproteins10−12 might also be shown by heme·DNAzymes.
In this paper, we show that GQs are indeed the only natural DNA/RNA higher-order structure to activate heme toward efficient carbene insertion chemistry. We explore the requirement for reducing and/or anaerobic environments for this catalysis; investigate the kinetics, substrate TONs, and the heme-dependence of heme·DNAzymes. Given that our chosen substrate, styrene, is hydrophobic, we explore the impact of organic cosolvents on this catalysis. We compare the performance of heme·DNAzymes with that of a cationic iron porphyrin in the presence of different folds of DNA. We go on to explore the stereoselectivity of different heme·DNAzymes, and finally, set out a clear set of long-term prospects for this unique class of biocatalysts.
Results and Discussion
Complex of Fe(II) Heme with a DNA GQ Catalyzes Carbene Insertion into Styrene
Hemin–GQ complexes have been extensively studied for their robust 1e– and 2e– mimicking activities and their versatile applications. Travascio et al. optimized the conditions for the optimal oxidative catalysis by the FeIII-heme·DNAzymes.1 A buffer pH of 8.0; potassium salts; a nitrogenous buffer such as NH4–HEPES or collidine (such amines participate in general acid–base chemistry to enhance the overall catalysis15); and a low concentration (>critical micelle concentration) of a nonionic detergent X-100 to help solubilize and disaggregate the added hemin,1,2 were key elements of an optimally catalytic solution. Using these same experimental conditions as a starting point, we investigated whether a heme·DNAzyme could catalyze a carbene transfer reaction (in comparison to heme alone or in the presence of other DNA folds) using the benchmark substrate (styrene) and ethyl diazoacetate (EDA) as the carbene donor. The gas chromatography (GC) elution profiles and retention time values of the various cyclopropane (“cPr”) products (diastereomers; and enantiomers in a chiral analysis) have been well characterized and standardized;11 it was therefore useful for us to attempt this reaction, to take advantage of facile identification of any products formed.
First, we used UV–vis spectroscopy to measure the binding affinity of FeII-heme to an intramolecularly folded DNA GQ (“G4”). Figure S2 shows the titration of a fixed concentration (0.5 μM) of FeII-heme with 0–16 μM “G4”. A dissociation constant (Kd) of 1.3 μM was computed from these data.
Next, we investigated the impact of heme in the presence of a molar excess of intramolecularly folded “G4” on the carbene transfer reaction [against controls of disaggregated hemin alone or in the presence of excess “double-stranded DNA (dsDNA)” (salmon sperm genomic DNA); or excess single-stranded “scrambled G4”]. The reaction was carried out in the usual heme·DNAzyme reaction buffer (40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 10 mM Na2S2O4, 1% dimethylformamide (DMF), 0.05% Triton X-100) and the reactants were added to final concentrations of 1 mM styrene, 7 mM EDA, 17 μM hemin, and 83 μM DNA. This carbene transfer reaction requires the participation of FeII-heme (rather than FeIII-heme); it is therefore carried out most logically under anaerobic conditions (deoxygenated buffer, under argon, in the presence of a reducing agent).11,12Figure 2 shows that under such an inert atmosphere, FeII-heme with excess G4 generates, after 1 h of reaction, a high yield of the cPr products (∼26-fold higher than the heme-alone control and the heme-dsDNA control, and ∼7-fold higher than the heme and scrambled G4 control).
Figure 2.
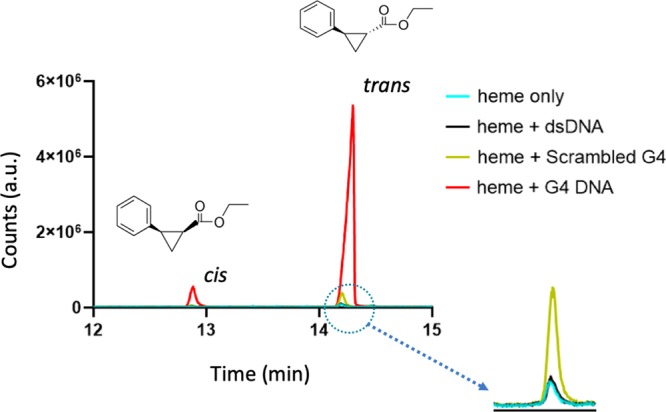
Gas chromatogram of cis- and trans-cPr, products obtained using hemin (Fe-III) heme as a catalyst in the absence and presence of DNA, namely, of excess salmon sperm “dsDNA”; “scrambled G4 DNA”; and “G4”, respectively. Reactions were carried out for 2 h at 22 °C, under anaerobic conditions, with 83 μM DNA, 30 μM hemin, 1 mM styrene, 7 mM EDA, and 10 mM Na2S2O4.
A recent publication on this reaction catalyzed by a nonheme iron cationic porphyrin in the presence of dsDNA has reported moderate yields of the product in the absence of both reducing agent and argon.13 However, we thought it useful to systematically examine the effect of these reagents/conditions upon the heme·DNAzyme-catalyzed carbene transfer reaction under our experimental conditions (Figure 3). Table S1 summarizes the detailed results. Under optimal conditions, with both sodium dithionite and argon present, the cPr product yield with the heme·DNAzyme was 75% after 1 h of reaction. In the absence of the reducing agent but in the presence of argon, the yield was 36%; this is because EDA itself is able to act to a degree as a reducing agent;16 however, in the presence of the reducing agent but in the absence of argon, the yield was 8%. In the absence of both reducing agent and argon, the yield was only 4%. Clearly, then, the effectiveness of this nucleic acid-based catalytic system shares the requirements with hemoproteins for an anaerobic atmosphere and a reducing agent to maintain the FeII state of the DNA-complexed heme.
Figure 3.
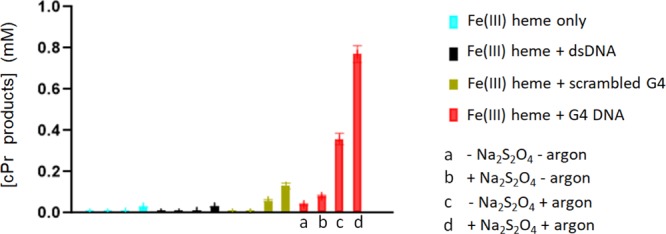
Impact of a reducing agent (sodium dithionite) and an anaerobic atmosphere (presence of argon) on the catalyzed production of cPr products by heme in the presence of different DNA folds. The generic reaction conditions were: 83 μM DNA (“dsDNA”, “scrambled G4”, or “G4”), 30 μM hemin, 1 mM styrene, 7 mM EDA, 10 mM Na2S2O4 in reaction buffer: 40 mM HEPES–NH4OH, pH 8.0, 20 mM KCl, 1% DMF, and 0.05% Triton X-100. For each set of data (blue, black, green, and red), the first bar (“a”) shows reaction in the absence of both dithionite and argon; “b” shows reaction in the presence of dithionite but in the absence of argon; “c” shows reaction in the absence of dithionite but in the presence of argon; and “d” shows the reaction in the presence of both dithionite and argon.
Kinetics of Carbene Insertion Reaction
To obtain an insight into individual reagent concentrations that might limit the optimal kinetics and yield of the heme·DNAzyme-catalyzed cPr products, we prepared five identical, standard reaction solutions in the reaction buffer (83 μM “G4” DNA, 17 μM hemin, 1 mM styrene, 7 mM EDA, 10 mM Na2S2O4 in a buffer of 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, 0.05% Triton X-100, under anaerobic conditions at 21 °C). Each reaction proceeded for 1 h, following which, 10 μL of supplementations of one single reagent (heme; styrene; EDA; or “G4” DNA) were made to individual reaction vials under anaerobic conditions. The different reactions now had, variously, heme to a final concentration of 33 μM; styrene to a final of 2 mM; EDA to a final 13 mM; or “G4” DNA to a final 116 μM. One of the reaction vials had no supplementation made to it, as a negative control. Following the various supplementations, the reactions were allowed to proceed for one further hour prior to termination and extraction using ethyl acetate. Figure S3 shows that relative to the negative control, no increase in cPr product formation was observed with either EDA or G4 DNA supplementations. By contrast, the product yield increased by 25–30% with both the styrene and hemin supplementations. Therefore, under our experimental conditions, heme and styrene were limiting reagents.
In order to evaluate the kinetics of product formation, eight separate samples were prepared identically for each DNA variant, and the reaction was carried out at different time points. Figure 4a shows the product yields obtained for experiments using 30 μM hemin in the presence of, variously, no added DNA; 80 μM “G4” DNA; “scrambled G4” DNA; and duplex (“dsDNA”). The product yield obtained with G4, after 1 h, was 77%. By contrast, with the other DNAs, the yields obtained after 2 h were 3% (heme only/no DNA) 3% (heme with dsDNA), and 11% (heme with scrambled G4 DNA).
Figure 4.
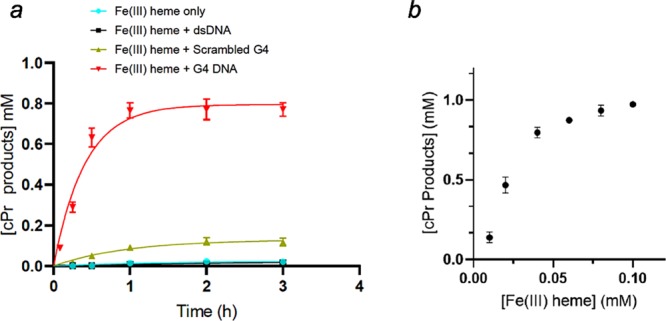
Kinetics of carbene insertion. (a) Carbene insertion reaction kinetics with fixed heme and DNA concentrations. Each reaction consisted of 80 μM DNA, 30 μM hemin, 1 mM styrene, 7 mM EDA, 10 mM Na2S2O4 in a reaction buffer consisting of 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, 0.05% Triton X-100, under anaerobic conditions at 21 °C. (b) Heme-dependence for the formation of cPr products by the heme–“G4” complex (heme·DNAzyme). Reactions were carried out for 1 h, under anaerobic condition, with a fixed concentration of “G4” DNA (83 μM) paired with varying concentrations (10, 20, 40, 60, 80, and 100 μM) of heme. The reaction solutions contained, in addition to the above, 1 mM styrene, 7 mM EDA, and 10 mM Na2S2O4 in a buffer solution consisting of 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, and 0.05% Triton X-100.
We investigated the importance of hemin concentration (0–100 μM) for the G4–heme DNAzyme under the above conditions. Figure 4b shows that with all other reactant concentrations being held constant (83 μM “G4” DNA; 1 mM styrene; and 7 mM EDA), the cPr product yield after 1 h rose monotonically to heme concentrations up to ∼40 μM (where the heme–G4 DNA ratio was 0.5), and plateaued beyond that; 10, 20, 40, 60, 80, and 100 μM heme (in combination with 83 μM G4) correspond, respectively, to 1, 2 4, 6, 8, and 10 mol % enzyme loading with respect to the styrene substrate. At the G4–heme ratio of 2:1, where the product yield starts to plateau, there was 83% cPr product yield and a TON of 21. At a 1:1 ratio (8 mol % catalyst), a 98% product yield and a TON of 12 were obtained. Depending on requirements for optimized yield, reaction rate, or indeed TON in this catalytic system, clearly appropriate concentrations of the heme·DNAzyme can be rationally chosen. We chose to use a concentration of 30 μM heme·DNAzyme for our next set of experiments, so as to ensure product yields of >50% and allow improvement of the TON through the optimization of styrene concentration.
Impact of an Organic Cosolvent
Ideally, a high-quality catalyst should not be consumed via side reactions distinct from the main reaction being catalyzed and should enable both high TONs as well as yield. Earlier studies on the 1e– and 2e– oxidation properties of heme·DNAzyme have shown that depending on the oxidative chemistry being catalyzed, the heme moiety gradually deactivates over time. Perhaps, the same drawback also applies for the carbene insertion reaction. Our initial choice of styrene concentration (1 mM) was based on maintaining phase homogeneity, given the poor solubility of styrene in aqueous buffers. In order to obtain the most optimal combination of reaction rate, product yield, and TON, we carried out a series of experiments that featured relatively high heme and styrene (>1 mM) concentrations by incorporating increasing concentrations of methanol as cosolvent. In an earlier study, Canale and Sen17 demonstrated that unlike most protein hemoenzymes, the peroxidase activity of heme·DNAzymes was strongly enhanced in 20–30% v/v methanol; indeed, the DNAzymes were still active in solutions containing as high as 80% methanol.17
As described above, under our initial reaction conditions (1 mM styrene and 7 mM EDA) good substrate yields (77%) with modest TON were observed. We therefore examined the possibility of enabling the use of higher styrene concentrations (while maintaining a constant EDA concentration of 7 mM) by introducing different concentrations of methanol as cosolvent.
Figure 5 shows time courses for usage of 10 or 20 mM styrene using 30 μM heme (0.4% of enzyme loading with respect to EDA), 83 μM “G4”, and 7 mM EDA. Product yields of 75 and 79% (with respect to EDA), respectively, were obtained using 10 and 20 mM styrene and 5% methanol (v/v) as cosolvent. The initial rate of reaction was 1.5-fold faster with 20 mM styrene than with 10 mM styrene (128 and 87 μM min–1, respectively). Increasing the methanol content to 30% (v/v) in the reaction buffer for the 20 mM styrene reaction, however, did not significantly impact either the initial reaction rate or product yield relative to the reaction carried out in the 5% methanol reaction buffer (v/v). In all cases, the reaction was complete within 1 h.
Figure 5.
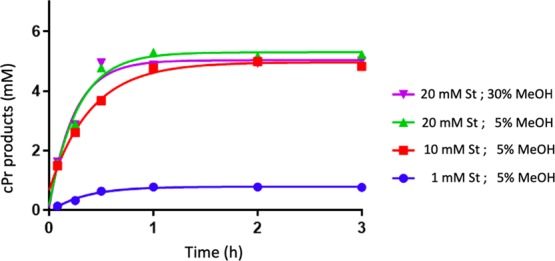
Effect of styrene concentration and organic solvent percentage on carbene insertion reaction kinetics. The reaction conditions were as follows: 83 μM “G4” DNA, 30 μM hemin, 7 mM EDA, 10 mM Na2S2O4 in a buffer: 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, 0.05% Triton-100X and under anaerobic conditions.
Impact of Non-G Residues at the 3′-Ends of Intramolecular GQs
“G4” (or “G4-AAA”) DNA, which is an intramolecularly folding GQ widely used for studying and implementing the oxidative activities of heme·DNAzymes, has three unpaired adenines at its 3′end. It has been shown that for H2O2-based DNAzyme oxidative activities, the presence of at least one (or more) of these adenines enhances the kinetics of catalysis.18 Indeed, these authors found that the presence of even one unpaired 3′ A residue gave DNAzyme initial rates >5-fold higher than having n (where n = 1–8) 3′ T residue(s) in place of A.18 The A residue(s) appear to contribute in an acid–base capacity toward generation of the activated heme species within oxidative heme·DNAzymes, mimicking the activity of soluble bases such as ammonia or collidine.15 To investigate whether the kinetics of carbene transfer catalysis of heme·DNAzymes is comparably impacted, three further GQ DNAs containing one 3′-adenine (“G4-A”), one thymine (“G4-T”), as well as three thymine residues (“G4-TTT”) were tested under the same reaction conditions used for the standard “G4” (“G4-AAA”) DNAzyme. Table 1 shows that “G4” and “G4-TTT” manifest essentially indistinguishable product yields and TONs; Table S2 further iterates that “G4-A” and “G4-T” likewise generate product yields indistinguishable from that of “G4”. For the “G4” DNAzyme, to examine whether the potassium ion concentration we used was sufficient to maximize its folding into a GQ, we varied the concentration of potassium chloride concentrations (0, 4, 20 100, 500, 750 mM and 1 M, respectively) and measured circular dichroism (CD) spectra of the folded “G4” DNA. Figure S4 shows that although the characteristic CD spectrum of a parallel-stranded GQ was reached at 20 mM KCl, full spectral saturation was seen at 500 mM KCl. We therefore compared the cPr product yields by all the GQs used in our study at both 20 and 500 mM KCl concentrations (Table S2). In all cases, use of the higher concentration of potassium chloride did not lead to any notable change in cPr product yield.
Table 1. Yields, TONs, and Diastereomeric Ratios of Carbene Insertion Reaction of Styrene with EDA One of Two Iron Porphyrins (FeIII-Heme or FeIII-TMPyP4) in the Presence and Absence of DNA: “dsDNA”; “Scrambled G4”; and the GQs: “G4-AAA”, “G4-TTT”, and “(dA4G5A4)4”a.
| catalyst | DNA type | yield % (1 mM styrene) | TON (1 mM styrene) | TON (20 mM styrene) | cis/trans |
|---|---|---|---|---|---|
| FeIII-heme | no DNA | 3 ± 0.4 | 1 | 7 | 10:90 |
| FeIII-heme | dsDNA | 3 ± 0.6 | 1 | 7 | 10:90 |
| FeIII-heme | scrambled G4 | 11 ± 1.3 | 5 | 9 | 9:91 |
| FeIII-heme | G4-AAA | 77 ± 3 | 26 | 177 | 8:92 |
| FeIII-heme | G4-TTT | 80 ± 2 | 27 | 182 | 8:92 |
| FeIII-heme | *(dA4G5A4)4 | 51 ± 5 | 17 | 116 | 15:85 |
| FeIII-TMPyP4 | no DNA | 3 ± 0.2 | 1 | 14:86 | |
| FeIII-TMPyP4 | dsDNA | 4 ± 0.1 | 1 | 13:87 | |
| FeIII-TMPyP4 | scrambled G4 | 8 ± 1.7 | 3 | 13:87 | |
| FeIII-TMPyP4 | G4-AAA | 11 ± 2 | 4 | 12:88 |
Reaction conditions were as follows: 80 μM DNA; 30 μM of either catalyst porphyrin, 1 and 20 mM styrene, 7 mM EDA, and 10 mM Na2S2O4 in the reaction buffer: 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, 0.05% Triton X-100 under anaerobic conditions at 21 °C [the asterisk notes that in the case of *(dA4G5A4)4, 80 μM total dA4G5A4 DNA was used, consistent with only ≤20 μM of the GQ, “(dA4G5A4)4”]. Yields, TON, and diastereomeric ratios were determined from GC analysis. Yields are based on the available limiting reagent (styrene or EDA).
Heme Compared to a Cationic Iron Porphyrin (Fe-TMPyP4)
Rioz-Martínez and co-workers have examined the catalytic activities of a series of cationic iron porphyrins in the presence of dsDNA for the carbene transfer reaction.13 It is known that cationic porphyrins with multiple positive charges as well as metallated versions of the same show strong binding affinities to DNA, and to GQs in particular. We examined whether a cationic porphyrin, Fe-meso-tetra(4-N-methylpyridyl)porphine (Fe-TMPyP4), also showed GQ DNA-enhanced catalysis of carbene insertion. Under our experimental conditions, free Fe-TMPyP4 showed a low level of (∼4% yield after 1 h) carbene insertion activity (Figure 6), comparable to that of uncomplexed heme (Figure 4). However, only very modest increases in cPR production were observed with Fe-TMPyP4 in the presence of “dsDNA”, “scrambled G4”, and “G4”—yielding 5, 8, and 11% yields of cPr products, respectively. First, to investigate whether Fe-TMPyP4 does interact with the three different folds of DNA used in our study, UV–vis spectra of 2 μM Fe-TMPyP4 were taken (Figure S1) in the absence of DNA and in the presence of 10 μM “dsDNA”; “scrambled G4”; or “G4” DNA (both G4, or G4-AAA, and its variant, G4-TTT, were examined). Unlike the major spectral changes seen with FeII and FeIII-heme binding to GQ DNA, a modest but defining red shift (from 422 to 431 nm) is seen in the Soret band of TMPyP4 in the presence of “G4-AAA” and “G4-TTT” but not in the presence of “dsDNA” or “scrambled G4”. Such a relatively small change in the spectrum of the strongly positively charged porphyrin in the presence of the different strongly negatively charged DNAs likely reflects mainly coulombic interactions between the two, which do not impact in major ways the porphyrin’s absorption spectrum. The consistent occurrence of the Soret band red shifts, however, is consistent with the binding of the Fe-TMPyP4 to the three GQ DNAs (G4-AAA, G4-TTT, and (dA4G5A4)4).
Figure 6.
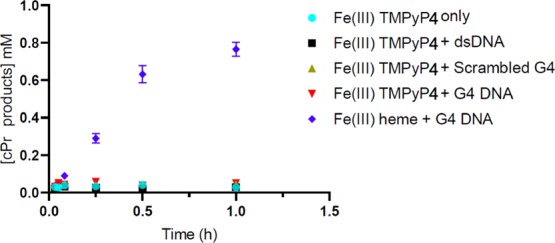
Carbene insertion reaction kinetics using FeIII-TMPyP4. Reactions conditions were as follows: 83 μM DNA, 30 μM porphyrin, 1 mM styrene, 7 mM EDA, 10 mM Na2S2O4 in reaction buffer (40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, and 0.05% Triton X-100) under anaerobic conditions.
Table 1 lists a number of key “performance” parameters for both heme and Fe-TMPyP4, both in reactions where these porphyrins do not have any added DNA, and where they do. It can be seen that (a) only the three GQ folded DNAs, “G4-AAA”, “G4-TTT”, and “(dA4G5A4)4” in conjunction with heme, generate high TONs as well as overall reaction yields; heme with no added DNA, or added single-stranded (scrambled GQ) or dsDNA are poor catalysts for the carbene insertion reaction. (b) It is only heme that shows this unique catalytic synergy with GQs toward carbene insertion; the positively charged, highly DNA-interactive Fe-TMPyP4, by contrast, is not an efficient catalyst for this reaction, at least under these conditions, either by itself or in the presence of the different DNAs.
Stereoselectivity
In the styrene carbene insertion reaction discussed here, the cis and trans diasteromeric cPr products each consist of a pair of enantiomers. Although many of the wild-type hemoproteins (cytochromes P450 and metmyoglobin) examined for this reaction do not impart significant stereoselectivity to the cPr products generated, a great triumph of directed evolution experiments carried out by Arnold11 and others12 is that they have generated a variety of mutant hemoproteins capable of distinctive diastereoselective (i.e., cis/trans ratios) as well as enantioselective production of the products. Such high stereoselectivity in the relevant mutant hemoproteins is made possible by the structures of those enzymes, most notably by the complex spatial and chemical environments of their active sites on the distal sides of their heme moieties.
Heme has been shown to stack on the terminal quartets of parallel-stranded or mixed-orientation GQs, with preference for binding upon the 3′-most G-quartet.5−8 In such a complex, the G-quartet makes up the “proximal” side of the heme, whereas the “distal” side of the heme, at least in unimolecular, propeller-looped GQs such as “G4-AAA” and “G4-TTT”—Figure 7a), is merely solvent, wholly lacking the kind of asymmetric spatial architecture that may reasonably impart selective stereoselectivity to the cPr products obtained. Nevertheless, we used a GC fitted with a HP-chiral-20B column to investigate any level of enantiometric excess (ee) within both the trans and cis cPr products catalyzed by the GQ heme·DNAzyme. All intramolecular GQ variants (“G4-A”, “G4-T”, “G4-AAA”, and “G4-TTT”) were tested to see if the pendant purine or pyrimidine bases in those GQs (Figure 7a) influenced ee of the generated products. In reactions carried out at 22 or 4 °C, ee values of the enantiomers making up the major, trans cPr product (Figure 8b) were found to be very small (∼1%) (Figure 8a). Such a lack of enantioselectivity from such structurally simpler, intramolecular GQ DNAzymes was consistent with earlier results on oxidative catalysis by the “G4-AAA” DNAzyme, where the hydrogen peroxide-mediated oxidation of thioanisole likewise showed little enantioselectivity.19
Figure 7.
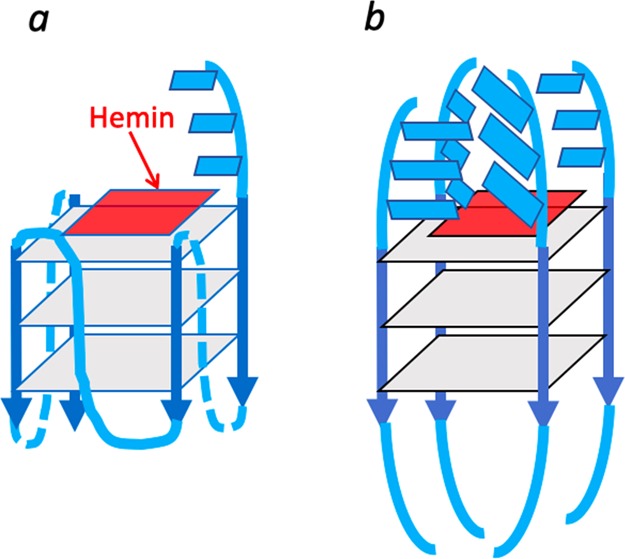
Visualizing the “active site” of heme·DNAzymes (GQ–heme complexes), where the heme end-stacks, preferentially, on the 3′-most guanine-quartet (guanine-quartets are shown as gray squares); (a) “G4-AAA” and “G4-TTT” oligonucleotides both form intramolecular, propeller looped GQs, with the 3′ terminal AAA or TTT overhanging (shown as blue rectangles). (b) An intermolecularly assembled GQ such as “(dA4G5A4)4” is expected to have a complex “distal” environment above the stacked heme, potentially imparting stereoselectivity to reactions catalyzed by the DNA-bound heme.
Figure 8.
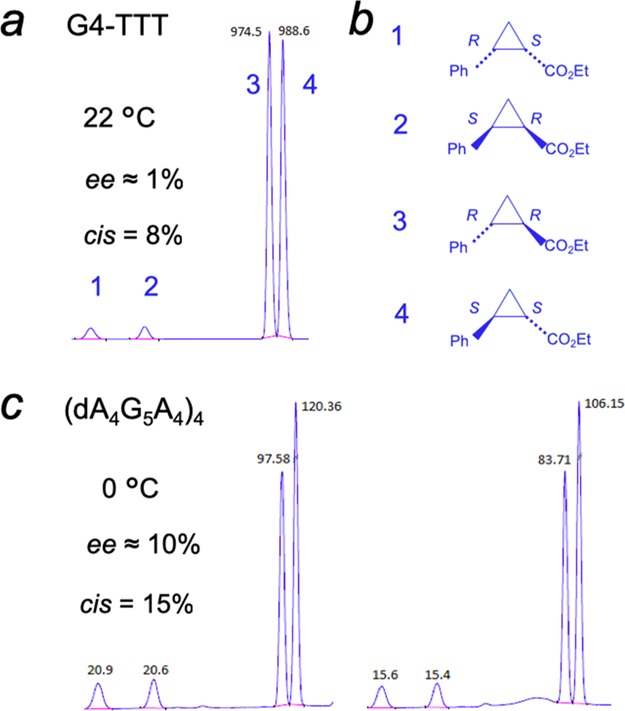
Chiral gas chromatogram of the cPr product of the carbene insertion reaction using (FeIII)-hemin as the catalyst in the presence of (a) folded “G4-TTT” at 22 °C and (c) “(dA4G5A4)4” GQ at 0 °C. Reaction conditions were as follows: 83 μM DNA, 30 μM hemin, 1 mM styrene, 7 mM EDA, 10 mM Na2S2O4 in a buffer: 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, 0.05% Triton X-100, under anaerobic conditions. The identity of the resolved cPr peaks (b) is as described in (11).
Our results this far showed that under our reaction conditions, heme in both the absence and presence of three forms of DNA used (single-stranded, double-stranded, and GQ) clearly favored the formation of the trans cPr product (92%), with both cis and trans products made up of racemic mixtures of enantiomers. Thus, these intramolecularly folded DNAzymes catalyzed the formation of cPr products with high rate and yield (98% using 8% mol DNAzyme), yet showed poor stereoselectivity. To try and improve stereoselectivity, two further strategies were examined. First, we examined a different class of parallel-stranded GQ—the unique, intermolecularly assembled, four-stranded (dA4G5A4)4 (Figure S4). As shown schematically in Figure 7b, the large number of pendant adenines on both the 5′ and 3′ sides of the central GQ core of “(dA4G5A4)4” potentially offer structurally more complex distal side environments to the bound heme. Second, we lowered the reaction temperature of the reaction down to 0 °C with the aim of reducing the thermal mobility of any structural complexity offered by the adenine overhangs. The temperature change in itself had little impact on the stereoselectivity properties of our original GQs (“G4-AAA” and “G4-TTT”)—no major change in diastereo- or enantioselectivity was observed for them between 22 and 0 °C. However, promisingly, “(dA4G5A4)4” gave a 3% ee for the dominant trans product at room temperature; this figure enhanced to 10% ee at 0 °C (Figure 8c). These results gave promise that structurally yet more complex RNA or DNA GQs are likely to offer even higher levels of stereoselectivity to heme·DNAzyme/ribozyme-catalyzed carbene transfer reactions. It is worth noting that our initial use of 80 μM total of “dA4G5A4” (Table 1) generated only ≥20 μM of the four-stranded GQ, (dA4G5A4)4, and gave a correspondingly lower yield of cPr product relative to the 80 μM concentrations of intramolecular GQ DNAzymes such as “G4”. However, increasing the concentration of starting dA4G5A4 in the reaction to 320 μM, prior to incubation to form the GQ, [(dA4G5A4)4], led to a dramatic increase in cPr product formation, consistent with those obtained from the other heme·DNAzymes (Table S2).
Conclusions
Herein, we report that heme·DNAzymes, long valued as versatile oxene-transfer catalysts to alkenes such as styrene1,2 and for catalyzing other useful oxidative reactions, are, additionally, vigorous catalysts for carbene insertion into a model alkene, styrene. Once again, it is the unique combination of heme and DNA GQs that shows this new catalytic activity; disaggregated heme by itself or in combination with other folds of DNA folds does not display this catalysis. Carbene insertion is thus both a new and distinct catalytic activity of heme·DNAzymes—one, moreover, that is a net synthetic reaction (forms new carbon–carbon bonds). Carbene transfer is also the first bona fide catalytic activity to be demonstrated for the ferrous (FeII) form of heme·DNAzymes (in contrast with the well-established 1e– and 2e– oxidative catalytic activities of the FeIII-DNAzymes).
The ability of heme·DNAzymes to catalyze carbene insertion is characterized by high rates of reaction and high TONs (i.e., the number of reaction cycles completed by a given molecule of heme·DNAzyme). With structurally simpler DNAzymes, lacking a complex “distal side” to their bound hemes, a shortcoming is evident of lack of interesting stereoselectivity. However, we report that with (dA4G5A4)4, a structurally more complex GQ that offers a crowded “distal side” environment, both a distinctive cis/trans ratio of products as well as a nontrivial ee are obtained, especially when the catalysis is carried out at 0 °C.
It should be noted that a cationic iron porphyrin, Fe-TMPyP2 (though not its isomers, such as Fe-TMPyP3) has been shown to bind to the negatively charged duplex DNA; and the complex so formed showed significant catalysis of carbene insertion as well as DNA-enhanced stereoselectivity—presumably owing to a complex structural environment found in the duplex DNA grooves, Fe-TMPyP3’s presumed locus of binding.13 By contrast, what distinguishes heme·DNAzymes from the above duplex DNA-cationic porphyrin complexes is, first, the tight and highly specific complex formed between the slightly anionic FeII-heme and the highly negatively charged DNA GQs. Clearly, this interaction is not charge-based and, above all, permits a sixfold coordination of the bound heme’s iron center. Our prior work1,2,19 as well as what we report here demonstrate that heme·DNAzymes are able to support at least two distinct classes of reaction: oxene and carbene insertions (in addition to purely peroxidase-type 1e– oxidations). That these are indeed mechanistically distinct reactions has been pointed out recently by Wei et al.,20 who state that “the increasingly useful C=C functionalizations mediated by heme carbenes feature an FeII-based, nonradical, concerted nonsynchronous mechanism, with early transition-state character. This mechanism differs from the FeIV-based, radical, stepwise mechanism of heme-dependent monooxygenases”.20 With regard to the oxidative activity of heme·DNAzymes, we and others have found the importance of general acid–base catalysis in the activation of heme by H2O2.1,17−19,21−26 Thus, G4-AAA and G4-CCC show notably enhanced oxidation rates relative G4-TTT or G4 lacking any pendant 3′ bases. Here, however, we have shown that G4-AAA and G4-TTT are indistinguishably efficient in their catalysis of carbene insertion.
GQs, both of genomic and of artificial origins, constitute a large and polymorphic family of nucleic acid folds; indeed, some structurally very complex GQs have recently been reported, including such fluorogenic RNA aptamers as RNA Mango and RNA Spinach.27−29 Hence, it should be possible to evolve (via rational design, directed evolution, or quasi-evolutionary methods such as in vitro selection) yet more structurally complex quadruplexes—using strategies analogous to the celebrated directed evolution of pre-existing heme protein enzymes toward novel functions, as reported by Arnold and others.10,11 We expect that successive generations of more structurally complex GQs will constitute new heme·DNAzymes capable of high product yields, high TONs, as well as major product stereoselectivity. Indeed, there is already a precedent of GQ DNA complexed with a cationic copper porphyrin enabling asymmetric catalysis of a Diels–Alder reaction.30
It is important to emphasize that unlike many other metalloporphyrins used for catalysis, heme is both an intrinsically biological and bioavailable metalloporphyrin, ubiquitous in all living systems and present in every living cell. RNA and DNA GQs have also been shown to form in living cells.31 Though rare in bacteria, GQs persist when expressed within them.32 Therefore, the potential exists for heme·ribozymes expressed within bacteria to catalyze “green” carbene transfer reactions on a large scale, just as evolved cytochrome P450 enzymes have been enabled to catalyze carbene transfer within living cells.33 Styrene as a promising alkene substrate has limited water-solubility; nevertheless, it has been used successfully for utilization for carbene insertion in living cells.33
Indeed, as macromolecular catalysts for in vitro utility, heme·DNAzyme and ribozymes enjoy a number of advantages over their much larger protein counterparts—such as wild-type as well as mutated/evolved hemoproteins such as metmyoglobin and cytochrome P450s. Constituted solely from small DNA or RNA oligomers and heme, these catalysts are much smaller than protein enzymes and are chemically robust, as well as highly functional in organic cosolvents relative to the average hemoprotein. They are cheap to synthesize and have long shelf-lives. Future generations of heme·DNAzymes and ribozymes likely hold significant promise as catalysts for carbon–carbon bond formation in vitro, and perhaps also on a useful, “green” scale in vivo.
Materials and Methods
Materials
All DNA oligonucleotides were purchased from Integrated DNA Technologies (IDT-Canada) and Core DNA Services of the University of Calgary. Sequences used in this study were: “G4” (or “G4-AAA”): 5′-dTGGGT AGGGC GGGTT GGGAA A-3′; “G4-TTT”: 5′-dTGGGT AGGGC GGGTT GGGTT T-3′, “scrambled G4”: 5′-dGTGAG GGAGT GCGTG GTGAGA G-3′, “G4-A”: 5′-dTGGGT AGGGC GGGTT GGGA-3′, “G4-T”: 5′-dTGGGT AGGGC GGGTT GGGT-3′, “A4G5A4”: 5′-dAAAAG GGGGA AAA-3′, and “RNA G4-AAA”: 5′-rUGGGU AGGGC GGGUU GGGAA A-3′. Hemin [FeIII–protoporphyrin XI] and FeIII-meso-tetra(N-methyl-4-pyridyl)porphine pentachloride [Fe(T4-MePyP)Cl5 or Fe-TMPyP4] were purchased from Frontier Scientific, Inc. Salmon testis DNA (“dsDNA”), methyl phenyl sulfoxide, trans-1-carbethoxy-2-phenylcyclopropane(ethyl 2-phenylcyclopropane-1-carboxylate), styrene, and EDA were purchased from Sigma-Aldrich. All of the above materials were used without further purifications.
A 5 mM hemin stock was prepared in DMF, aliquoted, and stored in the dark at −20 °C. Hemin was freshly diluted from this stock to the required final concentration for each experiment. Other stock solutions used were 1.2 M styrene in MeOH; 400 mM EDA in MeOH; and 60 mM methyl phenyl sulfoxide in MeOH.
UV–Visible Spectroscopy
A Varian Cary 300 bio UV–visible spectrophotometer was used, at room temperature, with a 10 mm quartz cuvette. 2 μM FeIII-heme (in the presence or absence of 10 mM of reducing agent) and 2 μM FeIII-TmPyP4·Cl5 solutions were prepared both in the absence and presence of the different DNAs (10 μM final concentration) in the following buffer: 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF (v/v), 0.05% Triton X-100 (w/v).
CD Spectroscopy
CD experiments were carried out in a JASCO J-810 spectropolarimeter at 25 °C, using 0.1 cm path-length cuvettes. CD spectra were recorded both in the absence and presence of FeIII-heme. The final concentration of different GQ DNAs was 10 μM in 40 mM HEPES, pH 8.0, 20 mM KCl, 1% DMF (v/v), 0.05% Triton X-100 (w/v). The final concentration of FeIII-heme was 0.5 μM. Spectra were recorded between 200 and 320 nm and were averaged from three scans.
Gas Chromatography
An Agilent 6890 gas chromatograph, equipped with a HP-5MS (30 m × 0.25 mm, 0.25 μm film) capillary column and interfaced to a 5973 N mass-selective detector was used, with the following temperature program: 100 °C for 5 min, 5 °C/min ramp up to 200 °C, 20 °C/min ramp up to 250 °C, then 250 °C for 5 min. Retention times using helium as the carrier gas (flow rate at 1.6 mL/min) were as follows: cis-cyclopropane products (12.84 min), trans cyclopropane products (14.3 min). The methods used were essentially the same as in ref (11), although the RF values are different owing to the use of a column different from that used in ref (11). Elution times using hydrogen as the carrier gas had the following values: cis-cyclopropane products (10.77 min), trans-cyclopropane products (12.14 min). For screening and quantification of the above cPr products, mass spectra were recorded in two modes; 115, 117, and 190 mass ions were chosen for the selected ion monitoring mode with a mass width of 0.2 Da and a scan time of 0.05 s.
Chiral analyses were carried out using GC–flame ionization detection, which was fitted with a HP-chiral-20B column (30 m × 0.25 mm × 0.25 μm film). Hydrogen was used as the carrier gas, with the following temperature program: oven temperature = 100 °C for 5 min; ramped by 1 °C/min to 135 °C; 135 °C for 10 min; ramped by 10 °C/min to 200 °C; 200 °C for 5 min. The following elution times were obtained: cis-cyclopropane products (39.48 and 40.19 min), and trans-cyclopropanes (41.89 and 42.12 min).
Procedure for Carbene Insertion Reaction under Anaerobic Conditions
In a GC vial, to 265 μL of 2× reaction buffer (1× reaction buffer is: 40 mM NH4–HEPES, pH 8.0, 20 mM KCl, 1% DMF, and 0.05% Triton X-100 was added 25 μL DNA (final concentration of 83 μM), followed by 10 μL of a FeIII-heme stock in DMF (to give final FeIII-heme concentration of either 17 or 30 μM). The GC vial was sealed, its contents allowed to mix and incubate at 21 °C for 5 min to enable proper DNA folding and complexation to FeIII-heme. The headspace of the sealed GC vial was made anaerobic by flushing argon over the DNA solution, without causing bubbles (3 min). Using a disposable syringe, 280 μL of Na2S2O4 in aqueous solution (deoxygenated by bubbling argon) was added to a final concentration of 10 mM and the reaction mixture was flushed with argon for another 2 min. Following removal of the gas line, styrene was added with a glass syringe to final concentrations of 1, 10, or 20 mM; the mixture was vortexed for 10 s to mix, following which 10 μL of 400 mM EDA stock in methanol was added (to a final concentration of ∼7 mM). The complete reaction mixture was mixed by vortexing and placed on a shaker for the appropriate reaction time. At the end of the designated reaction interval, the vial was opened and 10 μL of the internal standard, methyl phenyl sulfoxide (from a 60 mM stock in MeOH) was added. The reaction mixture was now extracted with 0.6 mL ethyl acetate. To determine the yield of the catalyzed reaction, first we calculated a correction factor between the internal standard (methyl phenyl sulfoxide) and the purchased standard of trans-cPr, both prepared in the same solution. The internal response factor (IRF) is defined from the chromatograms as the (integrated area of internal standard) × [cPr]/[IS] × (integrated area of cPr). The concentration of products in the reactions was calculated using the following equation: [cPr in a reaction] = [IS added to the reaction] × (area of cPr in the reaction) × IRF/(area of IS added to the reaction). Yield = [cPr]reaction/[cPr]max × 100.
Acknowledgments
We thank Professor Jeffrey Warren for his helpful discussions and Paul Saunders for excellent technical assistance. D.S. acknowledges funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) and from Simon Fraser University. H.I. was supported by the International Department at Simon Fraser University, and the generosity of Andrew Petter, Joy Johnson, Darren Schemmer, Shaheen Nanji, Mignon Alphonso and Peter Ruben is gratefully acknowledged. The Scholars at Risk (SAR) organization is gratefully acknowledged for offering the opportunity to H.I. to conduct this research.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02305.
UV–vis absorption spectra of 2 μm ferrous and ferric heme and ferric TMPyP4.5Cl; absorbance measurements of 404 nm of 0.5 μM ferrous heme; yields of cyclopropanation reaction; effect of different reagent supplementations on the yield of cPr products; yields of carbene insertion reaction of styrene with EDA; and native gel electrophoresis showing the formation of a unique intramolecular GQ product (PDF)
Author Contributions
H.I. and D.S. designed the research. H.I. and P.M. carried out the experiments. H.I. and D.S. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Travascio P.; Li Y.; Sen D. DNA-enhanced peroxidase activity of a DNA aptamer–hemin complex. Chem. Biol. 1998, 5, 505–517. 10.1016/s1074-5521(98)90006-0. [DOI] [PubMed] [Google Scholar]
- Sen D.; Poon L. C. RNA and DNA complexes with hemin [Fe(III) heme] are efficient peroxidases and peroxygenases: how do they do it and what does it mean?. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 478–492. 10.3109/10409238.2011.618220. [DOI] [PubMed] [Google Scholar]
- Grigg J. C.; Shumayrikh N.; Sen D. G-quadruplex structures formed by expanded hexanucleotide repeat RNA and DNA from the neurodegenerative disease-linked C9orf72 gene efficiently sequester and activate heme. PLoS One 2014, 9, e106449 10.1371/journal.pone.0106449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travascio P.; Bennet A. J.; Wang D. Y.; Sen D. A ribozyme and a catalytic DNA with peroxidase activity: active sites versus cofactor-binding sites. Chem. Biol. 1999, 6, 779–787. 10.1016/s1074-5521(99)80125-2. [DOI] [PubMed] [Google Scholar]
- Saito K.; Tai H.; Hemmi H.; Kobayashi N.; Yamamoto Y. Interaction between the heme and a G-quartet in a heme-DNA complex. Inorg. Chem. 2012, 51, 8168–8176. 10.1021/ic3005739. [DOI] [PubMed] [Google Scholar]
- Shimizu H.; Tai H.; Saito K.; Shibata T.; Kinoshita M.; Yamamoto Y. Characterization of the Interaction between Heme and a Parallel G-Quadruplex DNA Formed from d(TTAGGGT). Bull. Chem. Soc. Jpn. 2015, 88, 644–652. 10.1246/bcsj.20140374. [DOI] [Google Scholar]
- Shinomiya R.; Katahira Y.; Araki H.; Shibata T.; Momotake A.; Yanagisawa S.; Ogura T.; Suzuki A.; Neya S.; Yamamoto Y. Characterization of Catalytic Activities and Heme Coordination Structures of Heme-DNA Complexes Composed of Some Chemically Modified Hemes and an All Parallel-Stranded Tetrameric G-Quadruplex DNA Formed from d(TTAGGG). Biochemistry 2018, 57, 5930–5937. 10.1021/acs.biochem.8b00793. [DOI] [PubMed] [Google Scholar]
- Shinomiya R.; Katahira Y.; Araki H.; Shibata T.; Momotake A.; Yanagisawa S.; Ogura T.; Suzuki A.; Neya S.; Yamamoto Y. Structures and Catalytic Activities of Complexes between Heme and All Parallel-Stranded Monomeric G-Quadruplex DNAs. Biochemistry 2018, 57, 5938–5948. 10.1021/acs.biochem.8b00793. [DOI] [PubMed] [Google Scholar]
- Mergny J.-L.; Sen D. DNA Quadruple Helices in Nanotechnology. Chem. Rev. 2019, 119, 6290–6325. 10.1021/acs.chemrev.8b00629. [DOI] [PubMed] [Google Scholar]
- Wolf J. R.; Hamaker C. G.; Djukic J.-P.; Kodadek T.; Woo L. K. Shape and stereoselective cyclopropanation of alkenes catalyzed by iron porphyrins. J. Am. Chem. Soc. 1995, 117, 9194–9199. 10.1021/ja00141a011. [DOI] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Key H. M.; Dydio P.; Clark D. S.; Hartwig J. F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. 10.1038/nature17968. [DOI] [PubMed] [Google Scholar]
- Rioz-Martínez A.; Oelerich J.; Ségaud N.; Roelfes G. DNA-Accelerated Catalysis of Carbene-Transfer Reactions by a DNA/Cationic Iron Porphyrin Hybrid. Angew. Chem., Int. Ed. 2016, 55, 14136–14140. 10.1002/anie.201608121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioz-Martínez A.; Roelfes G. DNA-based hybrid catalysis. Curr. Opin. Chem. Biol. 2015, 25, 80–87. 10.1016/j.cbpa.2014.12.033. [DOI] [PubMed] [Google Scholar]
- Travascio P.; Sen D.; Bennet A. J. DNA and RNA enzymes with peroxidase activity—An investigation into the mechanism of action. Can. J. Chem. 2006, 84, 613–619. 10.1139/v06-057. [DOI] [Google Scholar]
- Salomon R. G.; Kochi J. K. Copper(I) catalysis in cyclopropanations with diazo compounds. Role of olefin coordination. J. Am. Chem. Soc. 1973, 95, 3300–3310. 10.1021/ja00791a038. [DOI] [Google Scholar]
- Canale T. D.; Sen D. Hemin-utilizing G-quadruplex DNAzymes are strongly active in organic co-solvents. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 1455–1462. 10.1016/j.bbagen.2016.11.019. [DOI] [PubMed] [Google Scholar]
- Li W.; Li Y.; Liu Z.; Lin B.; Yi H.; Xu F.; Nie Z.; Yao S. Insight into G-quadruplex-hemin DNAzyme/RNAzyme: adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Res. 2016, 44, 7373–7384. 10.1093/nar/gkw634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon L. C.-H.; Methot S. P.; Morabi-Pazooki W.; Pio F.; Bennet A. J.; Sen D. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 2011, 133, 1877–1884. 10.1021/ja108571a. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Tinoco A.; Steck V.; Fasan R.; Zhang Y. Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc. 2018, 140, 1649–1662. 10.1021/jacs.7b09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D.-M.; Xu J.; Shen H.-X. Positive Effects of ATP on G-Quadruplex-Hemin DNAzyme-Mediated Reactions. Anal. Chem. 2010, 82, 6148–6153. 10.1021/ac100940v. [DOI] [PubMed] [Google Scholar]
- Stefan L.; Denat F.; Monchaud D. Insights into How Nucleotide Supplements Enhance the Peroxidase-Mimicking DNAzyme Activity of the G-Quadruplex/Hemin System. Nucleic Acids Res. 2012, 40, 8759–8772. 10.1093/nar/gks581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C.; Zhang N.; Yan J.; Liu X.; Bing T.; Mei H.; Shangguan D. Activity Enhancement of G-Quadruplex/Hemin DNAzyme by Spermine. RSC Adv. 2014, 4, 1441–1448. 10.1039/c3ra45429k. [DOI] [Google Scholar]
- Chen J.; Guo Y.; Zhou J.; Ju H. The Effect of Adenine Repeats on G-Quadruplex/Hemin Peroxidase Mimicking DNAzyme Activity. Chem.—Eur. J. 2017, 23, 4210–4215. 10.1002/chem.201700040. [DOI] [PubMed] [Google Scholar]
- Chang T.; Gong H.; Ding P.; Liu X.; Li W.; Bing T.; Cao Z.; Shangguan D. Activity Enhancement of G-Quadruplex/Hemin DNAzyme by Flanking d(CCC). Chem.—Eur. J. 2016, 22, 4015–4021. 10.1002/chem.201504797. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhang Y.; Cheng M.; Guo Y.; Šponer J.; Monchaud D.; Mergny J.-L.; Ju H.; Zhou J. How Proximal Nucleobases Regulate the Catalytic Activity of G-Quadruplex/Hemin DNAzymes. ACS Catal. 2018, 8, 11352–11361. 10.1021/acscatal.8b03811. [DOI] [Google Scholar]
- Fernandez-Millan P.; Autour A.; Ennifar E.; Westhof E.; Ryckelynck M. Crystal structure and fluorescence properties of the iSpinach aptamer in complex with DFHBI. RNA 2017, 23, 1788–1795. 10.1261/rna.063008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner K. D.; Sjekloća L.; Song W.; Filonov G. S.; Jaffrey S. R.; Ferré-D’Amaré A. R. A homodimer interface without base pairs in an RNA mimic of red fluorescent protein. Nat. Chem. Biol. 2017, 13, 1195–1201. 10.1038/nchembio.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachman R. J. 3rd; Demeshkina N. A.; Lau M. W. L.; Panchapakesan S. S. S.; Jeng S. C. Y.; Unrau P. J.; Ferré-D’Amaré A. R. Structural basis for high-affinity fluorophore binding and activation by RNA Mango. Nat. Chem. Biol. 2017, 13, 807–813. 10.1038/nchembio.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilking M.; Hennecke U. The influence of G-quadruplex structure on DNA-based asymmetric catalysis using the G-quadruplex-bound cationic porphyrin TMPyP4·Cu. Org. Biomol. Chem. 2013, 11, 6940–6945. 10.1039/c3ob41366g. [DOI] [PubMed] [Google Scholar]
- Rhodes D.; Lipps H. J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. U.; Bartel D. P. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, aaf5371. 10.1126/science.aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins In Vivo. Nat. Chem. Biol. 2013, 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.